

Abstract
Immune monitoring is critical in settings of infection, autoimmunity, and cancer, but our understanding of the diversity of the antibody immune repertoire has been limited to selected target antigens and epitopes. Development of new vaccines requires monitoring of B cell immunity and identification of candidate antigens. As vaccines become more complex, novel techniques are required for monitoring the diversity of the B cell immune response. Since antibodies recognize both linear and conformational protein and glycoprotein epitopes, recent advances in proteomic and glycomic technologies for rapid display of antigenic structures are leading to methods for proteome-wide immune monitoring. Here, we review different approaches for protein display for immune monitoring, and provide methods for in situ protein display for the rapid detection and validation of antibody repertoires.
Keywords: B cell, Antibody, Immunity, Epitope, Protein display, Immune monitoring, Phage display, Protein arrays
1 Introduction
Antibodies were discovered in the last decade of the nineteenth century [1]. They were the first proteins that were described to be involved in a specific immune response and they are the most critical element of adaptive immunity for the majority of current vaccines. Methods to identify the recognition of specific antigens from pathogens and other immunogens by B lymphocytes remain an active field of research, primarily limited by methods of protein and glycoprotein production and analysis.
The earliest immunization strategies were based on simulating the course of natural infection through using inactivated or live attenuated infectious agents. Despite little knowledge of the immunological pathways and targets of the immune response, highly effective vaccines were developed that stimulate the body to produce durable B cell immunity against acute infections. Examples include vaccines against smallpox, cholera, anthrax, diphtheria, pertussis, and tetanus [2]. However, live attenuated vaccines pose a risk of reversion to virulence and cause complications in immunocompromised individuals. Inactivated vaccines limit this risk but are generally more expensive, not as immunogenic, and are liable to contamination [2].
A large proportion of successful vaccines in use today are pathogen subunits. These include bacterial toxoids, purified proteins, or purified polysaccharides. Of these, only a small number represent recombinant proteins such as vaccines against hepatitis B and HPV [3]. Pathogens with more complicated mechanisms of virulence require more than simple single-antigen vaccines [4]. More complex pathogens such as staphylococci, enterococci, and fungi have not yet been effectively targeted by immunization strategies [3].
In addition to vaccines against infections, cell-based vaccines [5, 6] and immune checkpoint inhibitors [7] have recently emerged as more complex immune modulation strategies for cancer. Progress of these promising novel strategies relies on deciphering immune signatures and surveillance of B cell immunity. However, identification of specific tumor-associated autoantibodies can be challenging. There are over 20,000 open reading frames in the human genome. When splice variation and polymorphism are considered, the number of potential antigens to which autoantibodies can be generated is enormous [8].
Identification of appropriate and promising target antigens for new vaccine development requires antibody-based assays [2, 9] since most current vaccines confer protection through stimulating B lymphocytes to produce neutralizing antibodies [10]. Antibodies are easily detectable, stable, and highly specific [11]. The first use of antibodies as reagents was in 1949 by Örjan Ouchterlony using the immunodiffusion assay [12]. Ten years later, the radioimmunoassay (RIA) was developed by Solomon Berson and Rosalyn Yalow for which Yalow was awarded the Nobel Prize [13]. Their invention paved the way for a variety of other immunoassays, permitting highly sensitive and specific detection of a multitude of proteins, and superseded many other bioassays including conventional pregnancy tests [14]. The main stumbling block for RIA was the need for purification of polyclonal antibodies in large quantities from animals [15], which was solved by the hybridoma method for production of monoclonal antibodies by Kohler and Milstein. To limit hazards and logistics of radiation, enzyme-linked reporters were developed [16] and the first paper on the modern ELISA was published in 1971 [17].
1.1 Proteomic Techniques for Monitoring of the Immune Response
One critical requirement for antibody-based assays is the efficient and reproducible expression, purification, and display of proteins. Sera are typically screened for antibodies to select antigens that are known to potentially be immunogenic or play a role in pathogenicity. This antigen selection does not measure the diversity of immune recognition [8]. To add complexity, proteome-wide immune monitoring requires the production of thousands of protein structures.
The need for tools to study proteins and the significant role they play in health and disease have led to the revolutionary advancements in the field of proteomics in the last 20 years. Effective targets of immunization and serological testing are best determined using a systems approach for monitoring the B cell immune response. Proteomic techniques that have been developed for epitope display are reviewed in [8, 11] and can be summarized as follows:
1.1.1 Phage Display
Phage display was first described in 1985 [18]. Candidate antigens are expressed in lambda phage from cDNA libraries constructed from a given pathogen or disease tissue. Phage-expressing proteins of interest are subsequently replicated onto nitrocellulose membranes and probed with patient sera. Phage display has been applied in antigen discovery in various pathogens such as hepatitis C virus [19], human cytomegalovirus [20], Mycoplasma pneumoniae [21], and Streptococcus pneumoniae [22]. Alternatively, solution-based phage display is used for autoantigen identification. Phage-displayed peptide libraries are subjected to affinity purification to isolate phage-carrying specific peptides. Autoantibody biomarkers of several cancers such as ovary and prostate have been identified using this technique [23–25]. However, because of the nature of cDNA cloned on the expression vectors, the major drawback of phage display is expression of proteins with truncations, frame shifts, and sometimes improper folding. In addition post-translational modifications (PTMs) are absent and abundant proteins are overrepresented.
1.1.2 Cellular Fractionation and Immunoblotting
In this strategy, candidate antigens from lysates of tissues or pathogens are separated by two-dimensional gel electrophoresis and serum reactivity is determined by immunoblotting or mass spectrometry. Next, bands are excised and proteins are identified by mass spectrometric analysis. This method has the advantage of using proteins with their relevant PTMs and it does not require cloning or expression procedures. However, proteins found in low concentrations may be masked by more abundant proteins.
1.1.3 Peptide Arrays
Peptides are displayed on a solid surface such as a glass or plastic slide. Relevant peptides that have overlapping sequences are determined bioinformatically so as to cover the whole ORFeome or a portion of the proteome. This circumvents difficulty with expression of full-length proteins, but conformational epitopes and posttranslational modifications are not detected. Recently, peptide arrays have been used to determine individual immunosignatures that can predict the protective efficacy of a given vaccine in mice [26].
1.1.4 Protein Arrays
Protein microarrays enable the display of thousands of proteins on the surface of a microscopic slide or in 96-well bead array format. A wide variety of protein expression systems are used including E. coli, yeast, or insect cells and then antigens are purified. However, these systems can be time consuming and unsuitable for high-throughput proteomic methods. Additionally, bacteria often fail to express most proteins with intact tertiary structures or PTMs, particularly those with high molecular weights or multiple domains [27, 28]. In vitro protein expression, on the other hand, diminishes the time required to obtain protein from DNA but adds the challenges of protein purity and reproducibility of expression [8].
