

Audet-Walsh et al. reveal the existence of a nuclear mTOR–androgen receptor transcriptional axis integral to the metabolic rewiring of prostate cancer cells.
Keywords: androgen receptor, CRPC, energy metabolism, nuclear receptor, steroid, ChIP-seq
Abstract
Androgen receptor (AR) signaling reprograms cellular metabolism to support prostate cancer (PCa) growth and survival. Another key regulator of cellular metabolism is mTOR, a kinase found in diverse protein complexes and cellular localizations, including the nucleus. However, whether nuclear mTOR plays a role in PCa progression and participates in direct transcriptional cross-talk with the AR is unknown. Here, via the intersection of gene expression, genomic, and metabolic studies, we reveal the existence of a nuclear mTOR–AR transcriptional axis integral to the metabolic rewiring of PCa cells. Androgens reprogram mTOR–chromatin associations in an AR-dependent manner in which activation of mTOR-dependent metabolic gene networks is essential for androgen-induced aerobic glycolysis and mitochondrial respiration. In models of castration-resistant PCa cells, mTOR was capable of transcriptionally regulating metabolic gene programs in the absence of androgens, highlighting a potential novel castration resistance mechanism to sustain cell metabolism even without a functional AR. Remarkably, we demonstrate that increased mTOR nuclear localization is indicative of poor prognosis in patients, with the highest levels detected in castration-resistant PCa tumors and metastases. Identification of a functional mTOR targeted multigene signature robustly discriminates between normal prostate tissues, primary tumors, and hormone refractory metastatic samples but is also predictive of cancer recurrence. This study thus underscores a paradigm shift from AR to nuclear mTOR as being the master transcriptional regulator of metabolism in PCa.
It is now well established that androgens play a central role in the development and progression of prostate cancer (PCa). Indeed, even at the metastatic level, androgens—and more precisely the androgen receptor (AR)—are well known to stimulate proliferation (Bolla et al. 2009; Ylitalo et al. 2017). However, androgen deprivation therapy (ADT) does not cure PCa, as the disease eventually evolves to castration-resistant PCa (CRPC). At this stage, several molecular mechanisms have been discovered, most of them leading to AR hyperactivation, including AR genomic amplification and mutation (Montgomery et al. 2008; Robinson et al. 2015; Watson et al. 2015; Schweizer and Yu 2017; Ylitalo et al. 2017). Consequently, further knowledge of downstream effectors of the AR is necessary to dissect the molecular mechanisms governing PCa progression and develop novel therapeutic approaches.
Reprogrammed metabolism has been recognized as a hallmark of cancer (Hanahan and Weinberg 2011; Pavlova and Thompson 2016), including in PCa. Metabolically, the prostate gland is unique in that epithelial prostate cells produce high levels of citrate due to a blunted mitochondrial tricarboxylic acid (TCA) cycle impeded by high zinc levels (Costello et al. 2005; Costello and Franklin 2017). Through the process of malignant transformation, citrate and zinc accumulation ceases, which is thought to be the consequence of restored mitochondrial function and increased lipid synthesis from citrate production (Costello et al. 2005; Costello and Franklin 2017). In PCa, androgens and their receptor have been shown to fuel cell metabolism by rewiring nutrients and altering citrate metabolism, notably by inducing mitochondrial activity, de novo lipogenesis, and aerobic glycolysis (Massie et al. 2011; Tennakoon et al. 2014; Audet-Walsh et al. 2017). mTOR is another critical regulator of metabolism and cancer cell growth (Menon and Manning 2008; Laplante and Sabatini 2013). The classic view of mTOR action is that microenvironmental inputs channel through a single mTOR hub to give rise to a diversity of outputs influencing anabolic processes such as protein and lipid synthesis. However, emerging evidence supports the concept of multiple mTOR hubs with distinct subcellular compartments that may have distinct functions and sensitivity to drugs (Goberdhan et al. 2016). Moreover, there is a growing number of reports from yeast to human cells supporting the notion that mTOR can localize to the nucleus, associate with chromatin, and directly regulate gene expression (Zhang et al. 2002; Bernardi et al. 2006; Li et al. 2006; Cunningham et al. 2007; Kantidakis et al. 2010; Betz and Hall 2013; Chaveroux et al. 2013). Nonetheless, very little progress has been made in investigating a potential role for nuclear mTOR in transcriptional regulation.
The mTOR signaling pathway is hyperactivated in PCa tumors compared with peri-tumoral or benign prostate tissues (Kremer et al. 2006; Evren et al. 2010; Sutherland et al. 2014). In fact, alterations leading to hyperactivation of mTOR and its upstream regulators (PI3K/Akt), such as loss of PTEN, are among the most frequent genomic alterations in metastatic CRPC (Taylor et al. 2010; Grasso et al. 2012; Robinson et al. 2015; Kumar et al. 2016). However, whether nuclear mTOR is directly implicated in PCa progression and/or in transcriptional cross-talk with AR in the regulation of gene programs is unknown.
In this study, we first demonstrate that activated AR enhances and reprograms mTOR chromatin-binding profiles and that nuclear mTOR activity is essential for androgen-mediated transcriptional reprogramming of metabolism in PCa cells. Significantly, we also observed a strong correlation between mTOR nuclear content and PCa aggressiveness and progression in clinical samples. Finally, we identified an mTOR-dependent transcriptional signature that can robustly discriminate between normal tissues, primary PCa, and CRPC metastatic tissues in patients and predict disease recurrence, thus underscoring the biological significance and clinical impact of the activity of nuclear mTOR in PCa.
Results
Activated AR induces mTOR nuclear localization and DNA binding
Cytoplasmic mTOR signaling has been shown previously to be activated by androgens and induced by the synthetic androgen R1881 (Xu et al. 2006; Massie et al. 2011). In agreement with these findings, R1881 increased activation marks of mTOR complex 1 (mTORC1), such as phosphorylation of S6 and S6K, and enhanced phosphorylation of Akt, an activation mark of mTORC2, in LNCaP and 22rv1 cells (Fig. 1A; Supplemental Fig. S1A). Accordingly, this process was impaired by anti-androgens (Fig. 1B). In addition, the PI3K inhibitor GDC-0941 also blunted AR-mediated activation of mTOR (Supplemental Fig. S1B). Interestingly, we noted increasing nuclear mTOR levels in a time-dependent manner following androgen stimulation, indicating the formation of an androgen-dependent mTOR nuclear hub (Fig. 1C; Supplemental Fig. S1C,D). Both genetic and pharmacological inhibition of AR abolished this effect (Fig. 1D; Supplemental Fig. S1E), demonstrating the direct involvement of the androgen signaling pathway on mTOR cellular localization. Knockdown of mTOR by siRNAs confirmed the antibody specificity for mTOR detection (Supplemental Fig. S1F). mTOR nuclear abundance was probably favored by increased mTOR total protein levels upon R1881 stimulation (Massie et al. 2011). In addition, immunofluorescence studies validated the androgenic stimulatory effects on nuclear mTOR abundance compared with its cytoplasmic compartment (Fig. 1E; Supplemental Fig. S1G). The mTORC1- and mTORC2-specific components RAPTOR and RICTOR, respectively, also showed increased nuclear levels upon R1881 stimulation (Fig. 1D; Supplemental Fig. S1H), consistent with previous reports describing their nuclear localization (Cunningham et al. 2007; Rosner and Hengstschlager 2008, 2012; Yadav et al. 2013).
Figure 1.

Cytoplasmic and nuclear mTOR activities are induced by androgen signaling. (A) Protein expression of components of the mTOR signaling pathway and AR in whole-cell lysates from LNCaP cells after a 48-h treatment with R1881 or vehicle. Tubulin is shown as a loading control. (B) Protein expression of components of the mTOR signaling pathway in whole-cell lysates from LNCaP cells after a 48-h treatment with R1881 and/or anti-androgens (bicalutamide [B] and enzalutamide [E]). Tubulin is shown as a loading control. (C) Western blot analysis of nuclear fractions of LNCaP cells treated with R1881 or vehicle for various amounts of time. Lamin B1 is shown as a loading control. (D) Western blot analysis of cytoplasmic and nuclear fractions of LNCaP cells transfected with control or anti-AR siRNA and treated with R1881 or vehicle for 48 h. Lamin B1 and tubulin are shown as controls for nuclear and cytoplasmic extracts, respectively. (E) Immunofluorescence showing increased mTOR (red) levels in the nucleus following a 48-h treatment with R1881 in LCNaP cells. Nuclei were stained with DAPI (blue). (F) Overlap between mTOR DNA-binding peaks in LNCaP cells treated for 48 h with R1881 or vehicle and heat maps of the signal intensity of mTOR genomic binding peaks in a window of ±2.5 kb. (G) Average ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) signal intensities normalized per reads for down-regulated, unregulated, and up-regulated mTOR peaks. (H) Examples of University of California at Santa Cruz Genome Browser graphical views of mTOR-binding peaks in LNCaP cells treated with R1881 or vehicle that are either unaffected (unregulated) or positively or negatively regulated by androgens. Genes and peaks (black boxes) are indicated below tag density graphs. ChIP-qPCR (ChIP combined with quantitative PCR) of mTOR in LNCaP (I) or LAPC4 (J) cells after 48 h of treatment with R1881 or vehicle. Relative fold enrichment was normalized over two negative regions and is shown relative to IgG (set at 1). Results are shown as the average of three independent experiments.
