

Abstract
Purpose
Presently, 52 independent nuclear single nucleotide polymorphisms (nSNPs) have been associated with age-related macular degeneration (AMD) but their effects do not explain all its variance. Genetic interactions between the nuclear and mitochondrial (mt) genome may unearth additional genetic loci previously unassociated with AMD risk.
Methods
Joint effects of nSNPs and selected mtSNPs were analyzed by two degree of freedom (2df) joint tests of association in the International AMD Genomics Consortium (IAMDGC) dataset (17,832 controls and 16,144 advanced AMD cases of European ancestry). Subjects were genotyped on the Illumina HumanCoreExome array. After imputation using MINIMAC and the 1000 Genomes Project Phase I reference panel, pairwise linkage disequilibrium pruning, and quality control, 3.9 million nSNPs were analyzed for interaction with mtSNPs chosen based on association in this dataset or publications: A4917G, T5004C, G12771A, and C16069T.
Results
Novel locus TRPM1 was identified with genome-wide significant joint effects (P < 5.0 × 10−8) of two intronic TRPM1 nSNPs and AMD-associated nonsynonymous MT-ND2 mtSNP A4917G. Stratified analysis by mt allele identified an association only in 4917A (major allele) carriers (P = 4.4 × 10−9, odds ratio [OR] = 0.90, 95% confidence interval [CI] = 0.87–0.93). Intronic and intergenic ABHD2/RLBP1 nSNPs demonstrated genome-wide significant joint effects (2df joint test P values from 1.8 × 10−8 to 4.9 × 10−8) and nominally statistically significant interaction effects with MT-ND5 synonymous mtSNP G12771A. Although a positive association was detected in both strata, the association was stronger in 12771A subjects (P = 0.0020, OR = 2.17, 95% CI = 1.34–3.60).
Conclusions
These results show that joint tests of main effects and gene–gene interaction reveal associations at some novel loci that were missed when considering main effects alone.
Keywords: age-related macular degeneration (AMD), genetics, risk, mitochondrial DNA, genome-wide interaction study (GWIS), joint effect
Age-related macular degeneration (AMD) is a common retinal disease of public health importance and is the most common cause of blindness in older adults in developed countries.1 AMD, as a complex disorder, is characterized by genetic and nongenetic factors. Currently, 34 genetic loci containing 52 independent single nucleotide polymorphisms (SNPs) have been associated with advanced AMD risk.2 However, these associations do not comprise all the genetic risks for advanced AMD. In addition to genetic factors from the nuclear genome, mitochondrial (mt) SNPs also are associated with AMD risk. A number of studies relate mtDNA variation to AMD through haplogroup classification, which is based on a set of defining mtSNPs and traces maternal lineages of different human populations. The mt haplogroups J, U, and T have been associated reproducibly with increased risk of AMD, whereas haplogroup H has been associated negatively with AMD risk in populations of European ancestry.3–7 A natural follow-up to these studies was to examine whether mitochondrial variation influenced risk of AMD by itself, or through interactions with other nuclear-encoded risk factors associated with AMD.
Mitochondrial genetic variation may influence AMD risk by modulating expression of nuclear-encoded genes or vice-versa. Expression of known genetic risk factors, such as CFH and C3, in cybrid RPE cell lines differs by mt haplogroup.3 Nuclear-encoded genes involved in drusen makeup, reactive oxygen species (ROS) regulation, and responses to oxidative stress (all processes that characterize AMD or engage in the manifestation of AMD) demonstrated significant changes in gene expression in response to mtDNA damage, including two genes (TIMP3, downregulated, and VEGFA, upregulated) that contain variants associated with AMD risk.8,9 These studies suggest that consideration of interactions between genetic variations in the nuclear and mitochondrial genomes might reveal novel genetic risk factors for AMD.
Consequently, this study included mtSNP × nuclear SNP (nSNP) statistical interactions in a genome-wide search for novel AMD risk factors. Our genome-wide interaction study (GWIS) of the large, phenotypically well characterized International AMD Genomics Consortium (IAMDGC) dataset 2 considered mtSNP × nSNP statistical interactions on AMD risk with the objective of identifying novel genetic loci associated with advanced AMD risk.
Methods
Description of the Dataset
A total of 33,976 participants (14,352 males and 19,624 females) of European ancestry from the IAMDGC primary dataset2 were included in the GWIS analyses. As described previously, individuals were evaluated clinically across 26 sites by fundus photography, with the exception of one site, and characterized by the presence of drusen, geographic atrophy (GA), and choroidal neovascularization (CNV); a number of sites (18 of the 26 sites) discerned between the advanced AMD subtypes through optical coherence tomography (OCT) or fluorescein angiography.2 The resulting dataset contained 17,832 controls (category “No AMD”; mean age 70.7 years, 56% female) and 16,144 advanced AMD cases (categories “GA Only,” n = 3,235; “CNV Only,” n = 10,749; and “Mixed GA/CNV,” n = 2,160); mean age, 76.8 years; 60% female; and at least 50 years old at the time of diagnosis). For protection of human subjects regarding data procurement, each site upheld the tenets outlined in the Declaration of Helsinki.2 These phenotype data and the genetic data amassed by the IAMDGC have been added to the database of Genotypes and Phenotypes (dbGaP) with accession code phs001039.v1.p1.2
Genotype Imputation
All individuals were genotyped at the Center for Inherited Disease Research (CIDR) with a custom-made Illumina HumanCoreExome genotyping array featuring, after quality control, 521,950 total variants (in which the total number of genotyped variants, excluding monomorphic variants, across all autosomal chromosomes was 434,054).2 Genotyping quality control criteria called for variants with less than 98.5% call rates and those with significant departures from Hardy-Weinberg equilibrium (P < 10−6) in controls to be dropped.2 Samples with <98.5% call rates were removed from analysis.2 MINIMAC and the 1000 Genomes Project Phase I reference population genotypes (version 3, SHAPEIT2 Reference) were used for imputation against the framework SNPs in the HumanCoreExome panel.2 A total of 27,602,838 nSNPs were imputed before imputation quality control, described in the following sections.
