

Abstract
Ductal Carcinoma In Situ (DCIS) is the most frequently diagnosed early stage breast cancer. Only a subset of patients progress to invasive ductal carcinoma (IDC), and this presents a formidable clinical challenge for determining which patients to treat aggressively and which patients to monitor without therapeutic intervention. Understanding the molecular and genomic basis of invasion has been difficult to study in DCIS cancers due to several technical obstacles, including low tumour cellularity, lack of fresh-frozen tissues, and intratumour heterogeneity. In this review we discuss the role of intratumour heterogeneity in the progression of DCIS to IDC in the context of three evolutionary models: independent lineages, evolutionary bottlenecks, and multiclonal invasion. We examine the evidence in support of these models and their relevance to the diagnosis and treatment of patients with DCIS. We also discuss how emerging technologies, such as single cell sequencing, STAR-FISH and imaging mass spectrometry are likely to provide new insights into the evolution of this enigmatic disease.
Keywords: DCIS, Ductal-Carcinoma-In-Situ, Intratumor Heterogeneity, Breast Cancer Genomics, Breast Cancer, Next Generation Sequencing, Single Cell Sequencing
Introduction
The most common type of malignant breast cancer, invasive ductal carcinoma (IDC), is the presumed endpoint in a progression initiated by ductal hyperplasia (DH) [1]. The simplicity of sequential progression from DH to IDC (Figure 1A) is appealing, however, much epidemiological evidence shows atypical ductal hyperplasia (ADH) and ductal carcinoma in situ (DCIS) often never progress to IDC [2–4]. DCIS is termed a non-obligatory precursor to IDC [1,5–7], and some experts question the use of the term ‘carcinoma’ to label this disease [8,9]. The difficulty in predicting progression is a major clinical challenge to the diagnosis and treatment of patients [4,10–13]. When treating patients with DCIS, clinicians are faced with the difficult decision of ‘active surveillance’ or aggressive therapy without knowing which patients are likely to progress to IDC. Traditionally, most oncologists have erred on the side of caution, leading to over-treatment of many patients with DCIS [11–13].
Figure 1. Progression of breast cancer and the histopathology of DCIS.
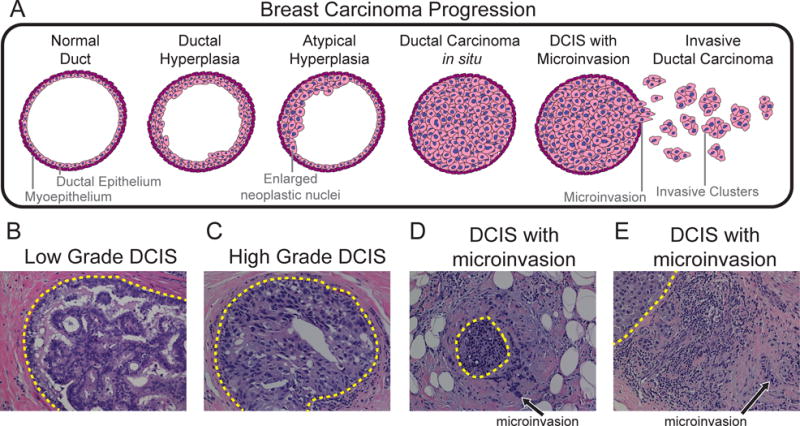
(A) Progressive stages of high-grade breast cancer as defined by histopathology (B–E) H&E stained tissue sections of low-grade and high-grade DCIS at 200× magnification: (B) low to intermediate grade cribriform type DCIS, (C) cross section of a duct involved by high grade solid type DCIS, (D–E) synchronous high grade DCIS and IDC with evidence of microinvasion.
Despite efforts to identify DCIS-IDC progression markers, few useful prognostic biomarkers have been discovered [14–16]. Genomic biomarker studies mainly applied gene expression microarrays or array copy genomic hybridization (aCGH) [17–24]. Many of these studies reported highly similar copy number profiles and gene expression signatures of synchronous DCIS-IDC regions [20,25–28]. With the development of next-generation sequencing (NGS) technologies, studies have begun to apply higher resolution methods to study invasive-specific mutations and copy number events in patients with synchronous DCIS-IDC [27–31]. Many of these studies have identified concordant and discordant mutations in patients with synchronous DCIS-IDC [26–28,30,31]. However, these initial genomic studies faced several technical obstacles, including low tumour purity, the unavailability of fresh-frozen tissues, and intratumour heterogeneity (ITH). Consequently, the genomic and molecular basis of invasion in DCIS breast cancers remains poorly understood.
Clinical Features of DCIS
Histopathology of DCIS material using H&E staining has identified different classes of patients. Some patients show evidence of pure DCIS, in which tumour cells are constrained by ductal boundaries (Figure 1B–C); others show evidence of synchronous DCIS-IDC, with adjacent microinvasion or distant invasion within the same patient (Figure 1D–E). In about twenty percent of IDC cases both in situ disease and invasive subpopulations are observed in isolated regions without evidence of microinvasion [32]. Patient samples with synchronous DCIS-IDC provide a golden opportunity to study the genomic and molecular basis of invasion without confounding effects of inter-patient heterogeneity.
Long-term follow up studies of patients with DCIS have shown a substantial difference in the progression of low-grade versus high-grade DCIS, with only 35% of low-grade DCIS patients progressing to have IDC over 50 years, while 50% of high-grade DCIS progressed to IDC over 3 years [2,33]. Previous reviews have discussed differences between low and high grade DCIS in detail [34,35]. Low-grade DCIS are more often ER+/PR+/HER2- with fewer copy number aberrations (CNAs) than high-grade DCIS [34,36–38]. A High-grade DCIS has atypical nuclei and is more often ER- and PR- [34,39]. High-grade DCIS usually has more genome-wide CNAs, including frequent events in 1q+, 5p+, 8p−, 8q+, 11q−, 13q−, 14q−, and 17q+ and focal amplifications on 6q22, 8q22, 11q13, 17q12, 17q22–24, and 20q13 [22,34,37,38,40,41]. Mutational markers of IDC include mutations in TP53 [27] and PTEN [31], amplifications of chromosome 17 and 11q [18,27,42], and loss of PIK3CA mutation [27,42]. In the United States, DCIS is routinely tested for oestrogen receptors (ER) and progesterone receptors (PR) to determine whether hormone receptor based therapy should be recommended [43]. Analysis of hormone receptor status shows patients with ER+ and PR+ disease often have low-grade DCIS tumours and are less likely to progress than patients with ER- and PR- disease and high-grade tumours [34,36–38]. While new a study showed that intratumour heterogeneity in the HER2 receptor was also associated with poor prognosis in DCIS patients [44], currently DCIS patients are not routinely tested for HER2 (ERBB2) amplification [45,46]. Gene expression tests, such as OncotypeDX, use a 21-gene panel to double predictive accuracy and have been utilized as prognosis markers for DCIS progression to IDC[47]. In summary, there are few useful clinical markers that predict which patients will progress from DCIS to IDC, outside of histopathological grade.
DCIS and Intratumour Heterogeneity
Intratumour heterogeneity (ITH) is frequently reported in invasive breast cancers [31,48–53] and in DCIS studies profiling DNA, RNA and protein levels [17,26,37,54–61]. ITH complicates diagnosis and treatment, but is beneficial for evolutionary studies since it provides a ‘permanent record’ of mutations during tumour growth [62]. Assuming mutational complexity increases over time, and using phylogenetic inference, several studies showed clonal lineages and evolutionary histories can be inferred from a single time-point tumour sample [48,49,63]. This experimental approach is important for evolutionary studies of DCIS, where often only a single time point sample can be obtained [26,28].
