

Abstract
ADAR RNA editing enzymes (adenosine deaminases acting on RNA) that convert adenosine bases to inosines were first identified biochemically 30 years ago. Since then, studies on ADARs in genetic model organisms, and evolutionary comparisons between them, continue to reveal a surprising range of pleiotropic biological effects of ADARs. This review focuses on Drosophila melanogaster, which has a single Adar gene encoding a homolog of vertebrate ADAR2 that site-specifically edits hundreds of transcripts to change individual codons in ion channel subunits and membrane and cytoskeletal proteins. Drosophila ADAR is involved in the control of neuronal excitability and neurodegeneration and, intriguingly, in the control of neuronal plasticity and sleep. Drosophila ADAR also interacts strongly with RNA interference, a key antiviral defense mechanism in invertebrates. Recent crystal structures of human ADAR2 deaminase domain–RNA complexes help to interpret available information on Drosophila ADAR isoforms and on the evolution of ADARs from tRNA deaminase ADAT proteins. ADAR RNA editing is a paradigm for the now rapidly expanding range of RNA modifications in mRNAs and ncRNAs. Even with recent progress, much remains to be understood about these groundbreaking ADAR RNA modification systems.
Keywords: ADAR, Drosophila melanogaster, RNA editing, dsRNA, RNA modification, epitranscriptome
INTRODUCTION
ADARs: promiscuous and site-specific RNA editing
ADARs (adenosine deaminases acting on RNA) were discovered in Xenopus laevis oocytes when single-stranded antisense RNAs were injected to silence target genes by forming double-stranded (ds) RNA. When injection was performed after germinal vesicle breakdown, when nuclear components are exposed to the cytoplasm, the dsRNA was destabilized. Initially, this was thought to be due to RNA helicase activity; however, the RNA strands had a much reduced capacity to reanneal after heat denaturation and cDNA sequences contained adenosine (A) to guanosine (G) sequence changes. Subsequently, this was found to be because of an enzymatic deamination of approximately half of all the adenosines in the dsRNA to inosine (Bass and Weintraub 1988; Wagner et al. 1989). The resulting inosine–uracil wobble base pairs (Pan et al. 1998) are less stable, leading to substantial strand unwinding (Serra et al. 2004). When the edited RNA is reverse-transcribed, inosine forms a Watson–Crick base pair with cytosine, so that G replaces A at the edited position.
There are many other enzymatic modifications of RNA. However, ADAR-mediated adenosine to inosine RNA modification has been the most widespread RNA modification detected by standard RNA-seq. Therefore, studies on ADAR RNA editing began much earlier and they now also lead the way toward understanding the effects of a range of other enzymatic modifications that have been found more recently in mRNA (O'Connell et al. 2015). Research into this intriguing ADAR RNA editing process over the past 30 years has revealed a wide and still expanding range of very fundamental biological roles for ADARs (Fig. 1). The first major finding was that, in addition to promiscuous editing in long dsRNAs like those formed in Xenopus antisense injections, ADARs also efficiently and site-specifically edit individual adenosines within short RNA hairpins formed in pre-mRNAs (Higuchi et al. 1993). Efficient site-specific ADAR RNA editing can lead to recoding of open reading frames because inosine decodes as guanosine.
FIGURE 1.

Summary of Drosophila Adar mutant phenotypes discussed here, with main references.
ADARs are primarily nuclear proteins that bind to dsRNA via two or more dsRNA-binding domains (dsRBDs) at the amino termini of the proteins and a larger carboxy-terminal deaminase domain about 400 amino acids long (Keegan et al. 2001). The deaminase active site contains a catalytic zinc atom chelated by two cysteines and one histidine residue within three deaminase motifs conserved between ADARs (Macbeth et al. 2005). These deaminase motifs are also present in the tRNA adenosine deaminases from which ADARs evolved (ADAT1, ADAT2, and ADAT3) (Gerber and Keller 2001), and in the much more distantly related cytosine deaminases acting on RNA and DNA (CDARs) (Hogg et al. 2011). The deaminase domain flips the target adenosine out of the dsRNA structure and into the active site (Matthews et al. 2016). ADAR catalytic activity depends on a molecule of inositol hexakisphosphate bound within the deaminase domain and forming a chain of hydrogen bonds through to the active site (Macbeth et al. 2005).
Vertebrate ADARs
This review will concentrate on invertebrate ADARs and especially on genetic studies on the biological roles of the Drosophila ADAR protein. However, it is helpful to place these studies within the context of studies on the vertebrate ADARs. Vertebrate ADAR1 is responsible for most of the promiscuous editing of long dsRNA (Riedmann et al. 2008). ADAR1 is widely expressed in many tissues, including white blood cells and central nervous system (CNS), but not in skeletal muscles (O'Connell et al. 1995). The constitutive nuclear ADAR1p110 isoform is essential for embryo viability (Hartner et al. 2004; Pestal et al. 2015), and it promiscuously edits inverted repeats of Alu elements (IR Alus), that are embedded in pre-mRNA introns and UTRs and in noncoding RNAs (Levanon et al. 2004). The effects of ADAR1 editing on dsRNA are attributed to structural alterations in the dsRNA after editing. I–U wobble pairs form and cause local dsRNA structure distortion to give unpaired bubbles or partial melting of the dsRNA (Serra et al. 2004; Mannion et al. 2014). Innate immune sensors detect perfectly paired dsRNA formed by virus replication and are not aberrantly activated by edited self-dsRNA. Endogenous dsRNA is generated mostly by hairpin-forming sequences or pairing of different copies of repetitive element sequences such as inverted-repeat IR Alus. The resulting endogenous dsRNAs are often somewhat imperfectly paired. However, IR Alus may also require RNA editing to prevent aberrant innate immune induction. The longer interferon (IFN)-inducible cytoplasmic ADAR1p150 isoform is induced late in the interferon response and edits all dsRNA transcripts in the cytoplasm, which then prevents or reverses aberrant innate immune responses to dsRNA (Mannion et al. 2014).
Vertebrate ADAR2 is less widely and less highly expressed than ADAR1 (Wu et al. 2009). ADAR2 is most highly expressed in CNS and appears to be primarily involved in site-specific editing of CNS transcripts encoding ion channel subunits (Higuchi et al. 2000). There is also an ADAR3 protein expressed in brain central ganglia that is closely related to ADAR2 (Melcher et al. 1996); ADAR3 does not edit any known target sites, although it does interfere with editing by ADAR2 (Chen et al. 2000).
Mutations in ADAR genes are associated with various conditions; Aicardi–Goutières syndrome (AGS) is a fatal childhood encephalopathy with interferon overexpression caused by ADAR1 mutation (Rice et al. 2012). ADAR2 (Hideyama et al. 2010) and ADAR3 (Donnelly et al. 2013) have been linked to glutamate excitotoxicity in amyotrophic lateral sclerosis (ALS). Both the long and short isoforms of ADAR1 show increased expression in many cancers and there are also large increases in ADAR editing in tumor transcripts (Fumagalli et al. 2015; Han et al. 2015; Paz-Yaacov et al. 2015). The roles of ADARs in cancer and other diseases have been reviewed elsewhere (Galeano et al. 2012; Gallo and Locatelli 2012). Two more distantly related conserved mammalian germline ADAR-like proteins (ADADs, also called Tenr) (Connolly et al. 2005) are not predicted to have any editing activity and may act solely as RNA-binding proteins, but they do indicate that ADARs or ADAR-like proteins also have roles in the germline.
