

This review by McKinnon focuses on the etiology of DNA damage in the nervous system and the multiple DNA repair pathways that maintain genome stability and prevent human neurologic disease.
Keywords: DNA damage, nervous system, genome stability, neurodevelopment, neurologic disease
Abstract
Multiple DNA repair pathways maintain genome stability and ensure that DNA remains essentially unchanged over the life of a cell. Various human diseases occur if DNA repair is compromised, and most of these impact the nervous system, in some cases exclusively. However, it is often unclear what specific endogenous damage underpins disease pathology. Generally, the types of causative DNA damage are associated with replication, transcription, or oxidative metabolism; other direct sources of endogenous lesions may arise from aberrant topoisomerase activity or ribonucleotide incorporation into DNA. This review focuses on the etiology of DNA damage in the nervous system and the genome stability pathways that prevent human neurologic disease.
Genome stability is critically important for human health. This is apparent from a myriad of inherited human syndromes characterized by defective DNA damage responses (McKinnon 2013; Madabhushi et al. 2014). These syndromes reveal that the nervous and immune systems are particularly susceptible to the consequences of DNA damage. Cancer is also a frequent feature of DNA repair deficiency syndromes. Defects in genome integrity are also increasingly being linked to broader health issues, including age-related degenerative events that mar cognitive ability and quality of life (Qiu et al. 2014; Chow and Herrup 2015; Suberbielle et al. 2015; Hill et al. 2016; Sepe et al. 2016; Barzilai et al. 2017). Clearly, understanding the mechanistic connections between faulty DNA damage signaling and human disease is of fundamental biomedical importance.
The nervous system is an integrated and expansive collection of different cell and tissue types. More than other organ systems, this tissue has a multitude of levels of control, including homeostasis within the brain, and controlling roles in fundamental aspects of human physiology, including breathing, heart function, endocrine control, and integration of environmental cues (Stiles and Jernigan 2010). Consideration of how DNA damage impacts neural function encompasses several areas, including neurodevelopment, where neurogenesis involves widespread cellular proliferation and, subsequently, migration, differentiation, and maturation, as well as synaptic connectivity to establish the functional nervous system. After birth, some ongoing neurogenesis occurs, but, for the bulk of this tissue, genome maintenance is needed in long-lived nonreplicating neurons. Over the life of an organism, comprehensive genome maintenance machinery is in place to prevent and correct DNA lesions that frequently occur.
An early appreciation of the potential importance of DNA repair pathways in the brain came from the study of human syndromes such as ataxia telangiectasia (A-T), which is characterized by hypersensitivity to ionizing radiation and neurodegeneration, implying a connection between these two features. Subsequently, a firm link identifying specific mutation of DNA damage response factors that cause neurodegenerative or neurodevelopmental syndromes has emerged during the last few decades. These human diseases are direct indicators of the importance of specific DNA repair pathways, as they reveal the resultant neuropathology arising when DNA repair is defective. Details of the physiologic impact of DNA damage have been greatly aided by the development of disease models that recapitulate many (although not all) of the effects of defective genome maintenance observed in humans. Thus, based on human inherited diseases and various mammalian experimental systems with engineered defects in DNA damage signaling, it is abundantly clear that the neural genome is constantly under threat from endogenous events that damage DNA.
The molecular details of genome maintenance processes are well understood in the immune system, where they involve controlled rearrangements via RAG1/2 such as V(D)J recombination or class switch recombination, which drives T-cell and B-cell maturation to form a functional immune system (Helmink and Sleckman 2012; Alt et al. 2013; Tubbs and Nussenzweig 2017). Early studies identified expression of RAG1 transcripts in the murine nervous system (Chun et al. 1991), although the implication of this finding is unclear, as evidence for recombination processes similar to those in T cells and B cells in the immune system have not been identified in the brain. While the nervous system is markedly impacted by DNA repair deficiency, far less is known in this tissue about the actual mechanisms causing neurodegenerative syndromes, and, in this setting, much of the etiology leading to persistent genome damage that derails normal cellular homeostasis remains unclear.
This review considers the causative agents that lead to neuropathology in various human genome instability syndromes and details the functional relevance of DNA repair and response factors in the nervous system. This includes the key neurodevelopmental events at threat from DNA damage and the central genome maintenance pathways that preserve the integrity of DNA in the mature nervous system. As the bulk of the mature nervous system is nonreplicating, the maintenance factors during development (replicating and immature, nonreplicating) and those critical in the adult (mature, nonreplicating) are different.
Endogenous DNA damage in the nervous system
To understand how DNA repair-deficient syndromes result in genome instability and neuropathology, two broad unanswered questions exist: (1) What are the responsible endogenous pathogenic lesions? (2) How do these selectively impact the nervous system in a cell type-specific or tissue-specific manner? For instance, why is the cerebellum so often a prevalent site of neuropathology? Is it due to increased oxidative metabolism, aspects of postnatal development, or transcriptional activity in certain cerebellar cell types that drive genome instability?
The lesions that cause neurologic disease in DNA repair deficiency syndromes can be partitioned between those in the developing or mature nervous system. During neurogenesis, where proliferation is a driving force for neural development, replication stress (abnormal replication fork progression) is a major source of DNA damage that can perturb neural development. Other sources of DNA damage are likely to be more pertinent in the mature nervous system but can also occur during neurogenesis. These include transcriptional disruption or oxidative damage, where reactive species chemically modify DNA, or free radicals that generate DNA strand breaks. Finally, other metabolic products may also be genotoxic, such as aldehydes (Langevin et al. 2011). Most DNA repair deficiency syndromes present relatively early in childhood, tacitly implying a developmental component of the disease. Thus, a reasonable assumption for many syndromes is that the impact of DNA damage happens during development. There is also likely to be a progressive phase, where cumulative genome damage worsens the presentation of the disease. Mouse models with targeted mutations in various DNA repair factors important for maintaining the genome—such as XRCC1, which suppresses single-strand break (SSB) accumulation, or those necessary to prevent DNA double-strand breaks (DSBs; e.g., TOPBP1 or DNA ligase IV)—show that endogenous DNA damage is a frequent event in the nervous system (Barnes et al. 1998; Gao et al. 1998; Lee et al. 2009, 2012b; Shimada et al. 2015).
Replication stress in the nervous system
An early source of DNA damage in the developing nervous system is replication-associated damage during neurogenesis (Lee et al. 2012b; McKinnon 2013; Magdalou et al. 2014; Zeman and Cimprich 2014; O'Driscoll 2017). Replication stress is connected to a variety of syndromes that have neurologic involvement (Table 1; Harley et al. 2016; O'Driscoll 2017; Reynolds et al. 2017). A key responder to replication stress is the DNA damage response serine/threonine kinase ATR (A-T and rad3-related) (Nam and Cortez 2011; Marechal and Zou 2013). ATR is an essential kinase that prevents DNA damage accumulation during replication, and hypomorphic mutation of this kinase can result in ATR-Seckel syndrome, a developmental disorder that affects multiple organs, including the nervous system (O'Driscoll et al. 2003). Many of the DNA damage response factors that prevent replication-associated lesions are essential for proliferation, and mutations causing dysfunction in these factors are generally hypomorphic, thereby maintaining some level of function in the defective protein (Baple et al. 2014; O'Driscoll 2017). ATR is activated by replication protein A (RPA)-coated stretches of ssDNA that accumulate at stalled replication forks (Marechal and Zou 2013). ATR signaling depends on the RAD9–RAD1–HUS1 (9-1-1) complex that recognizes DNA termini adjacent to RPA-bound ssDNA (Delacroix et al. 2007). TOPBP1 (topoisomerase II-binding protein 1) is required for ATR activation in conjunction with the ATR-interacting protein (ATRIP), which targets ATR to RPA-coated ssDNA (Kumagai et al. 2006; Wardlaw et al. 2014). The ATR-dependent DNA damage response involves multiple components, including those that activate ATR to phosphorylate downstream substrates such as CHK1 that modulate cell cycle progression (Nam and Cortez 2011; Marechal and Zou 2013; Zeman and Cimprich 2014).
Table 1.