Early studies of ITH used cytological and histopathological methods. These methods included fluorescence in situ hybridization (FISH) to measure DNA copy number of targeted genes or loci and immunohistochemistry (IHC) to measure protein levels across tissue sections. A number of DNA-FISH studies reported ITH in DNA copy number states of single tumour cells in the ducts of DCIS patients [19,23,61,64–68]. Multiple studies have reported ITH in known receptors such as HER2 in DCIS [44,61,64,68]. Heterogeneity in protein levels and targeted genes have also been reported using cytological and histological methods [48,54,61,69,70]. Allred et al. used IHC to stain specific proteins in DCIS and revealed spatial ITH in protein levels of p53 and HER2 [54]. However, these methods were often qualitative and limited to single targeted genes or proteins.
NGS methods provided quantitative measurements of thousands of mutations and CNAs in parallel. Three different experimental NGS approaches have been developed to resolve ITH: 1) deep-sequencing, 2) multi-region sequencing, and 3) single cell DNA sequencing. Deep-sequencing involves sequencing bulk tumour at high coverage depths to cluster mutation frequencies and identify clonal subpopulations. This approach has been applied to study ITH and clonal evolution in invasive breast cancer patients [51,71,72]. Multi-region sequencing involves spatially sampling different macroscopic regions of tumour mass, and sequencing each region independently to resolve geographic heterogeneity [31,73,74]. These methods enable the reconstruction of phylogenetic lineages to understand clonal evolution in breast cancer patients [31]. Single cell sequencing (SCS) methods can measure genome-wide copy number profiles [63,75], exomes [76,77], genomes [49,78] or targeted gene panels [79] in single cells. SCS methods can fully resolve ITH by reporting genomic information on individual tumour cells, but are more susceptible to sampling bias [80]. By sequencing and comparing multiple tumour cells, several studies have delineated the clonal substructure and evolutionary lineages of invasive breast cancers [48,49,63]. However, these NGS approaches have not yet been applied to DCIS.
Models of Invasion
In this review, we discuss three models of invasion during progression of DCIS to IDC: 1) independent evolution, 2) evolutionary bottlenecks and 3) multiclonal invasion (Figure 2). The independent evolution model posits that two different initiating cells (N1, N2) in normal breast tissue give rise separately to DCIS and IDC subpopulations (Figure 2A). The independent lineage model is in contrast to the direct lineage models (evolutionary bottlenecks and multiclonal evolution), which assume a single normal breast cell (N1) gave rise to both DCIS and IDC populations. The main difference between the direct lineage models is that the evolutionary bottleneck model posits that a clone in the ducts is selected during invasion and migrates into adjacent tissue, forming the invasive tumour (Figure 2B). In contrast, the multiclonal model posits invasion occurs through escape of multiple clones from the duct, through a coordinated process or a stochastic escape after the degradation of basement membrane (Figure 2C).
Figure 2. Evolutionary models of invasion in DCIS.
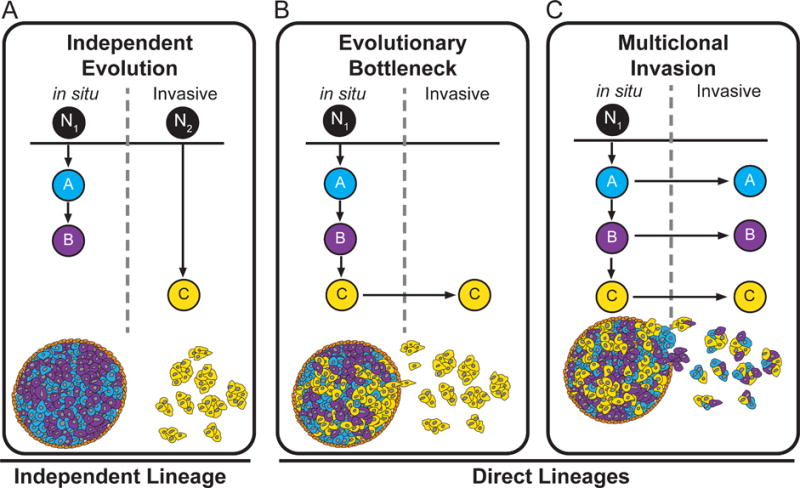
(A) Independent Evolution Model shows the in situ and invasive subpopulations evolving from independent lineages that originated from two different normal cells (N1, N2) in the breast. (B) Evolutionary Bottleneck Model shows the evolution of three clonal subpopulations from a single ancestral (N1), from which a single clone is selected during invasion and expands to form the invasive carcinoma. (C) Multiclonal Invasion Model shows the evolution of three clonal subpopulations from a single normal cell (N1), in the breast. In this model all three clones escape the duct and co-migrate into the adjacent tissues to establish the invasive carcinoma.
Independent Genomic Lineages
In the independent lineage model, two independent normal cells are assumed to give rise to in situ and invasive subpopulations. This model assumes the evolution of DCIS and IDC are independent, and cell lineages do not share any overlapping mutations or CNAs. The data supporting this model comes from histopathological sections, in which about 20% of cases have DCIS and IDC in different regions of the breast [32], and from a number of single marker studies showing discordance between synchronous in situ and invasive subpopulations[18,54]. Often, spatially distant synchronous DCIS-IDC cases show genetic and histopathological ITH, and are sometimes classified as different grades [20,31,54,60,81–83].
The emergence of multiple tumour lineages may be explained by cancer field effects giving rise to multiple tumour-initiating cells. Cancer field effects have been reported in tumours with external mutagens, such as UV exposure in eye lid cancers [84] and cigarette smoke in lung adenomas [85], as well as in tumours such as breast cancer, with no known external mutagens [86]. Cancer field effects may also occur in patients with germline mutations (eg. BRCA1, BRCA2, TP53) predisposed to cancer, often giving rise to multifocal tumours [87–89].
Support for independent lineages mostly comes from single targeted genes or protein studies, where targeted markers are discordant within a patient [20,90]. A study sequenced PIK3CA mutations in patients with matched IDC and DCIS reported only 30% concordance between in situ and invasive regions [90]. Deep-sequencing of the mitochondrial D-loop in DCIS patients suggested 61% of tumours were of non-clonal, or independent, origins [20]. Mathematical modeling studies also support parallel development of DCIS and IDC in some tumours [91]. In summary, the experimental support for the independent lineages model is based mainly on single marker studies that may miss general concordance due to limited profiling of other genomic mutations.
Direct Genomic Lineages
Direct lineage models provide an alternative explanation for the evolution of the DCIS and IDC subpopulations, through a single initiating cell (N1) in normal breast tissue (Figure 2B–C). Direct lineage models are supported by numerous genomic studies, using aCGH [26,42,92–97] and NGS [27,28,30,31] to profile synchronous DCIS-IDC. These studies report a high correlation in copy number profiles of in situ and invasive subpopulations in synchronous DCIS patients [19,23,27,42,93,97–99], and many concordant point mutations [26–28,100]. A recent meta-analysis pooled data from 38 studies and examined the relationship between in situ and invasive breast cancers, and found that 67% of the studies showed strong direct lineage [101]. Concordant mutations between DCIS and IDC are often referred to as ‘truncal’ mutations by evolutionary biologists, because they can be traced back to the last common ancestor in the tumour. However, many genomic studies also report discordant mutations and CNAs arising at later stages of evolution, specific to either in situ or invasive subpopulations [26–28,30,31,42,92–97,102]. These data suggest some DCIS cancers may evolve through an evolutionary bottleneck and with selection of minor clones of invasive phenotypes, while others may evolve through migration of multiple dominant clones. Distinguishing between direct lineage models is challenging, since most bulk genomic data has limited ability to resolve ITH and trace clonal lineages of invasive subpopulations.