DROSOPHILA ADAR FUNCTIONS IN THE NERVOUS SYSTEM
Neuronal excitability and neurodegeneration
The single Adar gene in Drosophila (Palladino et al. 2000a) (Palladino et al. 2000b) encodes an ADAR2-type protein (Keegan et al. 2004). The Adar gene is on the distal part of the X chromosome so male Adar mutant flies are usually studied. In embryos the expression of Adar is highest in the CNS, but it is also expressed at a lower level outside the CNS (Palladino et al. 2000b). Adar transcripts are expressed from two promoters: the stronger −4a promoter activated at metamorphosis and the constitutive −4b promoter active at all developmental stages (Fig. 2). Different ADAR protein isoforms are expressed in embryos and adults due to alternative splicing of exons −4a and −4b in the 5′ UTR and to alternative exons 0 and −1 containing alternative translation initiation codons. In adults the ADAR 3/4 spliced transcript predominates, and transcripts from the Adar 4a promoter have less of an alternative exon 3a extension between the two dsRBDs (Fig 2). Adult transcripts from the Adar 4a promoter also have increased editing of a specific codon that changes a serine residue to glycine in the deaminase domain (Palladino et al. 2000a), giving unedited ADAR3/4 S and edited ADAR3/4 G isoforms (Fig. 2). There are also some indications of ADAR expression outside CNS in larvae; data from modENCODE reveals that expression of Adar in haltere disc ML-DmD17-c3 and tumorous blood cell mbn2 cell lines is higher than in CNS ML-DmBG1-c1 and CNS ML-DmBG2-c2 cell lines derived from Drosophila larval central nervous system. However, the role of Adar in these tissues is unknown (Roy et al. 2010).
FIGURE 2.
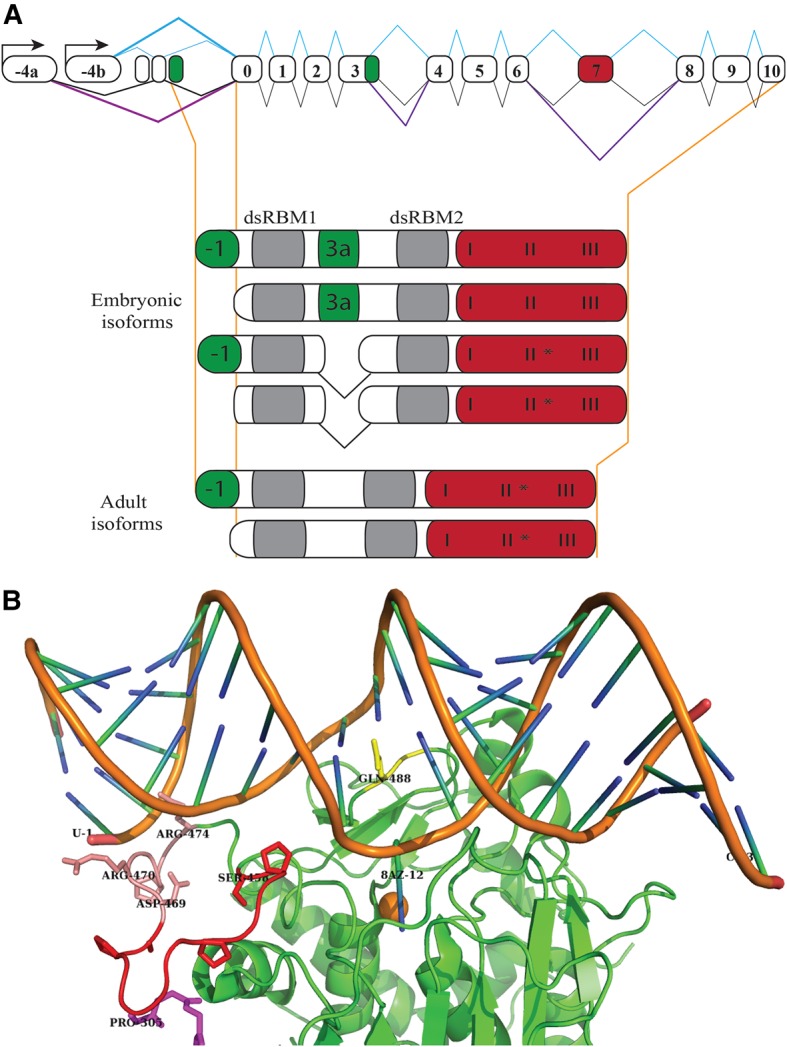
Drosophila ADAR isoforms and effects of deaminase domain S458G editing. (A) Drosophila Adar gene structure, embryonic splicing pattern (below the gene) and adult splicing pattern (above the gene), and ADAR protein isoforms expressed in embryos and adults. (B) Structure of human ADAR2 E488Q–Bdf2 RNA complex (PDB: 5E1). The edited RNA strand runs from 5′ U1 on the left to 3′ C23 on the right. The 8-azanebularine replacing the edited A at position 12 (marked 8AZ-12) is inserted into the deaminase domain active site near the catalytic zinc atom (orange sphere), and held there as a covalent reaction intermediate analog. The serine 458 loop (red), the 5′ RNA contacting region (salmon), the amino terminus of the deaminase domain (magenta), and the GQG base flipping loop (yellow) are highlighted on the deaminase domain protein structure. Several proline residues that cause the protein backbone in the loop to make changes in direction are also shown.
Under good conditions Adar5G1 null mutant flies develop into normal adults. However, they display some neurobehavioral defects such as uncoordinated locomotion and age-dependent neurodegeneration (Fig. 1). AdarIF4, another characterized mutant that has a deletion in the promoter not affecting the coding region, is phenotypically similar to Adar5G1, but has some residual expression of the Adar transcript at a low level (Palladino et al. 2000b). As these male Adar mutants are unable to mate, it is not possible to cross them to a heterozygous mutant female to produce a female homozygous Adar mutant. When the male Adar mutant has a Y chromosome with a translocation of the distal X chromosome that includes the Adar gene, fertility is restored and the male can transmit the Adar mutant X chromosome to his progeny, allowing a homozygous Adar mutant female to be produced. Such homozygous Adar mutant females have been generated to confirm that their mutant phenotypes are similar to those seen in males (Palladino et al. 2000b). However, homozygous female Adar mutants are not routinely studied.
Overexpression of the adult ADAR3/4 S isoform ubiquitously using the Actin 5c-GAL4 driver or in muscle using the Mef 2-GAL4 driver is lethal in embryos and larvae (Keegan et al. 2005). Lethality is probably due to hyperediting as Ca-α1D transcripts encoding the pore-forming α subunit of the muscle voltage-gated calcium channel are edited to a higher than normal level, for the larval stage, in larvae with Mef 2-GAL4 driven overexpression (Li et al. 2014). Interestingly, once past the embryo and larval stages, ADAR3/4 S overexpression is not lethal in adult flies; many sites in Ca-α1D and other transcripts that are edited at low levels in larvae become more fully edited in adult flies. A genetic screen was performed to identify chromosomal deletions (DroDel deficiencies) (Ryter and Schultz 1998), which rescue the lethality caused by the overexpression of the ADAR3/4 S (Li et al. 2014). The aim of the screen was to find genetic modifiers that affect the level or activity of the ADAR3/4 S protein or play an important role in the pathway downstream from ADAR3/4 S. Overexpression of ADAR3/4 S was under the control of the Actin 5c-GAL4 driver, combined with tub-GAL80ts. This allowed ADAR3/4 S overexpression to be prevented during embryonic development to maintain and to collect virgins from the stock carrying three transgenes at 25°C. Virgin females were then crossed to males of DrosDel deletion stocks at 29°C to test whether the DrosDel deletion suppresses Adar OE lethality.
