

Significance
B lymphocytes produce antibodies that provide protection from infections. Such antibodies evolve from precursors via pathogen-driven affinity maturation. Affinity maturation involves introduction of somatic hypermutations (SHMs) into antibody genes followed by selection of B lymphocytes producing antibodies that better neutralize the pathogen. Some HIV-1–infected humans develop broadly neutralizing antibodies (bnAbs) that recognize diverse HIV-1 strains. VRC01 is a potent bnAb that binds a crucial portion of HIV-1. Development of vaccine strategies to elicit VRC01-class antibodies is difficult due to the high SHM levels associated with their maturation. We report contributions of sequence-intrinsic mechanisms to the SHM patterns of a VRC01-class bnAb and its precursors in mice. Our findings provide insights into roles of antibody gene sequences in guiding bnAb maturation.
Keywords: broadly neutralizing antibodies, HIV-1, somatic hypermutation, intrinsic mutability, activation-induced cytidine deaminase
Abstract
Variable regions of Ig chains provide the antigen recognition portion of B-cell receptors and derivative antibodies. Ig heavy-chain variable region exons are assembled developmentally from V, D, J gene segments. Each variable region contains three antigen-contacting complementarity-determining regions (CDRs), with CDR1 and CDR2 encoded by the V segment and CDR3 encoded by the V(D)J junction region. Antigen-stimulated germinal center (GC) B cells undergo somatic hypermutation (SHM) of V(D)J exons followed by selection for SHMs that increase antigen-binding affinity. Some HIV-1–infected human subjects develop broadly neutralizing antibodies (bnAbs), such as the potent VRC01-class bnAbs, that neutralize diverse HIV-1 strains. Mature VRC01-class bnAbs, including VRC-PG04, accumulate very high SHM levels, a property that hinders development of vaccine strategies to elicit them. Because many VRC01-class bnAb SHMs are not required for broad neutralization, high overall SHM may be required to achieve certain functional SHMs. To elucidate such requirements, we used a V(D)J passenger allele system to assay, in mouse GC B cells, sequence-intrinsic SHM-targeting rates of nucleotides across substrates representing maturation stages of human VRC-PG04. We identify rate-limiting SHM positions for VRC-PG04 maturation, as well as SHM hotspots and intrinsically frequent deletions associated with SHM. We find that mature VRC-PG04 has low SHM capability due to hotspot saturation but also demonstrate that generation of new SHM hotspots and saturation of existing hotspot regions (e.g., CDR3) does not majorly influence intrinsic SHM in unmutated portions of VRC-PG04 progenitor sequences. We discuss implications of our findings for bnAb affinity maturation mechanisms.
Antibodies are the secreted form of B-cell antigen receptors (BCRs) and are composed of Ig chains. Exons that encode antigen-binding variable regions (“V(D)J exons”) of Ig heavy (IgH) chains are assembled in developing B lymphocytes from V (VH), D, and J (JH) gene segments by V(D)J recombination. Two antigen-contacting complementarity determining regions (CDRs) are encoded in the germline VH, whereas CDR3 is created somatically via V(D)J junctional assembly (1). Within the VH(D)JH exon, CDRs are embedded within framework regions (FWRs) that are relatively invariant in sequence because they encode requisite variable region structural features (2). About two-thirds of IgH V(D)J rearrangements occur in a “nonproductive” translational reading frame and do not produce functional IgH proteins. B cells often have a nonproductive IgH V(D)J exon in addition to the productive IgH allele that supports B-cell development (3). In response to antigen activation, mature B lymphocytes in germinal centers (GCs) of peripheral lymphoid organs undergo somatic hypermutation (SHM) of Ig V(D)J exons. Cycles of SHM and selection of clones with SHMs that increase BCR-antigen affinity lead to antibody affinity maturation (4). SHM is initiated by activation-induced cytidine deaminase (AID), which deaminates cytosines to uracils that are processed to generate mutations (5). SHM occurs on both DNA strands of productive and nonproductive alleles (5, 6). Transcription is required for SHM of V(D)J exons (5).
Within V(D)J exons predominant AID-targeting sites are at cytidines of short DGYW motifs (D = A/G/T, Y = C/T, W = A/T), with the palindromic AGCT motif representing a canonical example (7). However, whereas DGYW motifs in CDRs often are highly targeted, the same motifs in adjacent FWRs often are not (6, 8). The basis for differential AID targeting of identical hotspot motifs within a transcribed V(D)J exon is unknown. SHM of certain V(D)J hotspots has been proposed to influence subsequent AID targeting of others within the same sequence (9–11). However, experimental testing of this notion led to divergent conclusions, with transgene studies suggesting that V exon SHM hotspot targeting suppresses that of other hotspots (9) and cell line studies suggesting that hotspot targeting activates additional SHM hotspots (10, 11). Further clarification of the influence of specific SHMs, or SHMs in general, on subsequent targeting of other V exon sequences is important for elucidating mechanisms by which particular V(D)J sequences might influence the course of antibody affinity maturation.
Broadly neutralizing antibodies (bnAbs) against HIV-1 arise in a subset of HIV-1–infected patients (12). VRC01-class bnAbs bind the highly conserved CD4 binding site of HIV envelope gp120 as a structural mimic of CD4 and are among the most potent HIV-1 bnAbs (13). All VRC01-class bnAbs use the human VH1-2*02 segment (or the highly homologous VH1-46*01) and contain very high numbers of SHMs (up to 32% of VHDJH exon nucleotides) (13, 14). During the development of HIV-1 bnAbs in an infected individual, SHMs of unmutated germline precursor VH exon accumulate in affinity maturation intermediates that can be connected to the fully mutated mature antibody (12). Many SHMs in VH1-2*02–using bnAb sequences occur in VH1-2*02–using V(D)J exons of non-HIV-1–infected patients, suggesting they result from targeting of sequences intrinsically prone to SHM (15). However, critical SHMs also may occur in VH1-2*02–using V(D)J exons at sequences with intrinsically low SHM, requiring high overall SHM levels to achieve them. In support of this notion, only a fraction of mature VRC01-class bnAb SHMs are required for broad neutralizing activity (16–18).
We recently developed a mouse V(D)J exon passenger allele system to assay patterns and levels of intrinsic SHMs of desired test sequences in GC B cells (6). We now use this approach to assay intrinsic SHM across passenger substrates that represent various stages in the maturation of a human VRC01-class bnAb lineage. We identify potentially rate-limiting SHMs and reveal how SHM-targeting frequencies change during this VRC01-class bnAb maturation process.
Results
Overview of V(D)J Passenger Allele System.