Genotyped mt Variants Chosen for mtSNP × nSNP Joint Analyses
In the GWIS, joint effects with four mtSNPs (A4917G, T5004C, G12771A, and C16069T) were evaluated. The mtSNPs were selected, as described in more detail below, based on prior published associations with AMD risk or nominally statistically significant genetic main effects in the IAMDGC dataset. An additional criterion, mtSNP rare allele frequency overall of at least 0.005 (0.5%), was imposed for sufficient statistical power, given consideration with a common nuclear variant, to detect moderate to strong interaction effects. Some mtSNPs associated in prior studies (including several haplogroup-defining SNPs) were not available on the HumanCoreExome array. All four mtSNPs were statistically independent (data not shown).
Association of mtSNPs and advanced AMD was evaluated using logistic regression models implemented in the PLINK software package (version 1.07),10 adjusting for whole genome amplification (yes/no), which may introduce bias from generation of DNA product in specific nucleotide-rich or -poor regions, and population structure (first two variables from principal components analysis).2 mtSNP names reflect the base change and position based on the revised Cambridge reference sequence (rCRS). For the 265 available genotyped mt variants (of which nine were monomorphic), association between the advanced AMD phenotype and mt variants (or genotype) was nominally statistically significant for mtSNPs T5004C (minor allele frequency [MAF] = 0.017, P = 0.03, odds ratio [OR] = 0.83, 95% confidence interval [CI] = 0.70–0.99) and G12771A (MAF = 0.0054, P = 0.0008, OR = 1.69, 95% CI = 1.25–2.30). mtSNP A4917G was not associated significantly with advanced AMD risk (MAF = 0.10, P = 0.41, OR = 1.03, 95% CI = 0.96–1.10) in the current dataset, but was included in the GWIS based on its prior association with AMD risk in the study of Canter et al.4 The fourth mtSNP, C16069T (MAF = 0.0985, P = 0.15, OR = 0.95, 95% CI = 0.88–1.02) was not statistically significant in this dataset but was associated previously with advanced AMD in a set of 200 CNV patients and 385 controls (P < 0.05, OR = 1.74, 95% CI = 1.0–2.9).7 Due to missing mt genotypes, each GWIS sample size was slightly different from the total: 16,122 cases and 17,810 controls for mtSNP A4917G; 15,407 cases and 16,350 controls for mtSNP T5004C; 15,413 cases and 16,351 controls for G12771A; and 16,042 cases and 17,734 controls for C16069T.
Featured nSNPs in mtSNP × nSNP Interaction Analyses
To reduce computational time, the dataset of 27,602,838 imputed and genotyped nSNPs was pruned by eliminating SNPs that were correlated strongly with nearby SNPs. PLINK was used to determine the linkage disequilibrium (LD) values (r2) between each pair of variants in a window of 100 variants; LD was calculated using a 5-variant sliding window. If a variant pair's r2 value was greater than 0.8, one of the two SNPs was eliminated randomly. This LD level was chosen to ensure that nSNPs in strong LD were removed while permitting retention of nSNPs that were representative of the genomic regions across all autosomal chromosomes. After LD pruning, the total number of variants remaining was 11,697,015, and those variants with imputation quality scores (represented by R2 values) were retained for analysis according to previously established criteria:2 (1) common variants – MAF in controls (CAF) ≥ 0.01 and R2 > 0.30 and (2) rare variants – CAF < 0.01 and R2 > 0.80.
Consequently, the final total number of filtered nSNPs considered in each genome-wide interaction analysis was 3,866,946 variants (of which 57,862 were genotyped).
Genome-wide Interaction Analyses
Kraft's two degree of freedom (2df) joint test of genetic main effects and gene-environment interaction11 compares two logistic regression models: (1) a full model containing the mtSNP and nSNP main effects, a mtSNP × nSNP interaction term, and (2) a partial model lacking the nSNP main effect and the mtSNP × nSNP interaction term. Both models were adjusted for population stratification (two principal components) and whole genome amplification (WGA) status. Sensitivity analyses evaluated the effect of WGA status in models rerun for significant novel loci (see Discussion). In addition, selection of covariates for inclusion in regression models, particularly in assessing effects of sex and age (such as differences in applying a minimum age requirement for controls versus unspecified age restriction concerning controls), was decided based on sensitivity analyses that were used in primary analyses by the IAMDGC.2 A joint test statistic was created by taking the difference of the −2 log likelihoods for the two models; this statistic follows a 2df χ2 distribution. A significant joint test statistic signals one of three possible scenarios: a strong nSNP genetic main effect, a strong mtSNP × nSNP interaction effect, or a combination of both. The traditional GWAS significance threshold of P < 5 × 10−8 was used to identify statistically significant joint effects, and genome-wide significant loci were filtered further by nominal evidence of gene–gene interaction (interaction term P < 0.01). Regression models were constructed using the R programming language (version 3.0.1).12 The 2df joint test P value and test statistic were calculated with the lrtest function from the R package epicalc (version 2.15.1.0).13
Calculation of Genomic Inflation Factor (λgc) from 2df Joint Tests
For each GWIS, the genomic inflation factor λgc was expressed as the quotient of the median 2df observed and median 2df expected χ2 test statistics (with the latter value equal to 1.386).14 Only genotyped and imputed nuclear variants meeting quality control criteria and falling beyond the genomic boundaries of the 34 known AMD risk associated regions were included in calculations.2 By omitting variants in the 34 known AMD risk loci, this evaluates whether there still is notable deviation from the null hypothesis of the overwhelming majority of tested genetic loci being unassociated with advanced AMD; such deviation, if present, would imply systematic bias.15 Genomic boundaries for all 34 known risk loci are detailed in Supplementary Table S5 from the study of Fritsche et al.2
Conditional Analyses for Single nSNPs With Significant Joint and Interaction Effects
Variants that met the statistical significance thresholds for joint effects and interaction effects were included in conditional analyses. This step identified nSNPs with significant joint effects that are “shadow effects” of known AMD risk loci and are not independently associated with AMD risk. To account for potential effect modification by mtSNP, conditional analyses were performed stratified by mtSNP allele. Therefore, in each group (reference allele carriers versus alternate allele carriers), the logistic regression model contains nSNP allele dosage, known AMD risk variant allele dosage, and covariates for population stratification and whole genome amplification. At each AMD locus, the known AMD risk variants were selected from the list of the 52 independently associated risk variants identified by IAMDGC (see Supplementary Table S4 from the study of Fritsche et al.2).