Human neurologic syndromes resulting from defects in the DNA damage response
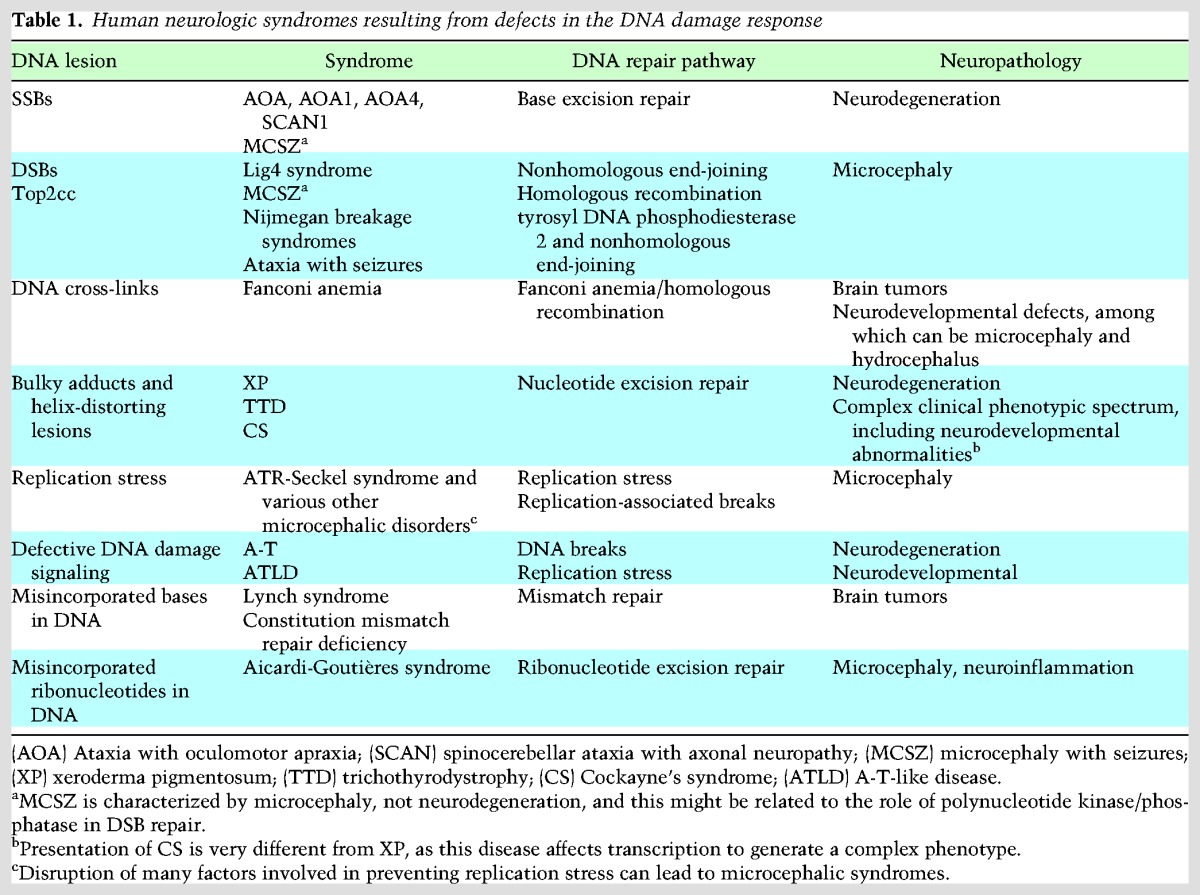
Mouse models of ATR deficiency further highlight the essential requirement for this DNA damage response kinase (Brown and Baltimore 2000; de Klein et al. 2000; Ruzankina et al. 2007; Murga et al. 2009). Deletion of ATR in cortical progenitors results in a reduced cortical size, although with the six-layer lamination (that reflects functional compartmentation in the cortex) still present, suggesting a generalized cellular attrition (Lee et al. 2012c). In contrast, deletion of ATR in the developing cerebellum has little apparent effect until embryonic day 16, a stage when granule neuron progenitor numbers rapidly increase via sonic hedgehog-driven proliferation from the rhombic lip (Hatten and Heintz 1995; Leto et al. 2016). At this point in cerebellar development, ATR-null granule neuron expansion ceases, and cerebellar development is stalled (Lee et al. 2012c). This indicates the critical need for ATR to monitor replication stress during this phase of rapid granule neuron progenitor proliferation. Furthermore, in the neocortex, ATR can modulate apoptosis of DNA-damaged progenitors in the proliferating ventricular zone and is also essential for establishment of a G2 cell cycle checkpoint after DNA damage in these progenitors (Enriquez-Rios et al. 2017). Thus, not all neural tissues respond identically when ATR is defective, and the impact of ATR loss and subsequent defective replication stress signaling can vary between brain regions.
TOPBP1 is critical for signaling replication stress, and this function occurs via its activation of ATR (Nam and Cortez 2011; Marechal and Zou 2013). While ATR activation is a key role of TOPBP1, the consequences of inactivation of this gene in the nervous system are substantially different from that of ATR loss (Lee et al. 2012b,c). Comparative analysis of ATR or TOPBP1 loss specifically in cortical progenitors (where deletion is driven by EMX1-cre) shows that, whereas ATR loss results in a smaller cortex, loss of TOPBP1 causes a complete ablation of this structure, suggesting that TOPBP1 has a broader role in signaling replication stress. Moreover, p53 inactivation substantially rescues this cortical deletion, indicating that while TOPBP1 loss promotes DNA damage-induced p53-dependent apoptosis and that TOPBP1 functions as a damage transducer, it is nonetheless dispensable for proliferation per se (Lee et al. 2012b).
Furthermore, frequent stochastic events such as DNA SSBs can occur during neurogenesis. If these encounter a replication fork, they can form DSBs, a potentially catastrophic lesion that can result in apoptosis, to which neural progenitors are particularly susceptible (McKinnon 2009). Transcriptional complexes can also collide with the replication process, eliciting a repair response similar to replication fork collapse (Hamperl and Cimprich 2016). Thus, endogenous replication-associated damage can have catastrophic effects in the nervous system, and this is likely to be a main etiology involved in neurodevelopmental disease. In contrast to many other DNA lesions, replication stress is a generalized genotoxic insult, compared with the relatively nervous system-selective effects of other types of DNA damage, such as those involving oxidative or transcription-associated damage.
Ribonucleotide (rNTP) incorporation into DNA
While DNA strand breaks and chemical modifications (e.g., base damage, DNA cross-links, etc.) cover a broad group of well-studied types of DNA damage (De Bont and van Larebeke 2004; Helleday et al. 2014), there are also other common DNA lesions that have been recognized recently to substantially impact genome stability (Fig. 1). Among these, rNTP incorporation into DNA, genome damage from transcription-associated R-loop formation (hybridization of nascent primary RNA transcripts to the transcribed DNA strand), and aberrant topoisomerase activity are potential threats to the neural genome (McKinnon 2016; Williams et al. 2016).
Figure 1.

Endogenous DNA damage relevant to diseases of the nervous system. Multiple types of endogenous lesions can impact the nervous system at all stages of development and maturity. Replication stress primarily affects proliferating neural progenitors. rNTPs can become incorporated into the DNA via DNA polymerases during neurogenesis. While this substantially impacts neural progenitors, it is also likely to occur throughout the developing and mature nervous system during DNA repair processes. In the mature nervous system, transcription-associated damage and aberrant topoisomerase activity will be a constant source of potential DNA damage. Oxidative damage can also impact immature cells but will be an ongoing threat to the mature nervous system.
The incorporation of rNTPs into DNA may be one of the most frequent mistakes that occur during DNA replication, with estimates suggesting that on the order of >1,000,000 rNTPs are incorporated during replication in a mammalian cell (Nick McElhinny et al. 2010a; Potenski and Klein 2014; Wallace and Williams 2014; Cerritelli and Crouch 2016; Williams et al. 2016, 2017). In fact, this lesion may be the most common type of endogenous DNA damage encountered by a proliferating cell. A main source of rNTP incorporation is the activity of DNA polymerases during replication (Nick McElhinny et al. 2010a,b; Potenski and Klein 2014; Crespan et al. 2016). While polymerase proofreading to detect base mismatches during DNA synthesis is very efficient, detection of rNTPs is less so, and thus many rNTPs are erroneously incorporated in each cycle of replication. The mismatch repair pathway can work in conjunction with DNA polymerases to remove both mismatched bases in DNA and also rNTP incorporation into DNA (Ghodgaonkar et al. 2013; Lujan et al. 2013). rNTP incorporation into DNA is problematic for DNA integrity, as the defining 2′-OH moiety on the rNTP sugar is particularly reactive and can cause strand breaks via hydrolysis, generating an aberrant 5′-OH end or nucleophilic attack forming a 2′–3′-cyclic phosphate (Williams et al. 2016). Incorporated rNTPs can also promote helical distortions, which can be sensed as DNA damage (Wallace and Williams 2014).
Removal of rNTPs incorporated into DNA is termed rNTP excision repair (RER), and a central player in this process is RNase H2, a multimeric enzyme complex composed of three separate polypeptides: RNase H2A, H2B, and H2C (Sparks et al. 2012; Potenski et al. 2014; Cerritelli and Crouch 2016; Williams et al. 2017). RNase H2 eliminates rNTPs in DNA via an initial 5′ incision to the rNTP followed by flap endonuclease activity prior to ligation (Sparks et al. 2012). These enzymes can also process/remove Okazaki RNA primers remaining after replication (Cerritelli and Crouch 2016). Inactivation of murine RNase H2B results in early embryonic lethality, which is linked to p53 signaling and genome instability, confirming the in vivo importance of RNase H2 during mammalian development (Hiller et al. 2012; Reijns et al. 2012). Hypomorphic mutations in any of the three subunits of human RNase H2 can lead to the human neurologic/neuroinflammatory syndrome known as Aicardi-Goutières syndrome (AGS) (Crow et al. 2006b; Reijns et al. 2011; Cerritelli and Crouch 2016). This syndrome is characterized by severe neurologic issues, including microcephaly (clinically defined as a head circumference three or more standard deviations below the mean for age and gender), and is considered to arise because of failed nucleic acid clearance, resulting in an interferon response leading to severe neuroinflammation (Crow and Manel 2015).
Mutations in other nucleases also give rise to AGS, including the major 3′ exonuclease TREX1 (Crow et al. 2006a). Studies with TREX1 knockout mice show elevated cytoplasmic DNA and inflammatory defects and chronic ATM (A-T, mutated)-dependent cell cycle checkpoint activation, suggesting that a prime TREX1 function is to activate DNA damage signaling (Morita et al. 2004; Yang et al. 2007). Since RNase H2 loss is also linked to a DNA damage response, the characteristic microcephaly in AGS may reflect elevated DNA damage during development, as it does in other DNA repair deficiency syndromes (McKinnon 2013). Thus, it is possible that DNA damage-induced cell loss (independent of the nucleic acid-induced inflammatory response) accounts for important aspects of the clinical presentation of AGS. Although the RER pathway is essential for genome stability during neurogenesis, its activity and contribution to neural homeostasis in post-mitotic neurons in the mature brain is unclear. Nonetheless, as neurons are long-lived, the failure to prevent accumulation of rNTPs during repair processes may also pose a threat in the mature brain.
While RNase H activity is essential for most rNTP removal activity, another enzyme that processes rNTPs in DNA is topoisomerase I (Top1). This function is in addition to the essential role that Top1 plays in the modulation of torsional stress during DNA transactions, including transcription (Pommier et al. 2016). Top1 possesses an endonuclease activity, and this activity has been shown to act on rNTP–DNA substrates (Sekiguchi and Shuman 1997). Recent studies in yeast reveal that, in the absence of RNase H2, rNTPs incorporated in DNA can be excised by TopI (Williams et al. 2013; Sparks and Burgers 2015; Huang et al. 2017). However, there appears to be some selectivity in the rNTP excision by Top1, as not all genomic rNTPs appear to be targets (Kim et al. 2011). Much of these data derive from yeast studies, and, in this organism, cells lacking RNase H2 do not exhibit growth defects, suggesting that cells must have other pathways allowing them to replicate rNTP-containing chromosomes. This is in contrast to mammalian cells, where loss of RNase H2 activity compromises proliferation (Hiller et al. 2012; Reijns et al. 2012). Therefore, assessment of the real impact of misincorporated rNTPs toward human neurologic disease, particularly in the mature nervous system, will require analysis of mammalian models to reveal the impact of compromised RER.
