

Abstract
Understanding how membrane proteins interact with detergents is of fundamental and practical significance in structural and chemical biology as well as in nanobiotechnology. Current methods for inspecting protein-detergent complex (PDC) interfaces require high concentrations of protein and are of low throughput. Here, we describe a scalable, spectroscopic approach that uses nanomolar protein concentrations in native solutions. This approach, which is based on steady-state fluorescence polarization (FP) spectroscopy, kinetically resolves the dissociation of detergents from membrane proteins and protein unfolding. For satisfactorily solubilizing detergents, at concentrations much greater than the critical micelle concentration (CMC), the fluorescence anisotropy was independent of detergent concentration. In contrast, at detergent concentrations comparable with or below the CMC, the anisotropy readout underwent a time-dependent decrease, showing a specific and sensitive protein unfolding signature. Functionally reconstituted membrane proteins into a bilayer membrane confirmed predictions made by these FP-based determinations with respect to varying refolding conditions. From a practical point of view, this 96-well analytical approach will facilitate a massively parallel assessment of the PDC interfacial interactions under a fairly broad range of micellar and environmental conditions. We expect that these studies will potentially accelerate research in membrane proteins pertaining to their extraction, solubilization, stabilization, and crystallization, as well as reconstitution into bilayer membranes.
Keywords: FhuA, Single-molecule electrophysiology, Nanopore, Protein engineering, Protein-detergent complex interactions
TOC Graphic
Introduction
Understanding the protein-detergent complex (PDC)1 interface remains a ubiquitous problem in the extraction, solubilization, stabilization, and crystallization of membrane proteins. The PDCs were traditionally examined by numerous techniques, including small-angle X-ray scattering (SAXS),2 circular dichroism (CD) spectroscopy,3 size-exclusion chromatography,4 and tryptophan fluorescence quenching.5 These approaches were used to determine a variety of physical properties of the proteomicelles, such as their size, shape, and distributions. In the past few years, there has been an increased interest in examining the details of the interfacial forces that govern the PDC formation as well as their effects on the protein structure, stability, and dynamics. CD spectroscopy was frequently used to determine the secondary structure and thermostability of membrane proteins under a variety of detergent solubilization conditions.5,6 Recently, the Brouillette group has developed differential scanning calorimetry (DSC) approaches for the inspection of the interactions between detergents and extramembranous water-soluble domains of the human cystic fibrosis transmembrane conductance regulator (hCFTR).7,8 Isothermal titration calorimetry (ITC) was also employed for the determination of thermodynamic phase diagrams of ternary lipid-detergent-protein systems and their deviation from data obtained in protein-free lipid-detergent mixtures.9 Moreover, nuclear magnetic resonance (NMR) spectroscopy was extensively used for the examination of the interactions of membrane proteins with detergent micelles.10,11 In particular, solid-state NMR is a versatile approach for the investigation of membrane proteins in various detergent and lipid environments, including cell membranes.12 For example, Frey and colleagues (2017) were able to characterize the internal backbone and side chain flexibility of the outer membrane protein X (OmpX) in micelles, bicelles, and nanodiscs using NMR relaxation in a broad range of timescales, from picoseconds to milliseconds.13 Such a versatility and time resolution of the NMR spectroscopy often facilitates direct correlations of the collected data with membrane protein folding14 and function.12
However, current approaches require high concentrations of proteins. Such a problem is especially critical to membrane proteins, because the yield is unpredictable due to limited expression levels and unproductive aggregation. In addition, the signal-to-noise ratio of the most spectroscopic and calorimetric techniques is severely deteriorated by protein aggregation. As a result, these approaches cannot be readily expanded to a high-throughput format for the concurrent inspection of a broad array of environmental conditions and interacting partners. These problems restrain opportunities for acquiring a better quantitative and mechanistic information on the PDC interactions.
Here, we developed a single-fluorophore, 96-well plate-reader approach for obtaining a fast and scalable readout of the PDC interactions at low nanomolar concentrations of the protein. This assay relied on the use of fluorescence polarization (FP) spectroscopy.15–18 In the past, FP spectroscopy was employed for inspecting the PDC interactions with either mild19 or harsh detergents20–22 and water-soluble proteins. In contrast to this work, prior sodium dodecyl sulfate (SDS)-protein interaction studies were focused on the mechanistic understanding of harsh detergent-induced protein unfolding22 and resistance of proteins to denaturation21 under diverse environmental conditions.
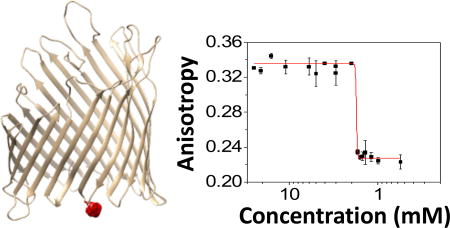
The steady-state fluorescence polarization (FP) spectroscopy facilitates the examination of the rotational mobility of a labeled protein. This analysis can be conducted by exciting a chemically attached fluorophore with plane-polarized light. If the labeled protein binds to a detergent micelle, then a slowed rotational diffusion of the PDC will be accompanied by an increased emission in the plane parallel to the polarized light and a decreased emission in the plane perpendicular to the polarized light. This emission change is measured and analyzed by a ratio between the numbers of free and bound proteins.16 We postulated that the dynamics of the dissociation of the PDC at detergent concentrations below the critical micelle concentration (CMC) can be examined by the steady-state fluorescence anisotropy and kinetic readouts. As a test case, we explored the detergent desolvation-induced unfolding of protein nanopores using ferric hydroxamate uptake component A (FhuA),23 a monomeric outer membrane β-barrel protein of Escherichia coli. We employed FhuA ΔC/Δ5L, an extensive truncation mutant (Fig. 1),24 so that FhuA was converted from a non-ion conducting transmembrane protein25 to a large-conductance nanopore. Therefore, this redesigned protein nanopore enabled parallel inspections of the anisotropy readout of detergent-refolded FhuA in solution and its ion transport activity, as judged by single-molecule electrophysiology in planar lipid membranes.