Post Hoc Power Analysis of Genome-Wide Significant Joint Effects
Power at α = 5 × 10−8 for the 2df joint tests of TRPM1 variants and mtSNP A4917G was estimated post hoc with QUANTO.16 This power then was compared to that calculated for only TRPM1 nSNP genetic effects on advanced AMD risk and the 1df interaction test. Population disease risk was set as 0.002. The proportion of individuals with the mtSNP risk allele was specified as 0.10 (equivalent to 4917G frequency). The TRPM1 nSNP allele frequency was specified as 0.42 (corresponding to the MAF), and the effect was modeled as log-additive. Using effect estimates from the original dataset, power was estimated for a joint test effect size of 1.18 (given the logistic regression model, or full model, containing nSNP, mtSNP, and mtSNP × nSNP interaction terms; refer to Supplementary Table S1), mtSNP marginal effects of 1.03, and TRPM1 nSNP marginal effect of 0.92 (given the regression model with only the TRPM1 nSNP term and covariates adjusting for WGA status and population stratification).
For the ABDH2/RLBP1 nSNPs and mtSNP G12771A, calculated power at α = 5 × 10−8 for the 2df joint test was contrasted with calculated power for the 1df interaction test and ABHD2/RLPB1 nSNP genetic main effects on advanced AMD risk. For 15,413 cases and 16,351 controls, the mtSNP allele frequency selected was 0.0054 (equivalent to 12771A frequency) while the nSNP ABHD2/RLBP1 allele frequency chosen was 0.50. Other parameters include the marginal effect of the mtSNP G12771A (1.69), marginal effect of the ABHD2/RLBP1 nSNPs (1.09) on advanced AMD, and interaction effects of 2.00 (corresponding effect size for the mtSNP G12771A × ABHD2/RLBP1 nSNP interaction term; refer to Supplementary Table S1).
Results
Novel Loci Detected When Considering Main and Interaction Effects
Four GWIS analyses were conducted using the 2df joint test of main effects and nSNP × mtSNP interaction in the overall dataset of 16,144 cases and 17,832 controls, identifying two novel loci. Results from mtSNP A4917G-nSNP (Fig. 1) and mtSNP G12771A-nSNP (Fig. 2) interaction analyses revealed, respectively, two novel loci TRPM1 and ABHD2/RLBP1 on chromosome 15. The incorporation of the mtSNP main effect and the pairwise interaction term enabled the detection of these loci with genome-wide significance, whereas main effects results in the previous GWAS2 and current analysis were just under that significance threshold (also refer to Table 1; Supplementary Table S2).
Figure 1.

Joint test results for mtSNP A4917G with 17,810 controls versus 16,122 advanced AMD cases. This Manhattan plot graphs the −log of the 2df joint test P values against the genomic position on each chromosome. Known AMD-associated regions with genome-wide significant joint effects and nominally significant interaction effects are labeled on the plot. Additionally, this mtSNP A4917G-nSNP GWIS detected novel locus TRPM1 on chromosome 15 with genome-wide significant joint effects.
Figure 2.

Joint test results for mtSNP G12771A with 16,351 controls versus 15,413 advanced AMD cases. This Manhattan plot summarizes the 2d joint test results for mtSNP G12771A-nSNP GWIS analyses by graphing the −log of the 2df joint test P value against the genomic position on each chromosome. The genome-wide statistical significance threshold is marked by the solid red line (P < 5 × 10−8). Known AMD-associated regions with genome-wide significant joint effects and nominally significant interaction effects are labeled on the plot. Additionally, novel locus ABHD2/RLBP1 on chromosome 15 was detected with genome-wide significant joint effects.
Table 1.
Joint Test Results (Abridged) for mtSNP A4917G-TRPM1 and mtSNP G12771A-ABHD2/RLBP1 Nuclear SNPs (nSNPs) Interaction Analyses

Joint Effects at Known AMD Risk Loci
In each mtSNP × nSNP GWIS (considering the joint effects of each nSNP and one of four mtSNPs: A4917G, T5004C, G12771A, or C16069T), as expected, genome-wide significant joint effects were detected for most known AMD risk loci identified in the IAMDGC dataset by Fritsche et al. 2 In general, these results are in accord with the main IAMDGC paper's primary analyses.2 Three of the 34 known risk loci for mtSNP T5004C-nSNP interaction analyses and four of the 34 known risk loci for mtSNP G12771A-nSNP interaction analyses did not have genome-wide significant joint effects.
Joint Test Results and 1df Interaction Test Results for Each GWIS
Figures 1 through 4 display the 2df joint test P values by chromosome for each GWIS. Since mtSNP × nSNP interaction term P values are not indicated on these plots, known AMD risk loci with genome-wide statistically significant joint effects and nominally significant interaction effects (P < 0.01) are labeled with their respective names in italics. Supplementary Figures S1 through S4 display plots of interaction term P values by chromosome and show no genome-wide significant 1df interaction tests. In analyses conditioned on known AMD risk SNPs in each region, none of the previously identified AMD loci contain new variants independently associated with risk (data not shown). Quantile-quantile (qq) plots for each GWIS (Supplementary Figs. S5–S8) demonstrated expected amounts of variance inflation once known AMD loci were removed from the analysis.