R loops and transcriptional damage
Another endogenous lesion that is potentially detrimental for neural genome stability is transcription-associated R loops (Aguilera and Garcia-Muse 2012; Groh and Gromak 2014; Skourti-Stathaki and Proudfoot 2014; Santos-Pereira and Aguilera 2015; Sollier and Cimprich 2015). During transcription, RNA polymerase generates forward positive torsional stress that impedes further DNA unwinding, and this is relieved by topoisomerase activity (Pommier et al. 2016). In contrast, negative torsion occurs in the wake of the polymerase and can lead to DNA strand separation and opening of the duplex. The resulting ssDNA region formed during transcription can base-pair with the nascent RNA transcript, generating an RNA–DNA duplex and an unpaired nontemplate DNA strand, giving rise to the term “R loop” for such structures. Other features such as high GC content (termed “GC skew”) can also predispose to R-loop formation (Ginno et al. 2012). Because R loops can form frequently and lead to genome instability, there are multiple DNA damage response factors that function to suppress or relieve R loops (Mischo et al. 2011; Bhatia et al. 2014; Sollier et al. 2014; Garcia-Rubio et al. 2015; Hatchi et al. 2015; Santos-Pereira and Aguilera 2015).
Some of the factors required to prevent R loops from becoming genotoxic, including RNase H2 and senataxin (SETX; a putative DNA/RNA helicase), are directly implicated in neurologic disease (Moreira et al. 2004; Mischo et al. 2011; Groh and Gromak 2014; Bennett and La Spada 2015). Inactivation of SETX, which has been linked to resolution of 3′ R loops during transcription, can result in ataxia with oculomotor apraxia 2 (AOA2) or amyotrophic lateral sclerosis, depending on whether it is an inactivation or a gain-of-function mutation (Skourti-Stathaki et al. 2011; Groh and Gromak 2014; Bennett and La Spada 2015). R loops have also been implicated in fragile X-associated tremor/ataxia syndrome, where they occur at the FMR1 locus and may affect gene function, leading to neuronal toxicity (Loomis et al. 2014). RNase H1 and H2 are also important for R-loop resolution in addition to their roles in rNTP removal from DNA (Chon et al. 2013; Amon and Koshland 2016; Williams et al. 2016). Importantly, if a main physiologic function of SETX is R-loop resolution, then this implicates R loops as a major source of endogenous genomic damage in the nervous system. However, in Setx−/− mice, R loops have been identified in testis but were not observed in the mature brain, perhaps implicating R-loop regulation by SETX as a pathogenic event primarily during neural development (Yeo et al. 2014). The way R loops generate genome instability remains unclear, although the displaced DNA loop might be susceptible to DNA breaks or the action of the APOBEC (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like) family of cytidine deaminase enzymes, and transcription-coupled DNA repair could also potentially generate breaks (Sollier and Cimprich 2015). It is also possible that R-loop formation could promote transcriptional disruption without generating frank DNA strand breaks.
DNA breaks in the nervous system
In contrast to neural development, sources of endogenous damage in the mature brain no longer include ongoing replication-associated lesions. Rather, the high oxygen consumption of the brain (Barzilai 2007) leads to increased respiration, oxidative lesions, and free radical production, which can result in oxidative DNA base damage and breakage of the DNA phosphodiester linkage. Furthermore, the high transcriptional activity of large neurons and their requirement to function for the life of the organism proportionally increase the probability of genotoxic events arising from normal cellular activities (Marguerat and Bahler 2012; Padovan-Merhar et al. 2015).
An additional prime source of endogenous damage in both developing and mature brains is the activity of topoisomerases. These enzymes function to introduce controlled breaks into DNA, and any aberrant activity can lead to DNA damage accumulation with pathogenic consequences (McKinnon 2016). All three classes of topoisomerase have been directly or indirectly linked to neurologic disease, including defects in enzymes that remove trapped Top1 and Top2 (Takashima et al. 2002; Gomez-Herreros et al. 2014); perturbation of long-gene expression, including genes linked to autism after Top1 or TOP2 inhibition (King et al. 2013); and Top 3b loss, which has been directly linked to schizophrenia (Stoll et al. 2013). Additional details concerning the role of topoisomerase-induced breaks in the nervous system are discussed later in the section on A-T and base excision repair (BER).
DNA strand break repair deficiency leads to neurologic disease
While the endogenous threats outlined above constantly occur to the genome, the real pathologic impact of these insults is revealed when the cellular response mechanism needed to alleviate the DNA damage is disabled. For many inherited neurologic diseases, the impact on the nervous system is startling in severity. Surprisingly, germline mutations in many ubiquitous DNA repair factors (particularly those responsible for preventing oxidative damage or the accumulation of DNA nicks) seem to affect only the nervous system (McKinnon 2013). In some cases, the biochemical function of the defective enzymes in inherited neurologic disease is to specifically process modified 3′ or 5′ DNA ends. However, the disease neuropathology indicates that these lesions, often associated with DNA nicks, are of profound systemic consequence to the nervous system and therefore must be a relatively frequent event needing timely resolution. Furthermore, the fact that individual enzymatic defects result in such profound neuropathology indicates that DNA repair redundancy is unavailable. This might be viewed as somewhat surprising given that the early onset of many of these syndromes suggests an overlap with development/neurogenesis, where some redundancy between repair pathways is potentially available, particularly in the cerebellum, a tissue that continues to develop postnatally for a period of ∼2 yr (Leto et al. 2016).
The following sections consider specific disease scenarios that illuminate the signaling pathways dealing with the diverse types of DNA damage encountered in the nervous system and how a failure to limit this damage leads to pathogenicity. Defects in all five major DNA repair pathways have been directly implicated in neurologic disease, and, although their detailed biochemistry is not discussed here, comprehensive recent reviews are available: nucleotide excision repair (Scharer 2013), the Fanconi anemia pathway (Walden and Deans 2014; Michl et al. 2016), homologous recombination (Jasin and Rothstein 2013), nonhomologous end-joining (Lieber 2010; Williams et al. 2014), mismatch repair (Kunkel and Erie 2015), and BER (Dianov and Hubscher 2013; Krokan and Bjoras 2013; Abbotts and Wilson 2017).
A-T and ATM: a paradigm for DNA damage-related neurodegeneration
The first inklings that DNA damage was a prominent etiologic agent in neurologic disease came from the realization that the childhood neurodegenerative syndrome A-T was intimately linked to radiosensitivity (Taylor et al. 1975). Subsequently, as the genetic basis for A-T was shown to be mutation of a DNA damage-responsive kinase, ATM (Savitsky et al. 1995), it became clear that genome instability was driving pathology in this neurodegenerative syndrome. The extreme radiation sensitivity of A-T suggested that defective DNA damage responses underpinned this disease and, accordingly, that the neurodegenerative phenotype reflected aspects of DNA damage (McKinnon 2012). Despite a multitude of systemic defects that includes the immune system, germinal tissues (meiosis is defective, and individuals with A-T are sterile) (Lange et al. 2011), and a predisposition to hematopoietic malignancy (T-cell and B-cell tumors), the hallmark and the most clinically intractable feature of A-T is neurodegeneration (Lavin 2008; McKinnon 2012; Shiloh and Ziv 2013; Rothblum-Oviatt et al. 2016). Cerebellar ataxia is apparent at a quite early age, and individuals with A-T are typically wheelchair-bound during their first decade of life. Characteristic neuropathology in A-T includes widespread loss of cerebellar granule and Purkinje neurons, white matter abnormalities (the loss of oligodendrocyte myelination), and a progressive nervous system-wide degenerative pathology (McKinnon 2012). Similar features also occur in many other DNA repair deficiency syndromes.
More compelling evidence that DNA damage underpins the neurodegeneration in A-T came from the discovery that, in a related neurologic syndrome called A-T-like disease (ATLD), the causative mutations were found in the DNA damage sensor MRE11, a component of a DNA damage-dependent ATM activation complex (Stewart et al. 1999; Stracker and Petrini 2011; Paull 2015). Although ATLD has an early onset ataxia similar to A-T, it is less severe, and this disease also lacks the extraneurologic features of A-T (Taylor et al. 2004, 2015; Palmeri et al. 2013).
Mre11 together with RAD50 and NBS1 form the MRN complex, which activates ATM after DNA damage (Stracker and Petrini 2011; Paull 2015). MRE11 and RAD50 cooperate to tether broken DNA ends and, together with NBS1, localize activated ATM to DNA damage (Hopfner et al. 2002; Kitagawa et al. 2004; Lee and Paull 2004; Falck et al. 2005; Wiltzius et al. 2005; Williams et al. 2010). Mutations of NBS1 are causative in the Nijmegen breakage syndrome (NBS), another radiosensitivity disorder that is characterized by prominent microcephaly rather than neurodegeneration (Varon et al. 1998; Chrzanowska et al. 2012). NBS also has many phenotypic similarities to A-T, including chromosomal instability, immunodeficiency, and predisposition to hematopoietic malignancies (Shiloh 1997; Chrzanowska et al. 2012). Additionally, microcephaly has also been associated with RAD50 mutation (Waltes et al. 2009). In contrast to ATM, the disease-causing mutations in ATLD and NBS are hypomorphic, as the MRN complex is indispensable for completion of DNA replication and survival (de Klein et al. 2000; Zhu et al. 2001), and while they disable important MRN functions, they nonetheless retain essential activity, allowing overall development.