Protein microarrays are currently commercially available from several sources, and are provided either as purified, printed proteins or as printed cDNA that can be expressed using in vitro transcription and translation. At this time, the antigenic display on protein microarrays is primarily the protein backbone, so the diversity of displayed antigenic structures from posttranslational modifications is more limited. As the content of ORFeome collections and the cost of protein expression improve, proteome-wide screening of sera for antibody responses is becoming feasible both for human antigens and pathogens. Here, we discuss three methods and overall strategies for using in situ protein display for detection of antibody responses in human sera or plasma.
1.2 Nucleic Acid Programmable Protein Array
To improve both the cost of purification of recombinant proteins and the stability of displayed protein, the nucleic acid programmable protein array (NAPPA) technique was developed using printed expression plasmids with an anti-tag antibody on microscopic glass slides [29–31] (Fig. 1a). At the time of the assay, in vitro transcription and translation (IVTT) are used for in situ expression of tagged target proteins encoded by the arrayed plasmids. The use of a human-coupled IVTT system derived from the human cell line HeLa results in ten times higher protein yields, more robust reproducibility, and less background than the previously used rabbit reticulocyte lysate system [32]. For immune monitoring, slides are incubated with subject sera or plasma to permit binding of antibodies to their corresponding protein spot on the array. Signals are detected using either a fluorescently labeled or an HRP-labeled secondary antibody. NAPPA arrays are available from the Arizona State University protein array core, www.NAPPAproteinarray.org.
Fig. 1.
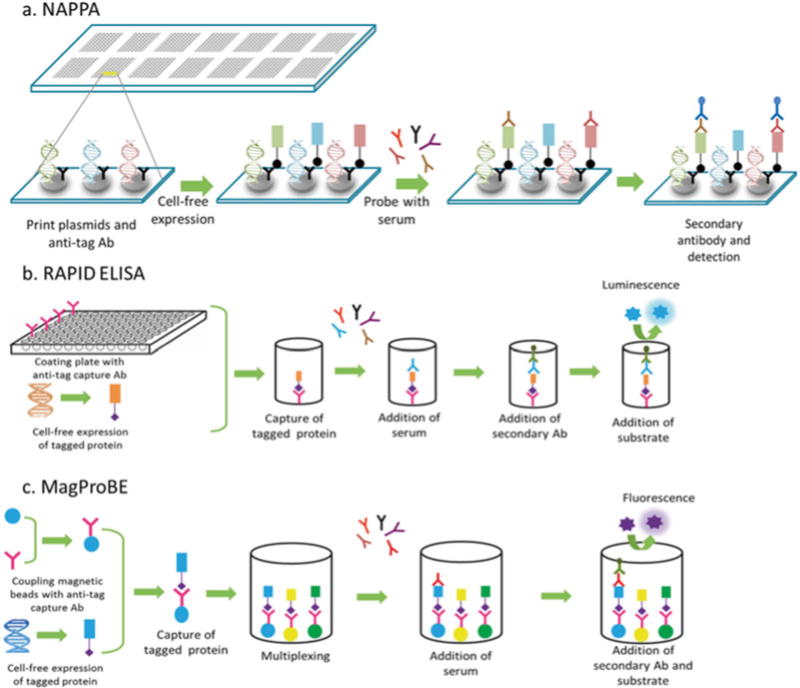
Different protein array techniques that use in situ protein display for detecting antibodies in human serum or plasma. (a) Nucleic acid programmable protein arrays (NAPPA): cDNA-containing plasmids are printed on microscopic slides along with an anti-tag antibody. In situ cell-free expression of tagged proteins generates protein microarrays that can be used to detect antibodies in serum using an appropriate secondary antibody. (b) Rapid antigenic protein in situ display ELISA (RAPID ELISA): 96-well plates are coated with an anti-tag antibody to capture freshly expressed tagged proteins. Displayed proteins are probed with serum and bound antibodies are detected using a secondary antibody. (c) Magnetic programmable bead ELISA (MagProBE): IVTT-expressed tagged proteins are captured onto anti-tag-coupled fluorescent beads and probed with serum. Multiplexing can be achieved by capturing different antigens on different-colored beads
Printing DNA on the arrays has several advantages over printing proteins. Unlike protein arrays, printed plasmids retain their activity following months of storage of the arrays under arid conditions. Since the production and purification of thousands of proteins are expensive, time consuming, and liable to protein unfolding over the multistep process of protein array production, the on-demand IVTT avoids these issues [33]. However, IVTT-derived proteins are produced with limited posttranslational modifications that are a significant component of the immune response.
A key advance in the field of protein microarrays has been the steady improvement in relevant ORFeome collections. The DNASU Plasmid Repository is the source of plasmid DNA used for NAPPA array production [34]. This plasmid collection was first started at the Harvard Institute of Proteomics in 2000 and is currently located at the Virginia G. Piper Center for Personalized Diagnostics in the Biodesign Institute (AZ, USA) [34]. The repository comprises and distributes a collection of over 200,000 plasmids containing the open reading frames (ORFs) of proteins from over 600 organisms, including 12,000 full-length human genes. The DNASU website, database, and physical repository (http://dnasu.asu.edu or http://dnasu.org) were designed to provide annotated and sequence-verified plasmids and online resources to the research community. All ORFs are cloned onto a master plasmid (pDONR), sequence verified, and stored in the DNASU repository. ORFs in DONR plasmids can be moved to a wide array of expression vectors using Gateway recombinational cloning.
NAPPA arrays have been used for the discovery of autoantibodies in cancer patient sera, such as autoantibodies to p53 in breast and ovarian cancer, BCL2 in prostate cancer, and ML-IAP in melanoma [35–37]. As an example of screening for infectious disease antigens, Pseudomonas aeruginosa outer membrane proteins printed on NAPPA have been used to screen sera from P. aeruginosa patients to identify immunogenic proteins [38].
1.3 Rapid Antigenic Protein In Situ Display (RAPID ELISA)
NAPPA protein microarrays are excellent tools for antigen discovery. However, validation requires methods for the analysis of a few antigens, but using thousands of sera. RAPID ELISA was developed as a robust tool that can be performed in most immunology laboratories using publicly available reagents. RAPID ELISA can be used to screen hundreds of sera rapidly and cost-effectively in order to confirm antibody biomarkers and immunogenicity of antigens discovered using protein microarrays [36, 39, 40]. As with NAPPA assays, tagged proteins are expressed using an in vitro transcription and translation system, but then captured in a 96-well plate through an anti-tag antibody (Fig. 1b). Sera are then incubated with the displayed proteins, and bound immunoglobulins are detected using secondary antibodies. To overcome the background problem encountered with human sera, an optimized serum blocking buffer consisting of E. coli lysate diluted 1:10 in PBST and 5 % milk was developed [41]. An eightfold increase in the relative light unit (RLU) ratio of antigen-specific IgG compared with control GST protein was observed with the use of this serum blocking buffer. Additionally, human HeLa cell lysate IVTT system and automation have further enhanced the efficiency, rapidity, and reproducibility of this technique.