As mTOR has been shown previously to directly interact with chromatin (Cunningham et al. 2007; Chaveroux et al. 2013), we next investigated whether androgens influence mTOR nuclear activity. To this end, we first performed chromatin immunoprecipitation (ChIP) coupled with high-throughput sequencing (ChIP-seq) of mTOR in LNCaP cells with or without R1881 stimulation. ChIP-seq analysis revealed increased mTOR–chromatin associations by androgens from ∼80,000–120,000 peaks (Fig. 1F; Supplemental Fig. S2A). Globally, 60,000 peaks were common between vehicle- and androgen-treated cells, with 17,136 peaks unique to basal conditions and 58,627 peaks specific to androgen treatment (Fig. 1F). Average binding intensities and representative University of California at Santa Cruz (UCSC) Genome Browser views of mTOR-binding profiles to both unregulated and androgen-regulated loci are shown in Figure 1, G and H, respectively. Unregulated peaks displayed the highest average signal intensity compared with R1881-sensitive peaks. Nevertheless, strong mTOR DNA binding was found either under basal conditions for “R1881-negatively regulated peaks” or in response to R1881 stimulation for “R1881-positively regulated peaks,” showing a clear enrichment of DNA binding compared with background (Fig. 1G–I). Of interest, the intensity of several binding events at genes known to be involved in PCa development, such as KLK3 (encoding PSA), TMPRSS2, UGT2B17, and MTOR itself, was modulated by R1881, as validated by ChIP combined with quantitative PCR (ChIP-qPCR) in LNCaP cells (Fig. 1I). mTOR genomic occupancy was also confirmed with a second antibody or using siRNAs against mTOR, validating the specific effects of R1881 on mTOR DNA binding (Supplemental Fig. S2B,C). ChIP-qPCR analysis in human LAPC4 cells that harbor wild-type PTEN confirmed the androgen responsiveness of mTOR binding to DNA (Fig. 1J). Furthermore, strong mTOR–chromatin associations were determined in the mouse prostate, indicating that mTOR binds DNA in both normal and PCa cells (Supplemental Fig. S2D). Taken together, our results demonstrate that AR activation not only leads to activation of the mTOR signaling pathway but also enhances mTOR nuclear levels and its interaction with the genome.
AR reprograms mTOR genomic binding profiles
The mechanism underlying mTOR–DNA interactions was next interrogated by DNA motif analysis that uncovered FOXA1 as the most enriched motif in both the absence and presence of R1881 (Supplemental Fig. S3A). FOXA1 is a pioneer transcription factor playing an important role in dictating nuclear receptor-binding profiles (Carroll et al. 2005; Laganière et al. 2005; Zaret and Carroll 2011), including AR in PCa cells (Jin et al. 2013; Pomerantz et al. 2015), as well as binding to non-AR targets involved in PCa cell cycle progression (Zhang et al. 2011). Furthermore, the androgen response element (ARE) was specifically enriched in mTOR-bound regions in the presence of androgens, suggesting targeted corecruitment of mTOR and AR to DNA (Fig. 2A; Supplemental Fig. S3A). These findings prompted us to cross-examine our mTOR ChIP-seq data set with a publicly available AR ChIP-seq data set (Massie et al. 2011). Strikingly, the mTOR–AR overlap in binding peaks increased from 23% to 81% following R1881 treatment, proportional to AR peaks (Fig. 2B; Supplemental Fig. S3B). ChIP-qPCR experiments confirmed co-occupancy of mTOR and AR at the same genomic loci following androgen treatment in LNCaP cells (Fig. 2C) and LAPC4 cells (Fig. 1J; Supplemental Fig. S3C). Additionally, AR knockdown using siRNAs impaired mTOR recruitment to androgen-sensitive peaks (Fig. 2D, left) without modifying mTOR recruitment at androgen-insensitive peaks (Fig. 2D, right). These results confirmed that AR drives mTOR DNA binding to specific loci following androgen stimulation and, together with the enrichment of ARE motifs at these mTOR-bound regions, suggested that mTOR and AR can associate in the same nuclear complex. Indeed, pharmacological inhibition of mTOR with rapamycin and torin 1 blunted the AR-mediated transcriptional activity of a luciferase reporter driven by two AREs (Fig. 2E). Additionally, sequential ChIP experiments (ChIP–reChIP) with AR and mTOR antibodies confirmed that both factors are corecruited in close vicinity to specific genes, including SLC26A3, FASN, HK2, and mTOR itself, following androgen stimulation (Fig. 2F; Supplemental Fig. S3D). Further validation was performed using coimmunoprecipitation (co-IP) experiments of AR and mTOR in LNCaP cells. In the absence of androgens, no interaction between AR and mTOR was detected (Fig. 2G). In contrast, in R1881-treated cells, AR was coimmunoprecipitated with mTOR (Fig. 2G, top), and, reciprocally, mTOR was coimmunoprecipitated with AR (Fig. 2G, bottom). Collectively, our data show that AR and mTOR can be part of the same complex and that AR drives mTOR DNA binding to specific loci.
Figure 2.

AR reprograms mTOR interaction with the genome of PCa cells. (A) Motif discovery analysis of mTOR ChIP-seq peaks identified from R1881-treated LNCaP cells revealed the ARE as the major motif enriched in response to androgen stimulation. (B) Overlap between AR and mTOR DNA-binding sites following R1881 or vehicle treatment. (C) ChIP-qPCR of mTOR and AR at the same binding sites in LNCaP cells following a 48 h of androgen treatment. (D) ChIP-qPCR analyses of AR and mTOR binding in LNCaP cells transfected with siControl (siC) or siAR and treated with vehicle or R1881 for 48 h. ChIP-qPCRs were performed for androgen-sensitive (left) and androgen-insensitive (right) mTOR-binding sites. (E) Luciferase reporter assay under the control of 2xAREs in LNCaP cells following a 24 h of treatment with R1881, rapamycin, torin 1, or vehicles, as indicated. Results are shown as the average of three independent experiments performed in triplicate. (F) ChIP–reChIP analysis shows corecruitment of AR and mTOR to the same genomic regions following androgen treatment. The significant enrichments at SLC26A3 and mTOR genes with IgG as a second antibody reflect the enrichment from the first ChIP, done with either AR or mTOR antibodies. Results are shown as the average of two independent experiments. (G) Co-IP of AR and mTOR in the absence or presence of androgens. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
mTOR is an essential component of AR transcriptional control
To assess the functional role of mTOR and AR DNA binding following R1881 treatment, we next studied how androgens regulate gene transcription in the presence or absence of a functional mTOR pathway. Importantly, both rapamycin and torin 1 inhibited mTOR activity even in the presence of R1881, as determined by the phosphorylation of S6K, S6, and AKT (Fig. 3A). This pharmacological response enabled the differentiation between AR-specific and AR–mTOR transcriptional regulation. Therefore, we next determined the transcriptome of LNCaP cells treated with R1881 and/or torin 1 to examine the functional consequence of loss of mTOR activity on androgen-mediated transcriptional control. While R1881 had a larger effect on global gene expression than torin 1, addition of torin 1 diminished the number of androgen-regulated genes by 35% (Supplemental Fig. S4A). Interestingly, although we observed that both PI3K and mTOR inhibitors led to an accumulation of mTOR in the nucleus, only PI3K inhibition diminished androgen-dependent mTOR recruitment to DNA, suggesting differential effects of these inhibitors on mTOR transcriptional activity (Supplemental Fig. S4B,C). At the mRNA level, androgen stimulation led to the significant modulation of several hundred genes, and cotreatment with torin 1 completely or severely impaired this hormonal response at about a third of the androgen-sensitive genes (36%) (Supplemental Fig. S4D,E). Functional pathway enrichment analysis of the subset of genes sensitive to both androgens and torin 1 identified cell cycle and metabolism as the two major biological functions codependent on AR and mTOR (Fig. 3B). Gene set enrichment analysis (GSEA) established that androgens stimulate metabolic gene signatures, including glycolysis and oxidative phosphorylation (OXPHOS), that were lost upon cotreatment with torin 1 (Fig. 3C,D). On the other hand, the “androgen response” gene signature was found to be mostly unaffected by torin 1 (Fig. 3E). The transcriptional responses of key genes from the identified signatures to the combination of R1881 and mTOR inhibitors were next validated by qRT–PCR. Some androgen-responsive targets, such as PFKFB2 and ADPGK, were essentially insensitive to mTOR inhibitors (Fig. 3F). In sharp contrast, the expression of several metabolic genes related to glycolysis (ENO1 and HK2), OXPHOS (ATP5L and NDUFA6), and lipogenesis (FASN) that was found to be significantly up-regulated by androgens was either lost or even reversed in response to mTOR inhibitors (Fig. 3G). Overall, these results demonstrate that mTOR is required for a specific subset of androgen-mediated gene programs predominantly in relation to metabolic functions.