Clonal Evolution
Population bottlenecks are frequently reported in the evolution of natural species through mechanisms such as allopatric speciation, where a small population of one species migrates to a distant geographical region and adapts to a new environment that selects for different traits [103]. In tumours, evolutionary bottlenecks have been reported in the context of metastatic dissemination [28,31,104] and in response to therapy [105–108]. Genomic analysis of synchronous DCIS-IDC has identified new mutations and CNAs in invasive subpopulations that were not detected in in situ ducts, suggesting that selection of minor clones may have occurred during invasion [27,28,30,31].
Several NGS studies sequenced synchronous DCIS-IDC regions to reconstruct phylogenetic lineages during invasion and study genome evolution (Table 1). Sidow and colleagues used whole-genome-sequencing (WGS) to analyse six breast cancer patients with matched longitudinal samples of atypical ductal hyperplasia (ADH), DCIS and IDC [30,109]. Lineage tracing using mutations, copy number changes and LOH identified truncal events that were concordant between DCIS and IDC early in the lineage, in addition to many specific CNAs and mutations that occurred later, consistent with an evolutionary bottleneck model. In another study, the authors used multi-region sequencing to perform lineage-tracing experiments in ER+/PR/HER2- synchronous DCIS patients by sampling from both in situ and invasive regions [31]. In one case, authors reported convergent evolution of two distinct PTEN mutations in invasive regions not present in in situ subpopulations, suggesting selection of a minor subclone during invasion [31]. In another study the authors reported a higher concordance in CNAs (80–90%) than point mutations (40%) in 6 patients with synchronous DCIS-IDC lesions [27]. The authors identified highly concordant mutations in TP53, while other invasive-specific mutations included those of FANCE, ATM, BCOR, PDGFRA, and PMS1. Reis-Filho and colleagues [26] performed aCGH profiling on 13 synchronous patients with DCIS and IDC and found 77% of patients had highly similar genome-wide copy number profiles, while others showed additional amplifications in invasive subpopulations (1q41, 2q24.2, 6q22.31, 7q11.21, 8q21.2 and 9p13.3), consistent with an evolutionary bottleneck [110].
Table 1. Genomic studies of DCIS breast cancers.
This table contains a list of genomic studies from next-generation sequencing, microarray CGH profiling and SNP array profiling of DCIS breast cancers. The columns are: primary method used in the paper, first author, year of publication, number of samples analysed, type of tissue, reference number and PubMed ID (PMID). The number of samples in each study are reported as DCIS-only, DCIS-IDC (synchronous samples) and IDC-only samples.
| Method | First Author | Year | Samples Analysed
|
Ref | PMID | |||
|---|---|---|---|---|---|---|---|---|
| DCIS | DCIS-IDC | IDC | Type | |||||
| NGS | Kim | 2015 | 6 | 5 | 0 | Frozen | [27] | 25831047 |
| Kroigard | 2015 | 0 | 1 | 0 | Frozen | [28] | 25730902 | |
| Yates | 2015 | 0 | 1 | 50 | FFPE | [31] | 26099045 | |
| Foschini | 2013 | 3 | 12 | 0 | FFPE | [20] | 23337025 | |
| Newburger | 2013 | 6 | 0 | 6 | FFPE | [30] | 23568837 | |
|
| ||||||||
| aCGH | Oikawa | 2015 | 1 | 0 | 5 | FFPE | [92] | 24402639 |
| Hernandez | 2012 | 0 | 13 | 0 | Frozen | [26] | 22252965 | |
| Liao | 2012 | 20 | 25 | 24 | FFPE | [93] | 22887771 | |
| Hwang | 2011 | 52 | 0 | 0 | FFPE | [94] | 21496874 | |
| Johnson | 2011 | 0 | 21 | 0 | FFPE | [42] | 22052326 | |
| Muggerud | 2010 | 31 | 42 | 36 | Frozen | [95] | 20663721 | |
| Iakovlev | 2008 | 6 | 15 | 0 | FFPE | [96] | 18628458 | |
| Yao | 2006 | 10 | 0 | 18 | Frozen | [97] | 16618726 | |
|
| ||||||||
| SNP-Arrays | Gorringe | 2015 | 31 | 18 | 0 | FFPE | [102] | 26321097 |
| Johnson | 2012 | 0 | 21 | 0 | FFPE | [42] | 22052326 | |
Other genomic studies did not perform evolutionary analyses, but instead reported concordance of mutations and CNAs in patients with synchronous DCIS-IDC. Studies using aCGH to profile microdissected DCIS and IDC regions often showed that while most CNAs are concordant, there are also many invasive-specific amplifications of oncogenes and deletions of tumour suppressors [26,42,92–97,111]. Similarly, sequencing and genotyping analysis of synchronous DCIS-IDC regions have reported patients with invasive-specific mutations consistent with an evolutionary bottleneck [18,19,26,27,30,31,42,60,96,99,112].
Collectively, these data are consistent with an evolutionary bottleneck model, in which a clone is selected during invasion, leading to the expansion of a minor genotype in the invasive carcinoma. An alternative explanation for discordant data has invasive clones continuing to evolve new mutations and CNAs after tumour cells escape from the ducts. To distinguish between these possibilities, higher resolution genomic methods are required to resolve ITH and perform lineage reconstruction, and to determine if the invasive genotype was pre-existing in the ducts in a minor subclone.
Multiclonal Invasion
Another direct lineage model is multi-clonal invasion. In this model, multiple clonal subpopulations in ducts co-migrate into invasive regions, after breakdown of the basement membrane, and together establish the invasive carcinoma (Figure 2C). This model assumes clonal subpopulations found in ducts also occur in invasive regions. Multiclonal invasion can be explained by two alternative scenarios. In one scenario, in situ clones cooperate through non-cell-autonomous paracrine or juxtacrine interactions to escape the basement membrane of ducts and invade surrounding tissues. This scenario implies selection of multiple clones and cooperative interactions between clones, and possibly the tumour microenvironment. Functional experiments in invasive breast cancer have shown non-cell-autonomous interactions of clones can promote tumour growth through secreted growth factors and cytokines [113] or through Wnt signalling in mouse models [114] to drive tumour progression.
Another scenario invokes a single ‘leader clone’ that is responsible for breaking down the basement membrane in ducts by genetic or non-genetic factors (eg. mechanical stress), allowing other ‘follower’ clones to escape the ducts and invade surrounding tissues. This scenario does not assume direct cooperative interactions between clones. Evidence for this process is supported by histopathological images showing a complete breakdown of basement membrane and myoepithelial layers in some DCIS cases. Irrespective of underlying mechanisms, both scenarios lead to similar mutations and variant allele frequencies (VAFs) in ductal and invasive regions due to similar proportions of clones. These data contrast with the population bottleneck model, in which a subset of mutations increases in frequency due to positive selection of a clone during invasion (Figure 2B).
Genomic evidence for a multiclonal invasion model comes from studies using aCGH, in which copy number profiles between in situ and invasive subpopulations were highly correlated [26,42,92–97,111]. Oikawa et al. reported a 97% concordance between DCIS and IDC genome-wide copy number profiles measure by aCGH [92]. Similarly, Hernandez et al. used aCGH to profile patients with synchronous DCIS-IDC, and identified 10 of 13 patients with highly similar copy number profiles. Johnson et al. used aCGH to profile 23 patients with synchronous DCIS-IDC and determined on average 83% of genomic CNAs were concordant, with many patients having highly similar copy number profiles [42]. NGS data also supports multiclonal invasion in cases where the concordance of mutations and subclonal mutation frequencies between in situ and invasive regions is very high [27,28,30]. However, while these data are consistent with a multiclonal invasion model, they are largely inferential and provide only indirect evidence.