1.4 Magnetic Programmable Bead ELISA (MagProBE)
A similar technique as RAPID ELISA for high-throughput serum screening is the magnetic programmable bead ELISA, MagProBE [39]. As with RAPID ELISA, tagged proteins are expressed by IVTT, but then expressed proteins are captured on anti-tag-coupled fluorescent magnetic beads (such as Luminex beads) in a 96-well plate (Fig. 1c). This is followed by steps of incubation with sera and then with a secondary antibody. Beads are coupled with the anti-tag antibody in advance and they are stable for at least 1 year. Coupling efficiency is confirmed using anti-Ig secondary antibodies. Chief among the advantages of MagProBE is the high reproducibility and automated washing. Additionally, bead array ELISA can be used for multiplex assays by capturing of different antigens on beads of different colors and then pooling them. Multiplexing saves both time and volume of serum, but the cost/antigen is higher than with RAPID ELISA. This technique has been used for multiplex detection of immunity to a panel of EBV antigens in healthy donor sera [42] and to investigate potential biomarkers of HPV-associated oropharyngeal carcinoma [43].
1.5 Recent Advances in Protein Display and Detection
Many immune-based biomarkers have clinical applications for early detection of disease. The applications require robust, reproducible, and cost-effective assays with improved limits of detection, multiplexing, and automation, all of which have substantially improved in the last decade. For example, chromogenic enzyme substrates have been the traditional reporter molecules for ELISAs. The more sensitive chemiluminescent substrates can now detect analyte concentrations in the picomolar range [15]. Ultrasensitive approaches, such as the single-molecule array technology, may allow detection of femtomolar concentrations of antibody through digital measurements of immunocomplexes. Nanoparticle-based ELISAs are reported to detect attograms of analytes [44].
Because routine laboratory diagnosis is costly and may not be accessible in resource-poor areas, point-of-care (POC) tests are emerging technologies for health screening, much of which currently depends on detection of antibodies. Affordable POC tests that give rapid and reliable results, require minimal training, and use no equipment are currently in use for HIV, syphilis, and malaria [45]. Integration of ELISA assays with microfluidics and molecular detection methods may transform vaccine monitoring and identification of at-risk individuals for clinical interventions.
2 Materials
2.1 In Vitro Protein Expression
DNA preparation systems (Nucleobond, Clontech, Mountain View, CA).
Plasmid with GST tag (DNASU, Tempe, AZ, USA).
1-Step Human Coupled IVTT Kit—DNA (Life Technologies, Carlsbad, CA, USA).
RNaseOUT (Thermo Fisher Scientific, Waltham, MA, USA).
EchoTherm Chill/Heat Incubator (Thermo Fisher Scientific, Waltham, MA, USA).
2.2 Serum Sample Preparation
E. coli DH5α.
Luria Broth (LB) media.
cOmplete, Mini Protease Inhibitor Cocktail Tablets (Roche, Basel, Switzerland).
Bovine serum albumin: BSA (Cell Signaling, Danvers, MA, USA).
Sonicator.
Centrifuge.
Shaking incubator.
2.3 Nucleic Acid Programmable Protein Array
NAPPA microarray slides obtained from the NAPPA Protein Array Core of DNASU (DNASU, Tempe, AZ, USA).
HybriWell gasket adhesive (Grace Bio-Labs, Bend, OR, USA).
Microarray Hybridization Chamber (Corning, Corning, NY, USA).
Modified pipette tip box with separators to wash up to four slides.
SuperBlock Buffer in PBS (Pierce Biotechnology, Rockford, IL, USA).
- Secondary fluorescence-tagged anti-human antibody (Ab) compatible with Tecan’s PowerScanner excitation lasers:
- Laser specification—red 2 635–63 nm diode laser, green 20 mW 532 nm solid-state laser.
- Emission filter wheel: 676/37 nm filter, 579/42 nm filter, two additional free positions for customer-defined filters.
PowerScanner microarray scanner (Tecan, Maennedorf, Switzerland).
Array-Pro Analyzer software (MediaCybernetics, Rockville, MD, USA).
2.4 Rapid Antigenic Protein In Situ Display (RAPID) ELISA
White opaque, flat-bottom, 96-well polystyrene high-bind hydrophobic-ionic microplate (Pierce Biotechnology, Rockford, IL, USA).
Sodium Bicarbonate (Sigma-Aldrich, St. Louis, MO, USA).
RNaseOUT (Thermo Fisher Scientific, Waltham, MA, USA).
HRP anti-human IgG antibody (Ab) (Jackson ImmunoResearch, West Grove, PA, USA).
HRP anti-mouse IgG Ab (Jackson ImmunoResearch, West Grove, PA, USA).
Anti-GST polyclonal Ab (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Anti-GST mouse monoclonal antibody (mAb) (Cell Signaling, Danvers, MA, USA).
Supersignal ELISA Femto Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA, USA).
Nunc Immuno Washer (Thermo Fisher Scientific, Waltham, MA, USA).
Titer plate shaker (Thermo Fisher Scientific, Waltham, MA, USA).
GloMax ®-96 Microplate Luminometer (Promega, Madison, WI, USA).
2.5 Magnetic Programmable Bead ELISA (MagProBE)
Monobasic Sodium Phosphate Anhydrous (MP Biomedicals, Santa Ana, CA, USA).
Sulfo-N-hydroxysulfosuccinimide (Sulfo-NHS) (Sigma-Aldrich, St. Louis, MO, USA).
1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) (Thermo Fisher Scientific, Waltham, MA, USA).
Bio-Plex Pro Magnetic Carboxylated 6.5 μm microspheres compatible with Luminex’s MAGPIX (Bio-Rad, Hercules, CA, USA) (see Note 3b).
2-(N-Morpholino)ethanesulfonic acid (MES) (Sigma-Aldrich, St. Louis, MO, USA).
BSA (Cell Signaling, Danvers, MA, USA).
Sodium Azide (Sigma-Aldrich, St. Louis, MO, USA).
Goat anti-GST polyclonal Ab (GE Healthcare Life Sciences, Pittsburgh, PA, USA).
Donkey anti-goat IgG-PE (Jackson ImmunoResearch, West Grove, PA, USA).
Biotin labeled anti-human IgG (Jackson ImmunoResearch, West Grove, PA, USA).