Figure 3.

Nuclear mTOR is essential for androgen-mediated transcriptional control of metabolic gene signatures. (A) Activation status of the mTOR signaling pathway following treatment with R1881 and/or cotreatment with the mTOR inhibitors rapamycin and torin 1. (B) Ingenuity Pathway Analysis (IPA) pathway enrichment of genes modulated by R1881 but blocked by mTOR inhibition in LNCaP cells. GSEA plots for androgen-stimulated glycolytic (C) and OXPHOS (D) gene signatures that are AR- and mTOR-dependent. “Core genes” are shown in the heat maps. (E) GSEA plots for the AR-dependent but mTOR-independent gene signatures associated with “androgen response.” Only genes identified as “core genes” are shown in the heat map. qRT–PCR analysis of metabolic genes modulated by androgens in an mTOR-independent (F) or mTOR-dependent (G) manner following a 48-h treatment with R1881 and/or cotreatment with rapamycin or torin 1. Values represent mean ± SEM of three independent experiments performed at least in duplicate. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
A functional nuclear mTOR–AR axis is required for the metabolic rewiring of PCa cells
Intersection of ChIP-seq and transcriptomic data sets shows that genes up-regulated by androgens and targeted by mTOR are significantly enriched for metabolic pathways and their associated cellular components such as mitochondria (Fig. 4A; Supplemental Fig. S5A; Supplemental Table S1). ChIP-qPCR assays confirmed that several metabolic genes identified as bona fide mTOR targets by ChIP-seq exhibited increased mTOR binding following R1881 treatment (Fig. 4B). To confirm the requirement of nuclear mTOR in transcriptional regulation of metabolic pathways, we next analyzed the effect of the mTOR–AR axis on the metabolic reprogramming of PCa cells. As expected, AR activation induced major metabolic readouts, such as glucose consumption, lactate production, mitochondrial biogenesis and activity, and lipid synthesis (Fig. 4C–G). Consistent with our functional genomics analyses (Fig. 3), loss of mTOR activity severely blunted the metabolic reprogramming induced by R1881 (Fig. 4C–G). Both glucose uptake and extracellular acidification rate (ECAR), an indicator of aerobic glycolysis, were induced by fivefold by R1881 but considerably impaired by rapamycin and torin 1 treatments in both LNCaP (Fig. 4C,D) and LAPC4 (Supplemental Fig. S5B) cells. Both mitochondrial respiration and mitochondrial content were also strongly induced by androgen treatment, and these effects were completely lost upon cotreatment with mTOR inhibitors (Fig. 4E,F). Similarly, mitochondrial respiration was induced by R1881 treatment, an effect completely lost upon mTOR inhibition in LAPC4 cells (Supplemental Fig. S5B). Additionally, inhibition of mTOR reversed androgen-stimulated de novo lipogenesis (Fig. 4G). Of note, a long-term exposure to the mTOR inhibitors rapamycin or torin 1 did not severely impact the viability of R1881-treated LNCaP cells (Supplemental Fig. S5C). Moreover, treatment of DU145 cells with mTOR inhibitors modestly impacted cell viability (Supplemental Fig. S5D). Reorganization of cellular nutrient utilization in PCa cells by AR activation is achieved in large part by altering the activity of mitochondria (Tennakoon et al. 2014; Audet-Walsh et al. 2017). In support of this notion, PCa cells treated with R1881 were found to be more sensitive to treatment with etomoxir, an inhibitor of fatty acid oxidation (FAO) (Fig. 4H). Importantly, mTOR inhibition abrogated the inhibitory effect of etomoxir on mitochondrial respiration in R1881-treated cells (Fig. 4H). Taken together, our results clearly show that AR activation reprograms PCa metabolism and nutrient usage through transcriptional regulation, which requires a functional mTOR pathway.
Figure 4.

Nuclear mTOR dictates androgen-dependent metabolic reprogramming. (A) Functional genomics analysis of AR–mTOR cross-talk in PCa cells. Genes positively regulated by AR activation and for which mTOR inhibition by torin 1 blocked this regulation are shown. Gene ontology (GO) component enrichment analysis of the subset of these genes containing at least one mTOR peak within ±20 kb of their transcription start sites is shown. (B) mTOR ChIP-qPCR analysis of metabolic genes in LNCaP cells treated for 2 d with R1881 or vehicle. Results are shown as the average of three independent experiments. (C) Glucose consumption of LNCaP cells after a 4-d treatment with R1881 and/or cotreatment with mTOR inhibitors. The ECAR (D), an indicator of lactate production, and the oxygen consumption rate (OCR) (E) were analyzed in LNCaP cells following a 3-d treatment with R1881 and/or cotreatment with mTOR inhibitors. (F) Relative mitochondrial/nuclear DNA content of LNCaP cells following a 96-h treatment with R1881 with or without mTOR inhibitors. (G) Triglyceride content of LNCaP cells treated for 96 h with R1881, mTOR inhibitors, or vehicles. (H) Oxidative capacity following inhibition of FAO by etomoxir in LNCaP cells following treatment with R1881 or vehicle. One representative experiment is shown. n = 5. (I) qRT–PCR analysis of metabolic gene expression following a 48-h treatment with androgens and/or α-amanitin. (J) Activation status of the mTOR signaling pathway following treatment with R1881 or vehicle with or without cotreatment with the RNA polymerase II inhibitor α-amanitin. Glucose consumption (K), lactate production (L), and OCR (M) of LNCaP cells after a 3-d treatment with or without androgens and cotreated with or without α-amanitin. Results from C–G, I, and K–M are shown as the average ± SEM of at least three independent experiments performed in triplicate, and metabolic data were further normalized for cell number. (**) P < 0.01; (***) P < 0.001.
To ascertain the necessity of nuclear mTOR in the metabolic rewiring induced by androgen signaling, we used the RNA polymerase II (Pol II) inhibitor α-amanitin to discriminate between cytoplasmic and nuclear mTOR functions. As expected, α-amanitin significantly abrogated the R1881-mediated induction of metabolic transcript levels (Fig. 4I). Importantly, α-amanitin did not impair the androgen-dependent activation of cytoplasmic mTOR signaling, as demonstrated by increased phosphorylation of S6K and S6 (Fig. 4J), an effect described previously by others (Xu et al. 2006). The androgen-mediated stimulation of aerobic glycolysis, as assessed by glucose consumption and lactate production, was strongly diminished following cotreatment with α-amanitin (Fig. 4K,L). Similarly, induction of oxygen consumption rate (OCR) by R1881 was completely lost upon α-amanitin cotreatment (Fig. 4M). Thus, although cytoplasmic mTORC1 alone is not sufficient to drive androgen-mediated metabolic rewiring in PCa, it likely contributes to nuclear mTOR transcriptional activity.
mTOR controls metabolic gene transcription in CRPC cells
Most androgen-dependent PCa cell lines are derived from metastatic and castration-resistant tumors, and, as such, most CRPCs in humans are still dependent on the AR activity for growth and survival (Taylor et al. 2010; Grasso et al. 2012; Robinson et al. 2015; Kumar et al. 2016). However, between 15% and 25% of these CRPCs become AR-negative and represent the most aggressive form of PCa (Taylor et al. 2010; Grasso et al. 2012; Robinson et al. 2015; Beltran et al. 2016; Kumar et al. 2016). As we determined an essential role of mTOR in the transcriptional control of metabolism by androgens, we hypothesized that targeting mTOR in AR-negative CRPC cells, where mTOR is hyperactivated in the absence of androgens, would have extensive beneficial effects on cellular growth and metabolism. In both PC3 and DU145 CRPC cell lines (see Supplemental Table S2 for cell line characteristics), the mTOR signaling pathway was indeed constitutively activated in the absence of androgens but nonetheless found to be sensitive to mTOR inhibitors (Fig. 5A). Importantly, we detected considerable nuclear mTOR levels in these cells in the absence of steroids (Fig. 5B), in line with the strong recruitment of mTOR to metabolic genes observed by ChIP-qPCR (Fig. 5C). Note that the nuclear mTOR levels observed are comparable with AR-positive LNCaP cells, which were originally derived from a lymph node metastasis (Horoszewicz et al. 1980, 1983). Inhibition of mTOR significantly impaired the mRNA expression of key genes related to glycolysis (HK2 and ENO1), mitochondrial function (ATP5L and NDUFA6), and lipid metabolism (FASN and ACACB) (Fig. 5D). Moreover, pharmacological blockade of mTOR markedly hindered glucose uptake and aerobic glycolysis with up to a 90% decrease in lactate production following torin 1 treatment (Fig. 5E). This metabolic impediment following mTOR inhibition was associated with decreased cellular proliferation and deterred migration in both PC3 and DU145 cells (Fig. 5F,G).