Clinical Implications of Models
The models of invasion have different implications for diagnosis and therapeutic treatment of DCIS patients. The independent lineage model assumes DCIS and IDC subpopulations are genetically unrelated, and DCIS subpopulations are unlikely to ever progress to IDC. Therefore, targeting any particular genetic aberration or protein marker in DCIS would unlikely have clinical relevance in terms of preventing formation of a subsequent invasive tumour. In contrast, both the evolutionary bottleneck and multiclonal evolution models support a direct genetic lineage between DCIS and IDC subpopulations. These models suggest targeting truncal mutations occurring early in tumour lineage, and subsequently inherited by both DCIS and IDC subpopulations, provide ideal targets for eliminating all tumour cells. Targeting truncal mutations has been proposed as a therapeutic strategy in treatment of advanced carcinomas [115,116] and is currently being investigated in a lung cancer clinical trial TRACERx [117]. The evolutionary bottleneck model suggests IDC populations have a number of invasive-specific mutations and CNAs can be targeted in early DCIS disease to prevent progression of invasive subclones. For example, in a multi-region sequencing study of a synchronous DCIS-IDC patient, the authors identified loss-of-function mutations in the PTEN tumour suppressor in invasive subpopulations not present in ducts [31]. This invasive-specific PTEN mutation could potentially be targeted with PIK3CA, AKT or mTOR inhibitors to treat the cancer [118]. The multiclonal invasion model has important clinical implications by suggesting cooperative interactions of clones may be exploited therapeutically to prevent invasion. This could be achieved by interfering with cooperative clonal interactions via drugs or antibodies targeting secreted factors or receptors that cells use in paracrine or juxtacrine interactions. However, such an approach requires a mechanistic knowledge of underlying cell interactions and signalling pathways used for cooperation, requiring detailed studies using in vitro or in vivo systems, such as xenografts. Direct lineage models also have important prognostic implications for measuring ITH using diversity indexes [52,63]. These models suggest DCIS patients with high diversity indexes (eg the Shannon index or Simpson’s Index) would be more likely to progress to IDC [61]. Studies of invasive breast cancers show that high diversity indexes can predict which patients’ tumours are more likely to metastasize or show poor therapy response [44,52,119]. Conversely, in a direct lineage model, a low genomic diversity would expect to predict a lower risk of invasion in DCIS patients.
Emerging Technologies
Several innovative technologies have recently been developed and have the potential to provide new insights into invasion in DCIS, including Single Cell Sequencing [80,120], specific-to-allele PCR–FISH [53] and Solid-phase Imaging Mass Cytometry [121] (Figure 3). Single Cell Sequencing methods were first reported for genomic copy number profiling [48] and transcriptome analysis [122] and have been applied widely to study ITH and clonal evolution in tumours [80,120]. In contrast to standard NGS methods, these methods have the ability to fully resolve ITH and report genomic information on rare clones. However a notable limitation is that most SCS methods require tissue to be dissociated into cell suspensions, in order to isolate single cells using methods such as flow-sorting, micromanipulation, microfluidics or microdroplets [123]. Unfortunately, such an approach is not ideal for studying DCIS and IDC subpopulations, because tumour cells are defined by spatial location in the duct or adjacent invasive regions in an H&E tissue section. Methods such as laser-capture-microdissection (LCM) can resolve spatial locations of single tumour cells in tissue sections, but have not been tested extensively in combination with SCS methods. Technical issues such as UV lasers damaging DNA and RNA prior to amplification or cells being cut in half during tissue sectioning using a cryomicrotome could lead to a substantial loss of DNA or RNA. Encouragingly, a recent study combined LCM with single cell RNA sequencing to compare single cell and bulk transcriptome profiles of motor neurons and dopamine neurons, suggesting technical feasibility for RNA profiling [124]. Data from our own group has shown genomic copy number profiling of single cells from breast tumour tissue sections is feasible by combining LCM with single-nucleus-sequencing to retain spatial information (Figure 3A).
Figure 3. Emerging technologies for investigating DCIS progression.
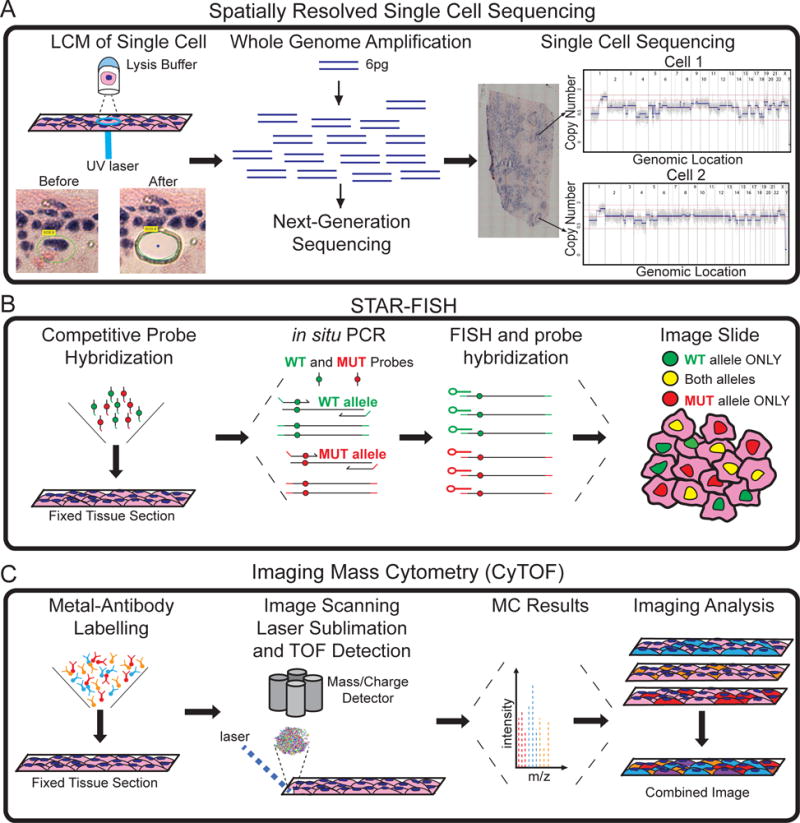
(A) Laser-Capture-Microdissection and single cell DNA sequencing to perform spatially resolved genomic profiling of individual tumour cells. (B) STAR-FISH profiling of targeted mutations in tissue sections. (C) Imaging CyTOF can perform proteomic analysis of up to one hundred proteins in thousands of single cells in tissue sections, while preserving their spatial context and organization.
Another emerging technology with potential for studying invasion in DCIS is Specific-To-Allele PCR–FISH (STAR-FISH) (Figure 3B) [53]. This method uses competitive probes to measure point mutations in thousands of single cells directly in tissue sections by in situ hybridization. STAR-FISH was used to measure both PIK3CA mutations and HER2 amplifications in the same single cells from HER2 positive breast cancer patients in response to trastuzumab treatment using FFPE tissue sections. Further development will enable genotyping multiple mutations concurrently using different fluorophore combinations. These methods are likely to have important applications for studying direct models of invasion, to determine if invasive subclones are rare subpopulations pre-existing in the ducts or represent dominant clones that co-invade surrounding tissues [53].