Figure 4.

Joint test results for mtSNP C16069T with 17,734 controls versus 16,042 advanced AMD cases. This Manhattan plot summarizes the 2df joint test results for mtSNP C16069T-nSNP interaction analyses by graphing the −log of the 2df joint test P value against the genomic position on each chromosome. The genome-wide statistical significance threshold is marked by the solid red line (P < 5 × 10−8). Known AMD-associated regions with genome-wide significant joint effects and nominally significant interaction effects are labeled on the plot. No novel loci were detected.
Figure 3.

Joint test results for mtSNP T5004C with 16,350 controls versus 15,407 advanced AMD cases. This Manhattan plot graphs the −log of the 2df joint test P values against the genomic position on each chromosome. The genome-wide statistical significance threshold is marked by the solid red line (P < 5 × 10−8). Known AMD-associated regions with genome-wide significant joint effects and nominally significant interaction effects are labeled on the plot. Novel regions were not detected in this mtSNP T5004C-nSNP GWIS.
MtSNP A4917G × nSNP Interaction on Chromosome 15
In the mtSNP A4917G GWIS, two intronic nuclear variants, rs6493454 and rs7182946, which are in the TRPM1 gene (transient receptor potential cation channel, subfamily M, member 1), generate genome-wide significant joint test results (2df joint test P value = 2.0 × 10−8 and 1.7 × 10−8, respectively) with nominally significant evidence of interaction (interaction term P value = 0.003; Table 1 or Supplementary Table S2). Both SNPs are in very strong linkage disequilibrium (LD, r2 = 0.98) with each other.
To determine the nature of the interaction between mtSNP A4917G and TRPM1 nSNPs, stratified analyses in the region were conducted including nSNPs originally removed by LD pruning (Table 2 or Supplementary Table S3; Fig. 5). The genome-wide significant association of TRPM1 SNPs with AMD (P values 4.4 × 10−9 [rs7182946] and 5.4 × 10−9 [rs6493454]) was restricted to carriers of the A allele at mtSNP 4917. The effect in carriers of the G allele was not significant (minimum P value = 0.25). Conditional analyses were used to determine if the associated nSNPs in TRPM1 are independent of nSNPs in LIPC associated previously with advanced AMD (rs2070895 and rs2043085).2 This novel association is independent of LIPC, which is located approximately 25 Mb away. Conditioning on the two LIPC variants in each group does not eliminate the effect at TRPM1 (from analyses for 4917G carriers, P values 0.24 [rs6493454] and 0.26 [rs7182946]; from analyses for 4917A carriers, P values 3.9 × 10−9 [rs6493454] and 3.2 × 10−9 [rs7182946]). A possible explanation for the lack of significant association of mtSNP A4917G in the IAMDGC dataset is this statistical interaction with the TRPM1 nSNPs. To explore this hypothesis, we stratified the IAMDGC dataset by TRPM1 nSNP, rs6493454, or rs7182946, genotype (combining heterozygotes and TRPM1 variant minor allele homozygotes), and detected a positive association with AMD and mt4917G (P = 0.028, OR = 1.10, 95% CI = 1.01–1.20) in TRPM1 minor allele carriers but not in TRPM1 major allele homozygotes (P = 0.075, OR = 0.89, 95% CI = 0.79–1.01).
Table 2.
Results for Association Between the TRPM1 nSNP, Which Was Kept After Linkage Disequilibrium Pruning, With Genome-Wide Significant Joint Effects and AMD Phenotype Stratified by mtSNP A4917G Genotype
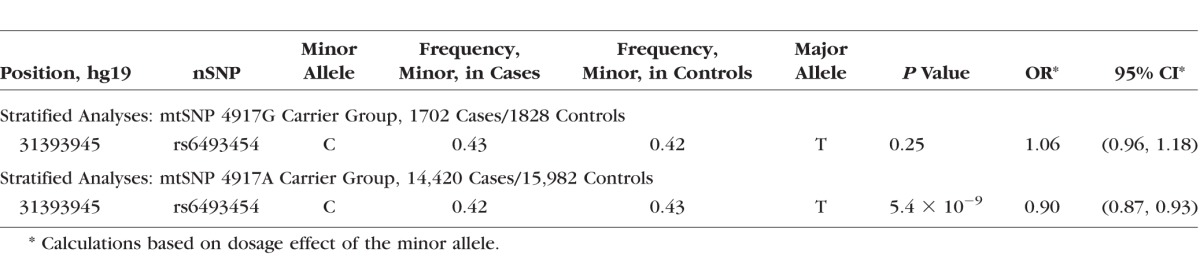
Figure 5.

Stratified analyses by mt allele for nSNPs in the TRPM1 region. The reference SNPs rs6493454 (purple diamond) and rs7182946, peak SNPs, are enclosed in the box on both plots. The genome-wide statistical significance threshold is marked by the solid dark line (P < 5 × 10−8; [A]) TRPM1 regional association plot in 4917G carriers (1702 cases and 1828 controls). No association (OR = 1.06, 95% CI = 0.96–1.18) was detected for these two SNPs or any other SNP in LD. (B) TRPM1 regional association plot in 4917A carriers (14,420 cases and 15,982 controls). Genome-wide significant association (OR = 0.90, 95% CI = 0.87–0.93) with AMD risk was detected at three nSNPs in strong LD.
MtSNP G12771A × nSNP Interaction on Chromosome 15
nSNPs in the ABHD2/RLBP1 region (abhydrolase domain containing 2 and retinaldehyde binding protein 1, respectively) demonstrate genome-wide significant joint effects (2df joint test P values from 1.8 × 10−8 to 4.9 × 10−8 ) and nominally statistically significant interaction effects (interaction term P values from 0.0059–0.0093) with mtSNP G12771A. As shown in Table 1 or Supplementary Table 2, all eight variants are in noncoding regions with annotations “intronic” or “intergenic.”