Figure 5.

Transcriptional control of metabolism by mTOR in AR-null PCa cells. (A) Status of the mTOR signaling pathway activity following treatment with vehicle (V), rapamycin (R), or torin 1 (T) in PC3 and DU145 cells in the absence of androgens. Tubulin is shown as a loading control. (B) Detection of nuclear mTOR by Western blotting in LNCaP, PC3, and DU145 cells. Lamin B1 and tubulin are shown as loading controls for nuclear and cytoplasmic extracts, respectively. (C) ChIP-qPCR assessment of mTOR recruitment to DNA regulatory regions of metabolic genes in PC3 and DU145 cells. Results are shown as the average of three independent experiments. (D) qRT–PCR assessment of metabolic gene expression related to glycolysis (left) or mitochondrial and lipid metabolism (right) following 48 h of treatment with vehicle, rapamycin (rapa), or torin 1. (E) Glucose consumption and lactate production measured from the media of PC3 and DU145 cultured cells following a 48-h treatment with rapamycin, torin 1, or vehicle (control). (F) Cell number determination of PC3 and DU145 cells following a 48-h treatment with rapamycin, torin 1, or vehicle (control). (G) Migration assay for PC3 and DU145 cells with or without treatment with the mTOR inhibitor torin 1. Values in D–G represent mean ± SEM of at least three independent experiments performed in triplicate. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
Nuclear mTOR levels correlate with PCa progression
To explore the clinical relevance of nuclear mTOR, we performed immunohistochemistry (IHC) analysis of mTOR using tissue microarrays (TMAs) of 232 human primary localized PCa samples and 30 peri-tumoral tissues (Table 1; Supplemental Fig. S6A–C). mTOR was detected mostly in the cytoplasm of epithelial cells from benign glands and cancer cells of tumor foci, with low or undetectable expression in stromal cells (Fig. 6A), which is consistent with previous studies (Dai et al. 2009; Evren et al. 2010; Sutherland et al. 2014). No difference in mTOR cytoplasmic staining intensity between peri-tumoral and tumor samples was found (Supplemental Fig. S6D). We also had access to primary tumors from patients with ADT: 12 cases of neo-adjuvant hormonal therapy (NHT) and 18 cases from CRPC (CRPC-ADT) as well as 12 invaded seminal vesicle specimens and 11 bone metastases. The cytoplasmic mTOR H-score index was significantly higher in CRPC-ADT and bone metastasis samples compared with peri-tumoral and primary untreated PCa specimens (Supplemental Fig. S6D). Notably, both aggressive PCa under ADT as well as extraprostatic tumor cells had a significantly higher mTOR nuclear H-score index compared with both peri-tumoral and primary untreated tumors, which generally displayed weak signals (1+) (Fig. 6B,C; Supplemental Fig. S6E). At least 50% of tumor samples exhibited >30% positive nuclear mTOR staining in patients under ADT or in metastatic samples compared with only 14% in primary untreated PCa tumors (P < 0.0001) (Fig. 6B; Supplemental Fig. S6F).
Table 1.
Clinical and pathological characteristics of the radical prostatectomy McGill cohort of hormone-naïve patients
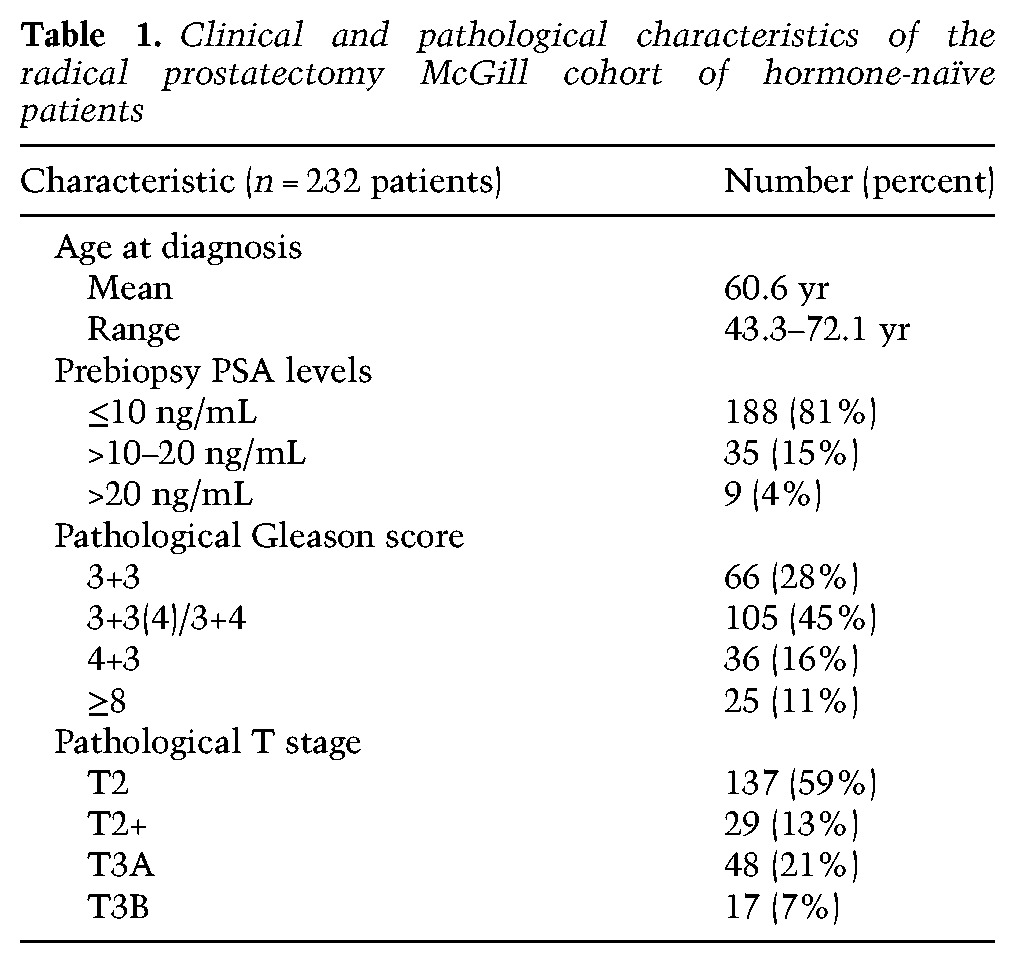
Figure 6.

Nuclear mTOR levels and activities are indicators of poor-outcome PCa. (A) Representative images from human prostate specimens immunostained for mTOR detection. Bar, 50 µm. (B) Histogram of nuclear mTOR distribution across patient samples. (C) Nuclear H-score of mTOR staining of peri-tumoral, primary PCa tumors, and metastatic lesions. (Peri-t) Peri-tumoral; (SV Inv.) seminal vesicle invasion; (Tx) treatment. Refer to Supplemental Figure S6F for cohort sizes. Significance is shown compared with both peri-tumoral and primary PCa tumors. (D) Unsupervised hierarchical clustering analysis with an mTOR targeted 622-gene signature in two independent clinical cohorts. In the data published by Lapointe et al. (2004), subtype colors denote sample classification as originally published. (N) Peri-tumoral tissues; (T) two tumors that clustered with peri-tumoral tissues in the original classification also clustered in a similar manner in the present study. In the data published by Tomlins et al. (2007), subtype colors discriminate between normal or peri-tumoral epithelial cells (N), stromal cells, primary localized PCa (primary T), and hormone-refractory metastatic PCa (MetHR). (E) A condensed mTOR targeted 24-gene signature capable of discriminating primary versus metastatic tumors and used to assess the risk of the biochemical recurrence rate (BCR) in patients. Kaplan-Meier biochemical recurrence-free survival analysis of patients from Taylor et al. (2010) (F) and The Cancer Genome Atlas (TCGA) provisional cohort (G) using a 24-gene mTOR targeted signature. Log-rank P-values are shown.