Recent advances in mass cytometry methods have led to the development of Solid-phase Imaging Mass Cytometry (IMC) techniques (Figure 3C). IMC can be applied to fixed tissue sections labelled with rare-earth metals conjugated to antibodies. The tissue sections are ionized by scanning lasers and mass-charge ratios are measured by a detector. The spatial intensity of each metal is quantified by mass cytometry by time of flight (CyTOF). Currently 32 antibodies measurements have been quantified across one tissue section at a 1μm resolution [121], with a theoretical limit of more than 100 antibodies [125]. However, a notable limitation of both IMC and STAR-FISH, is they are targeted approaches and require prior knowledge of genes or proteins being measured. This may be complicated to perform in DCIS patients, where inter-patient heterogeneity is extensive and common targets across patients, other than TP53 and PIK3CA, may be difficult to identify.
Conclusions
This review has discussed the genomic literature on DCIS breast cancers in the context of ITH and three models of invasion. Collectively, the genomic studies of synchronous DCIS-IDC patients suggest that an independent lineage model is uncommon, by showing most synchronous DCIS patients share a large number of concordant CNAs and mutations. Instead, genomic data support a direct lineage model and indicate evolution from a common origin: a single normal cell that gave rise to both DCIS and IDC subpopulations. However current data cannot distinguish between evolutionary bottleneck and multiclonal invasion models. Nevertheless, the concordance and discordance of mutations in DCIS and IDC regions of synchronous patients provide circumstantial evidence for distinguishing these models.
A number of reports are consistent with an evolutionary bottleneck model by showing DCIS cases in which additional CNAs and mutations were identified in IDC regions of synchronous DCIS patients that are absent in the DCIS regions [27,28,30,31,95,96]. These data are consistent with selection of a minor subclone in ductal regions that expanded during invasion. Another possibility is that invasive-specific mutations were acquired after invasion as the carcinoma expanded. Other genomic studies [26,42,92] report cases of synchronous DCIS-IDC patients with a high concordance of CNAs reported between DCIS and IDC regions, consistent with the multiclonal invasion model. These data could be explained by a single dominant clone migrating out of the ducts to establish the invasive carcinoma, but this is not supported by the observation of subclonal mutation frequencies. The central problem in distinguishing these models is that bulk genomic methods have limited ability to resolve ITH and cannot determine if invasive subclones were pre-existing in the ducts at low frequencies when cells migrated out of the ducts.
Future studies, using higher-resolution genomic methods, such as SCS and LCM, can overcome technical obstacles such as tumour purity and ITH. An important consideration is that non-genetic factors such as the tumour stroma may play an important role in invasion, as discussed in previous review articles [110,126]. Following genomic profiling, other techniques such as STAR-FISH and CyTOF will enable markers of invasive clones to be traced in thousands of cells in tissue sections. Combining these technologies can provide new insights into models of invasion in DCIS patients. To perform these studies high-quality fresh and frozen tissue sections will be needed from patients with synchronous DCIS-IDC, to directly compare DCIS and IDC subpopulations and understand invasion without confounding effects of inter-patient heterogeneity. This work is expected to lead to new clinical prognostics, and will have a major impact on preventing unnecessary treatments in patients where DCIS is unlikely to progress to IDC.
Acknowledgments
This study was supported by a grant from the Lefkofsky Family Foundation and from grants to N.N. from NCI (1RO1CA169244-01) and from the American Cancer Society (129098-RSG-16-092-01-TBG). N.N. is a T.C. Hsu Endowed Scholar, AAAS Wachtel Scholar and Andrew Sabin Family Fellow. The study is also supported by the Moonshot Knowledge Gap Award. Funding for M.E.E was provided by NCI (1RO1CA169244-01). A.K.C was supported by the Rosalie B. Hite Fellowship Fund for Cancer Research. We thank Tod Casasent for help with preparing the figures and reviewing the manuscript.
Footnotes
Conflicts of Interest Statement: No conflicts of interests.
Author contributions:
AC prepared figures and wrote the manuscript. NN and ME wrote the manuscript.
References
- 1.Allred DC. Ductal carcinoma in situ: terminology, classification, and natural history. J Natl Cancer Inst Monogr. 2010;2010:134–138. doi: 10.1093/jncimonographs/lgq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins LC, Tamimi RM, Baer HJ, et al. Outcome of patients with ductal carcinoma in situ untreated after diagnostic biopsy: results from the Nurses’ Health Study. Cancer. 2005;103:1778–1784. doi: 10.1002/cncr.20979. [DOI] [PubMed] [Google Scholar]
- 3.Francis A, Thomas J, Fallowfield L, et al. Addressing overtreatment of screen detected DCIS; the LORIS trial. Eur J Cancer. 2015;51:2296–2303. doi: 10.1016/j.ejca.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 4.Ernster VL, Barclay J. Increases in ductal carcinoma in situ (DCIS) of the breast in relation to mammography: a dilemma. J Natl Cancer Inst Monogr. 1997:151–156. doi: 10.1093/jncimono/1997.22.151. [DOI] [PubMed] [Google Scholar]
- 5.Wellings SR, Jensen HM. On the origin and progression of ductal carcinoma in the human breast. J Natl Cancer Inst. 1973;50:1111–1118. doi: 10.1093/jnci/50.5.1111. [DOI] [PubMed] [Google Scholar]
- 6.Page DL, Dupont WD, Rogers LW, et al. Continued local recurrence of carcinoma 15–25 years after a diagnosis of low grade ductal carcinoma in situ of the breast treated only by biopsy. Cancer. 1995;76:1197–1200. doi: 10.1002/1097-0142(19951001)76:7<1197::aid-cncr2820760715>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 7.Dupont WD, Page DL. Risk factors for breast cancer in women with proliferative breast disease. N Engl J Med. 1985;312:146–151. doi: 10.1056/NEJM198501173120303. [DOI] [PubMed] [Google Scholar]
- 8.Tavassoli FA. Ductal carcinoma in situ: introduction of the concept of ductal intraepithelial neoplasia. Mod Pathol. 1998;11:140–154. [PubMed] [Google Scholar]
- 9.McCaffery K, Nickel B, Moynihan R, et al. How different terminology for ductal carcinoma in situ impacts women’s concern and treatment preferences: a randomised comparison within a national community survey. BMJ Open. 2015;5:e008094. doi: 10.1136/bmjopen-2015-008094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alvarado M, Carter DL, Guenther JM, et al. The impact of genomic testing on the recommendation for radiation therapy in patients with ductal carcinoma in situ: a prospective clinical utility assessment of the 12-gene DCIS score result. J Surg Oncol. 2015;111:935–940. doi: 10.1002/jso.23933. [DOI] [PubMed] [Google Scholar]
- 11.Baxter NN, Virnig BA, Durham SB, et al. Trends in the treatment of ductal carcinoma in situ of the breast. J Natl Cancer Inst. 2004;96:443–448. doi: 10.1093/jnci/djh069. [DOI] [PubMed] [Google Scholar]
- 12.Jones JL. Overdiagnosis and overtreatment of breast cancer: progression of ductal carcinoma in situ: the pathological perspective. Breast Cancer Res. 2006;8:204. doi: 10.1186/bcr1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zujewski JA, Harlan LC, Morrell DM, et al. Ductal carcinoma in situ: trends in treatment over time in the US. Breast Cancer Res Treat. 2011;127:251–257. doi: 10.1007/s10549-010-1198-z. [DOI] [PubMed] [Google Scholar]