Figure 1. Side view of the FhuA ΔC/Δ5L protein,24 illustrating five truncated extracellular loops, L3, L4, L5, L10, and L11 of FhuA by top arrows.23.

The bottom arrow indicates the T7 β turn and site for protein labeling with Texas Red, which is marked by a red sphere.
We examined the interactions of this redesigned β-barrel protein nanopore with twelve detergents of diverse hydrophilic head groups and hydrophobic tails. This FP-based analytical approach enabled the determination of the apparent dissociation constants of the PDC, Kd, over a four-order of magnitude range. It should be noted that the detergent desolvation of a protein nanopore is intimately related to its unfolding owing to detergent depletion in its proximity. From a practical point of view, this approach facilitates the detection of low-affinity PDC interfacial interactions, whereas the signal-to-noise ratio is not significantly impaired by the extent of protein aggregation. Moreover, the ability to obtain quantitative information about specific detergent-membrane protein interactions in a high-throughput format will be valuable by identifying satisfactory solvation and crystallization conditions for structural studies of membrane proteins.26
Experimental Section
Refolding of FhuA ΔC/Δ5L
We employed a rapid-dilution refolding protocol27 to obtain FhuA ΔC/Δ5L. 40 µl of 6×His+-tag purified denatured protein was 50-fold diluted into 200 mM NaCl, 50 mM HEPES, pH 7.4 solutions at 4°C, which contained various detergents at concentrations above the CMC (Table S1). Different starting detergent concentrations were as follows (when multiple concentrations are given, the lower concentrations were needed to get dilutions with a low enough detergent concentration to cover the required range): (i) for n-decyl-β-d-maltopyranoside (DM), n-undecyl-β-d-maltopyranoside (UM), n-dodecyl-β-d-maltopyranoside (DDM), we used 5 and 20 mM starting detergent concentration; (ii) 50 mM 4-cyclohexyl-1-butyl-β-d-maltoside (CYMAL-4); (iii) 5 and 20 mM n-dodecyl-N,N-dimethylglycine (LD); (iv) 20 mM 1-lauroyl-2-hydroxy-sn-glycero-3-phosphocholine (LysoFos); (v) 0.2, 0.5, 1, and 25 mM 1-palmitoyl-2-hydroxy-sn-glycero-3-[phospho-rac-(1-glycerol)] (LPPG); (vi) 50 mM 3-[(3-cholamidopropyl)-dimethylammonio]-1-propane sulfonate] (CHAPS); (vii) 100 mM N,N'-bis-(3-d-gluconamidopropyl)cholamide (Big CHAP); (viii) 50 mM n-octyl-β-d-glucoside (OG); (ix) 50, 100, and 250 mM n-octyl-β-d-thioglucoside (OTG). All detergents were obtained from Anatrace (Maumee, OH), except LPPG, which was purchased from Avanti Polar Lipids (Alabaster, AL). All detergent solutions were freshly prepared to avoid hydrolysis and oxidation.28
Anisotropy measurements
We employed a SpectraMax® I3 plate reader (Molecular Devices, Sunnyvale, CA) equipped with the Paradigm® detection cartridge for Rhodamine FP spectroscopy, which features the excitation and emission wavelengths of 535 and 595 nm, respectively. We covalently attached a hydrophilic Texas Red fluorophore29 to a reactive cysteine sulfhydryl using an engineered flexible Gly/Ser-rich peptide loop within the T7 β turn (Fig. 1; Supplementary Information, Fig. S1). Similar fluorophore labeling approach was conducted using an engineered cysteine sulfhydryl on loop L6 of outer membrane protein G (OmpG D224C) (Supplementary Information, Fig. S2).30 We chose the labeling site on the aqueous phase-exposed regions of the proteins, because Texas Red is a hydrophilic compound. The primary advantage of Texas Red is its optical stability over a broad range of conditions.31 Measurements were taken on black flat bottom 96-well Costar assay plates (Corning Incorporated Kennebunk, ME). The fluorescence anisotropy was calculated using the parallel, Ip(t), and orthogonal, Io(t), time-dependent components of the emission intensity:31,32
| (1) |
where G is a sensitivity correction factor for the detection modes when emission polarizers are oriented vertically and horizontally. Thus,
| (2) |
where IHV is the intensity with the excitation and emission polarizers in a horizontal and vertical orientation, respectively, whereas IHH is the intensity with both the excitation and emission polarizers in a horizontal orientation. Because this study was conducted on a single instrument, G was assumed to be 1, and the numbers reported are uncalibrated anisotropy. The anisotropy measurements were conducted by taking the refolded protein sample and diluting it within individual wells with buffer containing various detergent concentrations. In this way, we gradually decreased the detergent concentration in individual wells, while keeping the final protein concentration constant at 28 nM. The anisotropy was determined for time periods in the range 30–60 minutes. Then samples were covered, placed at 4°C, and endpoints of the PDC reaction were collected 24 hours later for equilibrium determinations (Supporting Information, Fig. S3). Anisotropy traces presented throughout this article represent averages ±SD over a number of at least three independent data acquisitions.