Finally, we investigated the clinical relevance of the mTOR genomic and transcriptional multigene signature identified following androgen stimulation in vitro (Supplemental Table S1). First, we confirmed that this gene signature is sensitive to androgen modulation in vivo in PCa xenografts (Supplemental Fig. S6G). Next, unsupervised hierarchical clustering analysis was performed on a previously described expression-profiling study of tumor and matched benign prostate tissues that resulted in characterization of three different PCa subtypes (Lapointe et al. 2004). Remarkably, this mTOR targeted gene signature discriminated between benign and tumor tissues but also between the three different PCa subtypes with 95% concordance with the original characterization of these groups (Fig. 6D, left). Furthermore, in a second independent cohort (Tomlins et al. 2007), the functional mTOR gene signature discerned epithelial benign cells, stromal benign cells, and localized PCa but, more importantly, distinguished patients with CRPC metastatic tissues (Fig. 6D, right). Based on expression data of the mTOR targeted gene signature in these two cohorts, we next selected the top 24 genes based on clustering weights to discriminate between primary tumors and lymph node metastases in the Lapointe et al. (2004) cohort or metastatic CRPC in the Tomlins et al. (2007) cohort (Fig. 6E; Supplemental Fig. S6H; Supplemental Table S1). Combined expression data for these 24 genes effectively stratified patients based on their biochemical recurrence rate (BCR). First, this 24-gene expression signature was subdivided into three categories: low, intermediate, and aggressive signatures. In the cohort from Taylor et al. (2010), patients with the protective signature had only 13% BCR compared with 63% BCR in patients with the aggressive expression signature (Fig. 6F). We also validated this mTOR targeted signature in the cohort from The Cancer Genome Atlas (TCGA) consortium (Cerami et al. 2012; Gao et al. 2013), which represents a slightly less aggressive PCa cohort compared with the data set from Taylor et al. (2010). As such, patients with the aggressive signature had 22% BCR compared with patients with the protective and intermediate signatures, which had similar BCRs and were therefore combined (9% BCR) (Fig. 6G). Collectively, our work uncovers a significant correlation between nuclear mTOR activity and human PCa progression.
Discussion
In this study, we revealed a pivotal role of nuclear mTOR as an essential transcriptional effector of PCa cellular metabolism and a key integrator of the androgen signaling pathway. We first show that the key oncogenic driver of PCa development, AR, reprograms mTOR association with chromatin and that activation of mTOR-dependent metabolic gene networks is essential for the androgen-mediated metabolic reprogramming of cancer cells. In addition, this mTOR transcriptional regulatory activity is reactivated in AR-null cell lines despite the absence of androgen stimulation, possibly highlighting a novel mechanism involved in castration resistance. Furthermore, the identification of a functional mTOR targeted multigene signature could robustly discriminate between normal prostate tissues, primary tumors, and CRPC, which correlate with increased mTOR nuclear localization in CRPC tumors and metastases and its association with BCR. Taken together, our work unravels an important link between mTOR transcriptional control of cellular metabolism, its nuclear localization, and poor-outcome PCa.
The central role played by AR in the development and progression of PCa is well appreciated, including its role in reprogramming mitochondrial metabolism (Tennakoon et al. 2014; Audet-Walsh et al. 2017). Although AR is recruited to the regulatory regions of many of the genes involved in these metabolic processes, numerous androgen-sensitive genes are not direct AR transcriptional targets (Audet-Walsh et al. 2017) but were identified in our study as direct targets of mTOR (Fig. 4; Supplemental Fig. S5; Supplemental Table S1). Thus, our results highlight nuclear mTOR as a crucial node in androgen-mediated metabolic reprogramming of PCa cells and mechanistically explain why mTOR acts in a dominant manner over AR action (Supplemental Fig. S7). In addition, as the classic AR signature is comprised of AR direct targets (Fig. 3E), this probably explains why these genes are, in general, insensitive to mTOR blockade. Since AR is considered a master transcriptional regulator of PCa cells, our finding that mTOR has a dominant effect over AR on the control of metabolic gene transcription was unanticipated. This is of importance regarding the recent observation that OXPHOS and pyrimidine metabolism were shown to be the most dysregulated pathways in lethal PCa tumors (Kelly et al. 2016). Given that AR genomic binding profiles are reprogrammed in CRPC compared with untreated PCa tumors (Sharma et al. 2013; Stelloo et al. 2015), it would be interesting to also determine whether the mTOR genomic footprint is also reprogrammed during the establishment of castration resistance. Classic components of the mTOR complex, such as RAPTOR and RICTOR, were also observed to localize to the nucleus upon R1881 stimulation and were described previously to be nuclear in various cellular contexts (Cunningham et al. 2007; Rosner and Hengstschlager 2012; Alayev et al. 2016). DEPTOR, an mTOR component of both mTORC1 and mTORC2, also has been shown recently to be nuclear (Catena et al. 2016). However, the role, if any, that these classic mTOR partners play in transcriptional regulation remains to be determined. Interestingly, it was shown recently that DEPTOR can interact with KDM4A, a histone demethylase enzyme that contributes to the epigenetic regulation of the genome (Carbonneau et al. 2016). In that context, blockade of mTOR—either mTORC1 (rapamycin) or both mTORC1/2 (torin 1)—would be predicted to impair gene transcriptional regulation with potentially stronger effects from dual-complex inhibition, as observed in the present study with torin 1. The exact nature of the nuclear mTOR complex remains to be identified and fully characterized.
Loss of PTEN, a key upstream regulator of mTOR activity, is one of the most frequent genomic alterations in metastatic CRPC (Taylor et al. 2010; Grasso et al. 2012; Robinson et al. 2015; Kumar et al. 2016). In our study, we used PCa cellular models exhibiting genetic inactivation of PTEN (LNCaP and PC3 cells) as well as wild-type PTEN (LAPC4, 22rv1, and DU145 cells) and obtained similar conclusions toward mTOR transcriptional regulatory functions. Therefore, in both AR-positive and AR-null cells, mTOR can act as a metabolic transcriptional regulator in both the presence and absence of a functional PTEN. Interestingly, like what we observed for mTOR, PTEN can also be localized in the nucleus (Chen et al. 2017), and further understanding of its tumor suppressor functions, such as inhibiting mTOR nuclear activity, deserves further study.
Here, we established that nuclear mTOR is clearly linked to PCa evolution and aggressiveness and that androgens act as strong activators of both cytoplasmic and nuclear mTOR functions. This work adds a significant new dimension to the paradigm in which mTOR is mostly studied for its activation by growth factor-activated membrane receptors and nutrient availability (Menon and Manning 2008; Laplante and Sabatini 2013; Shimobayashi and Hall 2016). Furthermore, specific inhibition of RNA Pol II-driven transcription demonstrated that cytoplasmic mTOR activity alone is not sufficient for the androgen-mediated rewiring of PCa cell metabolism. Although α-amanitin inhibits global transcription, its effects on metabolic reprogramming support the requirement of specific gene signature programs associated with AR and mTOR transcriptional function to reprogram metabolism in PCa cells. While not being sufficient to rewire metabolism of PCa cells, classic mTORC1 signaling is surely required in this process along with mTOR transcriptional activity. Moreover, our findings uncovered that androgens promote the nuclear translocation of a significant portion of mTOR in actively proliferating PCa cells and that mTOR is significantly abundant in the nuclei of CRPC and cells invading extraprostatic tissues such as bone metastases in humans. Thus, nuclear mTOR immunostaining, shown in our study to associate with cancer aggressiveness, could potentially serve as a biomarker of tumor progression and an indicator of tumor sensitivity to various pharmacological treatments.