- 14.Bartlett JM, Nofech-Moses S, Rakovitch E. Ductal carcinoma in situ of the breast: can biomarkers improve current management? Clin Chem. 2014;60:60–67. doi: 10.1373/clinchem.2013.207183. [DOI] [PubMed] [Google Scholar]
- 15.Thompson A, Brennan K, Cox A, et al. Evaluation of the current knowledge limitations in breast cancer research: a gap analysis. Breast Cancer Res. 2008;10:R26. doi: 10.1186/bcr1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leonard GD, Swain SM. Ductal carcinoma in situ, complexities and challenges. J Natl Cancer Inst. 2004;96:906–920. doi: 10.1093/jnci/djh164. [DOI] [PubMed] [Google Scholar]
- 17.Aubele M, Mattis A, Zitzelsberger H, et al. Intratumoral heterogeneity in breast carcinoma revealed by laser-microdissection and comparative genomic hybridization. Cancer Genet Cytogenet. 1999;110:94–102. doi: 10.1016/s0165-4608(98)00205-2. [DOI] [PubMed] [Google Scholar]
- 18.Aubele M, Cummings M, Walsch A, et al. Heterogeneous chromosomal aberrations in intraductal breast lesions adjacent to invasive carcinoma. Anal Cell Pathol. 2000;20:17–24. doi: 10.1155/2000/930246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aubele M, Mattis A, Zitzelsberger H, et al. Extensive ductal carcinoma in situ with small foci of invasive ductal carcinoma: evidence of genetic resemblance by CGH. Int J Cancer. 2000;85:82–86. doi: 10.1002/(sici)1097-0215(20000101)85:1<82::aid-ijc15>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 20.Foschini MP, Morandi L, Leonardi E, et al. Genetic clonal mapping of in situ and invasive ductal carcinoma indicates the field cancerization phenomenon in the breast. Hum Pathol. 2013;44:1310–1319. doi: 10.1016/j.humpath.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 21.Luzzi V, Holtschlag V, Watson MA. Expression profiling of ductal carcinoma in situ by laser capture microdissection and high-density oligonucleotide arrays. Am J Pathol. 2001;158:2005–2010. doi: 10.1016/S0002-9440(10)64672-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reis-Filho JS, Lakhani SR. The diagnosis and management of pre-invasive breast disease: genetic alterations in pre-invasive lesions. Breast Cancer Res. 2003;5:313–319. doi: 10.1186/bcr650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Werner M, Mattis A, Aubele M, et al. 20q13.2 amplification in intraductal hyperplasia adjacent to in situ and invasive ductal carcinoma of the breast. Virchows Arch. 1999;435:469–472. doi: 10.1007/s004280050429. [DOI] [PubMed] [Google Scholar]
- 24.Westbury CB, Reis-Filho JS, Dexter T, et al. Genome-wide transcriptomic profiling of microdissected human breast tissue reveals differential expression of KIT (c-Kit, CD117) and oestrogen receptor-alpha (ERalpha) in response to therapeutic radiation. J Pathol. 2009;219:131–140. doi: 10.1002/path.2581. [DOI] [PubMed] [Google Scholar]
- 25.Ghazani AA, Arneson N, Warren K, et al. Genomic alterations in sporadic synchronous primary breast cancer using array and metaphase comparative genomic hybridization. Neoplasia. 2007;9:511–520. doi: 10.1593/neo.07301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernandez L, Wilkerson PM, Lambros MB, et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra-tumour genetic heterogeneity and clonal selection. J Pathol. 2012;227:42–52. doi: 10.1002/path.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim SY, Jung SH, Kim MS, et al. Genomic differences between pure ductal carcinoma in situ and synchronous ductal carcinoma in situ with invasive breast cancer. Oncotarget. 2015;6:7597–7607. doi: 10.18632/oncotarget.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kroigard AB, Larsen MJ, Laenkholm AV, et al. Clonal expansion and linear genome evolution through breast cancer progression from pre-invasive stages to asynchronous metastasis. Oncotarget. 2015;6:5634–5649. doi: 10.18632/oncotarget.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koboldt DC, Steinberg KM, Larson DE, et al. The next-generation sequencing revolution and its impact on genomics. Cell. 2013;155:27–38. doi: 10.1016/j.cell.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newburger DE, Kashef-Haghighi D, Weng Z, et al. Genome evolution during progression to breast cancer. Genome Res. 2013;23:1097–1108. doi: 10.1101/gr.151670.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yates LR, Gerstung M, Knappskog S, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21:751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lampejo OT, Barnes DM, Smith P, et al. Evaluation of infiltrating ductal carcinomas with a DCIS component: correlation of the histologic type of the in situ component with grade of the infiltrating component. Semin Diagn Pathol. 1994;11:215–222. [PubMed] [Google Scholar]
- 33.Sanders ME, Schuyler PA, Dupont WD, et al. The natural history of low-grade ductal carcinoma in situ of the breast in women treated by biopsy only revealed over 30 years of long-term follow-up. Cancer. 2005;103:2481–2484. doi: 10.1002/cncr.21069. [DOI] [PubMed] [Google Scholar]
- 34.Simpson PT, Reis-Filho JS, Gale T, et al. Molecular evolution of breast cancer. J Pathol. 2005;205:248–254. doi: 10.1002/path.1691. [DOI] [PubMed] [Google Scholar]
- 35.Simpson PT, Reis-Filho JS, Lakhani SR. Breast pathology: beyond morphology. Semin Diagn Pathol. 2010;27:91–96. doi: 10.1053/j.semdp.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 36.Fujii H, Szumel R, Marsh C, et al. Genetic progression, histological grade, and allelic loss in ductal carcinoma in situ of the breast. Cancer Res. 1996;56:5260–5265. [PubMed] [Google Scholar]
- 37.Buerger H, Mommers EC, Littmann R, et al. Ductal invasive G2 and G3 carcinomas of the breast are the end stages of at least two different lines of genetic evolution. J Pathol. 2001;194:165–170. doi: 10.1002/path.875. [DOI] [PubMed] [Google Scholar]
- 38.Roylance R, Gorman P, Harris W, et al. Comparative genomic hybridization of breast tumors stratified by histological grade reveals new insights into the biological progression of breast cancer. Cancer Res. 1999;59:1433–1436. [PubMed] [Google Scholar]
- 39.Simpson PT, Gale T, Reis-Filho JS, et al. Columnar cell lesions of the breast: the missing link in breast cancer progression? A morphological and molecular analysis. Am J Surg Pathol. 2005;29:734–746. doi: 10.1097/01.pas.0000157295.93914.3b. [DOI] [PubMed] [Google Scholar]
- 40.Shackney SE, Silverman JF. Molecular evolutionary patterns in breast cancer. Adv Anat Pathol. 2003;10:278–290. doi: 10.1097/00125480-200309000-00003. [DOI] [PubMed] [Google Scholar]
- 41.Buerger H, Otterbach F, Simon R, et al. Comparative genomic hybridization of ductal carcinoma in situ of the breast-evidence of multiple genetic pathways. J Pathol. 1999;187:396–402. doi: 10.1002/(SICI)1096-9896(199903)187:4<396::AID-PATH286>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 42.Johnson CE, Gorringe KL, Thompson ER, et al. Identification of copy number alterations associated with the progression of DCIS to invasive ductal carcinoma. Breast Cancer Res Treat. 2012;133:889–898. doi: 10.1007/s10549-011-1835-1. [DOI] [PubMed] [Google Scholar]
- 43.Harris L, Fritsche H, Mennel R, et al. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J Clin Oncol. 2007;25:5287–5312. doi: 10.1200/JCO.2007.14.2364. [DOI] [PubMed] [Google Scholar]
- 44.Kurozumi S, Padilla M, Kurosumi M, et al. HER2 intratumoral heterogeneity analyses by concurrent HER2 gene and protein assessment for the prognosis of HER2 negative invasive breast cancer patients. Breast Cancer Res Treat. 2016;158:99–111. doi: 10.1007/s10549-016-3856-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med. 2007;131:18–43. doi: 10.5858/2007-131-18-ASOCCO. [DOI] [PubMed] [Google Scholar]
- 46.Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007;25:118–145. doi: 10.1200/JCO.2006.09.2775. [DOI] [PubMed] [Google Scholar]