Results
Experimental rationale
We found that bringing the protein from a Gdm-HCl-denaturing condition to a detergent concentration above the CMC, via a fast-dilution refolding protocol,27 was accompanied by a maximum fluorescence anisotropy, rmax (Table S2). When the detergent concentration was brought to a value below the CMC, a reproducible and gradual time-dependent decrease in the fluorescence anisotropy was detected (Fig. 2A, Fig. 2B, and Fig. 2C). We interpret this decrease in the anisotropy as the dissociation of the detergent from the nanopore. This detergent desolvation-induced unfolding resulted in a decrease in the hydrodynamic radius of the protein nanopore, thereby leading to an increase in its local mobility. Such an interpretation was also supported by a decrease in the anisotropy endpoint (rmin) due to a gradually reduced final detergent concentration (c) within the well (Fig. 2D). Moreover, the equilibrium anisotropy endpoints showed a clear two-state transition between rmin and rmax. In conclusion, detergent-solubilized proteins were characterized by a maximum anisotropy, rmax, whereas detergent-desolvated proteins were featured by an absolute minimum anisotropy, rmin (Table S2).
Figure 2. Time-dependent anisotropy showing the protein-detergent complex (PDC) interfacial dynamics of FhuA ΔC/Δ5L.

The anisotropy data were acquired by adding overnight refolded protein to a bath of varying detergent concentration. All anisotropy measurements were carried out at room temperature in 200 mM NaCl, 50 mM HEPES, pH 7.4. The starting detergent concentrations were as follows: (A) 20 mM LysoFos; (B) 5 and 25 mM LD; and (C) 0.2, 0.5, and 1 mM LPPG. In (D), it is shown concentration-response anisotropy changes as a result of the PDC dissociation. The horizontal axis indicates individual dilutions of detergent concentrations, while keeping the final protein concentration constant at 28 nM (see Experimental Section). The anisotropy values on the vertical axis were collected 24 hours later for equilibrium determinations. The LPPG data points belonging to the maximum state (rmax = ~0.342) were obtained when the protein was refolded in either 0.5 or 1 mM LPPG. The orange horizontal line on the LPPG data points corresponds to a secondary maximum anisotropy value, rmax = 0.31, when the protein was refolded in 200 µM LPPG; (E) This panel shows a low anisotropy value, r1, which was recorded either in the presence of 40 mM SDS or 6 M Gdm-HCl. The top of each panel or vertical bars indicate the CMC (Table S1).
Specificity and sensitivity of the time-dependent anisotropy on detergent properties
LPPG exhibited a strong binding affinity to FhuA ΔC/Δ5L
Here, we show a clear distinction among the time-dependent anisotropy signatures acquired with anionic and zwitterionic detergents, such as LysoFos (Fig. 2A), LD (Fig. 2B), and LPPG (Fig. 2C). LPPG was able to maintain a uniform rmax-based PDC population when the protein was solubilized into a detergent concentration of 1 mM (Fig. 2C; Supporting Information, Fig. S4). To probe the LPPG-desolvation process of the protein, we solubilized FhuA ΔC/Δ5L in 0.5 and 0.2 mM LPPG. Indeed, an anisotropy decrease was noticed at very low final LPPG concentrations of 4 and 8 µM. For the sake of plot simplicity in Fig. 2C, we illustrated only a few time-dependent anisotropy traces (others are in Supporting Information, Fig. S5). The presence of an excess of denaturing agent (6 M Gdm-HCl) revealed a low anisotropy of ~0.17, most likely due to a drastic increase in mobility of the protein in the denatured state (Fig. 2E). Moreover, the observation of the absolute recorded minimum anisotropy, r1 (the dashed horizontal line), under denaturing conditions in the presence of 40 mM SDS, a harsh anionic surfactant, indicates that r1 corresponded to the highest tumbling rate of the protein nanopore.