It is tempting to speculate that the potential benefits of combining both current AR and mTOR inhibitors would lead to a better treatment of PCa. However, most clinical studies exploring the usefulness of mTOR inhibitors did not show any additive clinical benefits of such drugs in combination with first-generation anti-androgen or chemotherapy, as reviewed recently by Statz et al. (2017). In these clinical trials, rapalogs were found to only partially inhibit mTOR activities. Indeed, rapalogs such as rapamycin inhibit mTORC1, while new mTOR inhibitors such as torin 1 concomitantly inhibit both mTOR complexes. Accordingly, our results show a stronger inhibitory effect of torin 1 compared with rapamycin on androgen-mediated transcriptional metabolic reprogramming. As such, total blockade of mTOR or its positive upstream regulator, the PI3K/Akt signaling pathway, observed in our study to impair mTOR DNA binding (Supplemental Fig. S4C), would seemingly result in better clinical benefits. Indeed, preclinical models inhibiting both AR and the PI3K/Akt axis show promising results (Carver et al. 2011; Toren et al. 2015; Yadav et al. 2016). However, clinical pilot studies on anti-androgens and PI3K/Akt/mTOR inhibitors show high proportions of severe adverse effects and high rates of treatment discontinuation, with no clear indication that the PI3K/Akt/mTOR inhibitors reached and inhibited their targets in PCa tissues (Armstrong et al. 2017; Massard et al. 2017; Wei et al. 2017). Given the association of increased nuclear mTOR levels with disease evolution, specifically targeting the transcriptional function of mTOR could offer a novel therapeutic avenue for the management of poor-outcome PCa and perhaps other malignancies, with fewer side effects than global PI3K/Akt/mTOR inhibition.
Overall, our study uncovers a novel mechanism by which AR drives the metabolic reprogramming of PCa cellular metabolism and proliferation via modulation of nuclear mTOR function, therefore linking two major oncogenic drivers. Strikingly, the mTOR transcriptional axis was shown to be a dominant molecular component of AR signaling. This study thus reveals an unexpected function of nuclear mTOR and underscores a paradigm shift from AR to mTOR as being the master transcriptional regulator of cell metabolism during PCa progression. Given the positive association between nuclear mTOR abundance and PCa aggressiveness, nuclear detection of mTOR may be a potential biomarker for PCa progression and implicates the importance of designing effective inhibitors of mTOR (to abrogate its nuclear activity) as a therapeutic avenue for the management of this disease.
Materials and methods
Cell culture
LNCaP, LAPC4, PC3, DU145, and 22rv1 were originally obtained from American Type Culture Collection (ATCC). All cells were kept in culture for no more than 3 mo after resuscitation and were reauthenticated using the ATCC cell line authentication in July 2016. Cells were grown in phenol-red-free RPMI supplemented with 10% fetal bovine serum (FBS), penicillin, streptomycin, and sodium pyruvate at 37°C and 5% CO2. Cells were steroid-deprived in medium with 2% charcoal-stripped serum (CSS) for 48 h and then treated with 10 nM R1881 (Steraloids), 80 nM rapamycin (Calbiochem), 100 nM torin 1 (Toronto Research Medical), 2 µg/mL α-amanitin (Sigma), 1 µM GDC-0941 (gift from Dr William Muller, McGill University), or 10 µM enzalutamide (APExBIO). For siRNA transfections, cells were trypsinized, seeded in CSS medium, and transfected with either a nontargeted pool of siRNA or a pool of siRNA specific against AR or mTOR (SMARTpool) with Hiperfect transfection reagent per the manufacturer's instructions (Qiagen).
Metabolic analyses
Levels of metabolites consumed and produced by PCa cell lines in the culture medium were measured with a BioProfile Analyzer (Nova Biomedical). Briefly, cells were first seeded in medium containing CSS for 48 h to ensure steroid deprivation. Cells were then seeded at 400,000 cells per well for LNCaP and LAPC4 cells or 200,000 cells per well for other cell lines in 2 mL of medium containing CSS in six-well plates. For LNCaP and LAPC4 cells, cells were treated for a total of 4 d, with new treatments added after 48 h. Medium was then used for metabolite measurements, and cells were counted for normalization. For other cell lines, medium analysis was done after 48 h. For triglyceride quantification, LNCaP cells were treated with R1881, mTOR inhibitors, or vehicle for a total of 4 d, and the medium was replaced every 48 h. Lipids were then quantified using a triglyceride quantification assay kit (Abcam, ab65336).
The OCR and ECAR were measured using Seahorse XF instruments (Seahorse Bioscience) with samples prepared per the manufacturer's protocol. After steroid deprivation, cells were treated with medium containing vehicle, R1881, or mTOR inhibitors for 48 h and then seeded in Seahorse XF24 or XF96 plates. After 24 h, the medium was replaced by Seahorse XF assay medium, and cells were incubated in a CO2-free incubator for 1 h at 37°C to allow equilibration prior to loading into the XF instruments. XF24/XF96 assays consisted of cycles of sequential mix for 3 min, pause for 0–2 min, and measurement for 3–5 min, allowing determination of OCR/ECAR every 10 min. For each experiment, two to three measurements were taken at every step to allow OCR and ECAR quantification. After OCR/ECAR determinations, cells were counted for cell number normalization.
RNA and microarray analysis
RNA was extracted with the RNeasy minikit (Qiagen), and first strand cDNA synthesis was performed with ProtoScript II reverse transcriptase (New England Biolabs). cDNA expression was then quantified by SYBR Green-based qPCR techniques using the LightCycler 480 instrument (Roche). Relative expression was standardized to the expression of two housekeeping genes. The expression of 18S was also assessed in the context of RNA Pol II inhibition. Specific gene primers used for qRT–PCR are in Supplemental Table S3.
For microarray analysis, cells were seeded with or without treatments (R1881 and/or torin 1) in medium without steroids and harvested 48 h later. RNA was isolated as described above, and three samples per group were sent for microarray analysis at Génome Québec and McGill University Genome Centre (Illumina Human HT-12 Expression Beachip version 4). FlexArray software was used for microarray data normalization. Ingenuity Pathway Analysis (IPA) and GSEA were used to analyze pathway enrichment. For clustering analyses, data were processed with Gene Cluster 3.0 using the hierarchical linkage method followed by manual curating. Data were visualized using JavaTreeView (Saldanha 2004). Microarray data are available in the Gene Expression Omnibus (GEO): GSE93603.
Microarray data described by Lapointe et al. (2004) were analyzed as published previously. Six-hundred-sixty-two genes were included in the initial mTOR target gene lists, of which 161 genes (223 probes) were retrieved in the original data set of differentially expressed genes represented by 5153 probes. Microarray data from Sun et al. (2012) and Tomlins et al. (2007) were analyzed similarly.
Protein analysis
Cells were lysed with buffer K supplemented with protease and phosphatase inhibitors for whole-cell lysates or separated into nuclear and cytoplasmic fractions by differential centrifugation as described previously (Chaveroux et al. 2013). Co-IP protocols and antibodies used for Western blot are described in the Supplemental Material.
ChIP-qPCR and ChIP-seq
After steroid deprivation, human PCa cells were treated for 48 h with R1881 for AR (Santa Cruz Biotechnology, sc-816X) or mTOR ChIP (Abcam ab32028 for ChIP-seq and ChIP-qPCR; Santa Cruz Biotechnology N-19 sc1549R for ChIP-qPCR) assays. After protein/DNA cross-linking with 1% formaldehyde at room temperature, cells were lysed, nuclei were enriched by sequential centrifugations (Langlais et al. 2011), and ChIP was performed as described previously (Audet-Walsh et al. 2016).
For mouse prostate ChIP assays (Abcam, ab32028), a pool of three prostates per ChIP was used. For all ChIP-qPCR analyses, nontargeted rabbit IgG ChIP was used as a control for nonspecific antibody binding, and two to three negative regions were used for ChIP normalization between samples using a Roche LightCycler 480. Gene-specific primers used for human and mouse ChIP analyses are in Supplemental Tables S4 and S5, respectively.