Notably, mutations in MRE11 can influence disease phenotype, as specific MRE11 mutations can result in microcephaly rather than the neurodegeneration characteristic of ATLD (Matsumoto et al. 2011). The distinct phenotypes arising from various MRN hypomorphic alleles are likely to reflect the impact on replication in the case of microcephaly or on ATM activation when the outcome is neurodegeneration, probably via attenuating ATM-dependent apoptosis (Shull et al. 2009).
While the activation of ATM by DNA DSBs has been well established over the past decades (Lavin 2008; Shiloh and Ziv 2013; Paull 2015), it is less clear whether this is the lesion responsible for neurodegeneration in A-T. This is particularly so, as syndromes linked to DSB repair (DSBR) defects typically feature with microcephaly and not neurodegeneration. This is highlighted by (among others) ligase IV syndrome (Altmann and Gennery 2016) and the MRN-related diseases mentioned above. The connections between ATM and DSBs that might suggest a causative role in neurodegeneration include overall radiation sensitivity, defective DNA damage signaling after DSBs in the nervous systems of ATM-null mice (Herzog et al. 1998), and because the MRN complex activates ATM.
However, a spectrum of other possibilities for the underlying etiology in A-T has been proposed to explain the neurodegeneration that occurs early in A-T. While some of these also include DNA strand breaks, other suggestions involve oxidative stress, mitochondrial dysfunction, and altered ATM cytoplasmic (non-DNA damage) functions (Eaton et al. 2007; Guo et al. 2010; Valentin-Vega et al. 2012; Zhang et al. 2015; Fang et al. 2016). Currently, the important etiologic agents and the actual basis for cell loss that results in neurodegeneration remain uncertain. Despite this, strong clues exist as to what is likely to be the critical neuroprotective events requiring ATM, and these mostly involve DNA damage.
Aberrant regulation of Top1 has been identified recently as a potential endogenous pathogenic event in the brains of ATM-null mice (Katyal et al. 2014). Top1 covalently binds DNA and introduces a single-strand nick (in contrast, Top2 creates a DSB), and this activity is critical in a cell for multiple DNA transactions, including modulation of supercoiling and transcriptional regulation (Nitiss 2009; Pommier et al. 2016). Although the normal cellular activity of Top1 involves the covalent attachment of topoisomerases to DNA prior to cleavage, this transient DNA break is not detected as damage. However, if topoisomerase activity is perturbed, the enzyme can become trapped on DNA that activates DNA damage signaling (Pommier et al. 2006). Topoisomerases are critical for normal brain function, and dysregulation of all classes of topoisomerases have been strongly linked to a variety of neurologic diseases (McKinnon 2016).
ATM prevents the accumulation of endogenous Top1 cleavage complexes (Top1ccs) in mammalian brains and cultured cells by promoting the degradation of Top1 via ubiquitin/sumoylation, thereby minimizing the DNA damage generated by the Top1cc (Alagoz et al. 2013; Carlessi et al. 2014; Katyal et al. 2014). Thus, this Top1 complex represents the first identified endogenous pathogenic lesion in the mammalian brain attenuated by ATM (Katyal et al. 2014). As Top1cc generates DNA damage, this lesion may contribute importantly to the neurodegenerative phenotype of ATM. In nonreplicating cells, treatment with the Top1 inhibitor camptothecin results in transcription-associated DNA damage, as observed by γH2AX formation. This is ATM-dependent, as, in noncycling ATM-null cells, γH2AX is not formed after Top1 inhibition, indicating that Top1cc can specifically activate ATM (Sordet et al. 2009; Sakasai et al. 2010; Katyal et al. 2014). Although Top1 produces SSBs, if a Top1cc occurs in replicating cells (for instance, during postnatal cerebellar development), then they may become converted to DSBs, thus activating ATM via the MRN complex, potentially resulting in apoptosis (Shull et al. 2009).
ATM may also be important for preventing R-loop accumulation after spliceosome displacement at transcription-blocking lesions in noncycling primary cells (Tresini et al. 2015). This could be relevant to ATM regulation of Top1cc, as formation of these cleavage complexes can also result in increased R-loop formation and transcriptional disruption (Tuduri et al. 2009; El Hage et al. 2010). Because ATM prevents the accumulation of Top1ccs in the nervous system and because these lesions are potentially pathogenic, persistent Top1cc may be an important contributor to neurodegeneration in A-T. Notably, DNA SSBs associated with Top1cc accumulation in an ATM-null setting may be relevant to the neurodegeneration observed in inherited syndromes associated with defects in the BER pathway (see the next section). In fact, neurodegeneration in syndromes characterized by SSB repair (SSBR) defects have a neuropathology similar to that of A-T. For instance, the neurologic presentation of AOA1 (see below) is almost identical to that of A-T, leading to this distinct neurodegenerative disease initially being classified as an A-T variant (Aicardi et al. 1988).
Recently, accumulation of trapped Top2 (Top2cc) was found in various MRE11-deficient cells and also endogenously in the brains of Nbs1Nes-cre mice (Lee et al. 2012a; Aparicio et al. 2016; Deshpande et al. 2016; Hoa et al. 2016). Importantly, no Top1cc accumulation was found in response to specific defects in the MRN complex, consistent with ATM regulation of Top1 likely being independent of the MRN complex (Katyal et al. 2014). These data suggest distinct roles for ATM and MRN independently preventing Top1cc and Top2cc accumulation. Because these trapped topoisomerase complexes are pathogenic, it is possible that ATLD is a phenocopy of A-T, with both syndromes resulting from the aberrant regulation of different topoisomerases.
BER is critical in the nervous system
From a pathologic standpoint, defective DNA SSBR exclusively impacts the nervous system. While other types of DNA damage, such as bulky DNA adducts, DSBs (occurring during replication), and interstrand cross-links, impact the nervous system, they also affect other tissues throughout the body. As SSBs can be a product of oxidative metabolism and mitochondrial activity, it is likely that the substantial oxygen consumption of the brain underscores the critical need for BER (Attwell and Laughlin 2001; Barzilai 2007). Currently, germline mutations in four components of the BER pathway have been identified in neurologic disease, and, in all cases, these individuals are characterized by ataxia and exhibit pathology exclusively in the nervous system (Fig. 2).
Figure 2.

The BER pathway is essential in the nervous system. The BER system is required to correct SSBs and oxidative damage and is a critical main DNA repair pathway in the nervous system. The BER components listed in red have been identified in human neurodegenerative disease. The scaffold protein XRCC1 is key for efficient BER, and the repair enzymes aprataxin (APTX) and tyrosyl DNA phosphodiesterase-1 (TDP1) modify the 5′ and 3′ DNA ends, respectively, after damage to allow for religation/repair of the DNA break. Polynucleotide kinase/phosphatase (PNKP) has a dual kinase/phosphatase activity and can process both ends of a DNA break. Mutations in PNKP can result in different neurologic disease characterized by either microcephaly or neurodegeneration.
The BER pathway repairs DNA SSBs or damage to bases via insults such as oxidation. The repair of a damaged base initially involves the action of a lesion-specific DNA glycosylase followed by the apurinic/apyrimidinic endonuclease-1 to generate a DNA strand break (Krokan and Bjoras 2013; Li and Wilson 2014). Strand break repair uses XRCC1 and PARP (poly-ADP-ribose polymerase) to recruit repair factors involved in DNA end modification and DNA polymerase for gap filling and ligation via ligase III or ligase I (Caldecott 2008; Gao et al. 2011). Defects in this pathway are associated with multiple forms of AOA, including AOA1 and AOA4; spinocerebellar ataxia with axonal neuropathy (SCAN1) neurodegenerative syndromes; and, in certain cases, microcephaly with seizures (MCSZ) (Table 1; Date et al. 2001; Moreira et al. 2001; Takashima et al. 2002; Shen et al. 2010; Hoch et al. 2017).
Aprataxin (APTX), mutated in AOA1, is a member of the histidine triad superfamily of nucleotide hydrolases and possesses an AMP-lysine hydrolase activity, required for the repair of 5′-AMP intermediates that arise from failed DNA ligation reactions (Seidle et al. 2005; Ahel et al. 2006). APTX has also been implicated in the removal of rNTPs from DNA and can deadenylate RNA–DNA junctions to facilitate repair and removal of misincorporated rNTPs (Tumbale et al. 2014). Tyrosyl DNA phosphodiesterase 1 (TDP1) mutations can result in SCAN1. This enzyme repairs altered 3′ DNA ends arising from Top1–DNA covalent complexes or oxidative damage (El-Khamisy et al. 2005; Interthal et al. 2005; Zhou et al. 2005). TDP1-deficient neural cells have a pronounced defect in the repair of camptothecin-induced Top1ccs and hydrogen peroxide-induced SSBs (Hirano et al. 2007; Katyal et al. 2007). Although it is likely, but not certain, that the main role for TDP1 is repair of Top1cc damage in the brain, this is the case for TDP2, whose sole role appears to be repairing Top2cc (Cortes Ledesma et al. 2009; Nitiss and Nitiss 2013). Inactivating mutations in TDP2 leads to a neurologic disease associated with intellectual disability, seizures, and ataxia, underscoring the pathogenic impact of Top2cc in the nervous system (Gomez-Herreros et al. 2014; McKinnon 2016). As some gene expression regulated by Top2 involves DNA DSBs that activate DNA damage signaling (Ju et al. 2006; Bunch et al. 2015), in the brain, this activity is associated with specific types of gene expression programs (Madabhushi et al. 2015). Abnormal regulation of this process might contribute to tissue-specific DNA damage.
The pathology resulting from BER defects is associated with increased SSBs, a lesion known to activate PARP-mediated ADP ribosylation (Caldecott 2008). Increased PARP activity can deplete cellular NAD levels, leading to neurotoxicity (Gupte et al. 2017; Narne et al. 2017). This was confirmed in mice with XRCC1 deleted in the brain (Lee et al. 2009), in which the characteristic loss of cerebellar interneurons was rescued by coincident deletion of PARP1 (Hoch et al. 2017). These data implicate PARP1 hyperactivity as a major source of disease pathology induced by unrepaired SSBs in the brain. Pathology may also arise from the conversion of SSBs to lethal DSBs during cerebellar development, which can activate apoptosis or cell cycle arrest and could account for the additional rescue of affected XRCC1 tissues, such as cerebellar granule neurons, by p53 loss (Lee et al. 2009).