The concentration-response curves of the detergent-desolvation phases are Langmuir-Hill isothermal dissociation plots (Fig. 2D).33 These equilibrium concentration-response curves were fitted by a symmetrical four-parameter Hill equation, as previously conducted in other receptor-ligand binding assays:34
| (3) |
Such a fitting procedure implied the assumption that the solvent-accessible surface of the protein exhibits individual binding sites for specific detergents. Here, rmin and rmax indicate the above-mentioned minima and maxima of anisotropy, respectively (Supporting Information, Table S2). We also assumed that rmax corresponds to conditions in which all proteins (Ptot) are fully solvated (e.g., all binding sites are occupied), whereas rmin corresponds to conditions in which all proteins are desolvated (e.g., all detergent monomers are released; Supporting Information). p denotes the Hill coefficient. Assuming that all detergent molecules bind to protein surface with a similar binding affinity, p unambiguously indicates whether this binding occurs with a positive (p > 0) or a negative cooperativity (p < 0). It does not mean that p is equal to the exact number of binding sites of the protein surface for a certain detergent.33 All p values were greater than 1 (Supporting Information, Table S3), suggesting that several to many detergent monomers bind to protein surface with positive cooperativity. The Hill coefficients are non-integers, because they are likely affected by intermediate state(s) of detergent-protein bindings, which are weighted by their distribution fractions. The midpoint of the protein unfolding transition corresponds to a concentration c0, which is the apparent dissociation constant (Kd) (Table S3). The slope factor, q, is the slope of the unfolding transition at half saturation:
| (4) |
and is expressed in mM−1. q is also the steepness of the protein unfolding transition at half detergent saturation. When the protein was solubilized in 200 µM LPPG, we recorded data points corresponding to a substantially decreased rmax (the orange horizontal line, Fig. 2D). This was likely caused by the inability to completely solubilize a large fraction of the proteins present in solution, otherwise achievable in 1 and 25 mM LPPG. We expanded our FP measurements to the steroidal group-based detergents, which included the zwitterionic CHAPS and Big CHAP (Supporting Information, Fig. S6). CHAPS dissociated quickly from FhuA ΔC/Δ5L at a concentration of 2.1 mM, which is approximately 4-fold lower than the CMC. The long-lived fluctuations in the anisotropy signal were noticed when Big CHAP-solubilized FhuA ΔC/Δ5L was inspected, suggesting very weak PDC interactions (Table S3).
Neutral, maltoside-containing detergents
The same experimental approach was conducted in the case of four maltoside-containing detergents: DDM, UM, DM, and CYMAL-4 (Supporting Information, Fig. S7). The first three detergents have 12, 11, and 10 alkyl carbons, respectively, whereas CYMAL-4 is also a maltoside-containing detergent with a very short hydrophobic tail, which includes only 4 alkyl carbons as well as a cyclohexyl group. It is worth mentioning that these time-dependent kinetic reads reveal the timescale of the detergent desolvation-induced protein unfolding. For example, at a DM concentration of 0.45 mM, the dissociation of detergent from FhuA ΔC/Δ5L underwent three phases with lifetimes from a few minutes to tens of minutes.
Dependence of the detergent desolvation on the nanopore electrostatics
To further examine the specificity of the anisotropy readout on nanopore properties, we analyzed four proteins of closely similar structure, but of varying isoelectric point. These were wild-type OmpG35 and three FhuA derivatives, FhuA ΔC/Δ5L, FhuA ΔC/Δ5L_25N, and FhuA ΔC/Δ7L_30N (Fig. 3A).24 FhuA ΔC/Δ5L_25N is a FhuA ΔC/Δ5L-based nanopore, whose 25 negative residues were neutralized, whereas FhuA ΔC/Δ7L_30N features 30 such negative neutralizations and further truncation of four major extracellular loops (L4, L5, L7, and L8). For all FhuA-based nanopores, Texas Red was attached on T7 β turn. The isoelectric points of OmpG, FhuA ΔC/Δ5L, FhuA ΔC/Δ5L_25N, and FhuA ΔC/Δ7L_30N are 4.4, 5.7, 9.3, and 9.6, respectively; thereby, at physiological pH, the first two nanopores are acidic, whereas the other two are basic. Fig. 3B and Fig. 3C show the dose-response anisotropy following LysoFos and OG depletion in the chamber, respectively. We were able to refold both acidic nanopores in OG and noted the two-state protein unfolding transition. In contrast, experiments with basic nanopores showed low anisotropy endpoint values of ~0.17, because of very weak PDC interfacial interactions (Supporting Information, Fig. S8). Therefore, the anisotropy values continue to decrease even at concentrations well above the CMC. Similar trait, but with a different anisotropy signature was observed with the solubilization of FhuA ΔC/Δ5L in OTG, another neutral, glucoside-containing detergent. Remarkably, distinctive anisotropy signatures of OG and OTG were noted, although the only molecular difference between the two detergents is the replacement of an oxygen with a sulfur atom (Supporting Information, Fig. S9).
Figure 3. Concentration-response anisotropy changes recorded with protein nanopores of varying isoelectric point pI.

(A) Side views of the four protein nanopores inspected in this work, OmpG, FhuA ΔC/Δ5L, FhuA ΔC/Δ5L_25N, and FhuA ΔC/Δ7L_30N. Locations of fluorophore attachment are marked in yellow. Negative charge neutralizations with respect to FhuA ΔC/Δ5L are marked in red. For FhuA ΔC/Δ7L_30N, there are three additional lysine mutations in the β turns (marked in blue), out of which two are negative-to-positive charge reversals;44 The top of each cartoon indicates the nanopore abbreviated name and its respective isoelectric point. (B) Dose-response of the LysoFos depletion in the well; (C) Dose-response of OG depletion in the well. Vertical bars indicate the CMC (Table S1). The horizontal axis indicates individual dilutions of detergent concentrations, while keeping the final protein concentration constant at 28 nM (see Experimental Section). The anisotropy values on the vertical axis were collected 24 hours later for equilibrium determinations. All the other experimental conditions were the same as in Fig. 2.