Our passenger allele approach assays GC B-cell SHM-targeting frequencies of test sequences inserted into the passenger IgH V(D)J allele of a “VB1-8/passenger allele” ES cell line (6) (Fig. S1 A and B). The “productive” allele in this line encodes a B1-8 antibody IgH V(D)J (“VB1-8”) exon that supports B-cell development, whereas the passenger harbors a desired test sequence that cannot encode a protein and thus provides intrinsic SHM patterns (6). ES cell lines containing given passenger sequences are used for RAG-2–deficient blastocyst complementation (19) to generate chimeric mice in which all peripheral B cells derive from the modified passenger ES cell (6). Our current studies of VH1-2*02–related passenger alleles is facilitated by assaying Peyer’s patches (PP) GC B-cell intrinsic SHM patterns, because PP GC B-cell VB1-8 productive and passenger sequences have the same general patterns and levels of intrinsic SHM whether or not mice are immunized (6). To visualize hotspot emergence and study accumulation of SHMs on productive and passenger sequences we compare the SHM pattern of sequences in low (3–10 mutations per sequence), intermediate (11–20), and high (21–30) mutational strata, which allows a “pseudo-kinetic” analysis of the appearance of individual SHMs (6). This stratification enables identification of “primary” and “secondary” hotspots, respectively, as those that first appear in the low SHM strata and those that first appear in intermediate SHM strata (Fig. 1; discussed below). In the highest SHM strata, primary hotspots become saturated and SHMs accumulate in nonhotspot regions (6).
Fig. S1.

SHM targeting of VH1-2*02 attached to CDR3s with different SHM potentials. (A) VB1-8/passenger ES cells are generated from a parental ES cell line that contains a productive preassembled V(D)J allele encoding the IgH V region of the 4-hydroxy-3-nitrophenylacetyl-binding B1-8 antibody (VB1-8) inserted in place of the IgH DQ52 and J segments on one allele and a puromycin resistance gene cassette (PGK-PuroΔTK) inserted in the other allele. VB1-8/passenger ES cells with different test passenger sequences are generated by replacing the puromycin resistance cassette with a “passenger cassette” composed of a VB1-8 promoter and leader sequence containing a translation termination codon followed by test sequences, as previously described (6). Orange lines indicate homology arms of targeting vector. (B) VB1-8/passenger ES cells are injected into RAG-2–deficient blastocysts, generating chimeric mice in which all mature B cells carry a fixed VB1-8-productive allele and test passenger sequences in the V(D)J passenger allele. Pro, pas, V pro, and L represent productive allele, passenger allele, V promoter, and leader sequence, respectively. The red line across the leader sequence represents the termination codon. Productive allele-specific (white block arrows) and passenger allele-specific (red block arrows) first-round primer strategies are schematized (see Materials and Methods for PCR strategy and Table S2 for primer sequences). Passenger allele-specific regions are two nucleotide differences encoding the termination codon in leader, 33 bp from the subcloning construct upstream of passenger sequence and 38 bp from the subcloning construct downstream of the passenger sequence. (C and D) SHM frequencies, calculated over (C) the VH region or (D) each CDR/FWR region, for all mutated productive (blue bars) and passenger (white bars) allele sequences of VB1-8/VH1-2*02+CDR3VB1-8 and VB1-8/VH1-2*02+CDR3VRC-PG04 chimeras. Unpaired t tests comparing SHM frequency between the productive alleles of each genotype and between the passenger alleles of each genotype were performed. For D the only one of these comparisons that was significantly different (P < 0.05) is indicated. (E and F) SHM frequency at (E) position 293 and (F) position 246 at each mutational stratum.
Fig. 1.

SHM targeting of VH1-2*02 attached to CDR3s with different SHM potentials. (A) Schematic of VH1-2*02+CDR3VB1-8 and VH1-2*02+CDR3VRC-PG04 passenger test sequences. The VH1-2*02+CDR3VB1-8 passenger (Top) is the VH1-2*02 sequence attached to VB1-8 CDR3 (red box) and VB1-8 FWR4 sequences, followed by VB1-8 JH2 intron sequence. The VH1-2*02+CDR3VRC-PG04 passenger (Bottom) is composed of VH1-2*02 attached to CDR3 (blue box) and FWR4 of a hypermutated VRC-PG04 maturation intermediate (int16.5% in Fig. S5A). (B–D) Map of mutations (SHM profile) on VH1-2*02+CDR3VB1-8 (Top) and VH1-2*02+CDR3VRC-PG04 (Bottom) passenger sequences. y axis indicates mutation frequency plotted as the mean percent of sequences mutated at the indicated nucleotide ± SEM (green shading indicates top error bar). n = 9 VH1-2*02+CDR3VB1-8 chimeras and 8 VH1-2*02+CDR3VRC-PG04 chimeras, 3,000–5,000 mutated sequences per chimera. SHM profiles are shown separately for reads with (B) 3–10, (C) 11–20, or (D) 21–30 mutations within the VH. Blue and green asterisks/dotted lines indicate primary and secondary hotspot positions, respectively. Secondary hotspot positions are listed in Table S1.
CDR3 SHM Does Not Influence Intrinsic SHM Targeting of the Adjacent VH1-2*02 Segment.
To test the notion that SHMs of AID targets in a sequence may influence subsequent SHM of other targets (9–11) we assayed intrinsic SHM levels of the human VH1-2*02 germline passenger sequence attached to either the VB1-8 CDR3 that has not been subjected to SHM (6, 20) or to the CDR3 of an affinity maturation intermediate of VRC01-class bnAb VRC-PG04 that has undergone extensive SHM (21) (termed VH1-2+CDR3VB1-8 and VH1-2*02+CDR3VRC-PG04, respectively, Fig. 1A). In this regard, we hypothesized that the latter CDR3 may have saturated intrinsic hotspots and lack robust SHM targeting activity. We assayed nine VB1-8/VH1-2*02+CDR3VB1-8 and eight VB1-8/VH1-2*02+CDR3VRC-PG04 passenger chimeras. In these experiments, the VB1-8 productive alleles all had similar overall mutation levels and patterns (Figs. S1C and S2 A–C), indicating that the two distinct passenger sequences were subjected to comparable levels of AID activity and validating their direct comparison. In the low mutation stratum, seven VH1-2*02 primary hotspot positions were reproducibly mutated in >10% of sequences in both VH1-2*02+CDR3VB1-8 and VH1-2*02+ CDR3VRC-PG04 passengers (Fig. 1B, blue asterisks). These primary VH1-2*02 SHM hotspots include the G and C (positions 9 and 10) of AGCT in FWR1, the G (position 92) of GGCT in CDR1, the G and C (positions 102 and 103) of TGCA in CDR1, an A (position 179) in CDR2, and the G (position 246) of the AGCT in FWR3. We note that some of these primary hotspots are not predicted from computational models that defined the DGYW motif (7) or enrichment of targeting to CDRs (8), because they either are not at a DGYW motif or they occur in FWRs. At the intermediate mutational stratum, 31 additional secondary hotspot positions are reproducibly mutated in >10% of sequences of both passengers (Fig. 1C, green asterisks). At the highest mutational stratum (21–30 mutations per sequence), primary and secondary hotspot positions are still dominant in the SHM landscape, but SHMs of many other positions reproducibly rise above background (Fig. 1D). We conclude that individual VH1-2*02 nucleotides vary widely in intrinsic mutation frequency.
Fig. S2.

SHM profiles of productive allele sequences from VB1-8/VH1-2*02+CDR3VB1-8 chimeras and VB1-8/VH1-2*02+CDR3VRC-PG04 chimeras. (A–C) SHM profile of productive allele sequences from VB1-8/VH1-2*02+CDR3VB1-8 (top of each pair of profiles) and VB1-8/VH1-2*02+CDR3VRC-PG04 (bottom of each pair of profiles) chimeras at mutational stratum (A) 3–10, (B) 11–20, or (C) 21–30 mutations per sequence. (D) Mutation frequency of position 314 in productive and passenger alleles of VB1-8/VH1-2+CDR3VB1-8 at indicated mutational stratum.