While the functional relationships between XRCC1, APTX, and TDP1 during DNA repair offer a logical explanation for their roles in preventing neurodegeneration, a more enigmatic example of defects in this pathway leading to neurologic disease is that of the end-processing enzyme polynucleotide kinase/phosphatase (PNKP). Mutation of this enzyme can lead to either the neurodegenerative syndrome AOA4 or MCSZ. Notably, these are clinically distinct neurologic syndromes despite the fact that both can result from similar mutations in PNKP (Shen et al. 2010; Bras et al. 2015). Why similar gene mutations in PNKP lead to different diseases is unknown. In one family, siblings from consanguineous parents developed both microcephaly and ataxia but had mutations identical to those of individuals with MCSZ who did not develop ataxia; thus, it is likely that genetic modifiers influence the outcome of PNKP mutations (Poulton et al. 2013; Dumitrache and McKinnon 2017).
PNKP contains a C-terminal catalytic region with a fused bifunctional phosphatase and kinase domain and an N-terminal forkhead-associated (FHA) domain (Weinfeld et al. 2011). Correspondingly, PNKP has both a 3′ phosphatase and 5′ kinase activity for modifying the ends of a DNA break (Jilani et al. 1999; Weinfeld et al. 2011). Interestingly, this enzymatic activity of PNKP is used for both SSBR and DSBR (Whitehouse et al. 2001; Chappell et al. 2002; Koch et al. 2004; Karimi-Busheri et al. 2007; Zolner et al. 2011; Shimada et al. 2015). The FHA domain of PNKP is important for interaction with either the XRCC1 or XRCC4 scaffold proteins, which are required for assembling SSBR or DSBR (nonhomologous end-joining) components, respectively (Whitehouse et al. 2001; Koch et al. 2004; Bernstein et al. 2005; Ali et al. 2009; Zolner et al. 2011; Shimada et al. 2015). PNKP mutant mice also show both SSBR and DSBR defects in neural (and other) cells (Shimada et al. 2015). Biochemical studies showed that disease-causing mutations affecting PNKP resulted in compromised phosphatase activity, and MCSZ-derived lymphoid lines had very low PNKP protein expression that compromised enzymatic activity (Reynolds et al. 2012). Based on other syndromes, it seems likely that the DSBR defects account for the microcephaly in MCSZ and that the SSBR defect promotes ataxia in AOA4. However, if DSBR is an important function of PNKP, then why is the phenotype of MCSZ restricted to the nervous system?
Oxidative damage may also underpin expansion of triplet nucleotide repeats that appear in >40 human neurologic syndromes (McMurray 2010; Liu and Wilson 2012). In these diseases, somatic expansion of triplet repeats occurs in the nervous system until it reaches a threshold, after which neuropathology is apparent. The process that drives somatic expansion of these repeats appears to be linked to BER activity, and inactivation of various BER components has been shown to dramatically exacerbate triplet repeat expansion (McMurray 2010; Liu and Wilson 2012).
Maintenance of the mitochondrial genome is also critical for neural homeostasis, and the BER pathway has been directly implicated in mitochondrial DNA (mtDNA) repair (Sykora et al. 2011; Prakash and Doublie 2015). However, it appears that the key general event for widespread mtDNA maintenance involves DNA ligase 3 during mitochondrial genome replication (Gao et al. 2011). Moreover, although multiple BER factors have been identified in the mitochondria, clear evidence for mitochondriopathy do not feature in BER syndromes. It is possible that the heterogeneity and abundance of mitochondria make stochastic damage to mtDNA less consequential than nuclear DNA. Furthermore, other generalized processes have been identified that can clear damaged mtDNA, such as mitochondrial fusion (Chen et al. 2010).
Perspectives and future directions
Collectively, the details above indicate that the nervous system requires an armamentarium of factors to guard genome integrity. This is apparent both during neurogenesis, when replication is a driving force of development, and also in long-lived neural cells, where high oxidative metabolism and transcriptional activity cause a constant barrage of potentially deleterious DNA lesions.
Our knowledge regarding the physiologic importance of DNA repair factors derives from human syndromes and genetic manipulation in mouse models, although how the many potential endogenous lesions ultimately lead to neuropathology still remains unclear. A challenge in understanding disease etiology comes from disentangling the relative impact of DNA lesions during DNA replication in stem and progenitor cells, immature (noncycling) cells, or fully differentiated cells. Most DNA damage response defects are apparent early in childhood, suggesting a neurodevelopmental involvement to varying degrees. However, for many disease phenotypes that are restricted to the nervous system, a fundamental role for the defective protein in suppressing replication stress seems unlikely. Furthermore, syndromes resulting from DNA replication defects also do not show progressive degeneration in the nervous system. It is more likely that the impact of DNA repair defects in the nervous system is related to the profound physiologic change that happens after birth, where respiratory metabolism takes over from placental oxygen use. Notably, loss of DNA ligase III (a BER factor required for mtDNA maintenance) throughout the developing mouse nervous system has little consequence until after birth, where the sudden burst of respiration results in early lethality associated with widespread death throughout the nervous system (Gao et al. 2011). Consequently, in the perinatal period, neural tissue must deal with the impact of respiration and higher oxygen utilization. Thus, the genome maintenance factors needed at this stage differ dramatically from those required to maintain genomic integrity in rapidly proliferating neural tissues. The pathways most critical at this stage are those that deal with oxidative DNA damage; thus, defects in BER substantially impact the young nervous system. The progressive decline associated with most DNA repair diseases also indicates that critical maintenance functions are required in differentiated neural cells to offset the effects of a continual threat of different DNA lesions.
Mammalian genomes are also characterized by long interspersed element 1 (L1) retrotransposons distributed throughout the genome, accounting for up to 20% of human genomic sequences (Muotri et al. 2005; Erwin et al. 2014). Insertion of this DNA can result in genome diversity that could potentially influence individual cellular identity. As ATM signaling has been reported to restrict L1 transposition in human brains (Coufal et al. 2011), it is possible that there are pathogenic consequences for this retrotransposition event. Recently, the advent of single-cell genomic sequencing has revealed somatic copy number variation in a large portion of cortical neurons; among the changes in individual neuronal DNA are deletions and aneuploidy (McConnell et al. 2013). Notably, loss-of-function mutations associated with these single-nucleotide variants have been linked to autism spectrum disorder risk (D'Gama et al. 2015). Thus, these somatic mutations may have particular consequences associated with neurologic disease predisposition in the human population (McConnell et al. 2017). Although these genomic changes are prevalent in the brain, the underlying mechanism associated with DNA damage/repair resulting in altered individual genomes is presently unknown.
Other data integrating various genome transactions with DNA damage responses are also expanding our understanding relevant to genome maintenance in the nervous system. For instance, chromatin remodeling influences broad aspects of DNA repair, perhaps by regulating the accessibility of repair factors to chromatin (Price and D'Andrea 2013). Defective chromatin remodeling during development can also compromise genome stability of neural progenitors (Nechiporuk et al. 2016). Connecting DNA repair pathways and chromatin modification will continue to integrate more general cellular processes that respond to and facilitate DNA repair with modulation of chromosome integrity and cellular homeostasis.
The establishment of consortia to investigate uncharacterized neurologic diseases as well as the increased availability of genomics resources such as high-throughput sequencing will further reveal the importance of DNA damage response components not previously identified in disease, as was recently discovered for XRCC1 (Hoch et al. 2017). With the emergence of precision medicine, it is apparent that therapeutic intervention to ameliorate genome stability defects requires a detailed understanding of the central sources of genome damage and a greater understanding of how this impacts physiologically. Further elucidation of cellular strategies for genome maintenance in the nervous system will allow for the design of neuroprotective agents to prevent disease progression. Enhanced understanding of DNA maintenance pathways in a tissue-specific context could also identify potential cellular vulnerabilities that can be leveraged as anti-cancer strategies.
Acknowledgments
P.J.M. is supported by the National Institutes of Health (NS-37956 and CA-21765), the Cancer Center Support Grant (P30 CA21765), and the American Lebanese and Syrian Associated Charities of St. Jude Children's Research Hospital.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.301325.117.