The dataset then was stratified by mt allele at G12771A and analysis repeated in each subset (Table 3 or Supplementary Table S4; Fig. 6). nSNPs rs4932478 and rs4932480 (ABHD2 locus) plus rs11459118, rs875390, rs875391, rs2351006, rs144871045, and rs2070780 (ranging from genomic coordinates in ABHD2, ABHD2/RLBP1 [intergenic], and RLBP1) are in moderate to strong LD (r2 ≥ 0.70) with each other. For 12771A (mt minor/alternate allele) carriers, the effect size is large (minimum OR = 2.11) with the association between AMD phenotype and nSNP genotype nominally statistically significant (P < 0.05). In the 12771G carriers, the effect sizes are smaller (OR = 1.08, 95% CI = 1.05–1.12) with association close to genome-wide significant (P value = 3.1 × 10−7). With nonoverlapping 95% CIs, both effects are in the same directions, but the association appears stronger in the small number of 12771A carriers than in the 12771G carriers. Conditional analyses in both groups (LIPC variants identified by IAMDGC and the TRPM1 variants from nSNP × mtA4917G interaction analyses) did not eliminate the significant signal at these nSNPs associated with AMD risk (from analyses for 12771A carriers, P values from 0.0028 to 0.0048; from analyses for 12771G carriers, P values from 4.5 × 10−7 to 1.6 × 10−6; see Table 3 and Supplementary Table S4, which present P values in each subset regarding regression models without the known AMD risk variants in LIPC and associated TRPM1 variants from nSNP × mtA4917G interaction analyses, for comparison to the range of values listed here).
Table 3.
Two ABHD2/RLBP1 Variants That Were Kept After Linkage Disequilibrium Pruning and mtSNP G12771A Demonstrated Genome-Wide Significant Joint Effects and Nominally Statistically Significant Interaction Effects
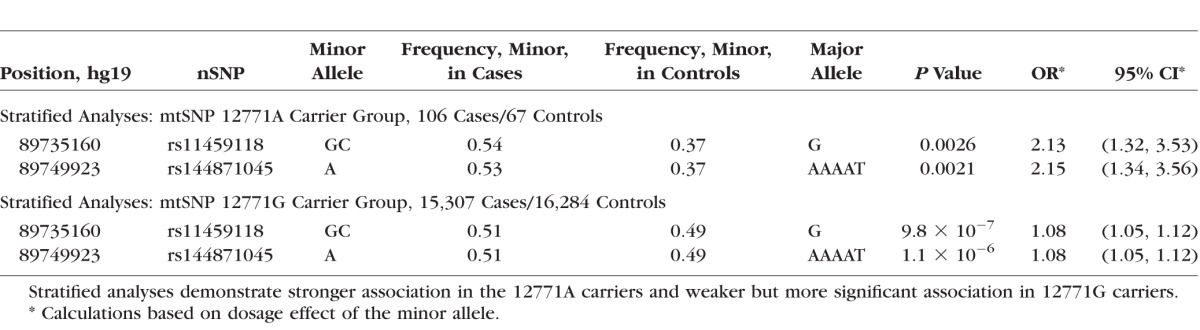
Figure 6.

Analysis of ABHD2/RLBP1 nSNP genotypes and AMD phenotype stratified by G12771A genotype. The nSNP rs4932480, which is selected as the reference SNP and notated on the plots as a purple diamond within a box, is in strong linkage disequilibrium (r2 ≥ 0.73) with the other seven nSNPS that are identified in this region to have strong joint effects and moderately significant interactions with mtSNP G12771A (see Table 1; Supplementary Table S2). Other markers are included in this region. The genome-wide statistical significance threshold is marked by the solid dark line (P < 5 × 10−8). (A) ABHD2/RLBP1 regional association plot in 12771A carriers (106 cases and 67 controls). Strong association (minimum OR = 2.11, 95% CI = 1.31-3.48) of ABHD2/RLBP1 SNPs was detected, and these 12771A carriers have a higher AMD risk in contrast to 12771G (major allele) carriers. (B) ABHD2/RLBP1 regional association plot in 12771G carriers (15,307 cases and 16,284 controls). ABHD2/RLBP1 SNPs were weakly associated (regarding effect size, or magnitude of the OR) with AMD (OR = 1.08, 95% CI = 1.05, 95% CI = 1.05–1.12).
Regulatory Potential of Identified nSNPs in Novel Loci on Chromosome 15
Both novel AMD loci on chromosome 15 are annotated as having possibly regulatory potential, indicating one mechanism by which these variants might influence development of AMD through altered gene expression. The UCSC Genome Browser was used to assess potential functional consequences of the two intronic TRPM1 SNPs and one intronic ABHD2 SNP, by examining annotations from the Encyclopedia of DNA Elements (ENCODE) Project.17,18 DNase I hypersensitivity sites (DHSs) overlap the genomic positions of peak markers rs6493454 (DHS coordinate range, 31393926–31394115) and rs7182946 (DHS coordinate range, 31394766–31395235), suggesting these regions may impact gene expression. Notably, the strongest DNase I signal peak at each site was obtained in the human RPE cell line, or cell type HRPEpiC (Supplementary Fig. S9). In the other locus, the ABHD2 SNP rs4932478 overlaps two transcription factor binding sites (RELA and RUNX3), which primarily influence expression in lymphoblastoid cell lines.