The predictive power of this analytical approach
We validated this FP-based analytical assay using two independent approaches. First, the unusually strong binding interactions between LPPG and FhuA ΔC/Δ5L (Fig. 2C, Fig. 2D; Supporting Information, Table S3) would normally result in a highly thermostable LPPG-solubilized membrane protein. This expectation was tested using temperature-dependent circular dichroism (CD) spectroscopy for identifying the presence of β-structure in solution. Second, we examined whether the zwitterionic and neutral detergents that solubilized well FhuA ΔC/Δ5L can be used for the reconstitution of uniform and stable protein nanopores into planar lipid membranes.
Fig. 4A confirmed that solubilizing detergents LPPG, LysoFos, UM, DM, and DDM facilitate the formation of β structure in solution (the method details are provided in Supporting Information).36 Protein samples solubilized with the other detergents did not exhibit a satisfactory CD signal under similar conditions. In Fig. 4B, we show a clear distinction in the temperature-dependent profile between LPPG and LysoFos. Protein refolded in LysoFos began losing CD signal by ~50°C, with measurements beyond 67°C rendered impossible by irreversible protein aggregation and visible precipitation in the cuvette. On the contrary, the CD signal recorded with LPPG-refolded FhuA ΔC/Δ5L continued to decrease slowly, while no visible precipitation was noted, suggesting a much more thermodynamically stable protein structure. These results are in excellent accord with the FP data, which indicated unusually strong binding interactions between LPPG and FhuA ΔC/Δ5L.
Figure 4. Unusual thermostability of LPPG-refolded protein nanopores and functional reconstitution of LysoFos-, DDM-, and OG-refolded protein nanopores.

(A) Wavelength circular dichroism scans of ~1 µM FhuA ΔC/Δ5L in 200 mM NaCl, 50 mM potassium phosphate, pH 7.4 with 20 mM of the specified detergent. In the negative control experiment, we used a buffer solution containing 6 M Gdm-HCl; (B) Temperature-dependent ellipticity θ225 of FhuA ΔC/Δ5L in either 20 mM LPPG or in 20 mM LysoFos. For DDM, UM, and DM, we could not achieve a sufficiently high aggregation-free protein concentration; (C) Representative step-wise insertions of single nanopores, over at least 6 distinct experiments, after the addition of DDM- (blue), OG- (black), or LysoFos-refolded (red) FhuA ΔC/Δ5L at an applied transmembrane potential of +40 mV. 40 µl pure and denatured 6×His+-tagged FhuA ΔC/Δ5L was 50-fold diluted into 29 mM DDM, 85 mM OG, or 16 mM LysoFos, containing 200 mM NaCl, 50 mM Tris.HCl, 1 mM EDTA, pH 8.0. The dilution ratio of the refolded protein within the bilayer chamber was ~ 1:1000. Therefore, the presence of detergent within the bilayer chamber did not affect the stability of the membrane;45 (D) The unitary-conductance histograms of DDM-, OG-, and LysoFos-refolded FhuA ΔC/Δ5L. The electrical recordings were collected using 1M KCl, 10 mM potassium phosphate, pH 7.4.
To validate the functional pore-forming properties of proteins solubilized with satisfactory detergents, we conducted single-molecule electrophysiology experiments (the method details are provided in Supporting Information).37,38 In these experiments, we used representative detergents of three different classes, in which a detergent desolvation-induced protein unfolding transition was noted (LysoFos, Fig. 3B; OG, Fig. 3C; DDM, Supporting Information, Fig. S7E). Indeed, we were able to confirm uniform single-channel insertions of LysoFos-, OG-, and DDM-refolded FhuA ΔC/Δ5L nanopores (Fig. 4C). The distributions of single-channel conductance values obtained with FhuA ΔC/Δ5L refolded in each detergent provided a similar average unitary conductance of ~4 nS (Fig. 4D). Relatively uniform-conductance channels were observed with the LysoFos-refolded protein, but ~15% and ~25% outliers of the average conductance were detected with DDM- and OG-refolded proteins, respectively. This finding indicates that the uniformity of the histogram peak of the single-channel conductance was impaired by the refolding detergent (Supporting Information, Fig. S10 and Fig. S11).
Discussion
In this work, we show that the FP spectroscopy can be used as a molecular reporter of the detergent desolvation-induced unfolding of a membrane protein. While the local flexibility of the fluorophore can indeed contribute to the steady-state anisotropy value, the unfolding transition only occurs at detergent concentrations comparable with or lower than the CMC. That supports the most likely possibility that the tumbling rate (e.g., rotational correlation time) of the protein nanopore is indeed affected by the detergent coat. On the contrary, at concentrations of satisfactorily solubilizing detergents much greater than the CMC, no significant change in fluorescence anisotropy was noticed. Therefore, the detergent-induced aggregation of several proteins in one large proteomicelle, further declining the rotameric mobility of the protein, is unlikely under these conditions. In some cases, especially at detergent concentrations below their corresponding CMC, we found either some small discrete changes in anisotropy or increased error bars (e.g., Fig. 2A, 0.5, 1 and 1.5 mM LysoFos). This variability resulted, most likely, from the coexistence of multiple sub-states of the protein nanopores during the desolvation process. These physicochemical alterations are perhaps induced by changes in the internal pressure created by the detergent layer around the protein, leading sometimes to more discrete changes in anisotropy (e.g., Fig. 2C, 0.008 mM LPPG).