The CDR3 portion of the VH1-2+CDR3VB1-8 passenger was highly targeted for SHM, whereas the CDR3 portion of the VH1-2*02+ CDR3VRC-PG04 passenger was not (Fig. 1 B–D and Fig. S1D). However, overall mutation frequencies of the VH1-2*02 segments in these two passengers were not significantly different, whether calculated over the entire VH (Fig. S1C) or over each CDR1, CDR2, or FWR region separately (Fig. S1D). We further compared SHM frequencies of the two passengers at each of the 294 nucleotide positions of the VH1-2*02 sequence and at each stratum (Materials and Methods). The only significant difference detected was at position 293, 2 bp upstream of CDR3, which was more highly targeted in VH1-2*02+CDR3VB1-8 versus VH1-2*02+CDR3VRC-PG04 (Fig. 1 B–D, arrow and Fig. S1E). However, no significant difference in SHM frequency was detected at position 246, which is the primary SHM hotspot (the G of the AGCT of FWR3) most proximal to CDR3 (Fig. S1F). These findings suggest that overall SHM of a rearranged VH1-2*02 segment occurs largely independently of the SHM status of the CDR3 to which it is fused. To further test independence of VH1-2*02 SHM from that of its associated CDR3 we examined SHM patterns of nonproductively rearranged, and therefore not selected, VH1-2*02-DJ exon sequences generated endogenously via V(D)J recombination in a VH1-2*02–rearranging mouse model that generates extremely diverse CDR3s (22). Even in the context of diverse CDR3s the SHM pattern of nonproductively rearranged VH1-2*02-DJ exon sequences was highly similar to that of the VH1-2*02 passenger at each tested SHM stratum (Pearson’s r = 0.87–0.90) (Fig. S3). The only exceptions were position 10, which is a primary hotspot in the VH1-2*02 passenger and a secondary hotspot in nonproductive VH1-2*02-DJ exon sequences, and positions 18 and 30, which are secondary hotspots in the VH1-2*02 passengers and not hotspots in nonproductively rearranged VH1-2*02 sequences (Fig. S3). Slightly increased SHM of promoter-proximal VH1-2*02 sequences in passenger sequence versus endogenously assembled V(D)J exons in VH1-2*02–rearranging mice may reflect the VH1-2*02 passenger’s being 33 bp more distal to the promoter than the endogenously assembled V(D)J exon, given that SHM is potentially diminished in the promoter-proximal region of transgenic V exons (23).
Fig. S3.

SHM profiles of VH1-2*02 passengers and nonproductive sequences of VH1-2*02–rearranging mouse model. (A–C) SHM profiles of passenger VH1-2*02+CDR3VB1-8 (Top; n = 9), passenger VH1-2*02+CDR3VRC-PG04 (Middle; n = 8), and nonproductive sequences from VH1-2*02–rearranging mouse model (22) (Bottom; n = 5), at mutational stratum (A) 3–10, (B) 11–20, or (C) 21–30 mutations per sequence. SHM profiles of passengers VH1-2*02+CDR3VB1-8 and VH1-2*02+CDR3VRC-PG04 are reproduced from Fig. 1 B–D. A t test was performed comparing the SHM frequency at each of 294 VH1-2*02 nucleotides between each VH1-2*02 passenger and the same nucleotide of nonproductive V(D)J exon sequences that use VH1-2*02. Significant differences P < Bonferroni-adjusted α of 0.05/294 are indicated with an asterisk.
Similar to what we previously described for the passenger mouse VB1-8 sequence (6), the human VH1-2*02 passengers had frequent deletions (Fig. 2 A and B, right) that are not found frequently on the productive VB1-8 VH(D)JH alleles (Fig. 2 A and B, left), due to their propensity to inactivate functional IgH chain expression (6). Deletion endpoints in VH1-2*02 passengers correlated with sites of high SHM (Pearson’s r = 0.71–0.73; Fig. 2 C–F). In accord with their relative intrinsic SHM levels, the CDR3 portion of VH1-2*02+CDR3VB1-8 was highly targeted for deletions (Fig. 2E), whereas the CDR3 portion of the VH1-2*02+ CDR3VRC-PG04 passenger was not (Fig. 2F). In this regard, we note that primary SHM hotspot position 314 in the AGCT sequence of the CDR3 of VH1-2*02+CDR3VB1-8 passenger allele is slightly dampened in level compared with that of the identical CDR3VB1-8 on the VB1-8 productive allele in the same experiments (Fig. S2D). This finding likely does not reflect dampened SHM levels per se but rather is caused by loss of B cells harboring AID-mediated V exon deletions on productive alleles due to impact of deletions on BCR expression and, thereby, B-cell viability (6).
Fig. 2.

Deletions in VH1-2*02 passengers. (A and B) Deletion frequency, calculated as the percent of mutated sequences that contain deletions of (A) VH1-2*02+CDR3VB1-8 and (B) VH1-2+CDR3VRC-PG04 passenger alleles, ± SD (C and D) Map of unique deletions in (C) VH1-2*02+CDR3VB1-8 and (D) VH1-2*02+CDR3VRC-PG04 passenger allele sequences. Deletions are represented by lines whose start and end indicate the start and end of the deletion. Deletions from each of the 9 VH1-2*02+CDR3VB1-8 and 8 VH1-2*02+CDR3VRC-PG04 chimeras are displayed with a different color. (E and F) The location of SHMs compared with the location of deletion endpoints. r is the Pearson correlation coefficient between SHM frequency and deletion endpoint frequency of each bin. n = 9 VB1-8/VH1-2*02+CDR3VB1-8 and 8 VB1-8/VH1-2*02+CDR3VRC-PG04 chimeras.
Etiology of Recurrently Mutated Nucleotides in VRC01-Class bnAbs.
Comparison of the VH1-2*02–encoded portions of six VRC01-class bnAb V(D)J exons revealed 10 positions, located in FWR1, CDR1, CDR2, and FWR3, that were “recurrently mutated” in each bnAb (Fig. 3 A–C). Recurrently mutated bnAb positions 56, 185, and 187 occurred at VH1-2*02 positions with low intrinsic SHM frequency that were neither primary nor secondary intrinsic SHM hotspots (Fig. 3C, yellow bars). Notably, a set of VH1-2*02 “silent” positions (at which SHMs do not change encoded amino acids) of intrinsic SHM frequency comparable to these three recurrently mutated positions were, on average, mutated in only one or two of the six VRC01-class bnAbs (Fig. 3 D and E). Based on the number of bnAbs mutated in each set of comparable silent positions, considered as a binomial distribution, we estimated the probability of SHMs occurring at positions 56, 185, and 187 in all six bnAb sequences to be low (P values ≤0.0019; Fig. 3E). Indeed this probability was significant when considered in the context of the rigorous Bonferroni-adjusted significance level (Fig. 3E). Therefore, SHMs are likely enriched among VRC01-class bnAbs at these three sites by positive selection. Positions 172 and 156 (Fig. 3C, white bars) also are not primary or secondary intrinsic hotspots but could not be statistically analyzed due to insufficient numbers of matched silent positions. In this regard, silent SHM VH1-2*02 positions have significantly lower intrinsic SHM frequencies than nonsilent SHM positions (Fig. S4F), possibly reflecting optimization of the V exon to target SHMs that change encoded amino acids. Five recurrently mutated VRC01-class bnAb positions occur in primary (position 102) or secondary (positions 230, 279, 170, and 98) VH1-2*02 intrinsic SHM hotspots (Fig. 3C, blue and green bars), making them candidates for being recurrently mutated mainly due to high intrinsic SHM levels. Accordingly, recurrent position 279 is silent (Fig. 3D). We conclude that our approach discriminates between recurrent SHMs in VRC01-class bnAbs that occur at dominant intrinsic hotspots versus those that occur at low intrinsic SHM positions and are likely recurrent due to positive selection.