DrosDel deficiencies on chromosome III were screened, and eight rescued ADAR3/4 S overexpression lethality in embryos and larvae. The rescuing effect of one of the deficiencies was mapped to a single gene, Rdl (Resistance to dieldrin); heterozygosity for the null allele Rdl1/+ rescued the Adar OE viability to 34% (Li et al. 2014). Rdl is an essential gene encoding the membrane pore subunit of the important inhibitory GABAA receptor in Drosophila. This suggests that overexpression of ADAR3/4 S leads to reduced neuronal excitability. Consistent with this idea, when Adar is knocked down by RNAi or overexpressed specifically in motor neurons, neuronal excitability is aberrantly increased or decreased, respectively (Li et al. 2014). Also, other mutations and drugs that reduce inhibitory GABA signaling rescue ADAR3/4 S overexpression lethality: for instance, heterozygous mutations in the Gad gene encoding the glutamate decarboxylase involved in the synthesis of GABA from glutamate or feeding larvae with GABA receptor antagonists (Li et al. 2014). Reciprocally, climbing defects in the Adar5G1 null mutant that show increased neuronal excitability are partially rescued by GABA modulators. The Rdl transcript is edited, but effects of expressing individual edited isoforms of RDL on ADAR3/4 S overexpression lethality or on Adar5G1 mutant phenotypes have not been tested due to toxic effects of UAS–Rdl construct expression.
Effects of ADAR S and ADAR G isoforms on editing and on CNS function
Drosophila ADAR edits the Adar transcript in adult flies to change amino acid 458 on the surface of the deaminase domain from a genome-encoded serine (S) to a glycine (G) (Palladino et al. 2000a). Embryos and larvae express primarily the genome-encoded ADAR S protein, which catalyzes site-specific editing on in vitro substrates eight times more efficiently than the adult ADAR G isoform (Keegan et al. 2005). This led to the suggestion that self-editing acts as a negative regulator of ADAR RNA editing activity (Keegan et al. 2005). Savva et al. (2012) used homologous recombination in Drosophila to add a carboxy-terminal HA epitope tag and then to knock in (hardwire) either serine or glycine codons at this site to make AdarS-HA and AdarG-HA strains. They confirmed that across the 100 sites most efficiently edited in dissected parts of flies, such as heads, thoraces, eyes, and antennae, if editing levels differ significantly from wild type at all, then the AdarS-HA strain shows higher levels of editing than the AdarG-HA strain. However, effects are much smaller than in vitro and many highly edited sites are insensitive to the change. The most conspicuous changes occur at sites with editing in wild type lower than 25%–30%, which are more difficult to quantitate. The AdarS-HA strain does however show some additional editing events not previously observed (Savva et al. 2012).
The AdarS-HA mutant has higher levels of single-fly open-field locomotion than the AdarG-HA protein (Savva et al. 2012), as previously observed when ADAR S and ADAR G are expressed from Adar cDNA constructs under the control of nervous system drivers (Keegan et al. 2005). Although detected changes in RNA editing between AdarS-HA and AdarG-HA strains across large body regions are mostly subtle, differential effects on behavioral phenotypes in adult flies are relatively strong. Climbing is a different assay in which fly movement is motivated by gravitaxis, a natural behavioral propensity of adult flies to move upward against gravity on the substratum when this is possible. AdarG-HA flies are less active in the climbing assay than AdarS-HA flies, and AdarG-HA flies also have more delay in beginning to court a virgin female, another motivated behavior, than AdarS-HA flies do (Savva et al. 2012). The benefit of combining the homologous recombination and GAL4/UAS-cDNA approaches should increase when behavioral differences can be assigned to smaller sets of neurons in which different levels of editing in a specific ion channel or other CNS transcripts appear in AdarS-HA and AdarG-HA strains. The AdarS-HA and AdarG-HA alleles expressed from the endogenous Adar locus are each produced as multiple ADAR protein isoforms with different amino termini and as exon 3a or exon 3/4 alternative spliced forms (Palladino et al. 2000a), and causative isoforms may be identified using UAS–cDNA construct lines.
Human ADAR2 deaminase domain–substrate RNA cocrystal structure
The structure of a human ADAR2 deaminase domain in a covalent complex with Bdf2 RNA from budding yeast was recently determined (Fig. 2B; Matthews et al. 2016). ADAR deaminase domains lacking dsRBDs do not normally edit dsRNA efficiently and budding yeast has no ADARs; the Bdf2 RNA was identified as a substrate for the human ADAR2 deaminase domain in a screen performed to discover the reason why human ADAR2 is toxic in yeast (Eifler et al. 2013). This substrate and others that are edited by the ADAR2 deaminase domain alone, combined with replacement of the edited adenosine in these substrates with 8-azanebularine to form a covalent adduct in the deaminase active site, allowed crystallization and has yielded important new structural information (Matthews et al. 2016). Unfortunately, the canonical GluR B Q/R site substrate did not prove favorable for crystallization. In the ADAR2 deaminase domain–Bdf2 RNA complex, the protein makes contacts with dsRNA 10 bp 5′ of the edited A (Fig. 2B). However, the GluR2 R/G substrate has only 5 bp of dsRNA 5′ to the edited A, and a similar major groove interaction and contact with the distant unedited strand will not be possible; the ADAR2 deaminase domain–Bdf2 RNA complex may differ in detail from the deaminase–RNA interaction in a canonical GluR B Q/R complex with full-length ADAR2.
The new data bring us closer to understanding the different effects of Drosophila ADAR S and ADAR G isoforms. A previous structure of the ADAR2 deaminase domain in the absence of RNA showed an 11-residue-long unstructured protein loop, beginning with serine 457, which is equivalent to the serine 458 residue in Drosophila ADAR (Macbeth et al. 2005). However, in the new human ADAR2 deaminase domain–Bdf2 RNA complex, this loop is structured and residues just after the loop make RNA contacts at the 5′ end of the binding site (Fig. 2B). The RNA dependence of folding the serine 457 loop in the human ADAR2 deaminase domain suggests that the precise RNA sequence and structure 5′ to the edited adenosine at different Drosophila editing sites could differentially affect binding of the ADAR S and ADAR G isoforms. In the present cocrystals, the RNA bends on the 5′ side of the edited A, and residues just after the ser 457 loop enter the major groove and there contact the more distant unedited RNA strand; this may not occur when full-length ADAR2 binds natural editing sites. A more relaxed binding arrangement might place the serine 457 loop over the minor groove only, with protein–RNA contacts limited to the edited strand no more than 5 bp 5′ of the edited A, as expected for the GluR2 R/G site. The side chain of serine 457 in ADAR2 does not interact with RNA but points inward and may instead help to fold the loop (Matthews et al. 2016). It seems likely that changing this residue to glycine will reduce the propensity of the serine 457 loop to fold, which could account for reduced editing activity of the ADAR G isoform. The ADAR2 serine 457 loop and the last linker residues before the deaminase domain are positioned immediately above the minor groove on the 5′ side of the deaminase domain, ideally placed to interact with any other protein making minor groove contacts on the 5′ side of the bound ADAR. Changing serine 457 to glycine by editing might affect potential ADAR interactions with other proteins, such as another ADAR protein or the Mle (Reenan et al. 2000), or Dicer (Ota et al. 2013) helicases.