To allow for the possibility that the relevant functional SNP in each locus is in linkage disequilibrium with the nSNPs most strongly associated with AMD, we used HaploReg, version 419 to evaluate regulatory potential of other SNPs in moderate-to-strong LD at the loci. Proximal variants in strong LD (r2 ≥ 0.80) with the two TRPM1 nSNPs, are all annotated as intronic while variants in moderate to strong LD (r2 ≥ 0.40) with the eight ABHD2/RLBP1 nSNPs, were intronic or in the 3′ untranslated region (3′UTR). Of the four variants in strong LD with the two TRPM1 SNPs, three were in promoter and enhancer histone marks, three overlapped DHSs in at least 1 tissue (maximum 5 tissues), and one (rs3809579) was located in 2 transcription factor binding sites, GATA2 and USF1 (Supplementary Fig. S10). Variants in LD with the ABHD2/RLBP1 nSNPs had annotations ranging from enhancer histone marks, DHSs, and corresponding transcription factors (such as GATA2, EBF1, and NFKB). In short, functional annotation of variants in these two loci are consistent with regulatory effects, suggesting that gene expression may differ by mtSNP genotype, influencing risk of AMD.
Discussion
Across the four GWIS, known AMD genetic factors were detected (as expected) and two novel AMD genetic risk factors, TRPM1 and ABHD2/RLBP1, were identified. A minority of the 34 known risk loci did not reach genome-wide significance for mtSNP T5004C-nSNP interaction and mtSNP G12771A-nSNP interaction analyses, possibly due to lower power of the 2df test to detect loci driven by main effects alone. At the two novel loci, TRPM1 and ABHD2/RLBP1, when restricting AMD cases to CNV only or GA only, the effect sizes were similar to those measures for the combined advanced AMD subtypes described in the Methods section (data not shown). Sensitivity analyses excluding subjects whose DNA source was from WGA did not change the effect sizes at these loci (data not shown). The large case–control data set with genome-wide genotyping from a single genotyping center provided substantial power to detect novel loci for advanced AMD by considering gene × gene interactions that might have obscured main effects in the original GWAS.2 Post hoc power calculations determined that the joint test in this dataset had 70% to 74% power at α = 5 × 10−8 to detect the two novel loci reported here, greater than power to detect main effects at these two loci (45%–49%) and the pairwise interactions (1%–2%). This increase in statistical power from using the 2df joint test may have allowed the detection of novel loci previously missed in the IAMDGC primary analyses.2 As testament to the project's large scale and ability to examine interactions involving rare alleles, joint effects of rare mtSNP G12771A (only 173 of the 31,764 genotyped individuals carry 12771A) and common ABHD2/RLBP1 SNPs were genome-wide statistically significant and met the interaction term significance threshold.
Both novel loci, TRPM1 and ABHD2/RLBP1, have functional connections to the visual system. Mutations in TRPM1 have been shown to cause autosomal recessive congenital stationary night blindness (CSNB)20–22 and to alter melanocyte function or melanin synthesis.23 In advanced AMD, photoreceptor death (particularly of rods, which are crucial in black and white vision) may produce night vision difficulties. According to the Complications of AMD Prevention Trial (CAPT) study involving 1052 subjects with multiple large drusen, positive association between night vision symptoms, as recorded from responses on a night vision questionnaire, and risk of GA or CNV was nominally statistically significant (CNV observed eyes [total = 1043], first quartile [worst night vision] versus fourth quartile [best night vision] – P = 0.01, relative risk (RR) = 1.92, 95% CI = 1.08–3.44; GA observed eyes [total = 988], first quartile versus fourth quartile – P = 0.001, RR = 4.60, 95% CI = 1.81–11.6).24 ABHD2's function is not well characterized and may possibly include immunological roles; for example, high ABHD2 expression was detected in macrophages situated in atherosclerotic lesions.25 In contrast, RLBP1 encodes a protein with a niche in the visual cycle, and variants in this gene have been associated with other retinal pathologies distinct from AMD, such as Newfoundland rod–cone dystrophy and retinitis punctata albescens.26,27 Retinitis punctata albescens (OMIM: 13688028) may be traced in families as following autosomal recessive patterns of inheritance; this clinical phenotype is distinct from AMD in that white specks in the eye are detected in patients' fundus photography, but drusen are not noted features.27 In contrast to retinitis punctata albescens and in stark contrast to AMD, vision deterioration is swifter for sufferers of Newfoundland rod–cone dystrophy (OMIM: 60747629); in fact, onset occurs before adolescence27 as observed by Eichers et al.,30 who also recorded patients' vision loss experiences occurring as a young adult or upon reaching middle-age. The mtSNPs that interact with these two loci, A4917G and G12771A, are in genes that encode NADH dehydrogenase (Complex I) subunits; A4917G is in MT-ND2, which encodes subunit 2, and G12771A is in MT-ND5, which encodes subunit 5. Complex I is a component of the electron transport chain, which participates in ATP production during oxidative phosphorylation. While mtSNP A4917G has been associated previously with AMD,4 G12771A has not been associated with AMD.
The mtSNPs A4917G and G12771A have not been implicated directly in mt damage or dysfunction; however, the following functional studies may offer biological connections between MT-ND5, the gene containing G12771A, and mt dysfunction in the context of AMD. In an investigation by Terluk et al.31 of 51 donor eyes (30 controls and 21 AMD cases or eyes with intermediate AMD pathology), a significant rise in RPE mtDNA damage in several regions of the mt genome in the macula was discovered; among the mt genomic regions found to have such a significant change in mtDNA damage when comparing cases to controls was MT-ND5 (P value = 0.03) where G12771A resides. Miceli and Jazwinski,8 comparing mtDNA-devoid ARPE-19 cells with mtDNA-containing ARPE-19 cells, recorded a lower mt membrane potential for those cells without mtDNA. Thus, mtDNA dysfunction, a characteristic of AMD pathology or physiology, may be accompanied by effects on membrane potential; as an illustration of a contributor to mt/mtDNA damage in RPE cells, oxidative stress emerging from exposure to peroxides can affect cytosolic and intracellular calcium (Ca2+) levels.32,33 Also, the fact that MT-ND5, a gene encoding a Complex I subunit, was one of the mt genomic regions to have a significant escalation in mtDNA damage suggests that vital AMD-defining processes impact bioenergetics. Such changes might alter the effect of nSNPs on risk of AMD, as suggested by the results of the current study.