For human mTOR ChIP-seq experiments, following mTOR ChIP with Dynabeads (Life Technologies) and purification of DNA with a purification kit (Qiagen), libraries of enriched DNA segments were prepared per the ChIP-seq library protocol by Illumina as described previously (Audet-Walsh et al. 2016) and subsequently sequenced at the McGill University and Génome Québec Innovation Centre. Sequences were aligned to the human genome database (Hg19) using BWA version 0.5.9 (Audet-Walsh et al. 2016). Peaks were called using MACS software version 1.4.1. Peak annotation, tag directory, bed file generation, and de novo motif discovery were performed with the HOMER package version 3.18. ChIP-seq data for AR in LNCaP cells were published previously and are publicly available (Massie et al. 2011). mTOR ChIP-seq data are available in the GEO database (GSE93845). To compare AR and mTOR ChIP-seq data sets, AR ChIP-seq peak data aligned to hg18 were first realigned to the hg19 genomic assembly using the online UCSC Lift Genome Annotations (LiftOver; https://genome.ucsc.edu/cgi-bin/hgLiftOver). Pathway enrichment analyses with IPA software (Ingenuity Systems) and gene ontology (GO) enrichment of cellular components were conducted using genes with binding peaks found within ±20 kb of the transcription start sites of the genes. To obtain the mTOR targeted multigene signature, all probes significantly modulated by R1881 with P < 0.01 were used for probe clustering analyses. Clustering weights were then used to select the best probes—allowing discrimination of the four groups (control, R1881, torin, torin + R1881)—and then manually curated. Finally, probes sensitive to both R1881 and torin 1 were annotated to specific genes and described as an mTOR target gene if there was an mTOR-binding site ±20 kb of their transcription start sites (the complete list of mTOR targeted genes is in Supplemental Table S1; heat maps of these genes following androgen or torin 1 treatment are in Supplemental Fig. S4D,E). From the mTOR targeted gene signature comprised of 662 genes, a 24-gene signature (Supplemental Table S1) was obtained and used to assess BCRs in patients as outlined in Supplemental Figure S6H. See the Supplemental Material for further details.
Luciferase assay
LNCaP cells were transfected with the firefly luciferase reporter under the control of 2xAREs using Fugene HD (Promega). Briefly, for six wells of a 24-well plate, 3 µg of DNA (2.4 µg of the firefly luciferase reporter and 50 ng of β-galactosidase expression vector as the internal control) were mixed with 150 µL of optiMEM medium (Invitrogen) and 9 µL of Fugene HD. The day after transfection, the medium was changed to medium containing CSS. Following steroid deprivation, cells were treated for 24 h with R1881 with or without mTOR inhibitors as indicated. Cell lysates were then analyzed for luciferase activity and β-galactosidase activity.
Human tissues, IHC, and clinical follow-ups
Study protocols were approved by the Ethics Board of the McGill University Health Centre Research Institute with informed consent obtained from all subjects. Formaldehyde-fixed paraffin-embedded (FFPE) archival blocks from PCa patients were collected from the Pathology Department. Primary prostate tumor tissue blocks were primarily from radical prostatectomy cases followed at the McGill University Health Centre between 1996 and 2007, most of whom were hormone-naïve or untreated prior to surgery (n = 232), except for 12 NHT cases who have had a 3- to 4-mo neo-adjuvant ADT treatment with LHRH agonists and anti-androgens. Radical prostatectomy blocks were reviewed to reassess the Gleason score and tumor stage of each case (tabulated in a pathological database), per the 2016 classification system (Epstein et al. 2016). The clinical database of radical prostatectomy cases was built separately and periodically updated through a retrospective search in medical records. For this study, complete follow-up data were available for the hormone-naïve radical prostatectomy cases (Table 1). Additional FFPE blocks were included to reflect more aggressive tumors in high-risk patients or advanced stages of disease: They consist of transurethral resections of the prostate (TURP; primary tumor) in patients who are castration-resistant (CRPC) and under palliative ADT (n = 18), seminal vesicles with evidence of invasion by prostate tumor cells when harvested at radical prostatectomy (n = 12), and bone metastases (n = 11). Representative tumor areas in radical prostatectomy, seminal vesicle, and bone metastasis FFPE blocks were punched (three 1-mm cancer cores) to build TMAs.
Antibodies, staining, and quantification of human clinical specimens are described in detail in the Supplemental Material.
Statistical methods
For in vitro experiments, P-values were generated using two-tailed Student's t-test (P < 0.05 [*], P < 0.01 [**], and P < 0.001 [***]). ANOVA analysis and post-hoc analysis (Tukey test) were used to assess significant differences in the TMA study. Post-hoc analyses (Games-Howell) were also performed and gave similar results.
Supplementary Material
Acknowledgments
We thank members of the V.G. laboratory for discussions, and Andrew Oestreich for technical assistance. This work was supported by a Terry Fox Research Institute Program Project Team Grant on Oncometabolism (116128 to V.G.), the Canadian Institutes of Health Research (CIHR; MOP-111144 to V.G.), the Consortium Québecois pour la Découverte de Médicaments (V.G.), the New Innovation Fund CFI 21875 (to V.G.), and the McGill Urology Research Funds (S.C.). E.A.-W. is recipient of a post-doctoral fellowship from CIHR and the Fonds de Recherche du Québec–Santé (FRQS) and was supported by a McGill Integrated Cancer Research Training Program (MICRTP) scholarship. T.Y. was a recipient of summer MICRTP studentship.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.299958.117.
References
- Alayev A, Salamon RS, Berger SM, Schwartz NS, Cuesta R, Snyder RB, Holz MK. 2016. mTORC1 directly phosphorylates and activates ERα upon estrogen stimulation. Oncogene 35: 3535–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong AJ, Halabi S, Healy P, Alumkal JJ, Winters C, Kephart J, Bitting RL, Hobbs C, Soleau CF, Beer TM, et al. 2017. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur J Cancer 81: 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audet-Walsh E, Papadopoli DJ, Gravel SP, Yee T, Bridon G, Caron M, Bourque G, Giguère V, St-Pierre J. 2016. The PGC-1α/ERRα axis represses one-carbon metabolism and promotes sensitivity to anti-folate therapy in breast cancer. Cell Rep 14: 920–931. [DOI] [PubMed] [Google Scholar]
- Audet-Walsh E, Yee T, McGuirk S, Vernier M, Ouellet C, St-Pierre J, Giguere V. 2017. Androgen-dependent repression of ERRγ reprograms metabolism in prostate cancer. Cancer Res 77: 378–389. [DOI] [PubMed] [Google Scholar]
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, et al. 2016. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 22: 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Simon MC, Rafii S, Pandolfi PP. 2006. PML inhibits HIF-1α translation and neoangiogenesis through repression of mTOR. Nature 442: 779–785. [DOI] [PubMed] [Google Scholar]
- Betz C, Hall MN. 2013. Where is mTOR and what is it doing there? J Cell Biol 203: 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolla M, de Reijke TM, Van Tienhoven G, Van den Bergh AC, Oddens J, Poortmans PM, Gez E, Kil P, Akdas A, Soete G, et al. 2009. Duration of androgen suppression in the treatment of prostate cancer. N Engl J Med 360: 2516–2527. [DOI] [PubMed] [Google Scholar]
- Carbonneau M, Gagne LM, Lalonde ME, Germain MA, Motorina A, Guiot MC, Secco B, Vincent EE, Tumber A, Hulea L, et al. 2016. The oncometabolite 2-hydroxyglutarate activates the mTOR signalling pathway. Nat Commun 7: 12700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, et al. 2005. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122: 33–43. [DOI] [PubMed] [Google Scholar]
- Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, et al. 2011. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19: 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catena V, Bruno T, De Nicola F, Goeman F, Pallocca M, Iezzi S, Sorino C, Cigliana G, Floridi A, Blandino G, et al. 2016. Deptor transcriptionally regulates endoplasmic reticulum homeostasis in multiple myeloma cells. Oncotarget 7: 70546–70558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. 2012. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaveroux C, Eichner LJ, Dufour CR, Shatnawi A, Khoutorsky A, Bourque G, Sonenberg N, Giguère V. 2013. Molecular and genetic crosstalks between mTOR and ERRα are key determinants of rapamycin-induced non-alcoholic fatty liver. Cell Metab 17: 586–598. [DOI] [PubMed] [Google Scholar]
- Chen M, Nowak DG, Narula N, Robinson B, Watrud K, Ambrico A, Herzka TM, Zeeman ME, Minderer M, Zheng W, et al. 2017. The nuclear transport receptor Importin-11 is a tumor suppressor that maintains PTEN protein. J Cell Biol 216: 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello LC, Franklin RB. 2017. Decreased zinc in the development and progression of malignancy: an important common relationship and potential for prevention and treatment of carcinomas. Expert Opin Ther Targets 21: 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello LC, Franklin RB, Feng P. 2005. Mitochondrial function, zinc, and intermediary metabolism relationships in normal prostate and prostate cancer. Mitochondrion 5: 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. 2007. mTOR controls mitochondrial oxidative function through a YY1–PGC-1α transcriptional complex. Nature 450: 736–740. [DOI] [PubMed] [Google Scholar]