There is a difficulty in combining the new information on the ADAR2 deaminase domain–RNA complexes with previous information on binding of dsRBD2 to the GluR B Q/R substrate. If the ADAR2 dsRBDs are positioned as on GluR2 Q/R substrate (Stefl et al. 2010), then dsRBD2 in the minor groove 3′ of the edited A will make an extremely close approach to the deaminase domain here, where gln 488 enters the dsRNA to compensate the unpaired base partner of the edited adenosine. Thomas and Beal (2017) suggested that the deaminase domain–dsRBD2 steric clashes involved are too great to overcome. Nevertheless, the complex the deaminase domain alone forms with RNA may differ from that formed in the full-length protein. Deaminase domain–dsRBD2 interaction might be possible by combined adjustments of protein domains and of the RNA helical structure. It remains to be determined whether dsRBD2 at the majority of editing sites maintains a fixed interaction with the deaminase domain at this point or if dsRBD2 is positioned differently on different editing sites. Consistent with a significant effect of the last dsRBD, dsRBD3 is the most important dsRBD for ADAR1 activity; amino terminally truncated or chimeric ADAR1 proteins retaining dsRBD3 retain some RNA editing activity in mammalian cell transfection assays (Liu and Samuel 1996; Liu et al. 2000). When ADAR1–ADAR2 hybrid proteins were made by fusions in the linker region between the last dsRBD and the deaminase domain, the resulting hybrid ADARs were functional and specificity for editing sites in cell transfection assays was largely determined by the deaminase domain (Wong et al. 2001). One explanation for this result would be that the ADAR1 and ADAR2 deaminase domain-proximal dsRBDs bind each RNA substrate similarly, and the ADAR1 deaminase domain–dsRBD3 interaction is exchangeable for the ADAR2 deaminase domain–dsRBD2 interaction.
The first half of the serine 457 loop also intriguingly folds up to touch the most amino-terminal residues of the deaminase domain (Fig. 2B). This raises a question about whether the ser 457 loop in a full-length ADAR2 protein might interact with residues in the linker immediately before the deaminase domain to affect the interaction of dsRBD2 with RNA or with the deaminase domain. Evidence from ADAR1-PKR1 chimeric proteins indicates that the lengths and sequences of linkers, particularly the linker before the deaminase domain, are important for activity in full-length ADAR1 (Liu et al. 2000). It will require structures of RNA complexes with full-length ADAR proteins or with deaminase domains retaining their proximal dsRBD to answer these questions.
ADAR effects on synaptic plasticity and sleep; neuromuscular junction defects in Adar mutants
Adar5G1 null mutant flies have increased numbers of aberrant boutons at neuromuscular junctions (NMJs) (Bhogal et al. 2011; Maldonado et al. 2013), with extreme aberrant accumulations of synaptic vesicles and of the synaptic vesicle proteins—synapsin, endophilin, and synaptotagmin (Maldonado et al. 2013), all proteins that are encoded by edited transcripts. Synaptic vesicle release dependent on calcium influx at the NMJ is reduced, possibly due to loss of editing in the synaptotagmin transcript and in the cacophony transcript that encodes the voltage-dependent calcium channel (Maldonado et al. 2013). Although the probability of release is reduced, the vesicles that are released are also larger and cause greater excitation of target muscles. This effect is still seen even though postsynaptic glutamate GluRIIA receptors on larval muscles are homeostatically down-regulated in Adar5G1 null mutant flies (Maldonado et al. 2013). It would be interesting to know whether the reciprocal effects of Adar depletion and Adar overexpression on motor neuron cell body excitability extend to similarly reciprocal effects on NMJs.
Robinson et al. (2016) demonstrated that flies with neuronal Adar knockdown by RNAi or the Adarhyp mutant that has 20% of normal editing levels have aberrantly increased sleep in continuous monitoring (Robinson et al. 2016). One theory on the purpose of sleep is that it allows a homeostatic reversal of accumulated effects of increased activity-dependent synaptic potentiation during waking. Robinson and coworkers therefore interpret the finding of increased sleep pressure in these Adar hypomorphs in terms of the Maldonado observations on the aberrant function of and glutamatergic vesicle accumulation at the NMJs in the Adar5G1 null mutant fly (Maldonado et al. 2013). The authors propose that the hypomorphic Adar mutants also have an accumulation of glutamatergic vesicles in unidentified brain neurons controlling pressure to sleep. The Adar mutants therefore fail to resolve increased potentiation at some critical, unidentified central synapses that drive the pressure to sleep. Consistent with this hypothesis, Robinson et al. (2016) demonstrate that the vesicular glutamate transporter protein is dramatically increased in heads of Adarhyp mutants and that heterozygous dvglut loss-of-function mutants with reduced vesicular glutamate transport or heterozygous glutamate receptor mutants rescue increased sleep in Adarhyp. The authors suggest a key role for synapsin, encoded by an edited transcript, in allowing synaptic vesicles to move from the reserve pool to the readily releasable pool. The loss of the protein kinase A site in synapsin by ADAR editing (Diegelmann et al. 2006) may regulate entry of vesicles into the readily releasable pool in Adarhyp and the stronger NMJ effects in the Adar5G1 null mutant.
The Adarhyp allele has an HA epitope tag on the ADAR carboxy terminus and a mini-white+ gene inserted in reverse orientation in intron 7 (Jepson et al. 2011). This interesting mutant allele was used in the sleep study and in other studies because it has only a 50% decrease in open-field locomotion, and this allows other behavioral phenotypes to be studied. The Adar5G1 null mutant or the other characterized Adar1F4 hypomorphic allele is far more severely affected, with negligible open-field locomotion and leg tremors and falling over not seen with Adarhyp. The Adarhyp allele produces 20% of wild-type levels of full-length ADAR-HA protein but it is less phenotypically defective than the Adar1F4 hypomorphic allele that also produces reduced Adar transcript levels. The Adarhyp mutant does not display the aberrant increased excitability in larval motorneurons that is observed with motorneuron-specific Adar RNAi knockdowns and in the Adar5G1 null mutant (Li et al. 2014; Robinson et al. 2016) so the better locomotion may be due to a reduced defect at the NMJ in the Adarhyp mutant. The Adarhyp allele may also produce ADAR proteins truncated in the deaminase domain and having the two dsRBDs. In mice, production of similar truncated proteins may ameliorate Adar1 mutant phenotypes (Wang et al. 2000; Pestal et al. 2015), so it is possible that truncated ADAR protein ameliorates the mutant phenotype in Adarhyp. The Drosophila DISCO Interacting Protein 1 (DIP1) (DeSousa et al. 2003) gene located in the X chromosome heterochromatin encodes a protein composed of just two dsRBDs related to those of Adar; it has not been determined whether Dip1 protein binds Adar substrates (Catanese and Matthews 2011) or if DIP1 mutations may exacerbate Adar mutant phenotypes.
DROSOPHILA ADAR AND RNAi
ADAR antiviral effects and cytoplasmic RNAi
Drosophila ADAR edits Sigma Virus dsRNAs (Carpenter et al. 2009) and transposon RNAs (Kawamura et al. 2008; Savva et al. 2013). The Drosophila Sigma virus is a neurotropic Rhabdovirus, distantly related to Rabies Virus. Sigma virus infects flies naturally or can be introduced by injection; it then becomes vertically inherited from parent to offspring (Carpenter et al. 2007). Sigma virus was first detected in wild Drosophila because infected flies are susceptible to killing by brief CO2 treatments used to anesthetize flies from examination and sorting. Sigma virus is a positive-strand RNA virus with a dsRNA replication intermediate and virus growth is inhibited by RNAi. It remains to be determined whether Adar mutant flies are more susceptible to Sigma virus infection; the DIP1 gene does contribute to virus resistance (Zhang et al. 2015).
In vertebrates the ADAR1 p150 isoform is encoded by a late IFN-inducible transcript, and the protein accumulates in the cytoplasm where it can edit cytoplasmic virus dsRNA at late stages of infection. This is the only ADAR protein that would be expected to inhibit RNAi phenomena in the cytoplasm, and tests using human ADARs expressed in Drosophila support this; ADAR1 p150 antagonizes RNAi directed by a polyadenylated, cytoplasmic white RNA hairpin, whereas catalytically inactive cytoplasmic hADAR1 E912A or nuclear ADAR1 p110, ADAR2, or Drosophila ADAR are less effective (Heale et al. 2009).