Streptavidin, R-Phycoerythrin Conjugate (SAPE) (Life Technologies, Carlsbad, CA, USA).
Greiner Microassay 96-Well Plate (Greiner Bio-One, Monroe, NC, USA).
DynaMag-2 Magnet (Thermo Fisher Scientific, Waltham, MA, USA).
Sonicator (Cole Parmer, Vernon Hills, IL, USA).
Rotator (Thermo Fisher Scientific, Waltham, MA, USA).
TC10 Cell Counter (Bio-Rad, Hercules, CA, USA).
Titer plate shaker (Thermo Fisher Scientific, Waltham, MA, USA).
ELx405 Microplate Washer (Bio-Tek, Winooski, VT, USA).
MAGPIX (Luminex, Austin, TX, USA).
2.6 NAPPA, RAPID ELISA, and Magnetic Bead Array ELISA Common Materials
Phosphate-buffered saline (PBS) 1× Powder Concentrate (Cole Parmer, Vernon Hills, IL, USA).
Powdered Milk (MP Biomedicals, Santa Ana, CA, USA).
Tween 20 (Sigma-Aldrich, St. Louis, MO, USA).
Copolymer microfuge tubes (USA Scientific, Ocala, FL, USA).
E. coli lysate.
Serum samples.
cDNA—pANT7cGST (DNASU, Tempe, AZ, USA).
5 % Milk PBS-Tween (PBST) 0.2 %.
3 Methods
3.1 In Vitro Transcription/Translation of Recombinant Proteins
High-purity DNA is required for the methods described in this chapter for optimum in vitro transcription/translation (IVTT). The plasmids used in this protocol are cDNA constructs with C-terminal-tagged GST fusion protein in pANT7_cGST. The GST tag allows the protein to be readily captured and displayed in situ after IVTT.
3.1.1 DNA Preparation
cDNA in pANT7_cGST is publicly available and can be ordered online through the plasmid repository of DNASU (https://dnasu.org/DNASU/Home.do).
To achieve high-purity DNA, column-based purification methods are used such as products available from Qiagen or Nucleobond.
3.1.2 In Vitro Transcription/Translation
Recombinant proteins should be expressed fresh on the day of the immunoassay.
Thaw the components of the 1-Step Human Coupled IVTT Kit on ice.
For RAPID ELISA and MagProBE, add RNASE Out to the reaction mix in addition to the IVTT components.
- Calculate the total volume needed for your assay (n + 1).
- NAPPA: Total volume of IVTT Master Mix based on the number of microarray slides being processed. Each slide requires the injection of 150 μL IVTT mixture.
- RAPID ELISA: For each antigen (Ag) per well 100 μL of diluted expressed antigen (Ag) is needed. The recombinant protein is first expressed and then after IVTT diluted 1:100 in 5 % Milk PBST-0.2 %.
- MagProBE: Per Ag and reaction (reaction includes technical replicate) a total of 24.5 μL of IVTT expression mixture is combined with 2 μL of DNA at 500 ng/μL.
-
1-Step Human Coupled IVTT components and DNA when performing RAPID ELISA and MagProBE are mixed as described below:
NAPPA
Component μL per slide
HeLa lysate 75
Accessory proteins 15
Reaction mix 30
Nuclease-free water 30
Total 150
RAPID ELISA
Component μL per well
HeLa lysate 0.57
Accessory proteins 0.11
Reaction mix 0.23
RNaseOUT 0.02
DNA (200 ng/μL) 0.24
Total 1.18
MagProBE
Component μL per two wells
HeLa lysate 5
Accessory proteins 2.5
Reaction mix 5
Nuclease-free water 3.5
PBS 1× 7.5
RNaseOUT 1
DNA (500 ng/μL) 2
Total 26.5
- Protein expression
- NAPPA: Slides are covered with HybriWell Gasket adhesive, DNA spots facing up, injected with 150 μL IVTT mixture from step 5 and placed into a chiller/heater incubator at 30 °C for 1.5 h followed by 15 °C for 0.5 h (see Note 1d).
- RAPID ELISA and MagProBE: Combine IVTT and DNA components in a polystyrene tube. Gently mix and place tubes into an incubator at 30 °C for 1.5 h.
- Assessment of protein expression: To ensure protein abundance, protein expression levels should be determined prior to use of the plasmids in the assay. Protein expression can vary from one plasmid to another and between different extraction batches. To assess protein expression, use an anti-GST Ab and its respective secondary detection Ab. If alternative plasmids with a different tag were used, use respective anti-tag Ab and appropriate secondary detection Ab (see Note 3c).
- NAPPA: Refer to the certificate of analysis provided with your order of NAPPA slides to review expression levels. All slide batches purchased will have undergone quality control to assess DNA and protein levels.
- RAPID ELISA: Protein expression should be above 5.0 × 108 RLUs, while the majority of the expression should be at 1.0 × 10 9 RLUs.
- MagProBE: Protein expression should be measured above 1000 median fluorescence intensity (MFI). The negative control wells without the expression plasmid should be well below 100 MFI.
3.2 Serum Sample Preparation
To prevent unspecific interactions from serum components to the detection Ab and maximize signal-to-noise ratios, serum samples are blocked prior to use in the assays.
3.2.1 E. coli Lysate Preparation and Utilization as a Blocking Agent
Blocking agents other than E. coli lysate can be used, such as 1–10 % BSA and 5 % milk in PBST-0.2 %. However, our laboratory uses E. coli lysate as a blocking agent which yields the lowest background and best signal-to-noise ratio.
Inoculate a preculture of E. coli DH5α into 2 mL LB liquid media and incubate at 37 °C for 7–8 h.
Transfer 500 μL of the preculture to 1 L of LB liquid media and grow overnight at 37 °C.
Pellet culture by centrifugation at 4 °C.
Add 20 mL of PBST to the first pellet for resuspension, then add the same 20 mL to the next pellet, and resuspend. Repeat until all pellets are resuspended.
Transfer suspension into a 50 mL tube, place on ice, and add three tablets of protease inhibitor. Vortex thoroughly until protease inhibitor tablets are completely dissolved.
Sonicate the bacteria at 500 W and 20 % amplitude for 10 min with a cycle of pulse on/off for 1 s using a microtip.
Aliquot lysate into tubes and boil at 99 °C for 10 min.
Spin down denatured precipitates at max speed for 5 min.
Collect supernatant and store at −20 °C for later use as a blocking agent.
3.2.2 Serum Sample Dilution
NAPPA
Optimal dilution in blocking reagent should be determined through optimization prior to use in the screening experiment.
For blocking serum, 50 % E. coli lysate in 5 % milk PBST-0.2 % is used.
Additionally, dilution ratios of the serum sample in blocking buffer can vary for optimal results. Typically, a sample-to-blocking buffer ratio of 1:50 is used in the assays.