The inverse association of TRPM1 intronic variants rs6493454 and rs7182946 and advanced AMD was restricted to the carriers of the common 4917A allele. The 4917G allele defines mt haplogroup T and was associated previously with increased risk of AMD.4,6 This pattern suggests heterogeneity of effects, where the inverse association of the minor alleles at TRPM1 SNPs is present only in individuals who lack the risk allele G at mt4917.
Although mtSNP A4917G in the IAMDGC dataset was not associated with (advanced) AMD risk, a finding supported by Tilleul et al.34 but one in contrast with previous reports of positive association,4,6 our results suggested a possible explanation for the mtSNP's nonsignificant main effects: varying degrees of interaction with alleles in the nuclear genome. As described in the stratified analyses on TRPM1 genotype (see Results), an association (positive) with AMD risk was present only in TRPM1 nSNP minor allele carriers. These results suggested that the interaction between mtSNP and nSNP genotype influences the detection of main effects at both loci and may explain the inconsistent results at mtSNP A4917G.
The patterns of association between variants in the ABHD2/RLBP1 region and advanced AMD were indicative of synergy between mtSNP and nSNP alleles. Associations with nSNPs in this region were stronger in the smaller subset carrying the 12771A allele ([minimum OR = 2.11] than the larger group carrying 12771G [minimum OR = 1.08]). The ORs for nSNPs in the mtSNP 12771A carrier group are greater than the OR for a combination of both carrier groups (reflected from main nSNP genetic effects where OR = 1.09). This suggests a potential synergistic interaction between the ABHD2/RLBP1 SNPs and mtSNP G12771A.
These two loci contain plausible candidate genes for AMD, based on their biological functions and prior association with other disorders of the visual system. The ion channel TRPM1 is expressed in the retina, specifically in depolarizing bipolar cells.35 The channel is a member of the metabotropic glutamate receptor 6 signaling pathway, a vision-related pathway in which interference with any signal transduction members may lead to blindness.35 Other studies have shown that TRPM1 regulates Ca2+ levels in melanocytes; molecular experiments demonstrate that disturbed TRPM1 expression incites a drop in intracellular Ca2+ concentration levels.36,37 Of factors common to regulatory mechanisms relevant to TRPM1 and RLBP1, expression of both genes is proposed to be controlled by microphthalmia-associated transcription factor (MITF), which may shape RPE and neural-crest derived melanocyte development, as supported by in vitro (ARPE-19 cell lines) and in vivo (mouse models) experiments.38−40
The consideration of gene × gene interaction in statistical models used by this study allowed the detection of two new loci for AMD. However, caution is needed in the interpretation of these results; statistical interactions are not necessarily indicative of physical, such as protein × protein, interactions in biological systems. As discussed earlier, the patterns of interaction detected may reflect genetic heterogeneity (in which effects of one locus are found in individuals lacking the risk allele at a second locus) or modest synergy (which may or may not imply biological interaction). The determination of the nature of such synergistic interactions depends on future functional genomic studies. Replication efforts should include examination of other racial and ethnic backgrounds, as the current study was limited to individuals of European ancestry.
This study demonstrated that considering interactions between the mitochondrial and nuclear genomes successfully identifies genetic risk factors not detected by GWAS studies that were focused on main effects of nuclear variation. The novel loci further the understanding of AMD pathophysiology and suggests potential synergy between mitochondrial and nuclear genetic variation in modulating risk of advanced AMD.
Supplementary Material
Acknowledgments
The authors thank the participants across all of the studies for their cooperation and time in allowing this research project to be accomplished. Resources were available from the computing centers of the University of Miami, University of Michigan, and University of Regensburg facilitated in the aforementioned analyses and data generation. For genotyping, the Center for Inherited Disease Research (CIDR) Program contract number is HHSN268201200008I.
Supported by 1X01HG006934-01 (GRA), R01 EYE022310 (JLH), EY012118 (MAP-V, WKS, JLH), and 5T32EY023194 (PJP), and by a National Health and Medical Research Council of Australia (NHMRC) Senior Research Fellowship 1028444 (PNB).