ADAR and nuclear heterochromatin gene silencing in Drosophila
Because Drosophila ADAR is nuclear, it should instead affect nuclear RNAi, which leads to transcriptional silencing of gene expression, heterochromatin formation, and position effect variegation (PEV) of transgenes inserted into repetitive heterochromatin regions of the genome (Pal-Bhadra et al. 2004). The mechanism by which nuclear RNAi leads to gene silencing is not well understood, and it is very interesting to study in Drosophila, which operates a conserved metazoan gene silencing mechanism even though Dipteran insects have largely dispensed with DNA CpG methylation during evolution.
To address the role of ADAR in RNAi-directed gene silencing, Savva and Reenan (Savva et al. 2013) expressed an epitope-tagged transgene (dADAR-HA) in the large larval salivary gland cells that produce glue protein to attach the larva to a surface where it pupates. Salivary gland cells have endopolyploidization of paired chromosomes to form polytene chromosomes with about 1000 copies of the euchromatic DNA accurately aligned. Salivary gland polytene chromosome squash preparations, partially fixed with formaldehyde to retain proteins on the chromatin, revealed dADAR-HA located at the distal tip of the small fourth chromosome. Localization of dADAR-HA to this site was sensitive to treatment of the chromosome squashes with dsRNA-specific nucleases (Savva et al. 2013). Two other epitope-tagged dsRNA-binding proteins—hmADAR-HA from Hydra magnipapillata, which has five dsRNA-binding domains, and FSH-B2-HA, a protein from flock house virus that inhibits DICER2—also localize to the tip of chromosome 4 when expressed in larval salivary glands, indicating that localization is targeted to a genome-expressed dsRNA (Savva et al. 2013). The most distal known gene on chromosome 4 contains multiple intronic transposons of the Hoppel type that are enriched on the fourth chromosome. Two of these intronic Hoppel elements are adjacent and inverted relative to each other and form a perfect fold-back dsRNA ∼2 kb long that is edited by ADAR. Drosophila transgenic lines made to express this Hoppel inverted repeat under GAL4 control in larval salivary glands localize dADAR-HA to the new UAS-Hoppel transgene insertion sites (Savva et al. 2013).
The authors showed that the inverted Hoppel repeat dsRNA generates endogenous siRNAs (esiRNAs) that silence the other Hoppels on the fourth chromosome by RNAi-directed heterochromatin formation. They used homologous recombination to delete the Hoppel repeats from this locus, which they call the Hoppel-killer (Hok+) locus. Two mutant alleles, Hokmwand HokloxP, are deletions of Hok that, respectively, leave a mini-white gene or only a loxP site, and no copies of Hoppel, at the Hok locus (Savva et al. 2013). In Hok+/Hokmw heterozygous flies or in Hok+ trans-heterozygotes with other mini-white transgene insertions close to Hoppel elements elsewhere on chromosome 4, position effect variegation (PEV) of the mini-white gene occurs. On the other hand, the Hokmw/Hokmw or Hokloxp/Hokmw heterozygotes lacking the Hoppel inverted-repeat dsRNA show a fully expressed mini-white gene that gives red eye color without variegation (Savva et al. 2013). This shows that the Hoppel-killer locus does produce the siRNAs that target heterochromatin silencing to other copies of the Hoppel transposon.
Hyperediting of dsRNA by ADARs in vitro has been shown to inhibit processing of dsRNA by DICER (Scadden and Smith 2001); the lower levels of ADAR editing typically found in vivo are expected to locally destabilize dsRNA structure and have been proposed to increase degradation of edited dsRNA (Scadden 2005). Therefore, ADAR editing should compete with DICER2 for dsRNA substrates produced by many types of transposons and by reducing siRNA production reduce PEV and heterochromatin formation caused by RNAi. ADAR certainly does antagonize RNAi; the Adar5G1 null mutant increases variegation of red eye color (increases RNAi), in Hok+/Hokmw and Hok+ trans-heterozygotes with other variegating mini-white transgenes on the fourth chromosome (Savva et al. 2013). The Adar5G1 mutation also decreases whole fly head extract levels of H3K4 trimethyl activating mark and increases H3K9 (monomethyl and dimethyl) silencing marks, indicating an impressive increase in overall head gene silencing. However, only three out of five tested transposon types show reduced expression. Decreased heterochromatin silencing and increased transposable element (TE) expression are also associated with aging in Drosophila; this has given rise to a transposon theory of aging (Wood and Helfand 2013), although transposon activation could be a correlate of impairments in a range of defense mechanisms with age. A recent publication shows that dietary restriction extends life span and prevents this decrease in heterochromatin silencing (Wood et al. 2016). Reduced expression of Adar or overexpression of Dicer2 also prevents the age-related increase in TE expression. Increased Dicer2 expression extends life span, but reduced Adar expression, which would cause decreases in expression of some TEs but may cause increases in others, was not shown to extend life span (Wood et al. 2016).
Another way that ADAR could affect DICER function would be by a direct ADAR–DICER2 protein–protein interaction. A very interesting observation by Savva is that the two ADAR isoforms; ADAR S-HA and ADAR G-HA, expressed from the endogenous Adar locus, have distinct effects on RNAi. The adult ADAR G-HA isoform, even though it is a less efficient RNA editing enzyme, suppresses RNAi strongly whereas ADAR S-HA does not suppress and may enhance RNAi (Savva et al. 2013). A difference between the isoforms is that the ADAR S-HA isoform is predominantly present in the nucleolus, whereas the ADAR G-HA is also located in unique spots within the nucleus (Savva et al. 2012). This suggestion of an isoform switch controlling Drosophila ADAR–DICER2 interactions is reminiscent of interactions described between vertebrate ADAR1 protein and vertebrate DICER (Ota et al. 2013). The DUF283 domain of DICER, which is related to dsRBDs, and dsRBD3 of ADAR1 are required for these interactions. dsRBD3 of ADAR1 has also been implicated in ADAR1 dimerization, suggesting that ADAR1 dsRBD3 chooses between interacting with another dsRBD3 or with the DUF283 domain of DICER. However, while Drosophila DICER2 is the ortholog of vertebrate DICER, Drosophila ADAR is the ortholog of vertebrate ADAR2, not of vertebrate ADAR1. Vertebrate ADAR2 is not known to interact with DICER, and it remains to be seen whether it does or whether Drosophila ADAR is more ADAR1-like in this respect. It would be interesting to determine whether vertebrate ADAR2 expressed in Drosophila mimics Drosophila dADAR effects on RNAi.
EVOLUTION OF ADARS
ADAR RNA editing in invertebrates
All metazoan ADARs share a common domain architecture: amino-terminal dsRBDs and a carboxy-terminal deaminase domain. Adar genes encoding proteins with ADAR1-type and ADAR2-type deaminase domains are distinguishable by sequence comparisons (Keegan et al. 2004). Both ADAR1 and ADAR2 genes are present in the genomes of the most basal metazoans, the Cnidaria, represented by the genome sequences of the starlet sea anemone Nematostella vectensis (Putnam et al. 2007) and other cnidarians and corals (Keegan et al. 2011). This is surprising, as it indicates that the single Adar gene in Drosophila does not correspond to an ancestor of both ADAR1 and ADAR2 in vertebrates. Instead, the evolutionary lineage between Nematostella and Drosophila lost ADAR1 (Keegan et al. 2011) and also about 1700 other protein domains, many more than have been lost between Nematostella and humans (Putnam et al. 2007). This gene loss appears to continue further in Caenorhabditis elegans. Arthropods and nematodes have been proposed to belong to an ecdysozoan group of animals that grow by molting (Aguinaldo et al. 1997). The physiology of the protostome group appears to have evolved away from Nematostella more dramatically than the deuterostomes (chordates, echinoderms, and mollusks) have. In octopus and squid, which are mollusks, genes encoding distinguishable ADAR1 and ADAR2 deaminases with dsRBDs are also present. ADAR1 currently appears to be absent from insects and crustaceans but present in arachnids and chelicerates (Keegan et al. 2011); analyses of further genomes may yet reveal a more complex pattern.