Fig. 3.

Recurrently mutated VH1-2*02 nucleotides among VRC01-class bnAbs. (A) Recurrently mutated VH1-2*02 positions among the IgH V region exon sequences of potent VRC01-class bnAbs VRC01 (14), VRC-PG04 (14), 3BNC117 (14), VRC-CH32 (21, 26), 12A21 (14), and N6 (31). (B) Locations within the VH1-2*02 sequence of recurrently mutated bnAb positions (red vertical lines) and of sequences that contribute to VRC01–gp120 contact sites [black squares (16)]. (C) Intrinsic mutation frequency of recurrently mutated bnAb positions calculated from VH1-2*02 passenger sequences of mutational stratum 21–30 mutations per sequence. Data shown as mean ± SEM of nine VB1-8/VH1-2*02+CDR3VB1-8 and eight VB1-8/VH1-2*02+CDR3VRC-PG04 chimeras. [s] indicates that position 279 is a silent SHM position. Blue and green bars indicate primary and secondary hotspots. Yellow bars indicate positions for which there are silent SHM positions of VH1-2*02 of comparable intrinsic mutation frequency. (D) Silent SHM positions of VH1-2*02. y axis indicates intrinsic mutation frequency, calculated as in C. Brackets indicate the set of silent SHM positions that are matched in intrinsic mutation frequency to each of positions 185, 187, and 56. (E) The number of VRC01-class bnAb sequences mutated at silent SHM positions matched in intrinsic mutation frequency to positions 185, 187, and 56. p is the probability of matched silent SHMs being mutated in six of six bnAbs, estimated by modeling the number of silent SHM positions mutated in each number of bnAbs as a binomial distribution. In C and E ** indicates positions with P values < Bonferroni-adjusted α of 0.05/3.
Fig. S4.

VH1-2*02 positions mutated in five or six out of six VRC01-class bnAbs. (A) VH1-2*02 positions that are mutated in five or six out of six IgH V region exon sequences of potent VRC01-class bnAbs VRC01 (14), VRC-PG04 (14), 3BNC117 (14), VRC-CH32 (21, 26), 12A21 (14), and N6 (31). (B) Locations within the VH1-2*02 sequence of positions mutated in five (purple vertical lines) or six (red vertical lines) out of six bnAb positions and of sequences that contribute to VRC01-gp120 contact sites [black squares (16)]. (C) Intrinsic mutation frequency of each VH1-2*02 position mutated in five or six out of six VRC01-class bnAbs. Intrinsic mutation frequency is as plotted in Fig. 3C. [s] indicates that positions 207 and 279 are silent SHM positions. Blue and green bars indicate primary and secondary hotspots, respectively. Yellow bars indicate positions for which there are silent SHM positions of VH1-2*02 of comparable intrinsic mutation frequency. (D) Silent SHM positions of VH1-2*02. y axis indicates intrinsic mutation frequency, plotted as in Fig. 3D. Brackets indicate the set of silent SHM positions that are matched in intrinsic mutation frequency to the indicated position. (E) The number of VRC01-class bnAbs mutated at silent SHM positions matched in intrinsic mutation frequency to the indicated positions. p indicates the probability of matched silent SHMs being mutated in five or six of six bnAbs, estimated by modeling the number of silent SHM positions mutated in each number of bnAbs as a binomial distribution. In C and E an asterisk indicates positions with P values <0.05. Two asterisks indicate positions with P values < Bonferroni-adjusted α of 0.05/12. (F) The mean intrinsic mutation frequency of silent (left) and nonsilent (right) positions of VH1-2*02, ± SD. P value from t test.
Influences of Affinity Maturation on Intrinsic SHM Targeting.
To examine how SHM-targeting patterns on VH1-2*02 exons change over the course of VRC01-class bnAb affinity maturation we assayed two VRC-PG04 bnAb lineage affinity maturation intermediates, termed int6.1% and int6.8% (21), as passengers and compared SHM patterns of their VH1-2*02–encoded portion to that of VH1-2*02. The int6.1% sequence diverges from VH1-2*02 by 18 “prior” nucleotide changes and the int6.8% by 20 prior changes (Fig. 4A, red lines below int6.1% and int6.8% profiles and Fig. S5A). Comparison of the intrinsic SHM frequency at each site of prior SHMs between the two intermediate and the germline VH1-2*02 revealed that the int6.1% and int6.8% sequences, respectively, had eight and five sites of prior SHMs that resulted in a significant difference in their intrinsic SHM frequency (Fig. 4A, asterisks). However, within sequence regions conserved between the intermediates and the germline VH1-2*02 sequence, SHM targeting patterns were highly similar (Fig. 4A). For example, the SHM frequency of a region of CDR1 and FWR2 nucleotides adjacent to a robustly targeted AGCA hotspot in the CDR2 of int6.1% were not significantly altered between int6.1% and VH1-2*02 (Pearson’s r = 0.98) (Fig. S5B).
Fig. 4.

SHM targeting of VRC-PG04 intermediates and mature VRC-PG04 passengers. (A) SHM profile of VH1-2*02 and FWR4 portions of VH1-2*02+CDR3VRC-PG04 (Top, reproduced from Fig. 1C), int6.1% (Middle), and int6.8% passengers (Bottom). Data from intermediate mutational stratum 11–20 mutations per sequence is shown. t tests were performed comparing mean SHM frequency between germline VH1-2*02 and int6.1% at the position of each of 18 prior SHMs, and comparing mean SHM frequency between VH1-2*02 and int6.8% at the position of each of 20 prior SHMs. Significant differences of P < Bonferroni-adjusted α of 0.05/18 for int6.1% or P < 0.05/20 for int6.8% are indicated by asterisks, and upward or downward arrows indicate whether SHM frequency of the intermediate was increased or decreased compared with VH1-2*02 at the position. n = 8 VH1-2*02+CDR3VRC-PG04, 6 int6.1%, and 5 int6.8% chimeras, 3,000–5,000 mutated sequences per chimera. (B) SHM profiles of VH1-2*02+CDR3VRC-PG04 (reproduced from Fig. 1 B and C) and mature VRC-PG04 passenger allele sequences at 3–10 (Top) and 11–20 (Bottom) mutations per sequence. There were few (<100) mature VRC-PG04 passenger sequences with 21–30 mutations per sequence, and thus the SHM profile of this mutational level is not shown. n = 8 VH1-2*02+CDR3VRC-PG04 chimeras and 6 mature VRC-PG04 chimeras, 3,000–5,000 mutated sequences per chimera. In A and B red lines indicate positions of prior SHMs within VH1-2*02 segment. CDR3 of unmutated common ancestor is not known for VRC-PG04 and therefore SHMs are not indicated for this region. Purple lines indicate the positions of two 3-bp insertions in CDR1 and CDR2. Productive allele mutation frequencies for these passengers are shown in Fig. S5D.