The total volume needed to incubate the NAPPA slide with the diluted serum is 2 mL using the hybridization chambers.
RAPID ELISA
To make serum blocking buffer, add milk powder to 5 % into E. coli lysate and dilute lysate into 5 % milk PBST-0.2 % in a ratio of 1:10.
Serum is diluted with serum blocking buffer in a ratio of 1:100.
The total volume needed for serum incubation is 100 μL of diluted serum per well.
MagProBE
Serum blocking buffer is mixed together by adding E. coli lysate containing 5 % BSA to PBS 1 % BSA in a ratio of 1:10.
Serum is diluted 1:80 into serum blocking buffer.
Per reaction (including one technical replicate), 50 μL of diluted serum is needed.
3.3 Nucleic Acid Programmable Protein Array
3.3.1 Ordering NAPPA Slides
Go to the http://nappaproteinarray.org/webpage. Custom arrays built for a specific project or predetermined collections are available for purchase. Pricing can be found here: http://nappaproteinarray.org/price.html.
Contact the NAPPA core manager to purchase arrays. All arrays are shipped along with a certificate of analysis (CofA) asserting the quality of the array. QC certificate will include quality confirmation of the DNA print by picogreen staining of the DNA spots, expression level analysis of all spots including positive and negative controls, and slide correlations within and, if applicable, between print batches (see Note 1b).
3.3.2 Assay Protocol
Block slides with SuperBlock for 1 h at room temperature (see Note 1a, c).
Rinse slides with ultrapure water and dry with filtered compressed air (see Note 1g).
With the DNA slide side facing up, apply HybriWell gasket to each slide. Seal by applying pressure with the supplied sticks on the sides of the gasket containing adhesive coating.
Prepare IVTT mixture as described in Subheading 3.1.2 and inject 150 μL into HybriWell Gasket injection port. To distribute the liquid throughout the entire gasket, gently massage/tap slides to push the liquid through while injecting. Tap bubbles out through one of the injection holes and then seal with supplied seal adhesives.
To express the proteins, incubate the slides in heater/chiller incubator for 1.5 h at 30 °C, followed by 0.5 h at 15 °C to facilitate binding of the recombinant protein with the conjugated anti-GST (anti-tag) Ab on the slide.
Prepare serum samples as described in Subheading 3.2.2 and incubate on a rotator for 2 h at RT.
After expression, remove HybriWells from slides and rinse/wash slides with PBST in modified pipette boxes with dividers.
Wash slides with 5 % milk in PBST-0.2 % three times for 5 min on a rocker.
Block with 5 % milk in PBST-0.2 % for 30 min.
Take slides out of the 5 % milk in PBST-0.2 %. Gently blot off some residual milk by tapping the edge of the slide on an absorbent paper and place in the hybridization chamber with the captured protein side up, without allowing the slide to dry completely.
Apply 2 mL of the diluted serum preparation on top of the slide. Close the hybridization chamber gently. Secure with the supplied metal clips and place in a rotator overnight at 4 °C.
Disassemble hybridization chambers carefully and place slides array side up into washing containers filled with 5 % milk in PBST-0.2 %. Wash three times for 5 min on a rocking shaker.
Prepare secondary fluorophore-tagged detection Ab in 5 % milk PBST-0.2 % (see Note 1e, f).
Place slides array side up into new hybridization chambers and apply 2 mL directly labeled secondary detection Ab (anti-human-fluorophore tagged). Close chambers gently and secure with metal frame clips. Incubate for 1 h on rotator at RT in the dark.
Wash slides three times for 5 min in PBST. After last wash rinse with ultrapure water and dry with compressed air. Store in dark slide box until scanning.
- Scan slides with previously determined scanner settings providing best signal-to-noise ratios in Tecan’s PowerScanner. If best scanner settings are not determined yet, scan slides with various laser intensity and gain settings. Following are typical laser intensity and gain settings normally used and depend on variations in serum dilution and secondary detection Abs of the experiment.
Cycle number Laser intensity Laser gain
1 10 10
2 25 25
3 50 50
4 75 75
Extract data using Array Pro Analyzer program.
3.4 RAPID ELISA
3.4.1 Protocol
Coat 96-well plates with 100 μL/well of 10 μg/mL anti-GST Ab (polyclonal) in 0.2 M sodium bicarbonate buffer overnight (pH 9.4) at 4 °C (see Note 3a).
Wash 96-well plate with PBST-0.2 % three times and blot after the last wash.
Block GST-coated 96-well plates with 5 % milk in PBST-0.2 %, 200 μL per well, for 1.5 h at RT.
Block diluted serum samples from Subheading 3.2.2 for at least 2 h at room temperature on a plate shaker.
Prepare recombinant proteins from IVTT preparation (see Subheading 3.1) and incubate for 1.5 h at 30 °C. After protein expression, dilute the proteins 1:100 by adding 5 % milk PBST-0.2 %.
Remove blocking buffer from the anti-GST-coated plate by dumping and blotting.
Add 100 μL/well of diluted expressed protein to the anti-GST-coated plate and incubate for 1 h at RT on a shaker to capture the GST-tagged recombinant protein.
Wash assay plate with PBST-0.2 %, 200 μL/well, using the NUNC immunowash 12. Let the wash buffer sit for 1 min before repeating the wash four more times. After the last wash, blot plates to completely remove wash buffer.
Add 100 μL diluted sera (see sample preparation step in Subheading 3.2.2) and primary control antibodies to the appropriate wells (see Note 2b). Shake plate at 500 rpm for 1 h at RT.
Wash plates. Dump sera out and wash plates with 200 μL/well of PBST-0.2 % using the NUNC immunowash 12 as described in step 8.
Add 100 μL/well of diluted secondary Ab (HRP goat-anti-human 1:10,000 in 5 % milk PBST-0.2 %, HRP-sheep-anti-human 1:6250 in 5 % milk PBST-0.2 %) to the appropriate wells. Use anti-human Ab for the wells which contain human sera and anti-mouse Ab for the control wells. Shake plate at 600 rpm for 1 h at RT.
Wash plate five times as described in step 8.
Mix equal volumes of each of solutions 1 and 2 from the SuperSignal ELISA Femto kit and add 100 μL/well.
Immediately read plate and detect chemiluminescence in the GloMax ®-96 Microplate reader per the manufacturer’s instruction (see Note 2c–g).
3.5 MagProBE
To screen for antibodies using a bead array, anti-GST capture Ab is first coupled to the beads and stored. Ag proteins are produced with IVTT similar to RAPID ELISA and then captured onto the beads with the capture Ab prior to use in the MagProBE assay (see Note 4.3a, d).