The evolutionary rationale for the expansion of site-specific editing in Drosophila and some other invertebrates is unclear. Drosophila belongs to the highly evolved dipteran insects, which are very successful and diversified, having evolved a complete metamorphosis between the legless larva and the two-winged adult dispersal stages. Drosophila ADAR RNA editing appears to be linked with the complete metamorphosis in Diptera; Adar expression and the number of edited sites in mRNAs peak in the pupa and adult when legs and wings are first formed and innervated (Graveley et al. 2011; Savva et al. 2012). For human health, the immune systems of biting insects in this advanced dipteran group are very important to understand, because they are vectors of major human diseases caused by protozoans, such as malaria and trypanosomiasis, and by viruses, such as dengue virus, Zika virus, West Nile virus, and many other arthropod-borne viruses (arboviruses). The even greater expansion of ADAR site-specific editing in octopus and squid (Liscovitch-Brauer et al. 2017) further suggests a link with evolution of the most sophisticated nervous systems and behaviors within different invertebrate groups. Studies on proteins from related fish and invertebrate species living at different ocean temperatures suggested that cold adaptation of enzymatic activity is facilitated by substituting smaller for larger amino acid side chains at key points of flexibility; ADAR RNA editing tends to do this, introducing many serines and glycines, and it has been proposed that editing facilitates cold adaptation of ion channels in octopuses (Garrett and Rosenthal 2012). Intriguingly, even the editing of the ser 458 loop in Drosophila ADAR conforms to this pattern, suggesting that the edited and unedited isoforms might have different temperature responses.
Other evolutionary approaches to mRNA editing
ADAR genes are not present in prokaryotic genomes. Prokaryotes have heterodimeric tRNA adenosine deaminases (TadA and TadB) and the catalytic activity resides in the TadB subunit. The TadA/TadB complex modifies adenosines at anticodon position 34, to give the inosine residues that participate in wobble decoding by certain tRNAs (Gerber and Keller 2001). The eukaryotic orthologs of TadA and TadB are the heterodimeric adenosine deaminases acting on tRNAs at position 34 (ADAT2 and ADAT3) that show some enlargement of the deaminase domain compared with TadA and TadB (Gerber and Keller 2001; Macbeth et al. 2005). Yeast and all other eukaryotes also have another monomeric ADAT1 enzyme that modifies position 37 in tRNAs, a position where tRNA base modifications prevent frameshifting. Yeast ADAT1 has a longer deaminase domain like metazoan ADARs, representing the first evolutionary appearance of the ADAR-type deaminase domain: ADAT1 has no dsRBDs (Gerber and Keller 2001). Comparative sequence analyses revealed that ADAT1 and ADARs probably evolved from a eukaryotic ADAT2-type protein, with loss of the ADAT3 dimer partner and further carboxy-terminal lengthening of the deaminase domain.
The fungus, Fusarium graminearum, which causes wheat head blight disease, was recently found to have meiosis-specific A to G changes in many mRNAs. Particularly common are stop-loss editing events that allow translation to read through stop codons in tRNA-resembling loops at the ends of target RNA hairpins. Stop-loss editing occurs in the transcript encoding the Tor homolog and in many other transcripts. Surprisingly, knockout of the ADAT1-like gene had no effect on this editing (Wang et al. 2016), suggesting that it may be due to the essential ADAT2 and ADAT3 proteins. Neurospora crassa also shows similar very extensive site-specific A to G editing during meiosis. The purpose of this editing during meiosis is unknown but it appears that in Fusarium, some proteins, such as TOR, are expressed as full-length proteins only after transcript editing. Similar meiosis-specific ADAT2/3 editing in other organisms, including multicellular organisms, might remain undetected.
It appears likely that ADAT1 proteins are also dsRNA-binding proteins, although there is no evidence yet that they edit any dsRNAs. From the structure of the ADAR2 deaminase domain–Bdf2 RNA complex and sequence comparisons to yeast ADAT1, it is clear that yeast ADAT1 has already evolved the whole domain that contacts dsRNA both 5′ of and 3′ of the edited A. The potential to form contacts on both sides of the edited A is unlikely to be required to edit tRNAs at position 37, because there is no more dsRNA 3′ of the edited A in the tRNA anticodon stem. Therefore, ADAT1 may also naturally recognize longer dsRNAs in yeast. It is possible that Bdf2 and other yeast RNAs found to bind ADAR2 are natural binding substrates of yeast ADAT1; if this is the case then the ADAR2 deaminase domain–Brf2 RNA complex (Matthews et al. 2016) could represent a more “ADAT1-type” mode of binding.
The evolution of the ADAT1/ADAR-type deaminase is associated, for unknown reasons, with binding of a molecule of inositol hexakisphosphate (called IHP or IP6) (Irvine 2005). The IP6 is required for RNA editing activity and almost entirely covered by the most evolutionarily recently acquired carboxy-terminal region of the deaminase domain (Macbeth et al. 2005). Yeast ADAT1 also binds IP6 though less tightly than ADAR2, allowing some exchange of IP6. It is now clear that a large number of nuclear and chromatin proteins, such as histone deacetylase complexes, bind IP4 and IP6 and depend on binding of these higher phosphoinositides for their activities (Watson et al. 2016). Further work is required to understand why ADAR-type deaminase domains evolved this interaction with IP6. Presumably, the IP6 was more readily exchangeable earlier in evolution and placed ADAR editing under control of IP6 signaling. ADAT1 appears to be present throughout the protists and only ADAT1, but no ADARs have been found in Trypanosoma. However, it will not be surprising if continuing analyses of genome sequences in the widely diverged protists yields further evolutionary intermediates between ADAT1 proteins and ADARs.
The ctenophores or comb jellies are a group of metazoans including swimming species that look somewhat like active cnidarians such as jellyfish. However, comb jellies move using ciliary combs in eight external rows rather than by muscle contraction as in jellyfish and have adhesive rather than stinging feeding organs on the paired tentacles. Ctenophores are distant from cnidarians and other metazoans based on sequence comparisons of ribosomal RNAs and proteins (Ryan et al. 2013). Ctenophores show expansions of ADAT1-like but not ADAR-like genes (Grice and Degnan 2015; Kohn et al. 2015). It remains to detect examples of RNA editing in transcripts encoding glutamate or acetylcholine receptors or other transcripts in organs where ADAT1-type transcripts are expressed, such as the aboral organ that forms the center of the CNS and links to the comb rows that control movement, and to show that the ADAT1-type proteins contribute to editing. An unusual type of RNA editing may be related to other features of ctenophore nervous systems, which appear highly derived compared with those of other metazoans, especially in presynaptic structures (Marlow and Arendt 2014). Ctenophores may have recruited an ADAT1 for editing, either independently of, or after divergence from, the standard Metazoan ADAR gene pattern.
CONCLUSION
ADAR RNA editing, unlike other modifications such as N6 methyladenosine modification, changes codon meaning. This recoding role is what makes ADAR functions significantly more complicated to disentangle than the effects of those other RNA modifications. Recent years have seen a considerable expansion in described effects of ADAR RNA editing on Drosophila CNS function. Previously, the Adar mutant alleles available severely affected locomotion, preventing investigation of more interesting behavioral effects. The recent study on sleep shows that new hypomorphic alleles of Adar with less impaired locomotion are very valuable (Robinson et al. 2016). It may soon be possible to assign aspects of the Adar mutant phenotype to loss of editing in certain sets of transcripts, such as synapsin, synaptotagmin, and cacophony transcripts in the case of NMJ defects, glutaminergic vesicle accumulations, synaptic plasticity, and sleep.