- Dai B, Kong YY, Ye DW, Ma CG, Zhou X, Yao XD. 2009. Activation of the mammalian target of rapamycin signalling pathway in prostate cancer and its association with patient clinicopathological characteristics. BJU Int 104: 1009–1016. [DOI] [PubMed] [Google Scholar]
- Epstein JI, Egevad L, Amin MB, Delahunt B, Srigley JR, Humphrey PA, Grading C. 2016. The 2014 International Society of Urological Pathology (ISUP) Consensus conference on Gleason grading of prostatic carcinoma: definition of grading patterns and proposal for a new grading system. Am J Surg Pathol 40: 244–252. [DOI] [PubMed] [Google Scholar]
- Evren S, Dermen A, Lockwood G, Fleshner N, Sweet J. 2010. Immunohistochemical examination of the mTORC1 pathway in high grade prostatic intraepithelial neoplasia (HGPIN) and prostatic adenocarcinomas (PCa): a tissue microarray study (TMA). Prostate 70: 1429–1436. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: p11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goberdhan DC, Wilson C, Harris AL. 2016. Amino acid sensing by mTORC1: intracellular transporters mark the spot. Cell Metab 23: 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. 2012. The mutational landscape of lethal castration-resistant prostate cancer. Nature 487: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- Horoszewicz JS, Leong SS, Chu TM, Wajsman ZL, Friedman M, Papsidero L, Kim U, Chai LS, Kakati S, Arya SK, et al. 1980. The LNCaP cell line—a new model for studies on human prostatic carcinoma. Prog Clin Biol Res 37: 115–132. [PubMed] [Google Scholar]
- Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, Mirand EA, Murphy GP. 1983. LNCaP model of human prostatic carcinoma. Cancer Res 43: 1809–1818. [PubMed] [Google Scholar]
- Jin HJ, Zhao JC, Ogden I, Bergan RC, Yu J. 2013. Androgen receptor-independent function of FoxA1 in prostate cancer metastasis. Cancer Res 73: 3725–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantidakis T, Ramsbottom BA, Birch JL, Dowding SN, White RJ. 2010. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc Natl Acad Sci 107: 11823–11828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RS, Sinnott JA, Rider JR, Ebot EM, Gerke T, Bowden M, Pettersson A, Loda M, Sesso HD, Kantoff PW, et al. 2016. The role of tumor metabolism as a driver of prostate cancer progression and lethal disease: results from a nested case-control study. Cancer Metab 4: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer CL, Klein RR, Mendelson J, Browne W, Samadzedeh LK, Vanpatten K, Highstrom L, Pestano GA, Nagle RB. 2006. Expression of mTOR signaling pathway markers in prostate cancer progression. Prostate 66: 1203–1212. [DOI] [PubMed] [Google Scholar]
- Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, Etzioni R, Bolouri H, Montgomery B, White T, et al. 2016. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med 22: 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganière J, Deblois G, Lefebvre C, Bataille AR, Robert F, Giguère V. 2005. Location analysis of estrogen receptor α target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc Natl Acad Sci 102: 11651–11656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais D, Couture C, Sylvain-Drolet G, Drouin J. 2011. A pituitary-specific enhancer of the POMC gene with preferential activity in corticotrope cells. Mol Endocrinol 25: 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. 2013. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci 126: 1713–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, Ferrari M, Egevad L, Rayford W, Bergerheim U, et al. 2004. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci 101: 811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Tsang CK, Watkins M, Bertram PG, Zheng XF. 2006. Nutrient regulates Tor1 nuclear localization and association with rDNA promoter. Nature 442: 1058–1061. [DOI] [PubMed] [Google Scholar]
- Massard C, Chi KN, Castellano D, de Bono J, Gravis G, Dirix L, Machiels JP, Mita A, Gonzalez BM, Turri S, et al. 2017. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur J Cancer 76: 36–44. [DOI] [PubMed] [Google Scholar]
- Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N, et al. 2011. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J 30: 2719–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Manning BD. 2008. Common corruption of the mTOR signaling network in human tumors. Oncogene 27: S43–S51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. 2008. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res 68: 4447–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. 2016. The emerging hallmarks of cancer metabolism. Cell Metab 23: 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz MM, Li F, Takeda DY, Lenci R, Chonkar A, Chabot M, Cejas P, Vazquez F, Cook J, Shivdasani RA, et al. 2015. The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet 47: 1346–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. 2015. Integrative clinical genomics of advanced prostate cancer. Cell 161: 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner M, Hengstschlager M. 2008. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum Mol Genet 17: 2934–2948. [DOI] [PubMed] [Google Scholar]
- Rosner M, Hengstschlager M. 2012. Detection of cytoplasmic and nuclear functions of mTOR by fractionation. Methods Mol Biol 821: 105–124. [DOI] [PubMed] [Google Scholar]
- Saldanha AJ. 2004. Java Treeview—extensible visualization of microarray data. Bioinformatics 20: 3246–3248. [DOI] [PubMed] [Google Scholar]
- Schweizer MT, Yu EY. 2017. AR-signaling in human malignancies: prostate cancer and beyond. Cancers 9: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, MacArthur S, Stark R, Warren AY, Mills IG, et al. 2013. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 23: 35–47. [DOI] [PubMed] [Google Scholar]
- Shimobayashi M, Hall MN. 2016. Multiple amino acid sensing inputs to mTORC1. Cell Res 26: 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statz CM, Patterson SE, Mockus SM. 2017. mTOR Inhibitors in castration-resistant prostate cancer: a systematic review. Target Oncol 12: 47–59. [DOI] [PubMed] [Google Scholar]
- Stelloo S, Nevedomskaya E, van der Poel HG, de Jong J, van Leenders GJ, Jenster G, Wessels LF, Bergman AM, Zwart W. 2015. Androgen receptor profiling predicts prostate cancer outcome. EMBO Mol Med 7: 1450–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, Chen D, Seo K, Modrusan Z, Gao WQ, et al. 2012. Androgen deprivation causes epithelial–mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res 72: 527–536. [DOI] [PubMed] [Google Scholar]
- Sutherland SI, Pe Benito R, Henshall SM, Horvath LG, Kench JG. 2014. Expression of phosphorylated-mTOR during the development of prostate cancer. Prostate 74: 1231–1239. [DOI] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. 2010. Integrative genomic profiling of human prostate cancer. Cancer Cell 18: 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, Zhang A, Xia X, Ilkayeva OR, Xin L, et al. 2014. Androgens regulate prostate cancer cell growth via an AMPK–PGC-1α-mediated metabolic switch. Oncogene 33: 5251–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, et al. 2007. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 448: 595–599. [DOI] [PubMed] [Google Scholar]
- Toren P, Kim S, Cordonnier T, Crafter C, Davies BR, Fazli L, Gleave ME, Zoubeidi A. 2015. Combination AZD5363 with enzalutamide significantly delays enzalutamide-resistant prostate cancer in preclinical models. Eur Urol 67: 986–990. [DOI] [PubMed] [Google Scholar]
- Watson PA, Arora VK, Sawyers CL. 2015. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer 15: 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei XX, Hsieh AC, Kim W, Friedlander T, Lin AM, Louttit M, Ryan CJ. 2017. A phase I study of abiraterone acetate combined with BEZ235, a dual PI3K/mTOR inhibitor, in metastatic castration resistant prostate cancer. Oncologist 22: 503–e543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Chen SY, Ross KN, Balk SP. 2006. Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res 66: 7783–7792. [DOI] [PubMed] [Google Scholar]
- Yadav RB, Burgos P, Parker AW, Iadevaia V, Proud CG, Allen RA, O'Connell JP, Jeshtadi A, Stubbs CD, Botchway SW. 2013. mTOR direct interactions with Rheb-GTPase and raptor: sub-cellular localization using fluorescence lifetime imaging. BMC Cell Biol 14: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav SS, Li J, Stockert JA, O'Connor J, Herzog B, Elaiho C, Galsky MD, Tewari AK, Yadav KK. 2016. Combination effect of therapies targeting the PI3K- and AR-signaling pathways in prostate cancer. Oncotarget 7: 76181–76196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylitalo EB, Thysell E, Jernberg E, Lundholm M, Crnalic S, Egevad L, Stattin P, Widmark A, Bergh A, Wikstrom P. 2017. Subgroups of castration-resistant prostate cancer bone metastases defined through an inverse relationship between androgen receptor activity and immune response. Eur Urol 71: 776–787. [DOI] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. 2011. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25: 2227–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shu L, Hosoi H, Murti KG, Houghton PJ. 2002. Predominant nuclear localization of mammalian target of rapamycin in normal and malignant cells in culture. J Biol Chem 277: 28127–28134. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wang L, Wu D, Chen H, Chen Z, Thomas-Ahner JM, Zynger DL, Eeckhoute J, Yu J, Luo J, et al. 2011. Definition of a FoxA1 cistrome that is crucial for G1 to S-phase cell-cycle transit in castration-resistant prostate cancer. Cancer Res 71: 6738–6748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.