We interpret that the two-state detergent desolvation-induced protein unfolding plots acquired in this work resulted from the average alterations in the overall cross-sectional size of the PDC due to detergent depletion within the well. Under conditions in which the detergent concentration is below the CMC, stochastic dissociation events of the detergent monomers from the proteomicelle are very likely,39 leading to ruptured proteomicelles containing misfolded or unfolded proteins (Supporting Information, Fig. S12, Table S2). The rotational diffusion coefficients of fully solvated nanopores, Drslow, for various inspected detergents, were in a broad range, 0.05 – 1.8 × 107 s−1, revealing greatly distinctive tumbling rates of diverse proteomicelles (details are provided in Supporting Information). In contrast, the rotational diffusion coefficients of unfolded proteins, Drfast, spanned a narrow range, between 2.9 and 6.8 × 107 s−1. At room temperature, a Dr = 3.0 × 107 s−1 corresponds to a rotational correlation time of 5.5 ns for an unfolded FhuA ΔC/Δ5L, which features a molecular weight of ~55 kDa. This is in good accord with the calculated rotational correlation time of 15.4 ns for a 50 kDa-protein at 20°C.40 Detergent desolvation-induced protein unfolding produced a change in the PDC hydrodynamic radius, ΔRh, within a broad range of 0.6 – 5.1 nm.
We were able to refold acidic nanopores in OG and the two-state unfolding transitions of these nanopores were noted under physiological conditions. In contrast, we acquired low anisotropy signals with the OG-refolded basic nanopores. Therefore, it is very unlikely that we can get functional membrane-inserted FhuA ΔC/Δ5L_25N and FhuA ΔC/Δ7L_30N nanopores under these denaturing conditions. This finding substantiates that the anisotropy readout is not primarily driven by the interaction of the fluorophore with a proteomicelle, but the protein-micelle interactions. In other words, even if there is some mobility restraint of the fluorophore dynamics by the detergent coat, the anisotropy readout pertaining to the desolvation kinetics and energetics is strongly dependent on the properties of the PDC interactions. Fig. 3C can be considered as a positive control experiment, in which all four anisotropy traces were collected under identical conditions, but of strongly varying PDC interfacial interactions.41
It is worth mentioning that FhuA ΔC/Δ5L interacted weakly with OG, as compared with LysoFos and DDM (Supporting Information, Table S3). A large number of unitary conductance outliers were noted with OG. This suggests that the insertion of many misfolded OG-solubilized proteins into bilayer were due to their residual detergent desolvation in aqueous phase (Fig. 4D). Although the Kd values for DDM and LysoFos were closely similar, some unitary conductance outliers observed with DDM, but not LysoFos, imply that a highly uniform conductance peak might be determined by other physicochemical factors as well (e.g., size and packing of the proteomicelles), as the protein insertion is preceded by demicellization. It is also true that the functional activity of membrane proteins is sometimes compromised in part by their reduced internal flexibility within detergent micelles, which is another reason for the appearance of some unitary conductance outliers.13
One consequence of the detergent dissociation from membrane proteins is their aggregation. This process is accompanied by changes in the interactions of waters with the protein surface. In the absence of detergent, the waters lose their capacity to form hydrogen bonds with the protein surface. Therefore, this unusual interaction is entropically unfavorable, leading to minimized interaction interfaces between water molecules and hydrophobic regions of the membrane protein. In this case, the waters are confined in small clusters surrounded by hydrophobically collapsed parts of the protein.
Our FP-based approach can be extended to other applications. For example, the inability to completely extract some lipids from membrane proteins during crystallographic studies is phenomenally interesting, indicating that the lipids have a required structural role for these proteins. With further developments, this FP method may be used to determine changes in the tumbling rate of membrane proteins under native (e.g., in lipid-bound detergent micelles) and denaturing (e.g., in the presence of Gdm-HCl) conditions. FP experiments with specific detergent-lipid solubilization mixtures9 might potentially generate an understanding of the structural role of these lipids in the stabilization of membrane proteins.
Despite obvious advantages of this FP-based analytical assay, there are a number of limitations. First, this approach is restrained to a low-molecular size fluorophore. Large-molecular size fluorophores (e.g., genetically engineered green fluorescence protein (GFP) and its derivatives) are prone to distort the flexibility, dynamics, and structure of the inspected protein. Second, proteins with multiple native cysteines cannot be used if the approach is extended to time-resolved anisotropy and time-dependent, steady-state FP studies, because individual fluorophore anisotropy spectra can complicate data interpretations due to their diverse residue and solvent environments during the desolvation process. Moreover, a hydrophilic fluorophore needs to be attached within the aqueous phase-exposed domains of the protein for a satisfactory anisotropy signal-to-noise ratio.
In summary, we examined the time-dependent detergent-desolvation of protein nanopores. To our knowledge, this is the first study on the kinetic read of desolvation of water-insoluble proteins at detergent concentrations well below the CMC. This approach can be readily expanded to a broad range of situations for identifying optimized interfacial interactions. These include ionic strength, temperature, osmotic pressure, viscosity, pH, binary and ternary mixtures of detergents as well as other non-classical detergent-like compounds, such as lipopeptides42 and amphipols.43 Future developments of this analytical approach will likely impact advancements in the synthetic chemistry of newly designed detergent-like molecules and membrane proteins.