Fig. S5.

SHM targeting of VRC-PG04 intermediates and mature VRC-PG04 passengers. (A) Nucleotide alignment of VH1-2*02+CDR3VRC-PG04 and VRC-PG04 intermediates. SHMs indicated in red. (B and C) SHM frequency of VH1-2*02+CDR3VRC-PG04 and int6.1% passengers in the (B) CDR1 and FWR2 regions or (C) at indicated nucleotide positions, from sequences of mutational stratum 11–20 mutations per sequence. P values from unpaired t test. (D) SHM frequency in VH region of productive and passenger alleles of indicated genotypes. Data from all mutated sequences. VB1-8/VH1-2*02+CDR3VRC-PG04 data reproduced from Fig. S1C. Unpaired t tests were performed comparing mutation frequencies of the VB1-8 productive allele of VB1-8/VH1-2*02+CDR3VRC-PG04 chimeras to that of VB1-8/int6.1% (P = 0.16), VB1-8/int6.8% (P = 0.45), and VB1-8/VRC-PG04 (P = 0.68) and comparing mutation frequencies of the passenger allele of VB1-8/VH1-2*02+CDR3VRC-PG04 to that of VB1-8/int6.1% (P = 0.90), VB1-8/int6.8% (P = 0.23), and VB1-8/VRC-PG04 (P = 0.0004). (E) Nucleotide alignment of mature VRC-PG04 VH region with germline VH1-2*02. AGCT, TGCA, and other DGYW motifs highlighted in orange, brown, and yellow, respectively. Blue and green asterisks indicate primary and secondary hotspots, respectively. Dotted red lines indicate DGYW motifs in the germline VH1-2*02 sequence converted by SHMs/insertions to non-DGYW motifs in the mature VRC-PG04 sequence.
All five prior SHMs of the VH1-2*02 portion of int6.8% that changed SHM targeting of a particular VH1-2*02 position resulted in decreases in intrinsic SHM frequency due to inactivation of SHM motifs (Fig. 4A, downward arrows). For int6.1%, five of eight prior SHMs that changed SHM-targeting frequency were associated with decreased intrinsic SHM frequency due to SHM motif inactivation (Fig. 4A, downward arrows), but three were associated with increases (Fig. 4A, upward arrows). The latter three int6.1% positions included position 83, where a C-to-G SHM in FWR1 converted ACCT to the AGCT AID targeting motif (4.2-fold increase); position 161, where an A-to-G SHM in CDR2 converted AACA to the AGCA AID targeting motif (17-fold increase); and position 166, where a G-to-A SHM in CDR2 converted TGG in VH1-2*02 to TAA but did not create an AID targeting motif (sevenfold increase) (Fig. S5C). We speculate that increased intrinsic SHM of position 166 most likely resulted via spreading from adjacent AID-targeted SHM hotspots to the newly generated polymerase eta TAA targeting motif (24). Notably, SHMs at positions 161 and 166 would produce amino acid changes at the VRC–PG04–gp120 interface (21). Thus, certain SHMs in a VRC-PG04-lineage intermediate promote further SHMs that may directly affect affinity maturation.
To assay how extensive affinity maturation influences the intrinsic SHM frequency of the V exon sequence we assayed the mature VRC-PG04 variable region sequence as a passenger and found that, compared with the VH1-2*02 passenger, it had substantially reduced intrinsic SHM frequency relative to the productive VB1-8 control allele (Fig. 4B and Fig. S5D). Accordingly, of the seven VH1-2*02 primary SHM hotspots (Fig. 1B) four were mutated in the final mature VRC-PG04 sequence, including one (position 92) that was the site of a 3-bp insertion (Fig. S5E). Another one (position 103) was inactivated as a DGYW by prior SHM of an adjacent primary hotspot (Fig. S5E). Notably, the G and the C at positions 9 and 10 within the FWR1 AGCT primary hotspot were not mutated and remained as SHM hotspots in the mature VRC-PG04 sequence (Fig. S5E), potentially reflecting negative selection against clones containing SHMs at these particular FWR sites. In addition, 21 of the 31 secondary hotspots identified in the 11–20 mutations per sequence stratum (Fig. 1C) were mutated in the VRC-PG04 sequence, including two (positions 181 and 182) that were the sites of another 3-bp insertion (Fig. S5E). Of the 10 secondary hotspot nucleotides that were not mutated in VRC-PG04, 9 were likely inactivated as hotspots via prior mutation of adjacent nucleotides that were in most cases themselves either primary or secondary hotspots (Table S1). Thus, the low intrinsic mutability of the mature VRC-PG04 variable region sequence derives, at least in substantial part, from SHM of primary or secondary SHM hotspots in the progenitor V(D)J exon into non-DGYW motifs over the course of affinity maturation.
Table S1.
Analysis of VH1-2*02 secondary hotspots not mutated in VRC-PG04
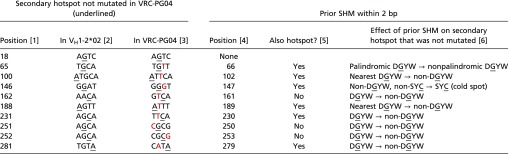 |
List of VH1-2*02 secondary hotspot positions (nucleotide positions): 18, 25, 30, 65, 66, 89, 96, 98, 100, 105, 132, 146, 147, 162, 170, 177, 181, 182, 188, 189, 197, 230, 231, 247, 251, 252, 255, 279, 281, 284, 285. The 12 VH1-2*02 secondary hotspots not mutated in VRC-PG04 are listed. Column 1 is the position of the unmutated hotspot. Columns 2 and 3 show the sequence context of the unmutated hotspot in VH1-2*02 (column 2) and VRC-PG04 (column 3). In column 3, the unmutated hotspot is underlined and the nearest prior SHM within 2 bp is labeled in red. The position of the nearest prior SHM is noted in column 4. Whether or not this prior SHM is itself a hotspot, and its potential effect on the hotspot that was not mutated in VRC-PG04, is noted in columns 5 and 6 respectively. A/T nucleotides undergo SHM by spreading of mutational activity from adjacent DGYWs, hence the relevance of the hotspot status of the “nearest” DGYW. SYC was defined as a cold spot motif by ref. 35.