3.5.1 Antibody Preparation for Bead Conjugation
The commercial anti-GST antibodies contain sodium azide, which needs to be removed first with dialysis.
Pre-wet dialysis cassette membrane with cold PBS.
Dilute Ab in 1 mL PBS.
Inject diluted Ab into cassette. Remove air.
Dialyze 3 times against 1 L cold PBS over a total of 12–24 h.
Remove Ab from dialysis cassette.
Determine concentration by absorbance (mg/mL = UV280/1.6).
Aliquot and store at 20 °C.
3.5.2 Antibody Coupling to Magnetic Microspheres
The assay is optimized for the use of anti-GST Ab. For other anti-tag Abs, the Ab/bead ratios should be titered. Volumes listed are for small scale (2.5 × 10 6 beads) and large scale [in brackets, 12.5 × 10 6 beads (1 ml)].
Equilibrate microspheres to RT and resuspend thoroughly (see Note 3g, h).
Transfer 2.5 × 10 6 microspheres to a microcentrifuge tube.
Wash microspheres by placing tube on a magnet. Wait for 1 min to allow capture of the beads by the magnet. When liquid is clear, remove supernatant and add 100 μL [200 μL] sterile water. Resuspend microspheres thoroughly. Place tube back onto magnet and remove supernatant after beads are captured (see Note 3f).
To activate microspheres, add 80 μL 100 mM monobasic sodium phosphate (pH 6.2) and resuspend thoroughly. Then add 10 μL [50 μL] 50 mg/mL sulfo-NHS to microspheres. Mix well and add 10 μL [50 μL] of 50 mg/mL EDC. Mix well. Incubate for 20 min at RT covered in foil. Vortex for 10 s every 10 min.
Wash microspheres twice with 250 μL [500 μL] of 50 mM MES (pH 5.0) by placing tube in the magnetic rack, allowing spheres to be captured before removing supernatant. Resuspend thoroughly in between washes in wash buffer MES.
For coupling the spheres with anti-GST, resuspend microspheres in 100 μL [200 μL] 50 mM MES (pH 5.0). Add 12.5 μg [62.5 μg] anti-GST capture Ab and add 50 mM MES to a volume of 500 μL [1 mL]. Vortex for 10 s and place on a rotator for 2 h at RT. Protect reagents from light by wrapping tubes in foil or rotating in the dark.
Remove supernatant on the magnetic rack and wash microspheres with 500 μL [1 mL] PBS-BN (PBS, 1 % BSA, 0.05 % azide, pH 7.4). Resuspend thoroughly and incubate for 30 min on a rotator. Exchange the PBS-BN with 1 mL PBS-0.05 % Tween (pH 7.4) and resuspend spheres well. Repeat these steps one more time.
Resuspend in 250 μL [1 mL] PBS-BN and count cells in hemocytometer.
Store anti-GST-coupled microspheres at 4 °C.
3.5.3 Confirmation of Antibody Coupling to Microspheres
Prior to use in ELISA assays, the amount of anti-GST Ab that is covalently attached to the microspheres is confirmed using an independent anti-GST mAb. Beads of any fluorescent color can be used; colors in adjacent fluorescent spectra can be readily distinguished, and cross-reactivity was not observed.
Resuspend coupled microspheres and aliquot enough of the microspheres into a copolymer tube. Per reaction, 5000 microspheres in PBS 1 % BSA in a total volume of 50 μL/well are required.
Mix coupled microspheres thoroughly and add 50 μL to appropriate wells into the microassay 96-well plate.
Dilute donkey anti-goat IgG-PE to 4 μg/mL in enough volume for 50 μL/well.
Add 50 μL detection Ab per well and incubate for 30 min at RT shaking at 750 rpm.
Wash wells twice with 100 μL PBS-1 % BSA-0.2 % Tween using magnetic handheld washer, dump, and blot.
Resuspend microspheres in 100 μL/well PBS-1 % BSA. Shake for a minimum of 5 min at 750 rpm.
Analyze with MAGPIX per the manufacturer’s instructions.
3.5.4 Capturing In Vivo-Generated Protein to Anti-GST-Coupled Microspheres: Day 1
The multiplexed bead array ELISA is a 2-day assay. Antigens are expressed with IVTT on the day of the reaction. For assay planning, sera are tested in duplicate, and protein capture onto the beads is tested in duplicate at the same time using separate wells, using anti-GST mAbs. Controls include the vector control or any control GST fusion protein, including GST alone.
Express protein antigens as described in Subheading 3.1.2.
Prepare working stock of anti-GST-coupled microspheres. Plan for enough microspheres for 2000 magnetic beads/reaction/antigen.
Add expressed protein directly to diluted beads and mix protein and beads for 2 h on a rotator.
3.5.5 Detection of Autoantibodies in Human Serum: Day 1
While beads and antigens are mixing, block serum as described in Subheading 3.2.2 for 2 h at RT on rotator or rocker.
After coupling of the Ag protein to the microspheres, aliquot microspheres into a tube for 2000 magnetic beads/reaction/antigen and resuspend thoroughly. To multiplex, pipette 10 μL of each 200 beads/μL normalized Ag-bound beads together into one tube (see Note 3e).
Wash beads with PBS-1 % BSA-0.2 % Tween.
Block beads by resuspending them in 50 μL/well of 10 % E. coli lysate with 5 % BSA, in PBS-1 % BSA. Seal with aluminum foil and shake at 750 rpm at RT for 1 h.
Add 50 μL of diluted and blocked serum to the bead resuspension from the previous step. Mix well and split serum-bead mixture to two wells, each containing 50 μL. Seal plate with tape, cover with lid, and wrap in foil. Incubate overnight at 4 °C while shaking at 750 rpm on an orbital shaker.
3.5.6 Detection of Autoantibodies in Human Serum: Day 2
Bring plate to RT by shaking it at 750 rpm for 30 min.
Wash wells twice with 100 μL PBS-1 % BSA.
Prepare detection Ab by diluting biotin-labeled goat anti-human Ab 1:6000 in PBS-1 % BSA. Add 50 μL to each well and incubate for 30 min at RT on a shaker at 750 rpm.
Wash beads twice with PBS-1 % BSA-0.2 % Tween.
Dilute streptavidin-R-PE 1:1500 in PBS-1 % BSA. Add 50 μL to each well, seal, and incubate for 30 min at RT on a shaker at 750 rpm.
Wash beads twice with PBS-1 % BSA-0.2 % Tween.
Resuspend microspheres in 100 μL PBS-1 % BSA per well. Shake plate until ready to analyze with MAGPIX. Shake for a minimum of 5 min.
Analyze plate with MAGPIX per the manufacturer’s instructions.