- 47.Solin LJ, Gray R, Baehner FL, et al. A multigene expression assay to predict local recurrence risk for ductal carcinoma in situ of the breast. J Natl Cancer Inst. 2013;105:701–710. doi: 10.1093/jnci/djt067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Waters J, Leung ML, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nguyen A, Yoshida M, Goodarzi H, et al. Highly variable cancer subpopulations that exhibit enhanced transcriptome variability and metastatic fitness. Nat Commun. 2016;7:11246. doi: 10.1038/ncomms11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–399. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Almendro V, Cheng YK, Randles A, et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep. 2014;6:514–527. doi: 10.1016/j.celrep.2013.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janiszewska M, Liu L, Almendro V, et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat Genet. 2015;47:1212–1219. doi: 10.1038/ng.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Allred DC, Wu Y, Mao S, et al. Ductal carcinoma in situ and the emergence of diversity during breast cancer evolution. Clin Cancer Res. 2008;14:370–378. doi: 10.1158/1078-0432.CCR-07-1127. [DOI] [PubMed] [Google Scholar]
- 55.Stratton MR, Collins N, Lakhani SR, et al. Loss of heterozygosity in ductal carcinoma in situ of the breast. J Pathol. 1995;175:195–201. doi: 10.1002/path.1711750207. [DOI] [PubMed] [Google Scholar]
- 56.Aubele M, Werner M. Heterogeneity in breast cancer and the problem of relevance of findings. Anal Cell Pathol. 1999;19:53–58. doi: 10.1155/1999/960923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hicks J, Krasnitz A, Lakshmi B, et al. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 2006;16:1465–1479. doi: 10.1101/gr.5460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Radford DM, Phillips NJ, Fair KL, et al. Allelic loss and the progression of breast cancer. Cancer Res. 1995;55:5180–5183. [PubMed] [Google Scholar]
- 59.Chin K, de Solorzano CO, Knowles D, et al. In situ analyses of genome instability in breast cancer. Nat Genet. 2004;36:984–988. doi: 10.1038/ng1409. [DOI] [PubMed] [Google Scholar]
- 60.Desmedt C, Fumagalli D, Pietri E, et al. Uncovering the genomic heterogeneity of multifocal breast cancer. J Pathol. 2015;236:457–466. doi: 10.1002/path.4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Park SY, Gonen M, Kim HJ, et al. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest. 2010;120:636–644. doi: 10.1172/JCI40724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Navin NE, Hicks J. Tracing the tumor lineage. Mol Oncol. 2010;4:267–283. doi: 10.1016/j.molonc.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao R, Davis A, McDonald TO, et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet. 2016 doi: 10.1038/ng.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burkhardt L, Grob TJ, Hermann I, et al. Gene amplification in ductal carcinoma in situ of the breast. Breast Cancer Res Treat. 2010;123:757–765. doi: 10.1007/s10549-009-0675-8. [DOI] [PubMed] [Google Scholar]
- 65.Murphy DS, McHardy P, Coutts J, et al. Interphase cytogenetic analysis of erbB2 and topoII alpha co-amplification in invasive breast cancer and polysomy of chromosome 17 in ductal carcinoma in situ. Int J Cancer. 1995;64:18–26. doi: 10.1002/ijc.2910640106. [DOI] [PubMed] [Google Scholar]
- 66.Munn KE, Walker RA, Varley JM. Frequent alterations of chromosome 1 in ductal carcinoma in situ of the breast. Oncogene. 1995;10:1653–1657. [PubMed] [Google Scholar]
- 67.Chen T, Sahin A, Aldaz CM. Deletion map of chromosome 16q in ductal carcinoma in situ of the breast: refining a putative tumor suppressor gene region. Cancer Res. 1996;56:5605–5609. [PubMed] [Google Scholar]
- 68.Park K, Han S, Kim HJ, et al. HER2 status in pure ductal carcinoma in situ and in the intraductal and invasive components of invasive ductal carcinoma determined by fluorescence in situ hybridization and immunohistochemistry. Histopathology. 2006;48:702–707. doi: 10.1111/j.1365-2559.2006.02403.x. [DOI] [PubMed] [Google Scholar]
- 69.Ellsworth RE, Vertrees A, Love B, et al. Chromosomal alterations associated with the transition from in situ to invasive breast cancer. Ann Surg Oncol. 2008;15:2519–2525. doi: 10.1245/s10434-008-0051-7. [DOI] [PubMed] [Google Scholar]
- 70.Navin N, Hicks J. Future medical applications of single-cell sequencing in cancer. Genome Med. 2011;3:31. doi: 10.1186/gm247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nik-Zainal S, Van Loo P, Wedge DC, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eirew P, Steif A, Khattra J, et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2014 doi: 10.1038/nature13952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang J, Fujimoto J, Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346:256–259. doi: 10.1126/science.1256930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gerlinger M, Horswell S, Larkin J, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–233. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Baslan T, Kendall J, Rodgers L, et al. Genome-wide copy number analysis of single cells. Nat Protoc. 2012;7:1024–1041. doi: 10.1038/nprot.2012.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leung ML, Wang Y, Waters J, et al. SNES: single nucleus exome sequencing. Genome Biol. 2015;16:55. doi: 10.1186/s13059-015-0616-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu X, Hou Y, Yin X, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148:886–895. doi: 10.1016/j.cell.2012.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zong C, Lu S, Chapman AR, et al. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338:1622–1626. doi: 10.1126/science.1229164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Leung ML, Wang Y, Kim C, et al. Highly multiplexed targeted DNA sequencing from single nuclei. Nat Protoc. 2016;11:214–235. doi: 10.1038/nprot.2016.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Navin NE. Cancer genomics: one cell at a time. Genome Biol. 2014;15:452. doi: 10.1186/s13059-014-0452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buerger H, Otterbach F, Simon R, et al. Different genetic pathways in the evolution of invasive breast cancer are associated with distinct morphological subtypes. J Pathol. 1999;189:521–526. doi: 10.1002/(SICI)1096-9896(199912)189:4<521::AID-PATH472>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 82.Fujii H, Marsh C, Cairns P, et al. Genetic divergence in the clonal evolution of breast cancer. Cancer Res. 1996;56:1493–1497. [PubMed] [Google Scholar]
- 83.Waldman FM, DeVries S, Chew KL, et al. Chromosomal alterations in ductal carcinomas in situ and their in situ recurrences. J Natl Cancer Inst. 2000;92:313–320. doi: 10.1093/jnci/92.4.313. [DOI] [PubMed] [Google Scholar]
- 84.Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jakubek Y, Lang W, Vattathil S, et al. Genomic landscape established by allelic imbalance in the cancerization field of a normal appearing airway. Cancer Res. 2016;76:3676–3683. doi: 10.1158/0008-5472.CAN-15-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Forsberg LA, Rasi C, Pekar G, et al. Signatures of post-zygotic structural genetic aberrations in the cells of histologically normal breast tissue that can predispose to sporadic breast cancer. Genome Res. 2015;25:1521–1535. doi: 10.1101/gr.187823.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang D, Khan S, Sun Y, et al. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA. 2011;306:1557–1565. doi: 10.1001/jama.2011.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Abdel-Fatah TM, Perry C, Arora A, et al. Is there a role for base excision repair in estrogen/estrogen receptor-driven breast cancers? Antioxid Redox Signal. 2014;21:2262–2268. doi: 10.1089/ars.2014.6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Crook T, Brooks LA, Crossland S, et al. p53 mutation with frequent novel condons but not a mutator phenotype in BRCA1- and BRCA2-associated breast tumours. Oncogene. 1998;17:1681–1689. doi: 10.1038/sj.onc.1202106. [DOI] [PubMed] [Google Scholar]