Discussion
Our studies show that local sequence context is a more dominant determinant of VRC01-class bnAb intrinsic mutation frequency than long-range influences of hotspots at other sites within a VDJ exon. Introducing SHM hotspots into a V exon caused a global increase in overall V exon SHM frequency in cell-line studies (10, 11). However, we show that in vivo association of VH1-2*02 with CDR3s that have high or low SHM potential did not markedly influence PP GC B-cell SHM frequency of VH1-2*02 sequences, including that of adjacent FWR3 sequences or that of the most CDR3-proximal primary hotspot position. The only increase in SHM frequency observed was at position 293, which is 2 bp upstream of the CDR3 and thus could have been influenced by adjacent sequence differences created by the VH-to-DJH junction. Our findings also contrast with those of transgene studies that indicated that introduced hotspots lead to compensatory decreases in SHM frequency elsewhere in the V(D)J exon (9), because we did not observe a change in VH1-2*02 SHM frequency in passengers containing CDR3s with high or low SHM potential. Likewise, we also found that SHM patterns of VH1-2*02 in association with diverse CDR3s of V(D)J exons assembled in vivo were highly similar to that of the VH1-2*02 passenger exons with fixed CDRs that had high or low SHM potential. Together, our studies suggest that neither introduction of new SHM hotspots nor saturation of existing SHM hotspots has a major general influence on SHM across V(D)J exons in GC B cells.
Certain SHMs in VH1-2*02–using antibodies may be intrinsically favored (15, 25). Our findings indeed demonstrate that a number of recurrently mutated VRC01-class bnAb positions occur at primary or secondary VH1-2*02 SHM hotspots. Of these, only one (position 170) encodes an amino acid at the VRC01–gp120 interface (16), and another (position 270) is silent, supporting the notion that most of these recurrent SHMs may occur due to high intrinsic SHM frequency in the absence of positive or negative selection. We also identified three recurrently mutated VH1-2*02 positions with quite low intrinsic SHM frequencies that, based on strict statistical analyses, are not predicted to occur recurrently. Thus, these SHMs are likely enriched by positive selection, a notion supported by two such positions (185 and 187) affecting the VRC01–gp120 interface (Fig. 3B). Our findings that V exon deletions also correlate with sites of high intrinsic SHM (6) (Fig. 2) suggest that intrinsic SHM frequency data should be applicable for assessing impact of intrinsic SHM frequency on deletions (or insertions) in VRC01-class bnAbs (26). By extending our matched silent SHM position-based analysis to positions mutated in five out of six bnAbs, we identified eight additional, nonsilent positions with intrinsic SHM frequencies (Fig. S4C) that had a relatively low probability of occurring without selection (P ≤ 0.0341; Fig. S4 C and E). However, none of these reached Bonferroni-adjusted significance (Fig. S4E). If additional VRC01-class bnAbs are identified and added to the six currently available, it may be possible to better clarify whether or not any of these eight potential additional positions become stronger candidates for positive selection. Overall, the methods we describe to identify recurrent bnAb SHMs (and deletions) that depend on strong positive selection, along with our recent mouse models (22), could contribute to studies aimed at designing immunogens to accelerate development of bnAb lineages (27).
Our finding that local sequence context affects intrinsic SHM frequency implies that different VDJ exon sequences, including intermediates along a particular lineage, will have different local potentials for affinity maturation. In this regard, the two VRC01-lineage affinity maturation intermediates that we assayed had different potentials to promote particular SHMs that would affect the bnAb–gp120 interface (Fig. 4A). Such findings again may be relevant to the design of sequential immunization strategies to drive B cells along desired bnAb maturation pathways (12, 28, 29), because leading a response through one intermediate versus another may influence the frequency with which particular SHMs are generated. However, overall intrinsic SHM levels of a highly mutated mature VRC-PG04 bnAb are very low due to loss of intrinsic SHM targets due to high intrinsic SHM hotspot mutability and/or by selection for SHMs that inactivate them (Fig. 4B). To further increase potency and breadth of such bnAbs for therapeutic goals, increased SHM potential might be gained by embedding AID targeting motifs into the mature bnAb V exon sequence, while preserving key interaction residues. Another approach might be to modify a germline bnAb precursor to contain the intrinsically low SHMs that are functionally implicated by recurrent presence in bnAbs lineages. These and related approaches again could be facilitated by our rapidly generated and more physiological V(D)J-rearranging vaccination mouse models that are designed to test HIV-1 immunization strategies (22).
Materials and Methods
Generation of Targeted ES Cells and Mouse Chimeras.
ES cell lines containing passenger alleles were generated as previously described (6). Briefly, an ES cell line containing a VB1-8 productive allele and a puromycin cassette in place of the V(D)J exon on the other allele was generated. ES cell lines containing passenger alleles were generated by replacing the puromycin cassette with the respective passenger test sequences. Finally, the modified ES cells were used to generate chimeric mice with totally ES-cell-derived B and T lymphocytes via our RAG-2 blastocyst complementation (19). See SI Materials and Methods for details.
GC B-Cell Isolation.
PP were collected from 8- to 12-wk-old passenger allele chimeras or VH1-2*02–rearranging mice (22) that were immunized with NP-CGG (N-5055E; Biosearch Technologies) for 10 d. This short-term immunization does not influence SHM patterns of either productive or passenger VB1-8 alleles in splenic or PP GC B cells (6). However, due to their higher level SHM levels, which occur even without immunization (6), we focused our analyses on PP GC B cells, which were isolated by flow cytometric sorting of B220+/Peanut Agglutininhi B cells.
PCR and Sequencing.
Productive and passenger alleles were amplified in separate PCR reactions using allele-specific primers (Table S2 for primer sequences and SI Materials and Methods for PCR conditions). The products were sequenced by Illumina MiSeq high-throughput sequencing (Illumina).
Table S2.
Primer sequences
| Allele | Round | Forward primer 5′ → 3′* | Reverse primer 5′ → 3′ |
| VB1-8 productive | First | CATGGGATGGAGCTGTATCAT | AAGGCTCTGAAATCGATACCGT |
| Second | TACACTCTTTCCCTACACGACGCTCTTCCGATCTXXXXXXCTTTCTCTCCACAGGTGTCC | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTAAGAGAGAGGTTGTAAGGACTCACC | |
| Third | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCT | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTAAGAGAGAGGTTGTAAGGACTCACC | |
| VH1-2+CDR3VB1-8 passenger | First | ATGGGATGGAGCTGACTCAT | TCTGTCGACGATATCGGATC |
| Second | TACACTCTTTCCCTACACGACGCTCTTCCGATCTXXXXXXATAGGCGCTATGGCGCGA | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTAAGAGAGAGGTTGTAAGGACTCACC | |
| Third | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCT | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTAAGAGAGAGGTTGTAAGGACTCACC | |
| VH1-2+CDR3VRC-PG04, int6.1%, int6.8% passengers | First | ATGGGATGGAGCTGACTCAT | CCGCTCTAGCCTCGAGTACGTAAG |
| Second | TACACTCTTTCCCTACACGACGCTCTTCCGATCTXXXXXXCTTTCTCTCCACAGGTGTCC | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTTCTGTCGACGATATCGGATC | |
| Third | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCT | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTTCTGTCGACGATATCGGATC | |
| int16.6%, VRC-PG04 passengers | First | ATGGGATGGAGCTGACTCAT | CCGCTCTAGCCTCGAGTACGTAAG |
| Second | TACACTCTTTCCCTACACGACGCTCTTCCGATCTXXXXXXCTTTCTCTCCACAGGTGTCC | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTTCTGTCGACGATATCGGATC | |
| Third | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCT | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTTCTGTCGACGATATCGGATC | |
| VH1-2–rearranging mice (22) (homozygous IGHV1-2*02/ ΔIGCR1) | First | TGGACCTGGAGGATCCTCTT | GGCTCTGAGATCCCTAGACAG |
| Second | TACACTCTTTCCCTACACGACGCTCTTCCGATCTXXXXXXTTCTCTCCACAGGAGCCCAC | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTTCTGTCGACGATATCGGATC | |
| Third | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCT | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACC | |
| VH1-2–rearranging mice (22) (IGHV1-2*02/ ΔIGCR1 and human JH2 knock-in on one IgH allele, JH deletion on other IgH allele) | First | TGGACCTGGAGGATCCTCTT3' | GGCTCTGAGATCCCTAGACAG |
| Second | TACACTCTTTCCCTACACGACGCTCTTCCGATCTXXXXXXTTCTCTCCACAGGAGCCCAC | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTTGGACAGAGAAGACTGGGAGG | |
| Third | AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCT | CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCTTGGACAGAGAAGACTGGGAGG |
XXXXXX are nucleotides that are unique to each library (barcodes).