Drosophila and other arthropods lack a homolog of the vertebrate ADAR1 protein that has been shown to play a very important role in innate immunity by controlling innate immune responses to dsRNA, through editing the RNA and also probably through protein interactions with innate immune sensor helicases and the evolutionarily related helicase DICERs. It will be very interesting to study roles of ADAR1 proteins in invertebrates that do have them, such as cephalopods or chelicerates. However, the absence of ADAR1 in flies and the lack of evidence that ADAR2 plays any role in vertebrate innate immunity make it more difficult to interpret the interesting new finding that Drosophila ADAR isoforms interact with heterochromatin gene silencing (Savva et al. 2013). Is this something that all ADAR2 proteins do or is it an arthropod-specific adaptation? Much of the work on ADAR2-type proteins now uses Drosophila, and the recent structure of an ADAR2 deaminase domain–RNA complex helps to explain the effect of editing in the Adar transcript itself to produce an isoform with quite distinct effects. The deaminase domain–RNA complex may also now identify ADAR residues likely to be involved in interactions with other proteins.
ADAR editing of adenosine to inosine in RNA is the leading example of an enzymatic RNA modification. If the more recently discovered examples of enzymatic mRNA modification have similarly diverse effects, then the Epitranscriptome field has much work to do. However, some effects of ADARs may act in parallel with effects of other RNA modifications, allowing the research on ADARs to guide the way (O'Connell et al. 2015). This might be true for the ADAR1 effects on dsRNA recognition by innate immune sensors in vertebrates and possibly in some invertebrates, such as cephalopods and chelicerates. Alterations in ADAR RNA editing are also significant in many cancer cell types; this too may involve the innate immune role of ADAR1. Other RNA modifications may also be abnormal and have significant effects in cancer cells. Of course, the new RNA modifications are also being found below the backbone and are being studied in Drosophila also (Haussmann et al. 2016; Lence et al. 2016), which will facilitate understanding their fundamental roles.
ACKNOWLEDGMENTS
This project has received funding from the European Union's Seventh Framework Programme for Research, Technological Development and Demonstration under grant agreement no. 621368.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.060921.117.
REFERENCES
- Aguinaldo AMA, Turbeville JM, Linford LS, Rivera MC, Garey JR, Raff RA, Lake JA. 1997. Evidence for a clade of nematodes, arthropods and other moulting animals. Nature 387: 489–493. [DOI] [PubMed] [Google Scholar]
- Bass BL, Weintraub H. 1988. An unwinding activity that covalently modifies its double-strand RNA substrate. Cell 55: 1089–1098. [DOI] [PubMed] [Google Scholar]
- Bhogal B, Jepson JE, Savva YA, Pepper ASR, Reenan RA, Jongens TA. 2011. Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat Neurosci 14: 1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter JA, Obbard DJ, Maside X, Jiggins FM. 2007. The recent spread of a vertically transmitted virus through populations of Drosophila melanogaster. Mol Ecol 16: 3947–3954. [DOI] [PubMed] [Google Scholar]
- Carpenter JA, Keegan LP, Wilfert L, O'Connell MA, Jiggins FM. 2009. Evidence for ADAR-induced hypermutation of the Drosophila sigma virus (Rhabdoviridae). BMC Genet 10: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catanese DJ Jr, Matthews KS. 2011. Disconnected Interacting Protein 1 binds with high affinity to pre-tRNA and ADAT. Biochem Biophys Res Commun 414: 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K. 2000. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 6: 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly CM, Dearth AT, Braun RE. 2005. Disruption of murine Tenr results in teratospermia and male infertility. Dev Biol 278: 13–21. [DOI] [PubMed] [Google Scholar]
- DeSousa D, Mukhopadhyay M, Pelka P, Zhao X, Dey BK, Robert V, Pélisson A, Bucheton A, Campos AR. 2003. A novel double-stranded RNA-binding protein, disco interacting protein 1 (DIP1), contributes to cell fate decisions during Drosophila development. J Biol Chem 278: 38040–38050. [DOI] [PubMed] [Google Scholar]
- Diegelmann S, Nieratschker V, Werner U, Hoppe J, Zars T, Buchner E. 2006. The conserved protein kinase-A target motif in synapsin of Drosophila is effectively modified by pre-mRNA editing. BMC Neurosci 7: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM. 2013. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80: 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eifler T, Pokharel S, Beal PA. 2013. RNA-Seq analysis identifies a novel set of editing substrates for human ADAR2 present in Saccharomyces cerevisiae. Biochemistry 52: 7857–7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli D, Gacquer D, Rothé F, Lefort A, Libert F, Brown D, Kheddoumi N, Shlien A, Konopka T, Salgado R, et al. 2015. Principles governing A-to-I RNA editing in the breast cancer transcriptome. Cell Rep 13: 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeano F, Tomaselli S, Locatelli F, Gallo A. 2012. A-to-I RNA editing: the “ADAR” side of human cancer. Semin Cell Dev Biol 23: 244–250. [DOI] [PubMed] [Google Scholar]
- Gallo A, Locatelli F. 2012. ADARs: allies or enemies? The importance of A-to-I RNA editing in human disease: from cancer to HIV-1. Biol Rev Camb Philos Soc 87: 95–110. [DOI] [PubMed] [Google Scholar]
- Garrett S, Rosenthal JJ. 2012. RNA editing underlies temperature adaptation in K+ channels from polar octopuses. Science 335: 848–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP, Keller W. 2001. RNA editing by base deamination: more enzymes, more targets, new mysteries. Trends Biochem Sci 26: 376–384. [DOI] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice LF, Degnan BM. 2015. The origin of the ADAR gene family and animal RNA editing. BMC Evol Biol 15: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HM, Eterovic AK, Yuan Y, et al. 2015. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell 28: 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. 2004. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 279: 4894–4902. [DOI] [PubMed] [Google Scholar]
- Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, Fray RG, Soller M. 2016. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 540: 301–304. [DOI] [PubMed] [Google Scholar]
- Heale BS, Keegan LP, McGurk L, Michlewski G, Brindle J, Stanton CM, Caceres JF, O'Connell MA. 2009. Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J 28: 3145–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideyama T, Yamashita T, Suzuki T, Tsuji S, Higuchi M, Seeburg PH, Takahashi R, Misawa H, Kwak S. 2010. Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J Neurosci 30: 11917–11925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Single FN, Köhler M, Sommer B, Sprengel R, Seeburg PH. 1993. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell 75: 1361–1370. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single F, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg P. 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406: 78–81. [DOI] [PubMed] [Google Scholar]
- Hogg M, Paro S, Keegan LP, O'Connell MA. 2011. RNA editing by mammalian ADARs. Adv Genet 73: 87–120. [DOI] [PubMed] [Google Scholar]
- Irvine RF. 2005. Inositide evolution - towards turtle domination? J Physiol 566: 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson JE, Savva YA, Yokose C, Sugden AU, Sahin A, Reenan RA. 2011. Engineered alterations in RNA editing modulate complex behavior in Drosophila: regulatory diversity of adenosine deaminase acting on RNA (ADAR) targets. J Biol Chem 286: 8325–8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Saito K, Kin T, Ono Y, Asai K, Sunohara T, Okada TN, Siomi MC, Siomi H. 2008. Drosophila endogenous small RNAs bind to Argonaute 2 in somatic cells. Nature 453: 793–797. [DOI] [PubMed] [Google Scholar]
- Keegan LP, Gallo A, O'Connell MA. 2001. The many roles of an RNA editor. Nat Rev Genet 2: 869–878. [DOI] [PubMed] [Google Scholar]