References
- Abbotts R, Wilson DM III. 2017. Coordination of DNA single strand break repair. Free Radic Biol Med 107: 228–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, Garcia-Muse T. 2012. R loops: from transcription byproducts to threats to genome stability. Mol Cell 46: 115–124. [DOI] [PubMed] [Google Scholar]
- Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC. 2006. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 443: 713–716. [DOI] [PubMed] [Google Scholar]
- Aicardi J, Barbosa C, Andermann E, Andermann F, Morcos R, Ghanem Q, Fukuyama Y, Awaya Y, Moe P. 1988. Ataxia-ocular motor apraxia: a syndrome mimicking ataxia-telangiectasia. Ann Neurol 24: 497–502. [DOI] [PubMed] [Google Scholar]
- Alagoz M, Chiang SC, Sharma A, El-Khamisy SF. 2013. ATM deficiency results in accumulation of DNA-topoisomerase I covalent intermediates in neural cells. PLoS One 8: e58239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali AA, Jukes RM, Pearl LH, Oliver AW. 2009. Specific recognition of a multiply phosphorylated motif in the DNA repair scaffold XRCC1 by the FHA domain of human PNK. Nucleic Acids Res 37: 1701–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. 2013. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 152: 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmann T, Gennery AR. 2016. DNA ligase IV syndrome; a review. Orphanet J Rare Dis 11: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon JD, Koshland D. 2016. RNase H enables efficient repair of R-loop induced DNA damage. Elife 5: e20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio T, Baer R, Gottesman M, Gautier J. 2016. MRN, CtIP, and BRCA1 mediate repair of topoisomerase II-DNA adducts. J Cell Biol 212: 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB. 2001. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21: 1133–1145. [DOI] [PubMed] [Google Scholar]
- Baple EL, Chambers H, Cross HE, Fawcett H, Nakazawa Y, Chioza BA, Harlalka GV, Mansour S, Sreekantan-Nair A, Patton MA, et al. 2014. Hypomorphic PCNA mutation underlies a human DNA repair disorder. J Clin Invest 124: 3137–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. 1998. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol 8: 1395–1398. [DOI] [PubMed] [Google Scholar]
- Barzilai A. 2007. The contribution of the DNA damage response to neuronal viability. Antioxid Redox Signal 9: 211–218. [DOI] [PubMed] [Google Scholar]
- Barzilai A, Schumacher B, Shiloh Y. 2017. Genome instability: linking ageing and brain degeneration. Mech Ageing Dev 161: 4–18. [DOI] [PubMed] [Google Scholar]
- Bennett CL, La Spada AR. 2015. Unwinding the role of senataxin in neurodegeneration. Discov Med 19: 127–136. [PubMed] [Google Scholar]
- Bernstein NK, Williams RS, Rakovszky ML, Cui D, Green R, Karimi-Busheri F, Mani RS, Galicia S, Koch CA, Cass CE, et al. 2005. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol Cell 17: 657–670. [DOI] [PubMed] [Google Scholar]
- Bhatia V, Barroso SI, Garcia-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A. 2014. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511: 362–365. [DOI] [PubMed] [Google Scholar]
- Bras J, Alonso I, Barbot C, Costa MM, Darwent L, Orme T, Sequeiros J, Hardy J, Coutinho P, Guerreiro R. 2015. Mutations in PNKP cause recessive ataxia with oculomotor apraxia type 4. Am J Hum Genet 96: 474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D. 2000. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 14: 397–402. [PMC free article] [PubMed] [Google Scholar]
- Bunch H, Lawney BP, Lin YF, Asaithamby A, Murshid A, Wang YE, Chen BP, Calderwood SK. 2015. Transcriptional elongation requires DNA break-induced signalling. Nat Commun 6: 10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott KW. 2008. Single-strand break repair and genetic disease. Nat Rev Genet 9: 619–631. [DOI] [PubMed] [Google Scholar]
- Carlessi L, Fusar Poli E, Bechi G, Mantegazza M, Pascucci B, Narciso L, Dogliotti E, Sala C, Verpelli C, Lecis D, et al. 2014. Functional and molecular defects of hiPSC-derived neurons from patients with ATM deficiency. Cell Death Dis 5: e1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerritelli SM, Crouch RJ. 2016. The balancing act of ribonucleotides in DNA. Trends Biochem Sci 41: 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC. 2002. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J 21: 2827–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. 2010. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141: 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon H, Sparks JL, Rychlik M, Nowotny M, Burgers PM, Crouch RJ, Cerritelli SM. 2013. RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res 41: 3130–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow HM, Herrup K. 2015. Genomic integrity and the ageing brain. Nat Rev Neurosci 16: 672–684. [DOI] [PubMed] [Google Scholar]
- Chrzanowska KH, Gregorek H, Dembowska-Baginska B, Kalina MA, Digweed M. 2012. Nijmegen breakage syndrome (NBS). Orphanet J Rare Dis 7: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun JJ, Schatz DG, Oettinger MA, Jaenisch R, Baltimore D. 1991. The recombination activating gene-1 (RAG-1) transcript is present in the murine central nervous system. Cell 64: 189–200. [DOI] [PubMed] [Google Scholar]
- Cortes Ledesma F, El Khamisy SF, Zuma MC, Osborn K, Caldecott KW. 2009. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 461: 674–678. [DOI] [PubMed] [Google Scholar]
- Coufal NG, Garcia-Perez JL, Peng GE, Marchetto MC, Muotri AR, Mu Y, Carson CT, Macia A, Moran JV, Gage FH. 2011. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc Natl Acad Sci 108: 20382–20387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespan E, Furrer A, Rosinger M, Bertoletti F, Mentegari E, Chiapparini G, Imhof R, Ziegler N, Sturla SJ, Hubscher U, et al. 2016. Impact of ribonucleotide incorporation by DNA polymerases β and λ on oxidative base excision repair. Nat Commun 7: 10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Manel N. 2015. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, et al. 2006a. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 38: 917–920. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, et al. 2006b. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet 38: 910–916. [DOI] [PubMed] [Google Scholar]
- Date H, Onodera O, Tanaka H, Iwabuchi K, Uekawa K, Igarashi S, Koike R, Hiroi T, Yuasa T, Awaya Y, et al. 2001. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. NatGenet 29: 184–188. [DOI] [PubMed] [Google Scholar]
- De Bont R, van Larebeke N. 2004. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19: 169–185. [DOI] [PubMed] [Google Scholar]
- de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. 2000. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 10: 479–482. [DOI] [PubMed] [Google Scholar]
- Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. 2007. The Rad9–Hus1–Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev 21: 1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande RA, Lee JH, Arora S, Paull TT. 2016. Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA adducts. Mol Cell 64: 593–606. [DOI] [PubMed] [Google Scholar]
- D'Gama AM, Pochareddy S, Li M, Jamuar SS, Reiff RE, Lam AT, Sestan N, Walsh CA. 2015. Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron 88: 910–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov GL, Hubscher U. 2013. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res 41: 3483–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrache LC, McKinnon PJ. 2017. Polynucleotide kinase-phosphatase (PNKP) mutations and neurologic disease. Mech Ageing Dev 161: 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, Shadel GS. 2007. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J Clin Invest 117: 2723–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hage A, French SL, Beyer AL, Tollervey D. 2010. Loss of topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev 24: 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. 2005. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 434: 108–113. [DOI] [PubMed] [Google Scholar]
- Enriquez-Rios V, Dumitrache LC, Downing SM, Li Y, Brown EJ, Russell HR, McKinnon PJ. 2017. DNA-PKcs, ATM, and ATR interplay maintains genome integrity during neurogenesis. J Neurosci 37: 893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin JA, Marchetto MC, Gage FH. 2014. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci 15: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. 2005. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434: 605–611. [DOI] [PubMed] [Google Scholar]
- Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, Shamanna RA, Kalyanasundaram S, Bollineni RC, Wilson MA, et al. 2016. NAD+ replenishment improves lifespan and healthspan in Ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab 24: 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, et al. 1998. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95: 891–902. [DOI] [PubMed] [Google Scholar]
- Gao Y, Katyal S, Lee Y, Zhao J, Rehg JE, Russell HR, McKinnon PJ. 2011. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 471: 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rubio ML, Perez-Calero C, Barroso SI, Tumini E, Herrera-Moyano E, Rosado IV, Aguilera A. 2015. The Fanconi anemia pathway protects genome integrity from R-loops. PLoS Genet 11: e1005674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodgaonkar MM, Lazzaro F, Olivera-Pimentel M, Artola-Boran M, Cejka P, Reijns MA, Jackson AP, Plevani P, Muzi-Falconi M, Jiricny J. 2013. Ribonucleotides misincorporated into DNA act as strand-discrimination signals in eukaryotic mismatch repair. Mol Cell 50: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F. 2012. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 45: 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Herreros F, Schuurs-Hoeijmakers JH, McCormack M, Greally MT, Rulten S, Romero-Granados R, Counihan TJ, Chaila E, Conroy J, Ennis S, et al. 2014. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat Genet 46: 516–521. [DOI] [PubMed] [Google Scholar]
- Groh M, Gromak N. 2014. Out of balance: R-loops in human disease. PLoS Genet 10: e1004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. 2010. ATM activation by oxidative stress. Science 330: 517–521. [DOI] [PubMed] [Google Scholar]
- Gupte R, Liu Z, Kraus WL. 2017. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 31: 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamperl S, Cimprich KA. 2016. Conflict resolution in the genome: how transcription and replication make it work. Cell 167: 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley ME, Murina O, Leitch A, Higgs MR, Bicknell LS, Yigit G, Blackford AN, Zlatanou A, Mackenzie KJ, Reddy K, et al. 2016. TRAIP promotes DNA damage response during genome replication and is mutated in primordial dwarfism. Nat Genet 48: 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, Kamieniarz-Gdula K, Dimitrov S, Pathania S, McKinney KM, Eaton ML, et al. 2015. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell 57: 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten ME, Heintz N. 1995. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci 18: 385–408. [DOI] [PubMed] [Google Scholar]
- Helleday T, Eshtad S, Nik-Zainal S. 2014. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 15: 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmink BA, Sleckman BP. 2012. The response to and repair of RAG-mediated DNA double-strand breaks. Annu Rev Immunol 30: 175–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. 1998. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 280: 1089–1091. [DOI] [PubMed] [Google Scholar]
- Hill SJ, Mordes DA, Cameron LA, Neuberg DS, Landini S, Eggan K, Livingston DM. 2016. Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc Natl Acad Sci 113: E7701–E7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A. 2012. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med 209: 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano R, Interthal H, Huang C, Nakamura T, Deguchi K, Choi K, Bhattacharjee MB, Arimura K, Umehara F, Izumo S, et al. 2007. Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation? EMBO J 26: 4732–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoa NN, Shimizu T, Zhou ZW, Wang ZQ, Deshpande RA, Paull TT, Akter S, Tsuda M, Furuta R, Tsusui K, et al. 2016. Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes. Mol Cell 64: 580–592. [DOI] [PubMed] [Google Scholar]