Sequencing Alignment and Analysis for Productive and Passenger Alleles.
Miseq sequencing data reads were demultiplexed using the fastq-multx tool from ea-utils (https://expressionanalysis.github.io/ea-utils/) and Illumina adaptor sequences trimmed using the SeqPrep utility (https://github.com/jstjohn/SeqPrep) and aligned to a reference sequence using Bowtie2 (30). A custom script was used to call mutations. A nucleotide differing from the reference sequence was counted as a mutation only if the Illumina Sequencing Quality Score was >20, corresponding to a probability of <0.01 that the base was called incorrectly. A custom pipeline was used to process the sequencing data and to call mutations. SHM frequencies are calculated as the percent of sequences containing a mutation at the indicated nucleotide(s). Duplicate sequences are retained. As described previously (6), we control for PCR repeats by analyzing at least five independent mouse replicates for each experiment and then averaging mutation frequency of a given nucleotide across replicates.
Detection of Differences Between SHM Profiles at Individual Nucleotides.
To detect whether the intrinsic SHM pattern of VH1-2*02+CDR3VB1-8 and VH1-2*02+CDR3VRC-PG04 differs significantly at any individual nucleotide we performed an unpaired t test comparing the mutation frequency of the two passengers at each of the 294 nucleotide positions of the VH1-2*02, at each mutational stratum. To correct for multiplicity of testing we applied a Bonferroni-adjusted P value cutoff for significance of 0.05/294. The SHM profile of the VH1-2*02–rearranging mice was compared with each of the VH1-2*02 passengers by the same method.
Detection of Positively Selected SHMs in VRC01-Class bnAbs.
We selected VRC01-class bnAb sequences with high breadth and potency for analysis, using the criteria that they neutralized >75% of tested HIV isolates with an IC50 <50 μg/mL. In addition, each bnAb analyzed came from a different donor, ensuring that all SHMs in a given bnAb assayed occurred independently of those in other assayed bnAbs. To determine a set of silent mutation positions of VH1-2*02 matched in intrinsic mutation frequency to each recurrently mutated bnAb position, for each recurrently mutated position we tested the significance of the difference of its mean intrinsic mutation frequency with that of each silent SHM position of VH1-2*02 (shown in Fig. 3D and Fig. S4D) by ANOVA with Fisher’s least significant difference post hoc test. A silent mutation position was considered to be of mutation frequency similar to that of the recurrently mutated position if P > 0.05. Nominal probabilities of matched silent SHM positions being mutated in each number of bnAbs were estimated by modeling the frequency distribution of silent SHM positions mutated in each number of bnAbs as a binomial distribution.
SI Materials and Methods
Generation of Targeted ES Cells and Mouse Chimeras.
As previously described (6), ES cell lines containing passenger alleles were generated from a parental ES cell line that contains a productive VB1-8 inserted in place of the IgH DQ52 and J segments on one allele (32) and a puromycin cassette PGK-puroΔTK (33) flanked by heterologous loxP sites inserted into the other allele. To generate ES cell lines containing different test passenger sequences, the puromycin cassette was exchanged with a passenger cassette containing a VB1-8 promoter and leader sequence containing a translation termination codon, followed by test sequences. The puromycin cassette was exchanged by either recombinase-mediated cassette exchange (RMCE) (33) or CRISPR/Cas9-mediated gene targeting. ES cell lines carrying VH1-2*02+CDR3VRC-PG04, int6.1%, and int6.8% passengers were generated by RMCE. Passengers are cloned into circular exchange constructs, and 100 μg of the construct is coelectroporated into ES cells with 7.5 μg of pMC-Cre plasmid. Transfected cells are selected with ganciclovir. ES cell lines carrying VH1-2*02+CDR3VB1-8 and VRC-PG04 were generated by CRISPR/Cas9-mediated gene targeting. Briefly, passengers are cloned into an IgH DQ52 and J region replacement targeting construct (32). The targeting vector and three single guide RNAs designed to target the puromycin cassette were conucleofected (P3 Primary Cell 4D-Nucleofector X Kit; Lonza) and selected with G418 and ganciclovir, as described in ref. 6. Correctly targeted ES cell clones were verified by Southern blotting and used to generate chimeric mice by RAG-2 blastocyst complementation (19).
PCR Amplification and High-Throughput Sequencing.
Productive and passenger alleles were amplified in separate PCR reactions using allele-specific primers. Sequencing adapters and barcodes were added to the end of DNA fragments by a second and third round of PCR. For the first PCR, a 50-μL reaction containing 100 ng DNA, 200 μM dNTP, 0.2 μM forward primer, 0.2 μM reverse primer, 5 μL of 10× Pfu PCR buffer, and 2.5 U Pfu Turbo DNA Polymerase was subjected to PCR with the following conditions: 95 °C for 10 min, 28 cycles of 95 °C for 30 s, 56 °C for 30 s, and 72 °C for 1 min and then 72 °C for 10 min. For the second PCR, a 50-μL reaction containing 2 μL PCR product from the first PCR, 200 μM dNTP, 0.2 μM P7-tagged primer, 0.2 μM barcode-specific primer, 5 μL of 10× Pfu PCR buffer, and 2.5 U Pfu Turbo DNA Polymerase was subjected to eight cycles of PCR with the same conditions as the first PCR. For the third PCR, a 50-μL reaction containing 2 μL PCR product from the second PCR, 200 μM dNTP, 0.2 μM P7 primer, 0.2 μM P5-tagged primer, 5 μL of 10× Pfu PCR buffer, and 2.5 U Pfu Turbo DNA Polymerase was subjected to 12 cycles of PCR with the same condition as the first PCR. See Table S2 for primer sequences. The products were sequenced by Illumina MiSeq high-throughput sequencing (Illumina).
Sequencing Alignment and Analysis for VH1-2*02–Rearranging Mice (22).
Sequences were analyzed by a customized pipeline as previously described (34), which can be downloaded at Bitbucket (https://bitbucket.org/adugduzhou/htgtsrep). Briefly, paired reads were preprocessed, joined, and filtered and then subjected to V, D, and J gene assignment by IgBLAST. Sequences were classified as “productive” if the V gene was in-frame and had the first complete coding triplet for the J genes and contained no stop codon or pseudo-VH. Otherwise, sequences were classified as “nonproductive.”