Acknowledgments
This study was supported by NCI UOI CA11734 (K.S.A.). We thank Marika Hopper for assistance in the preparation of this manuscript and Julia Cheng for techinical assistance. Dr. Anderson is a consultant and has stock options with Provista Diagnostics.
Footnotes
- NAPPA assay protocol can be fully automated using the HS 4800 Pro or 400 Pro Hybridization Station (Tecan, Maennedorf, Switzerland) for high throughput. Per our assay optimization studies, we recommend automating the steps after protein expression, beginning with the PBST wash, for best protein expression and detection results.
- Always handle slides carefully without touching the array surface.
- Slide blocking step with SuperBlock can be alternatively incubated overnight at 4 °C.
- Avoid any air bubbles/pockets on the array for even distribution of the reagent solutions.
- When handling fluorophores, keep them in the dark, wrapped in aluminum foil.
- For best results in your experiment, optimize the dilution level of the secondary detection Ab to maximize signal-to-noise ratio and minimize background.
- Slide drying steps can be done in a centrifuge instead by placing slides in a slide rack into a container with absorbent paper. Slides are spun at 230 × g for 1 min.
- All steps can be automated for high-throughput processing on liquid handlers such as the Biomek FX/NX (Beckman Coulter, Brea, CA, USA).
- An expression control and a secondary Ab control should be included for each antigen.
- Background ranges vary greatly between different serum samples used. The majority of the measurements between the 25–75 percentiles should be 5.0 × 10 7 to 8.9 × 10 7 RLUs. In average, the GST background is between 1.0 × 10 8 and 8.0 × 10 8 RLUs.
- Secondary control measurements should be below 10 6 RLUs for anti-mouse and below 10 7 for the anti-human HRP-labeled secondary detection Ab.
- Criteria for repeating samples: Samples are run in duplicates. If the duplicate samples give a signal difference greater than 50 %, this sample should be repeated.
- Negative response criteria: A serum sample is considered negative in its immune response to an antigen when the ratio of the measured RLU of the sample for that antigen to that of the same sample in the GST background well is below 2.
- Positive response criteria: A serum sample is positive in its immune response to an antigen if the ratio from the respective antigen to GST is above 2.
- MagProBE ELISA has comparable sensitivities, specificities, and limits of detection as standard protein ELISA for the detection of antibodies in sera to the viral antigen EBNA-1 and the tumor antigen p53 [39].
- There are different chemical compositions of Luminex microspheres. The assay described here uses the magnetic microspheres, but also works with SeroMap microspheres, with comparable sensitivities and specificities as the magnetic microspheres.
- Protein expression with IVTT can vary from batch to batch. These studies have been optimized for the pANT7-c-terminal GST vector. Over 100 tumor and viral antigens have been expressed and captured using this assay on Luminex beads, with over 90 % of the antigens expressed as determined by GST detection. Both high-quality DNA and careful use of RNase-free pipettes and filter tips are needed for efficient protein expression.
- The cost for MagProBE is $5.00/Ag/sample.
- Up to ten Ags have been multiplexed at one time using MagProBE. Theoretically, this assay could be used to multiplex up to 100 Ags but is limited only by the number of spectrally distinguishable microspheres.
- For manual wash steps of the beads, use handheld magnetic stands. After allowing the beads to collect on the side of the magnet, remove the liquid by discarding and gentle blotting of the plate with the magnet attached. Add 100 μL/well of wash buffer PBS-1 % BSA-0.2 % Tween. Allow beads to be captured and remove the buffer by discarding and gentle blotting. Repeat wash step one more time. All washing steps can be alternatively automated using a liquid handler such as the Bio-Tek ELx 405.
- All bead resuspension steps should be carried out in the following manner for thorough dispersion of the beads: vortexing for 10 s, sonicating for 20 s, and vortexing again for 10 s.
- Luminex microspheres are light sensitive. To maintain their integrity, protect them from light exposure.
References
- 1.Behring K. Ueber das zustandekommen der diphtherie-immunität und der tetanus-immunität bei thieren. Dtsch Med Wochenschr. 1890;16:1113–1114. [PubMed] [Google Scholar]
- 2.De Veer M, Meeusen E. New developments in vaccine research–unveiling the secret of vaccine adjuvants. Discov Med. 2011;12:195–204. [PubMed] [Google Scholar]
- 3.Meinke A, Henics T, Nagy E. Bacterial genomes pave the way to novel vaccines. Curr Opin Microbiol. 2004;7:314–320. doi: 10.1016/j.mib.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Berzofsky JA, Ahlers JD, Belyakov IM. Strategies for designing and optimizing new generation vaccines. Nat Rev Immunol. 2001;1:209–219. doi: 10.1038/35105075. [DOI] [PubMed] [Google Scholar]
- 5.FDA. Approval letter – Provenge 2010 Apr 29; [Google Scholar]
- 6.Le DT, Pardoll DM, Jaffee EM. Cellular vaccine approaches. Cancer J. 2010;16:304. doi: 10.1097/PPO.0b013e3181eb33d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qiu J, Anderson KS. Autoantibodies and biomarker discovery. In: Issaq HJ, Veenstra TD, editors. Proteomic and metabolomic approaches to biomarker discovery. Elsevier; Massachusetts, USA: 2013. [Google Scholar]
- 9.Silverstein AM. A history of immunology. Academic; New York: 2009. [Google Scholar]
- 10.Zinkernagel RM, Hengartner H. Protective ‘immunity’ by pre-existent neutralizing antibody titers and preactivated T cells but not by so‐called ‘immunological memory’. Immunol Rev. 2006;211:310–319. doi: 10.1111/j.0105-2896.2006.00402.x. [DOI] [PubMed] [Google Scholar]
- 11.Anderson KS, LaBaer J. The sentinel within: exploiting the immune system for cancer biomarkers. J Proteome Res. 2005;4:1123–1133. doi: 10.1021/pr0500814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ouchterlony O. Antigen-antibody reactions in gels. Acta Pathol Microbiol Scand A. 1949;26:507–515. doi: 10.1111/j.1699-0463.1949.tb00751.x. [DOI] [PubMed] [Google Scholar]
- 13.Berson SA, Yalow RS. Quantitative aspects of the reaction between insulin and insulin-binding antibody. J Clin Invest. 1959;38:1996–2016. doi: 10.1172/JCI103979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kricka LJ, Savory J. International year of Chemistry 2011. A guide to the history of clinical chemistry. Clin Chem. 2011;57:1118–1126. doi: 10.1373/clinchem.2011.165233. [DOI] [PubMed] [Google Scholar]