- Hoch NC, Hanzlikova H, Rulten SL, Tetreault M, Komulainen E, Ju L, Hornyak P, Zeng Z, Gittens W, Rey SA, et al. 2017. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 541: 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, Karcher A, Henderson B, Bodmer JL, McMurray CT, et al. 2002. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature 418: 562–566. [DOI] [PubMed] [Google Scholar]
- Huang SN, Williams JS, Arana ME, Kunkel TA, Pommier Y. 2017. Topoisomerase I-mediated cleavage at unrepaired ribonucleotides generates DNA double-strand breaks. EMBO J 36: 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, Champoux JJ. 2005. SCAN1 mutant Tdp1 accumulates the enzyme—DNA intermediate and causes camptothecin hypersensitivity. EMBO J 24: 2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, Rothstein R. 2013. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5: a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilani A, Ramotar D, Slack C, Ong C, Yang XM, Scherer SW, Lasko DD. 1999. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3′-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J Biol Chem 274: 24176–24186. [DOI] [PubMed] [Google Scholar]
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. 2006. A topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science 312: 1798–1802. [DOI] [PubMed] [Google Scholar]
- Karimi-Busheri F, Rasouli-Nia A, Allalunis-Turner J, Weinfeld M. 2007. Human polynucleotide kinase participates in repair of DNA double-strand breaks by nonhomologous end joining but not homologous recombination. Cancer Res 67: 6619–6625. [DOI] [PubMed] [Google Scholar]
- Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ. 2007. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J 26: 4720–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katyal S, Lee Y, Nitiss KC, Downing SM, Li Y, Shimada M, Zhao J, Russell HR, Petrini JH, Nitiss JL, et al. 2014. Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nature Neurosci 17: 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Huang SN, Williams JS, Li YC, Clark AB, Cho JE, Kunkel TA, Pommier Y, Jinks-Robertson S. 2011. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332: 1561–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IF, Yandava CN, Mabb AM, Hsiao JS, Huang HS, Pearson BL, Calabrese JM, Starmer J, Parker JS, Magnuson T, et al. 2013. Topoisomerases facilitate transcription of long genes linked to autism. Nature 501: 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. 2004. Phosphorylation of SMC1 is a critical downstream event in the ATM–NBS1–BRCA1 pathway. Genes Dev 18: 1423–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch CA, Agyei R, Galicia S, Metalnikov P, O'Donnell P, Starostine A, Weinfeld M, Durocher D. 2004. Xrcc4 physically links DNA end processing by polynucleotide kinase to DNA ligation by DNA ligase IV. EMBO J 23: 3874–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokan HE, Bjoras M. 2013. Base excision repair. Cold Spring Harb Perspect Biol 5: a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, Dunphy WG. 2006. TopBP1 activates the ATR-ATRIP complex. Cell 124: 943–955. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA. 2015. Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet 49: 291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange J, Pan J, Cole F, Thelen MP, Jasin M, Keeney S. 2011. ATM controls meiotic double-strand-break formation. Nature 479: 237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ. 2011. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475: 53–58. [DOI] [PubMed] [Google Scholar]
- Lavin MF. 2008. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 9: 759–769. [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT. 2004. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304: 93–96. [DOI] [PubMed] [Google Scholar]
- Lee Y, Katyal S, Li Y, El-Khamisy SF, Russell HR, Caldecott KW, McKinnon PJ. 2009. The genesis of cerebellar interneurons and the prevention of neural DNA damage require XRCC1. Nat Neurosci 12: 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KC, Padget K, Curtis H, Cowell IG, Moiani D, Sondka Z, Morris NJ, Jackson GH, Cockell SJ, Tainer JA, et al. 2012a. MRE11 facilitates the removal of human topoisomerase II complexes from genomic DNA. Biol Open 1: 863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Katyal S, Downing SM, Zhao J, Russell HR, McKinnon PJ. 2012b. Neurogenesis requires TopBP1 to prevent catastrophic replicative DNA damage in early progenitors. Nat Neurosci 15: 819–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Shull ER, Frappart PO, Katyal S, Enriquez-Rios V, Zhao J, Russell HR, Brown EJ, McKinnon PJ. 2012c. ATR maintains select progenitors during nervous system development. EMBO J 31: 1177–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto K, Arancillo M, Becker EB, Buffo A, Chiang C, Ding B, Dobyns WB, Dusart I, Haldipur P, Hatten ME, et al. 2016. Consensus paper: cerebellar development. Cerebellum 15: 789–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wilson DM III. 2014. Human apurinic/apyrimidinic endonuclease 1. Antioxid Redox Signal 20: 678–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. 2010. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79: 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wilson SH. 2012. DNA base excision repair: a mechanism of trinucleotide repeat expansion. Trends Biochem Sci 37: 162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis EW, Sanz LA, Chedin F, Hagerman PJ. 2014. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet 10: e1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Williams JS, Clausen AR, Clark AB, Kunkel TA. 2013. Ribonucleotides are signals for mismatch repair of leading-strand replication errors. Mol Cell 50: 437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Pan L, Tsai LH. 2014. DNA damage and its links to neurodegeneration. Neuron 83: 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC, et al. 2015. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161: 1592–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magdalou I, Lopez BS, Pasero P, Lambert SA. 2014. The causes of replication stress and their consequences on genome stability and cell fate. Semin Cell Dev Biol 30: 154–164. [DOI] [PubMed] [Google Scholar]
- Marechal A, Zou L. 2013. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5: a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marguerat S, Bahler J. 2012. Coordinating genome expression with cell size. Trends Genet 28: 560–565. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Miyamoto T, Sakamoto H, Izumi H, Nakazawa Y, Ogi T, Tahara H, Oku S, Hiramoto A, Shiiki T, et al. 2011. Two unrelated patients with MRE11A mutations and Nijmegen breakage syndrome-like severe microcephaly. DNA Repair 10: 314–321. [DOI] [PubMed] [Google Scholar]
- McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS, Vermeesch JR, Hall IM, et al. 2013. Mosaic copy number variation in human neurons. Science 342: 632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Moran JV, Abyzov A, Akbarian S, Bae T, Cortes-Ciriano I, Erwin JA, Fasching L, Flasch DA, Freed D, et al. 2017. Intersection of diverse neuronal genomes and neuropsychiatric disease: the brain somatic mosaicism network. Science 356: eaal1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ. 2009. DNA repair deficiency and neurological disease. Nat Rev Neurosci 10: 100–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ. 2012. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol 7: 303–321. [DOI] [PubMed] [Google Scholar]
- McKinnon PJ. 2013. Maintaining genome stability in the nervous system. Nat Neurosci 16: 1523–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ. 2016. Topoisomerases and the regulation of neural function. Nat Rev Neurosci 17: 673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray CT. 2010. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet 11: 786–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michl J, Zimmer J, Tarsounas M. 2016. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J 35: 909–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischo HE, Gomez-Gonzalez B, Grzechnik P, Rondon AG, Wei W, Steinmetz L, Aguilera A, Proudfoot NJ. 2011. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol Cell 41: 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonca P, Costa M, Barros J, Yanagisawa T, et al. 2001. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. NatGenet 29: 189–193. [DOI] [PubMed] [Google Scholar]
- Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, Tranchant C, Aubourg P, Tazir M, Schols L, et al. 2004. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet 36: 225–227. [DOI] [PubMed] [Google Scholar]
- Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, Daly G, Lindahl T, Barnes DE. 2004. Gene-targeted mice lacking the Trex1 (DNase III) 3′ → 5′ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol 24: 6719–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. 2005. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435: 903–910. [DOI] [PubMed] [Google Scholar]
- Murga M, Bunting S, Montana MF, Soria R, Mulero F, Canamero M, Lee Y, McKinnon PJ, Nussenzweig A, Fernandez-Capetillo O. 2009. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet 41: 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam EA, Cortez D. 2011. ATR signalling: more than meeting at the fork. Biochem J 436: 527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narne P, Pandey V, Simhadri PK, Phanithi PB. 2017. Poly(ADP-ribose)polymerase-1 hyperactivation in neurodegenerative diseases: the death knell tolls for neurons. Semin Cell Dev Biol 63: 154–166. [DOI] [PubMed] [Google Scholar]
- Nechiporuk T, McGann J, Mullendorff K, Hsieh J, Wurst W, Floss T, Mandel G. 2016. The REST remodeling complex protects genomic integrity during embryonic neurogenesis. Elife 5: e09584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Kumar D, Clark AB, Watt DL, Watts BE, Lundstrom EB, Johansson E, Chabes A, Kunkel TA. 2010a. Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol 6: 774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Watts BE, Kumar D, Watt DL, Lundstrom EB, Burgers PM, Johansson E, Chabes A, Kunkel TA. 2010b. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc Natl Acad Sci 107: 4949–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss JL. 2009. DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer 9: 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss JL, Nitiss KC. 2013. Tdp2: a means to fixing the ends. PLoS Genet 9: e1003370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Driscoll M. 2017. The pathological consequences of impaired genome integrity in humans; disorders of the DNA replication machinery. J Pathol 241: 192–207. [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. 2003. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501. [DOI] [PubMed] [Google Scholar]
- Padovan-Merhar O, Nair GP, Biaesch AG, Mayer A, Scarfone S, Foley SW, Wu AR, Churchman LS, Singh A, Raj A. 2015. Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms. Mol Cell 58: 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmeri S, Rufa A, Pucci B, Santarnecchi E, Malandrini A, Stromillo ML, Mandala M, Rosini F, De Stefano N, Federico A. 2013. Clinical course of two Italian siblings with ataxia-telangiectasia-like disorder. Cerebellum 12: 596–599. [DOI] [PubMed] [Google Scholar]
- Paull TT. 2015. Mechanisms of ATM Activation. Annu Rev Biochem 84: 711–738. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Barcelo JM, Rao VA, Sordet O, Jobson AG, Thibaut L, Miao ZH, Seiler JA, Zhang H, Marchand C, et al. 2006. Repair of topoisomerase I-mediated DNA damage. Prog Nucleic Acid Res Mol Biol 81: 179–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Sun Y, Huang SN, Nitiss JL. 2016. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 17: 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenski CJ, Klein HL. 2014. How the misincorporation of ribonucleotides into genomic DNA can be both harmful and helpful to cells. Nucleic Acids Res 42: 10226–10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenski CJ, Niu H, Sung P, Klein HL. 2014. Avoidance of ribonucleotide-induced mutations by RNase H2 and Srs2-Exo1 mechanisms. Nature 511: 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton C, Oegema R, Heijsman D, Hoogeboom J, Schot R, Stroink H, Willemsen MA, Verheijen FW, van de Spek P, Kremer A, et al. 2013. Progressive cerebellar atrophy and polyneuropathy: expanding the spectrum of PNKP mutations. Neurogenetics 14: 43–51. [DOI] [PubMed] [Google Scholar]