- 90.Miron A, Varadi M, Carrasco D, et al. PIK3CA mutations in in situ and invasive breast carcinomas. Cancer Res. 2010;70:5674–5678. doi: 10.1158/0008-5472.CAN-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sontag L, Axelrod DE. Evaluation of pathways for progression of heterogeneous breast tumors. J Theor Biol. 2005;232:179–189. doi: 10.1016/j.jtbi.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 92.Oikawa M, Yano H, Matsumoto M, et al. A novel diagnostic method targeting genomic instability in intracystic tumors of the breast. Breast Cancer. 2015;22:529–535. doi: 10.1007/s12282-013-0516-9. [DOI] [PubMed] [Google Scholar]
- 93.Liao S, Desouki MM, Gaile DP, et al. Differential copy number aberrations in novel candidate genes associated with progression from in situ to invasive ductal carcinoma of the breast. Genes Chromosomes Cancer. 2012;51:1067–1078. doi: 10.1002/gcc.21991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hwang ES, Lal A, Chen YY, et al. Genomic alterations and phenotype of large compared to small high-grade ductal carcinoma in situ. Hum Pathol. 2011;42:1467–1475. doi: 10.1016/j.humpath.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 95.Muggerud AA, Hallett M, Johnsen H, et al. Molecular diversity in ductal carcinoma in situ (DCIS) and early invasive breast cancer. Mol Oncol. 2010;4:357–368. doi: 10.1016/j.molonc.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Iakovlev VV, Arneson NC, Wong V, et al. Genomic differences between pure ductal carcinoma in situ of the breast and that associated with invasive disease: a calibrated aCGH study. Clin Cancer Res. 2008;14:4446–4454. doi: 10.1158/1078-0432.CCR-07-4960. [DOI] [PubMed] [Google Scholar]
- 97.Yao J, Weremowicz S, Feng B, et al. Combined cDNA array comparative genomic hybridization and serial analysis of gene expression analysis of breast tumor progression. Cancer Res. 2006;66:4065–4078. doi: 10.1158/0008-5472.CAN-05-4083. [DOI] [PubMed] [Google Scholar]
- 98.Cummings MC, Aubele M, Mattis A, et al. Increasing chromosome 1 copy number parallels histological progression in breast carcinogenesis. Br J Cancer. 2000;82:1204–1210. doi: 10.1054/bjoc.1999.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hwang ES, DeVries S, Chew KL, et al. Patterns of chromosomal alterations in breast ductal carcinoma in situ. Clin Cancer Res. 2004;10:5160–5167. doi: 10.1158/1078-0432.CCR-04-0165. [DOI] [PubMed] [Google Scholar]
- 100.Sakr RA, Weigelt B, Chandarlapaty S, et al. PI3K pathway activation in high-grade ductal carcinoma in situ–implications for progression to invasive breast carcinoma. Clin Cancer Res. 2014;20:2326–2337. doi: 10.1158/1078-0432.CCR-13-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Petridis C, Brook MN, Shah V, et al. Genetic predisposition to ductal carcinoma in situ of the breast. Breast Cancer Res. 2016;18:22. doi: 10.1186/s13058-016-0675-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gorringe KL, Hunter SM, Pang JM, et al. Copy number analysis of ductal carcinoma in situ with and without recurrence. Mod Pathol. 2015;28:1174–1184. doi: 10.1038/modpathol.2015.75. [DOI] [PubMed] [Google Scholar]
- 103.Wu CI, Ting CT. Genes and speciation. Nat Rev Genet. 2004;5:114–122. doi: 10.1038/nrg1269. [DOI] [PubMed] [Google Scholar]
- 104.Gundem G, Van Loo P, Kremeyer B, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–357. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kim H, Zheng S, Amini SS, et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015;25:316–327. doi: 10.1101/gr.180612.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Patch AM, Christie EL, Etemadmoghadam D, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–494. doi: 10.1038/nature14410. [DOI] [PubMed] [Google Scholar]
- 108.Landau DA, Clement K, Ziller MJ, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014;26:813–825. doi: 10.1016/j.ccell.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Weng Z, Spies N, Zhu SX, et al. Cell-lineage heterogeneity and driver mutation recurrence in pre-invasive breast neoplasia. Genome medicine. 2015;7:28. doi: 10.1186/s13073-015-0146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cowell CF, Weigelt B, Sakr RA, et al. Progression from ductal carcinoma in situ to invasive breast cancer: revisited. Mol Oncol. 2013;7:859–869. doi: 10.1016/j.molonc.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nyante SJ, Devries S, Chen YY, et al. Array-based comparative genomic hybridization of ductal carcinoma in situ and synchronous invasive lobular cancer. Hum Pathol. 2004;35:759–763. doi: 10.1016/j.humpath.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 112.Aubele MM, Cummings MC, Mattis AE, et al. Accumulation of chromosomal imbalances from intraductal proliferative lesions to adjacent in situ and invasive ductal breast cancer. Diagn Mol Pathol. 2000;9:14–19. doi: 10.1097/00019606-200003000-00003. [DOI] [PubMed] [Google Scholar]
- 113.Marusyk A, Tabassum DP, Altrock PM, et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514:54–58. doi: 10.1038/nature13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cleary AS, Leonard TL, Gestl SA, et al. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 2014;508:113–117. doi: 10.1038/nature13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013;108:479–485. doi: 10.1038/bjc.2012.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Alizadeh AA, Aranda V, Bardelli A, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21:846–853. doi: 10.1038/nm.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jamal-Hanjani M, Hackshaw A, Ngai Y, et al. Tracking genomic cancer evolution for precision medicine: the lung TRACERx study. PLoS Biol. 2014;12:e1001906. doi: 10.1371/journal.pbio.1001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dillon LM, Miller TW. Therapeutic targeting of cancers with loss of PTEN function. Curr Drug Targets. 2014;15:65–79. doi: 10.2174/1389450114666140106100909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Almendro V, Kim HJ, Cheng YK, et al. Genetic and phenotypic diversity in breast tumor metastases. Cancer Res. 2014;74:1338–1348. doi: 10.1158/0008-5472.CAN-13-2357-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Navin NE. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015;25:1499–1507. doi: 10.1101/gr.191098.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Giesen C, Wang HA, Schapiro D, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11:417–422. doi: 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]
- 122.Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 123.Wang Y, Navin NE. Advances and applications of single-cell sequencing technologies. Mol Cell. 2015;58:598–609. doi: 10.1016/j.molcel.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nichterwitz S, Chen G, Aguila Benitez J, et al. Laser capture microscopy coupled with Smart-seq2 for precise spatial transcriptomic profiling. Nat Commun. 2016;7:12139. doi: 10.1038/ncomms12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Levenson RM, Borowsky AD, Angelo M. Immunohistochemistry and mass spectrometry for highly multiplexed cellular molecular imaging. Lab Invest. 2015;95:397–405. doi: 10.1038/labinvest.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Allen MD, Marshall JF, Jones JL. alphavbeta6 Expression in myoepithelial cells: a novel marker for predicting DCIS progression with therapeutic potential. Cancer Res. 2014;74:5942–5947. doi: 10.1158/0008-5472.CAN-14-1841. [DOI] [PubMed] [Google Scholar]