Supplementary Material
Acknowledgments
The authors thank Bert van den Berg, Jack Gugel, Celina Hsieh, and Motahareh Ghahari Larimi for valuable feedback and stimulating discussions. We are grateful to Stephan Wilkens for his reagents during the early stage of this work. This research was supported by the US National Institutes of Health grants GM113299 (A.R.B.), GM115762 (to S.N.L.), GM115442 (to M.C.), and GM088403 (to L.M.).
Footnotes
Supporting Information. (i) Protein expression and purification under denaturing condition; (ii) Protein labeling of FhuA derivatives; (iii) Expression, purification, and labeling of OmpG D224C proteins; (iv) Contributions of anisotropy values to the Langmuir-Hill isothermal binding curves; (v) Secondary structure determination of the refolded FhuA ΔC/Δ5L protein in solution using circular dichroism; (vi) Single-channel and macroscopic electrical recordings using planar lipid bilayers; (vii) Acquiring equilibrium steady-state endpoints of the FP anisotropy at different concentrations of detergents of varying chemistry; (viii) Rotational motions of the protein nanopores under detergent solvation and desolvation conditions; (ix) Fluorescence anisotropy readout acquired with LPPG-refolded FhuA ΔC/Δ5L at final refolding detergent concentration of 25 mM; (x) Detailed time- and concentration-dependent anisotropy traces acquired with anionic and zwitterionic detergents; (xi) Steroidal group-containing detergents are weakly binding to FhuA ΔC/Δ5L; (xii) Dissociation of maltoside-containing detergents from FhuA ΔC/Δ5L; (xiii) Dependence of time-dependent, steady-state fluorescence anisotropy on proteins of closely similar structure, but varying isoelectric point; (xiv) Current-voltage relationship of FhuA ΔC/Δ5L refolded in detergents of varying chemistry; (xv) Stability of the open-state current of the refolded FhuA ΔC/Δ5L proteins at higher applied transmembrane potentials. These materials are available free of charge via the Internet at http://pubs.acs.org
References
- 1.Vergis JM, Purdy MD, Wiener MC. Anal. Biochem. 2010;407:1–11. doi: 10.1016/j.ab.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pollock NL, Satriano L, Zegarra-Moran O, Ford RC, Moran O. J. Struct. Biol. 2016;194:102–111. doi: 10.1016/j.jsb.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Grosse W, Psakis G, Mertins B, Reiss P, Windisch D, Brademann F, Burck J, Ulrich A, Koert U, Essen LO. Biochemistry. 2014;53:4826–4838. doi: 10.1021/bi500660q. [DOI] [PubMed] [Google Scholar]
- 4.Ehsan M, Du Y, Scull NJ, Tikhonova E, Tarrasch J, Mortensen JS, Loland CJ, Skiniotis G, Guan L, Byrne B, Kobilka BK, Chae PS. J. Am. Chem. Soc. 2016;138:3789–3796. doi: 10.1021/jacs.5b13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tulumello DV, Deber CM. Biochemistry. 2009;48:12096–12103. doi: 10.1021/bi9013819. [DOI] [PubMed] [Google Scholar]
- 6.Miles AJ, Wallace BA. Chem. Soc. Rev. 2016;45:4859–4872. doi: 10.1039/c5cs00084j. [DOI] [PubMed] [Google Scholar]
- 7.Yang Z, Wang C, Zhou Q, An J, Hildebrandt E, Aleksandrov LA, Kappes JC, DeLucas LJ, Riordan JR, Urbatsch IL, Hunt JF, Brouillette CG. Protein Sci. 2014;23:769–789. doi: 10.1002/pro.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Z, Brouillette CG. Methods Enzymol. 2016;567:319–358. doi: 10.1016/bs.mie.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 9.Jahnke N, Krylova OO, Hoomann T, Vargas C, Fiedler S, Pohl P, Keller S. Anal.Chem. 2014;86:920–927. doi: 10.1021/ac403723t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Columbus L, Lipfert J, Jambunathan K, Fox DA, Sim AY, Doniach S, Lesley SA. J. Am. Chem. Soc. 2009;131:7320–7326. doi: 10.1021/ja808776j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stanczak P, Horst R, Serrano P, Wuthrich K. J. Am. Chem. Soc. 2009;131:18450–18456. doi: 10.1021/ja907842u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker LA, Folkers GE, Sinnige T, Houben K, Kaplan M, van der Cruijsen EA, Baldus M. Methods Enzymol. 2015;557:307–328. doi: 10.1016/bs.mie.2014.12.023. [DOI] [PubMed] [Google Scholar]
- 13.Frey L, Lakomek NA, Riek R, Bibow S. Angew. Chem. Int. Ed. Engl. 2017;56:380–383. doi: 10.1002/anie.201608246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raschle T, Rios Flores P, Opitz C, Muller DJ, Hiller S. Angew. Chem. Int. Ed. Engl. 2016;55:5952–5955. doi: 10.1002/anie.201509910. [DOI] [PubMed] [Google Scholar]
- 15.Kwok KC, Cheung NH. Anal.Chem. 2010;82:3819–3825. doi: 10.1021/ac1002245. [DOI] [PubMed] [Google Scholar]