- Prakash A, Doublie S. 2015. Base excision repair in the mitochondria. J Cell Biochem 116: 1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price BD, D'Andrea AD. 2013. Chromatin remodeling at DNA double-strand breaks. Cell 152: 1344–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Lee S, Shang Y, Wang WY, Au KF, Kamiya S, Barmada SJ, Finkbeiner S, Lui H, Carlton CE, et al. 2014. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest 124: 981–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, Bubeck D, Gibson LC, Graham SC, Baillie GS, Jones EY, Jackson AP. 2011. The structure of the human RNase H2 complex defines key interaction interfaces relevant to enzyme function and human disease. J Biol Chem 286: 10530–10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, et al. 2012. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149: 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JJ, Walker AK, Gilmore EC, Walsh CA, Caldecott KW. 2012. Impact of PNKP mutations associated with microcephaly, seizures and developmental delay on enzyme activity and DNA strand break repair. Nucleic Acids Res 40: 6608–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JJ, Bicknell LS, Carroll P, Higgs MR, Shaheen R, Murray JE, Papadopoulos DK, Leitch A, Murina O, Tarnauskaite Z, et al. 2017. Mutations in DONSON disrupt replication fork stability and cause microcephalic dwarfism. Nat Genet 49: 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. 2016. Ataxia telangiectasia: a review. Orphanet J Rare Dis 11: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. 2007. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1: 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakasai R, Teraoka H, Takagi M, Tibbetts RS. 2010. Transcription-dependent activation of ataxia telangiectasia mutated prevents DNA-dependent protein kinase-mediated cell death in response to topoisomerase I poison. J Biol Chem 285: 15201–15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Pereira JM, Aguilera A. 2015. R loops: new modulators of genome dynamics and function. Nat Rev Genet 16: 583–597. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, et al. 1995. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268: 1749–1753. [DOI] [PubMed] [Google Scholar]
- Scharer OD. 2013. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol 5: a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidle HF, Bieganowski P, Brenner C. 2005. Disease-associated mutations inactivate AMP-lysine hydrolase activity of aprataxin. J Biol Chem 280: 20927–20931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi J, Shuman S. 1997. Site-specific ribonuclease activity of eukaryotic DNA topoisomerase I. Mol Cell 1: 89–97. [DOI] [PubMed] [Google Scholar]
- Sepe S, Milanese C, Gabriels S, Derks KW, Payan-Gomez C, van IJcken WF, Rijksen YM, Nigg AL, Moreno S, Cerri S, et al. 2016. Inefficient DNA repair is an aging-related modifier of Parkinson's disease. Cell Rep 15: 1866–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Gilmore EC, Marshall CA, Haddadin M, Reynolds JJ, Eyaid W, Bodell A, Barry B, Gleason D, Allen K, et al. 2010. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet 42: 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y. 1997. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet 31: 635–662. [DOI] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y. 2013. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14: 197–210. [PubMed] [Google Scholar]
- Shimada M, Dumitrache LC, Russell HR, McKinnon PJ. 2015. Polynucleotide kinase-phosphatase enables neurogenesis via multiple DNA repair pathways to maintain genome stability. EMBO J 34: 2465–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shull ER, Lee Y, Nakane H, Stracker TH, Zhao J, Russell HR, Petrini JH, McKinnon PJ. 2009. Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes Dev 23: 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Proudfoot NJ. 2014. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev 28: 1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Proudfoot NJ, Gromak N. 2011. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 42: 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, Cimprich KA. 2015. Breaking bad: R-loops and genome integrity. Trends Cell Biol 25: 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollier J, Stork CT, Garcia-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA. 2014. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell 56: 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordet O, Redon CE, Guirouilh-Barbat J, Smith S, Solier S, Douarre C, Conti C, Nakamura AJ, Das BB, Nicolas E, et al. 2009. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep 10: 887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, Burgers PM. 2015. Error-free and mutagenic processing of topoisomerase 1-provoked damage at genomic ribonucleotides. EMBO J 34: 1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM. 2012. RNase H2-initiated ribonucleotide excision repair. Mol Cell 47: 980–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, Raams A, Byrd PJ, Petrini JH, Taylor AM. 1999. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99: 577–587. [DOI] [PubMed] [Google Scholar]
- Stiles J, Jernigan TL. 2010. The basics of brain development. Neuropsychol Rev 20: 327–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Pietilainen OP, Linder B, Suvisaari J, Brosi C, Hennah W, Leppa V, Torniainen M, Ripatti S, Ala-Mello S, et al. 2013. Deletion of TOP3β, a component of FMRP-containing mRNPs, contributes to neurodevelopmental disorders. Nat Neurosci 16: 1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Petrini JH. 2011. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 12: 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suberbielle E, Djukic B, Evans M, Kim DH, Taneja P, Wang X, Finucane M, Knox J, Ho K, Devidze N, et al. 2015. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat Commun 6: 8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykora P, Croteau DL, Bohr VA, Wilson DM III. 2011. Aprataxin localizes to mitochondria and preserves mitochondrial function. Proc Natl Acad Sci 108: 7437–7442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, Mao Y, Quiocho FA, Roa BB, Nakagawa M, et al. 2002. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 32: 267–272. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Harnden DG, Arlett CF, Harcourt SA, Lehmann AR, Stevens S, Bridges BA. 1975. Ataxia telangiectasia: a human mutation with abnormal radiation sensitivity. Nature 258: 427–429. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Groom A, Byrd PJ. 2004. Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair 3: 1219–1225. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Lam Z, Last JI, Byrd PJ. 2015. Ataxia telangiectasia: more variation at clinical and cellular levels. Clin Genet 87: 199–208. [DOI] [PubMed] [Google Scholar]
- Tresini M, Warmerdam DO, Kolovos P, Snijder L, Vrouwe MG, Demmers JA, van IWF, Grosveld FG, Medema RH, Hoeijmakers JH, et al. 2015. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 523: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, Nussenzweig A. 2017. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168: 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuduri S, Crabbe L, Conti C, Tourriere H, Holtgreve-Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C, et al. 2009. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol 11: 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbale P, Williams JS, Schellenberg MJ, Kunkel TA, Williams RS. 2014. Aprataxin resolves adenylated RNA–DNA junctions to maintain genome integrity. Nature 506: 111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Vega YA, Maclean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, Cleveland JL, Green DR, Kastan MB. 2012. Mitochondrial dysfunction in ataxia-telangiectasia. Blood 119: 1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, Beckmann G, Seemanova E, Cooper PR, Nowak NJ, et al. 1998. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93: 467–476. [DOI] [PubMed] [Google Scholar]
- Walden H, Deans AJ. 2014. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys 43: 257–278. [DOI] [PubMed] [Google Scholar]
- Wallace BD, Williams RS. 2014. Ribonucleotide triggered DNA damage and RNA–DNA damage responses. RNA Biol 11: 1340–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltes R, Kalb R, Gatei M, Kijas AW, Stumm M, Sobeck A, Wieland B, Varon R, Lerenthal Y, Lavin MF, et al. 2009. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Hum Genet 84: 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw CP, Carr AM, Oliver AW. 2014. TopBP1: a BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair 22: 165–174. [DOI] [PubMed] [Google Scholar]
- Weinfeld M, Mani RS, Abdou I, Aceytuno RD, Glover JN. 2011. Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem Sci 36: 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, Weinfeld M, Caldecott KW. 2001. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 104: 107–117. [DOI] [PubMed] [Google Scholar]
- Williams GJ, Lees-Miller SP, Tainer JA. 2010. Mre11–Rad50–Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair 9: 1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Smith DJ, Marjavaara L, Lujan SA, Chabes A, Kunkel TA. 2013. Topoisomerase 1-mediated removal of ribonucleotides from nascent leading-strand DNA. Mol Cell 49: 1010–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GJ, Hammel M, Radhakrishnan SK, Ramsden D, Lees-Miller SP, Tainer JA. 2014. Structural insights into NHEJ: building up an integrated picture of the dynamic DSB repair super complex, one component and interaction at a time. DNA Repair 17: 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Lujan SA, Kunkel TA. 2016. Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat Rev Mol Cell Biol 17: 350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Gehle DB, Kunkel TA. 2017. The role of RNase H2 in processing ribonucleotides incorporated during DNA replication. DNA Repair 53: 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltzius JJ, Hohl M, Fleming JC, Petrini JH. 2005. The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nat Struct Mol Biol 12: 403–407. [DOI] [PubMed] [Google Scholar]
- Yang YG, Lindahl T, Barnes DE. 2007. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131: 873–886. [DOI] [PubMed] [Google Scholar]
- Yeo AJ, Becherel OJ, Luff JE, Cullen JK, Wongsurawat T, Jenjaroenpun P, Kuznetsov VA, McKinnon PJ, Lavin MF. 2014. R-loops in proliferating cells but not in the brain: implications for AOA2 and other autosomal recessive ataxias. PLoS One 9: e90219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. 2014. Causes and consequences of replication stress. Nat Cell Biol 16: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, Dere R, Tait-Mulder J, Lee JH, Paull TT, et al. 2015. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol 17: 1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Lee JW, Tatavarthi H, Lupski JR, Valerie K, Povirk LF. 2005. Deficiency in 3′-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (TDP1). Nucleic Acids Res 33: 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Petersen S, Tessarollo L, Nussenzweig A. 2001. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr Biol 11: 105–109. [DOI] [PubMed] [Google Scholar]
- Zolner AE, Abdou I, Ye R, Mani RS, Fanta M, Yu Y, Douglas P, Tahbaz N, Fang S, Dobbs T, et al. 2011. Phosphorylation of polynucleotide kinase/ phosphatase by DNA-dependent protein kinase and ataxia-telangiectasia mutated regulates its association with sites of DNA damage. Nucleic Acids Res 39: 9224–9237. [DOI] [PMC free article] [PubMed] [Google Scholar]