- 15.Wu AH. A selected history and future of immunoassay development and applications in clinical chemistry. Clin Chim Acta. 2006;369:119–124. doi: 10.1016/j.cca.2006.02.045. [DOI] [PubMed] [Google Scholar]
- 16.Lequin RM. Enzyme immunoassay (EIA)/enzyme-linked immunosorbent assay (ELISA) Clin Chem. 2005;51:2415–2418. doi: 10.1373/clinchem.2005.051532. [DOI] [PubMed] [Google Scholar]
- 17.Engvall E, Perlmann P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry. 1971;8:871–874. doi: 10.1016/0019-2791(71)90454-x. [DOI] [PubMed] [Google Scholar]
- 18.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 19.Santini C, Brennan D, Mennuni C, et al. Eficient display of an HCV cDNA expression library as C-terminal fusion to the capsid protein D of bacteriophage lambda. J Mol Biol. 1998;282:125–135. doi: 10.1006/jmbi.1998.1986. [DOI] [PubMed] [Google Scholar]
- 20.Beghetto E, De Paolis F, Spadoni A, Del Porto P, Buffolano W, Gargano N. Molecular dissection of the human B cell response against cytomegalovirus infection by lambda display. J Virol Methods. 2008;151:7–14. doi: 10.1016/j.jviromet.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Beghetto E, De Paolis F, Montagnani F, Cellesi C, Gargano N. Discovery of new Mycoplasma pneumoniae antigens by use of a whole-genome lambda display library. Microbes Infect. 2009;11:66–73. doi: 10.1016/j.micinf.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Beghetto E, Gargano N, Ricci S, et al. Discovery of novel Streptococcus pneumoniae antigens by screening a whole-genome λ-display library. FEMS Microbiol Lett. 2006;262:14–21. doi: 10.1111/j.1574-6968.2006.00360.x. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Yu J, Sreekumar A, et al. Autoantibody signatures in prostate cancer. N Engl J Med. 2005;353:1224–1235. doi: 10.1056/NEJMoa051931. [DOI] [PubMed] [Google Scholar]
- 24.Chatterjee M, Mohapatra S, Ionan A, et al. Diagnostic markers of ovarian cancer by high-throughput antigen cloning and detection on arrays. Cancer Res. 2006;66:1181–1190. doi: 10.1158/0008-5472.CAN-04-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen G, Wang X, Yu J, et al. Autoantibody profiles reveal ubiquilin 1 as a humoral immune response target in lung adenocarcinoma. Cancer Res. 2007;67:3461–3467. doi: 10.1158/0008-5472.CAN-06-4475. [DOI] [PubMed] [Google Scholar]
- 26.Legutki JB, Johnston SA. Immunosignatures can predict vaccine efficacy. Proc Natl Acad Sci U S A. 2013;110:18614–18619. doi: 10.1073/pnas.1309390110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stevens RC. Design of high-throughput methods of protein production for structural biology. Structure. 2000;8:R177–R185. doi: 10.1016/s0969-2126(00)00193-3. [DOI] [PubMed] [Google Scholar]
- 28.Jackson AM, Boutell J, Cooley N, He M. Cell-free protein synthesis for proteomics. Brief Funct Genomic Proteomic. 2004;2:308–319. doi: 10.1093/bfgp/2.4.308. [DOI] [PubMed] [Google Scholar]
- 29.Ramachandran N, Hainsworth E, Bhullar B, et al. Self-assembling protein microarrays. Science. 2004;305:86–90. doi: 10.1126/science.1097639. [DOI] [PubMed] [Google Scholar]
- 30.Ramachandran N, Raphael JV, Hainsworth E, et al. Next-generation high-density self-assembling functional protein arrays. Nat Methods. 2008;5:535–538. doi: 10.1038/nmeth.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramachandran N, Hainsworth E, Demirkan G, LaBaer J. On-chip protein synthesis for making microarrays New and emerging proteomic techniques. Springer; New York, NY: 2006. pp. 1–14. [DOI] [PubMed] [Google Scholar]
- 32.Festa F, Rollins SM, Vattem K, et al. Robust microarray production of freshly expressed proteins in a human milieu. Proteomics-Clin Appl. 2013;7:372–377. doi: 10.1002/prca.201200063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JR, Magee DM, Gaster RS, LaBaer J, Wang SX. Emerging protein array technologies for proteomics. Expert Rev Proteomics. 2013;10(1):65–75. doi: 10.1586/epr.12.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seiler CY, Park JG, Sharma A, et al. DNASU plasmid and PSI: biology-materials repositories: resources to accelerate biological research. Nucleic Acids Res. 2013;42(Database issue):D1253–D260. doi: 10.1093/nar/gkt1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson KS, Ramachandran N, Wong J, et al. Application of protein microarrays for multiplexed detection of antibodies to tumor antigens in breast cancer. J Proteome Res. 2008;7:1490–1499. doi: 10.1021/pr700804c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramachandran N, Anderson KS, Jv R, et al. Tracking humoral responses using self assembling protein microarrays. Proteomics Clin Appl. 2008;2:1518–1527. doi: 10.1002/prca.200800034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson KS, Cramer DW, Sibani S, et al. Autoantibody signature for the serologic detection of ovarian cancer. J Proteome Res. 2015;14:578–586. doi: 10.1021/pr500908n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montor WR, Huang J, Hu Y, et al. Genome-wide study of Pseudomonas aeruginosa outer membrane protein immunogenicity using self-assembling protein microarrays. Infect Immun. 2009;77:4877–4886. doi: 10.1128/IAI.00698-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson KS. Multiplexed Detection of Antibodies Using Programmable Bead Arrays. In: Wu Catherine J., editor. Protein microarray for disease analysis. Humana Press; 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson KS, Wong J, Vitonis A, et al. p53 autoantibodies as potential detection and prognostic biomarkers in serous ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2010;19:859–868. doi: 10.1158/1055-9965.EPI-09-0880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Barker K, Steel J, et al. A versatile protein microarray platform enabling antibody profiling against denatured proteins. Proteomics Clin Appl. 2013;7:378–383. doi: 10.1002/prca.201200062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong J, Sibani S, Lokko NN, LaBaer J, Anderson KS. Rapid detection of antibodies in sera using multiplexed self-assembling bead arrays. J Immunol Methods. 2009;350:171–182. doi: 10.1016/j.jim.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson KS, Wong J, D’Souza G, et al. Serum antibodies to the HPV16 proteome as biomarkers for head and neck cancer. Br J Cancer. 2011;104:1896–1905. doi: 10.1038/bjc.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de la Rica R, Stevens MM. Plasmonic ELISA for the ultrasensitive detection of disease biomarkers with the naked eye. Nat Nanotechnol. 2012;7:821–824. doi: 10.1038/nnano.2012.186. [DOI] [PubMed] [Google Scholar]
- 45.A study of a new candidate vaccine against Hepatitis C Virus (HCV) http://clinicaltrials.gov/ct2/show/NCT01070407.