- 16.Rossi AM, Taylor CW. Nat. Protoc. 2011;6:365–387. doi: 10.1038/nprot.2011.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turman DL, Nathanson JT, Stockbridge RB, Street TO, Miller C. Proc. Natl. Acad. Sci. U.S.A. 2015;112:5697–5701. doi: 10.1073/pnas.1505301112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stoddart LA, White CW, Nguyen K, Hill SJ, Pfleger KD. Br. J. Pharmacol. 2016;173:3028–3037. doi: 10.1111/bph.13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jutila A, Zhu K, Patkar SA, Vind J, Svendsen A, Kinnunen PK. Biophys. J. 2000;78:1634–1642. doi: 10.1016/S0006-3495(00)76715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersen KK, Oliveira CL, Larsen KL, Poulsen FM, Callisen TH, Westh P, Pedersen JS, Otzen D. J. Mol. Biol. 2009;391:207–226. doi: 10.1016/j.jmb.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 21.Fano M, van de Weert M, Moeller EH, Kruse NA, Frokjaer S. Arch. Biochem. Biophys. 2011;506:92–98. doi: 10.1016/j.abb.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 22.Naidu KT, Prabhu NP. J. Phys. Chem. B. 2011;115:14760–14767. doi: 10.1021/jp2062496. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson AD, Hofmann E, Coulton JW, Diederichs K, Welte W. Science. 1998;282:2215–2220. doi: 10.1126/science.282.5397.2215. [DOI] [PubMed] [Google Scholar]
- 24.Wolfe AJ, Mohammad MM, Thakur AK, Movileanu L. Biochim. Biophys. Acta. 2016;1858:19–29. doi: 10.1016/j.bbamem.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Udho E, Jakes KS, Buchanan SK, James KJ, Jiang X, Klebba PE, Finkelstein A. Proc. Natl. Acad. Sci. U.S.A. 2009;106:21990–21995. doi: 10.1073/pnas.0910023106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pusey ML. Cryst. Growth Des. 2011;11:1135–1142. doi: 10.1021/cg1013522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohammad MM, Iyer R, Howard KR, McPike MP, Borer PN, Movileanu L. J. Am. Chem. Soc. 2012;134:9521–9531. doi: 10.1021/ja3043646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linke D. Methods Enzymol. 2009;463:603–617. doi: 10.1016/S0076-6879(09)63034-2. [DOI] [PubMed] [Google Scholar]
- 29.Titus JA, Haugland R, Sharrow SO, Segal DM. J. Immunol. Methods. 1982;50:193–204. doi: 10.1016/0022-1759(82)90225-3. [DOI] [PubMed] [Google Scholar]
- 30.Fahie M, Chisholm C, Chen M. ACS nano. 2015;9:1089–1098. doi: 10.1021/nn506606e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gradinaru CC, Marushchak DO, Samim M, Krull UJ. Analyst. 2010;135:452–459. doi: 10.1039/b920242k. [DOI] [PubMed] [Google Scholar]
- 32.Jameson DM, Ross JA. Chem. Rev. 2010;110:2685–2708. doi: 10.1021/cr900267p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woodbury CP. Introduction to macromolecular binding equilibria. CRC Press, Taylor & Francis; Boca Raton: 2008. [Google Scholar]
- 34.Prinz H. J. Chem. Biol. 2010;3:37–44. doi: 10.1007/s12154-009-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang B, Tamm LK. Proc. Natl. Acad. Sci. U.S.A. 2007;104:16140–16145. doi: 10.1073/pnas.0705466104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Greenfield NJ. Nat. Protoc. 2006;1:2527–2535. doi: 10.1038/nprot.2006.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheneke BR, van den Berg B, Movileanu L. ACS Chem. Biol. 2015;10:784–794. doi: 10.1021/cb5008025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Couoh-Cardel S, Hsueh YC, Wilkens S, Movileanu L. Sci. Rep. 2016;6:24774. doi: 10.1038/srep24774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Borysik AJ, Robinson CV. Langmuir. 2012;28:7160–7167. doi: 10.1021/la3002866. [DOI] [PubMed] [Google Scholar]
- 40.Lakowicz JR. Principles of Fluorescence Microscopy. 2. Springer; New York: 2006. [Google Scholar]
- 41.Khao J, Arce-Lopera J, Sturgis JN, Duneau JP. Eur. Biophys. J. 2011;40:1143–1155. doi: 10.1007/s00249-011-0745-9. [DOI] [PubMed] [Google Scholar]
- 42.Prive GG. Curr. Opin. Struct. Biol. 2009;19:379–385. doi: 10.1016/j.sbi.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Kleinschmidt JH, Popot JL. Arch. Biochem. Biophys. 2014;564:327–343. doi: 10.1016/j.abb.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 44.Mohammad MM, Movileanu L. J. Phys. Chem. B. 2010;I114:8750–8759. doi: 10.1021/jp101311s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohammad MM, Tomita N, Ohta M, Movileanu L. ACS Chem. Biol. 2016;11:2508–2518. doi: 10.1021/acschembio.6b00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.