- Keegan LP, Leroy A, Sproul D, O'Connell MA. 2004. Adenosine deaminases acting on RNA (ADARs): RNA-editing enzymes. Genome Biol 5: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan LP, Brindle J, Gallo A, Leroy A, Reenan RA, O'Connell MA. 2005. Tuning of RNA editing by ADAR is required in Drosophila. EMBO J 24: 2183–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan LP, McGurk L, Palavicini JP, Brindle J, Paro S, Li X, Rosenthal JJ, O'Connell MA. 2011. Functional conservation in human and Drosophila of Metazoan ADAR2 involved in RNA editing: loss of ADAR1 in insects. Nucleic Acids Res 39: 7249–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn AB, Sanford RS, Yoshida MA, Moroz LL. 2015. Parallel evolution and lineage-specific expansion of RNA editing in ctenophores. Integr comp Biol 55: 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, Kreim N, Andrade-Navarro MA, Poeck B, Helm M. 2016. m6A modulates neuronal functions and sex determination in Drosophila. Nature 540: 242–247. [DOI] [PubMed] [Google Scholar]
- Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D, et al. 2004. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 22: 1001–1005. [DOI] [PubMed] [Google Scholar]
- Li X, Overton IM, Baines RA, Keegan LP, O'Connell MA. 2014. The ADAR RNA editing enzyme controls neuronal excitability in Drosophila melanogaster. Nucleic Acids Res 42: 1139–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscovitch-Brauer N, Alon S, Porath HT, Elstein B, Unger R, Ziv T, Admon A, Levanon EY, Rosenthal JJ, Eisenberg E. 2017. Trade-off between transcriptome plasticity and genome evolution in cephalopods. Cell 169: 191–202.e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Samuel CE. 1996. Mechanism of interferon action: functionally distinct RNA-binding and catalytic domains in the interferon-inducible, double-stranded RNA-specific adenosine deaminase. J Virol 70: 1961–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lei M, Samuel CE. 2000. Chimeric double-stranded RNA-specific adenosine deaminase ADAR1 proteins reveal functional selectivity of double-stranded RNA-binding domains from ADAR1 and protein kinase PKR. Proc Natl Acad Sci 97: 12541–12546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. 2005. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 309: 1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado C, Alicea D, Gonzalez M, Bykhovskaia M, Marie B. 2013. Adar is essential for optimal presynaptic function. Mol Cell Neurosci 52: 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow H, Arendt D. 2014. Evolution: ctenophore genomes and the origin of neurons. Curr Biol 24: R757–R761. [DOI] [PubMed] [Google Scholar]
- Matthews MM, Thomas JM, Zheng Y, Tran K, Phelps KJ, Scott AI, Havel J, Fisher AJ, Beal PA. 2016. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat Struct Mol Biol 23: 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, Seeburg PH. 1996. RED2, a brain specific member of the RNA-specific adenosine deaminase family. J Biol Chem 271: 31795–31798. [DOI] [PubMed] [Google Scholar]
- O'Connell MA, Krause S, Higuchi M, Hsuan JJ, Totty NF, Jenny A, Keller W. 1995. Cloning of cDNAs encoding mammalian double-stranded RNA-specific adenosine deaminase. Mol Cell Biol 15: 1389–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell MA, Mannion NM, Keegan LP. 2015. The epitranscriptome and innate immunity. PLoS Genet 11: e1005687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. 2013. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 153: 575–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal-Bhadra M, Leibovitch BA, Gandhi SG, Chikka MR, Bhadra U, Birchler JA, Elgin SC. 2004. Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science 303: 669–672. [DOI] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. 2000a. dADAR, a Drosophila double-stranded RNA-specific adenosine deaminase is highly developmentally regulated and is itself a target for RNA editing. RNA 6: 1004–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. 2000b. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell 102: 437–449. [DOI] [PubMed] [Google Scholar]
- Pan B, Nath S, Sun L, Hart D, Sundaralingam M. 1998. Crystal structure of an RNA octamer duplex r (CCCIUGGG) 2 incorporating tandem I· U wobbles. Nucleic Acids Res 26: 5699–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Yaacov N, Bazak L, Buchumenski I, Porath HT, Danan-Gotthold M, Knisbacher BA, Eisenberg E, Levanon EY. 2015. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep 13: 267–276. [DOI] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. 2015. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43: 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV, et al. 2007. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317: 86–94. [DOI] [PubMed] [Google Scholar]
- Reenan RA, Hanrahan CJ, Barry G. 2000. The mle(napts) RNA helicase mutation in Drosophila results in a splicing catastrophe of the para Na+ channel transcript in a region of RNA editing. Neuron 25: 139–149. [DOI] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. 2012. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 44: 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedmann EM, Schopoff S, Hartner JC, Jantsch MF. 2008. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA 14: 1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JE, Paluch J, Dickman DK, Joiner WJ. 2016. ADAR-mediated RNA editing suppresses sleep by acting as a brake on glutamatergic synaptic plasticity. Nat Commun 7: 10512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, Lin MF, et al. 2010. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330: 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JF, Pang K, Schnitzler CE, Nguyen A-D, Moreland RT, Simmons DK, Koch BJ, Francis WR, Havlak P; NISC Comparative Sequencing Program. 2013. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science 342: 1242592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter JM, Schultz SC. 1998. Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J 17: 7505–7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva YA, Jepson JEC, Sahin A, Sugden AU, Dorsky JS, Alpert L, Lawrence C, Reenan RA. 2012. Auto-regulatory RNA editing fine-tunes mRNA re-coding and complex behaviour in Drosophila. Nat Commun 3: 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savva YA, Jepson JE, Chang Y-J, Whitaker R, Jones BC, St Laurent G, Tackett MR, Kapranov P, Jiang N, Du G. 2013. RNA editing regulates transposon-mediated heterochromatic gene silencing. Nat Commun 4: 2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden AD. 2005. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat Struct Mol Biol 12: 489–496. [DOI] [PubMed] [Google Scholar]
- Scadden AD, Smith CW. 2001. RNAi is antagonized by A-->I hyper-editing. EMBO Rep 2: 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra MJ, Smolter PE, Westhof E. 2004. Pronounced instability of tandem IU base pairs in RNA. Nucleic Acids Res 32: 1824–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefl R, Oberstrass FC, Hood JL, Jourdan M, Zimmermann M, Skrisovska L, Maris C, Peng L, Hofr C, Emeson RB, et al. 2010. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell 143: 225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JM, Beal PA. 2017. How do ADARs bind RNA? New protein-RNA structures illuminate substrate recognition by the RNA editing ADARs. Bioessays 39 10.1002/bies.201600187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner RW, Smith JE, Cooperman BS, Nishikura K. 1989. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc Natl Acad Sci 86: 2647–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Khillan J, Gadue P, Nishikura K. 2000. Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 290: 1765–1768. [DOI] [PubMed] [Google Scholar]
- Wang C, Xu JR, Liu H. 2016. A-to-I RNA editing independent of ADARs in filamentous fungi. RNA Biol 13: 940–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PJ, Millard CJ, Riley AM, Robertson NS, Wright LC, Godage HY, Cowley SM, Jamieson AG, Potter BV, Schwabe JW. 2016. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat Commun 7: 11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SK, Sato S, Lazinski DW. 2001. Substrate recognition by ADAR1 and ADAR2. RNA 7: 846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Helfand SL. 2013. Chromatin structure and transposable elements in organismal aging. Front Genet 4: 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Jones BC, Jiang N, Chang C, Hosier S, Wickremesinghe P, Garcia M, Hartnett DA, Burhenn L, Neretti N, et al. 2016. Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc Natl Acad Sci 113: 11277–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW III, et al. 2009. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 10: R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhang L, Gao X, Qi S, Chang Z, Wu Q. 2015. DIP1 plays an antiviral role against DCV infection in Drosophila melanogaster. Biochem Biophys Res Commun 460: 222–226. [DOI] [PubMed] [Google Scholar]