Acknowledgments
We thank Pei-Yi Huang and Yuko Fujiwara for generating the chimeras and Kenneth Ketman and Natasha Barteneva for help with FACS sorting. This work was supported by NIH Grants R01AI077595 and CHAVI-ID 5UM1AI100645 (to F.W.A); the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases, NIH; NIH Grant F30AI114179- 01A1 (to J.K.H.); NIH Grant 1U19AI117892-02 (to T.B.K.); and Dana Farber/Harvard Cancer Center Core Grant 5P30 CA006516 (to D.N.). F.W.A. is an investigator and Z.D. is a postdoctoral fellow of the Howard Hughes Medical Institute. L.-S.Y. was a Cancer Research Institute postdoctoral fellow.
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequence reported in this paper has been deposited in the National Center for Biotechnology Information Sequence Read Archive (accession no. SRP111486).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1709203114/-/DCSupplemental.
References
- 1.Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152:417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amzel LM, Poljak RJ. Three-dimensional structure of immunoglobulins. Annu Rev Biochem. 1979;48:961–997. doi: 10.1146/annurev.bi.48.070179.004525. [DOI] [PubMed] [Google Scholar]
- 3.Mostoslavsky R, Alt FW, Rajewsky K. The lingering enigma of the allelic exclusion mechanism. Cell. 2004;118:539–544. doi: 10.1016/j.cell.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 4.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 5.Hwang JK, Alt FW, Yeap LS. Related mechanisms of antibody somatic hypermutation and class switch recombination. Microbiol Spectr. 2015;3:MDNA3-0037-2014. doi: 10.1128/microbiolspec.MDNA3-0037-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeap LS, et al. Sequence-intrinsic mechanisms that target AID mutational outcomes on antibody genes. Cell. 2015;163:1124–1137. doi: 10.1016/j.cell.2015.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rogozin IB, Diaz M. Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. J Immunol. 2004;172:3382–3384. doi: 10.4049/jimmunol.172.6.3382. [DOI] [PubMed] [Google Scholar]
- 8.Dörner T, et al. Analysis of the frequency and pattern of somatic mutations within nonproductively rearranged human variable heavy chain genes. J Immunol. 1997;158:2779–2789. [PubMed] [Google Scholar]
- 9.Goyenechea B, Milstein C. Modifying the sequence of an immunoglobulin V-gene alters the resulting pattern of hypermutation. Proc Natl Acad Sci USA. 1996;93:13979–13984. doi: 10.1073/pnas.93.24.13979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baughn LB, et al. Recombinase-mediated cassette exchange as a novel method to study somatic hypermutation in Ramos cells. MBio. 2011;2:e00186-11. doi: 10.1128/mBio.00186-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei L, et al. Overlapping hotspots in CDRs are critical sites for V region diversification. Proc Natl Acad Sci USA. 2015;112:E728–E737. doi: 10.1073/pnas.1500788112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mascola JR, Haynes BF. HIV-1 neutralizing antibodies: Understanding nature’s pathways. Immunol Rev. 2013;254:225–244. doi: 10.1111/imr.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kwong PD, Mascola JR. Human antibodies that neutralize HIV-1: Identification, structures, and B cell ontogenies. Immunity. 2012;37:412–425. doi: 10.1016/j.immuni.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheid JF, et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science. 2011;333:1633–1637. doi: 10.1126/science.1207227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonsignori M, et al. NISC Comparative Sequencing Program Maturation pathway from germline to broad HIV-1 neutralizer of a CD4-mimic antibody. Cell. 2016;165:449–463. doi: 10.1016/j.cell.2016.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou T, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329:811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burton DR, Mascola JR. Antibody responses to envelope glycoproteins in HIV-1 infection. Nat Immunol. 2015;16:571–576. doi: 10.1038/ni.3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jardine JG, et al. Minimally mutated HIV-1 broadly neutralizing antibodies to guide reductionist vaccine design. PLoS Pathog. 2016;12:e1005815. doi: 10.1371/journal.ppat.1005815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J, Lansford R, Stewart V, Young F, Alt FW. RAG-2-deficient blastocyst complementation: An assay of gene function in lymphocyte development. Proc Natl Acad Sci USA. 1993;90:4528–4532. doi: 10.1073/pnas.90.10.4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cumano A, Rajewsky K. Structure of primary anti-(4-hydroxy-3-nitrophenyl)acetyl (NP) antibodies in normal and idiotypically suppressed C57BL/6 mice. Eur J Immunol. 1985;15:512–520. doi: 10.1002/eji.1830150517. [DOI] [PubMed] [Google Scholar]
- 21.Wu X, et al. NISC Comparative Sequencing Program Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science. 2011;333:1593–1602. doi: 10.1126/science.1207532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian M, et al. Induction of HIV neutralizing antibody lineages in mice with diverse precursor repertoires. Cell. 2016;166:1471–1484 e1418. doi: 10.1016/j.cell.2016.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rada C, Yélamos J, Dean W, Milstein C. The 5′ hypermutation boundary of kappa chains is independent of local and neighbouring sequences and related to the distance from the initiation of transcription. Eur J Immunol. 1997;27:3115–3120. doi: 10.1002/eji.1830271206. [DOI] [PubMed] [Google Scholar]
- 24.Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, Kunkel TA. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat Immunol. 2001;2:530–536. doi: 10.1038/88732. [DOI] [PubMed] [Google Scholar]
- 25.Sheng Z, et al. NISC Comparative Sequencing Program Gene-specific substitution profiles describe the types and frequencies of amino acid changes during antibody somatic hypermutation. Front Immunol. 2017;8:537. doi: 10.3389/fimmu.2017.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kepler TB, et al. Immunoglobulin gene insertions and deletions in the affinity maturation of HIV-1 broadly reactive neutralizing antibodies. Cell Host Microbe. 2014;16:304–313. doi: 10.1016/j.chom.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonsignori M, et al. Antibody-virus co-evolution in HIV infection: Paths for HIV vaccine development. Immunol Rev. 2017;275:145–160. doi: 10.1111/imr.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haynes BF, Kelsoe G, Harrison SC, Kepler TB. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol. 2012;30:423–433. doi: 10.1038/nbt.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Escolano A, et al. Sequential immunization elicits broadly neutralizing anti-HIV-1 antibodies in Ig knockin mice. Cell. 2016;166:1445–1458 e1412. doi: 10.1016/j.cell.2016.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang J, et al. Identification of a CD4-binding-site antibody to HIV that evolved near-pan neutralization breadth. Immunity. 2016;45:1108–1121. doi: 10.1016/j.immuni.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonoda E, et al. B cell development under the condition of allelic inclusion. Immunity. 1997;6:225–233. doi: 10.1016/s1074-7613(00)80325-8. [DOI] [PubMed] [Google Scholar]
- 33.Han L, Masani S, Yu K. Overlapping activation-induced cytidine deaminase hotspot motifs in Ig class-switch recombination. Proc Natl Acad Sci USA. 2011;108:11584–11589. doi: 10.1073/pnas.1018726108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin SG, et al. Highly sensitive and unbiased approach for elucidating antibody repertoires. Proc Natl Acad Sci USA. 2016;113:7846–7851. doi: 10.1073/pnas.1608649113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424:103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]