Disclosure: P.J. Persad, None; I.M. Heid, None; D.E. Weeks, P; P.N. Baird, None; E.K. de Jong, None; J.L. Haines, P; M.A. Pericak-Vance, P; W.K. Scott, P
Appendix
The following lists all International AMD Genomics Consortium (IAMDGC) members and their affiliated institutions:
Gonçalo R. Abecasis,1 Anita Agarwal,2 Jeeyun Ahn,3 Rando Allikmets,4,5 Isabelle Audo,6–9 Paul N. Baird,10 Elisa Bala,11 Mustapha Benchaboune,12 Hélène Blanché,13 John Blangero,14 Frédéric Blond,6–8 Alexis Boleda,15 Milam A. Brantley Jr.,2 Jennifer L. Bragg-Gresham,1,16 Caroline Brandl,17–19 Kari E. Branham,20 Murray H. Brilliant,21 Matthew Brooks,20 Alexander Brucker,22 Gabriëlle H. S. Buitendijk,23,24 Kathryn P. Burdon,25 Melinda S. Cain,10 Peter Campochiaro,26,27 Albert Caramoy,28 Daniel Chen,29 Emily Y. Chew,30 David Cho,31 Itay Chowers,32 Valentina Cipriani,33,34 Ian J. Constable,35 Jessica N. Cooke Bailey,36 Monique D. Courtenay,37 Jamie E. Craig,38 Angela J. Cree,39 Christine A. Curcio,40 Margaret DeAngelis,41 Eiko K. de Jong,42 Jean-François Deleuze,13,43 Anneke I. den Hollander,42,44 Bal Dhillon,45 Kimberly F. Doheny,46 Lebriz Ersoy,28 Lindsay A. Farrer,47–51 Sascha Fauser,28 Henry Ferreyra,29 Ken Flagg,29 Johanna R. Foerster,1 Lars G. Fritsche,1 Linn Gieser,15 Bamini Gopinath,52 Michael B. Gorin,53,54 Mathias Gorski,1,7 Srinivas V. Goverdhan,39 Felix Grassmann,18 Michelle Grunin,32 Robyn H. Guymer,10 Shira Hagbi-Levi,32 Stephanie A. Hagstrom,55 Jonathan L. Haines,36,56 Janette Hall,38 Michael A. Hauser,57–59 Caroline Hayward,60 Scott J. Hebbring,21 John R. Heckenlively,20 Iris M. Heid,17 Alex W. Hewitt,10,25,35 Joshua D. Hoffman,61 Frank G. Holz,62 Carel B. Hoyng,42 David J. Hunter,63,64 Wilmar Igl,17 Robert P. Igo Jr.,36 Timothy Isaacs,35 Sudha K. Iyengar,36 Yingda Jiang,65 Jie Jin Wang,52 Matthew P. Johnson,14 Nicholas Katsanis,66–68 Jane C. Khan,35,69,70 Ivana K. Kim,71 Terrie E. Kitchner,21 Caroline C. W. Klaver,23,24 Barbara E. K. Klein,72 Michael L. Klein,73 Ronald Klein,72 Jaclyn L. Kovach,74 Alan M. Kwong,1 Stewart Lake,38 Thomas Langmann,28 Reneé Laux,36 Yara T. E. Lechanteur,42 Kristine E. Lee,72 Thierry Léveillard,6–8 Mingyao Li,75 Helena Hai Liang,10 Gerald Liew,52 Danni Lin,29 Andrew J. Lotery,39 Hongrong Luo,29 David A. Mackey,10,25,35 Guanping Mao,29 Tammy M. Martin,73 Ian L. McAllister,35 J. Allie McGrath,61 Joanna E. Merriam,4 John C. Merriam,4 Stacy M. Meuer,72 Paul Mitchell,52 Saddek Mohand-Saïd,6–8,12 Anthony T. Moore,33,34,76 Emily L. Moore,72 Denise J. Morgan,41 Margaux A. Morrison,41 Chelsea E. Myers,72 Matthias Olden,17 Anton Orlin,77 Mohammad I. Othman,20 Hong Ouyang,29 Kyu Hyung Park,78 Neal S. Peachey,11,55 Margaret A. Pericak-Vance,37 Eric A. Postel,57 Rinki Ratnapriya,15 Christina A. Rennie,79 Andrea J. Richardson,10 Jane Romm,46 Guenther Rudolph,80 José-Alain Sahel,6–8,12,81–83 Nicole T. M. Saksens,42 Rebecca J. Sardell,37 Debra A. Schaumberg,63,84,85 Tina Schick,28 Hendrik P. N. Scholl,26,62 Matthew Schu,47–51 Stephen G. Schwartz,74 William K. Scott,37 Sebanti Sengupta,1 Humma Shahid,70,86 Giuliana Silvestri,87 R. Theodore Smith,4,88 Eric Souied,89 Emmanuelle Souzeau,38 Dwight Stambolian,31 Chloe M. Stanton,60 Klaus Stark,17 Zhiguang Su,90 Anand Swaroop,15 Ava G. Tan,52 Barbara Truitt,36 Evangelia E. Tsironi,91 Cornelia M. van Duijn,24 Claudia N. von Strachwitz,92 Brendan J. Vote,25 Katherina Walz,37 Bernhard H. F. Weber,18 Daniel E. Weeks,65,93 Cindy Wen,29 Armin Wolf,80 Zhenglin Yang,94,95 John R. W. Yates,33,34,70 Donald Zack,26,27,96–98 Xiaowei Zhan,1,99,100 Kang Zhang.29,90
1Center for Statistical Genetics, Department of Biostatistics, University of Michigan, Ann Arbor, MI, USA; 2Department of Ophthalmology and Visual Sciences, Vanderbilt University, Nashville, TN, USA; 3Department of Ophthalmology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul, Republic of Korea; 4Department of Ophthalmology Columbia University, New York, NY, USA; 5Department of Pathology & Cell Biology, Columbia University, New York, NY, USA; 6INSERM, U968, Paris, F-75012, France; 7UPMC Univ Paris 06, UMR_S 968, Institut de la Vision, Department of Genetics, Paris, F-75012, France; 8CNRS, UMR_7210, Paris, F-75012, France; 9Department of Molecular Genetics, Institute of Ophthalmology, London, UK; 10Centre for Eye Research Australia, Department of Surgery (Ophthalmology) University of Melbourne, Royal Victorian Eye and Ear Hospital, East Melbourne, Victoria, Australia; 11Louis Stokes Cleveland VA Medical Center, Cleveland, OH, USA; 12Centre Hospitalier National d'Ophtalmologie des Quinze-Vingts, INSERM-DHOS CIC 503, Paris, France; 13CEPH Fondation Jean Dausset, Paris, France; 14South Texas Diabetes and Obesity Institute, University of Texas Rio Grande Valley School of Medicine, Brownsville, TX, USA; 15Neurobiology Neurodegeneration & Repair Laboratory (N-NRL), National Eye Institute, National Institutes of Health, Bethesda, MD, USA; 16Kidney Epidemiology and Cost Center, Department of Biostatistics, Department of Internal Medicine - Nephrology, University of Michigan, Ann Arbor, MI, USA; 17Department of Genetic Epidemiology, University of Regensburg, Germany; 18Institute of Human Genetics, University of Regensburg, Germany; 19Department of Ophthalmology, University Hospital Regensburg, Regensburg, Germany; 20Department of Ophthalmology and Visual Sciences, University of Michigan, Kellogg Eye Center, Ann Arbor, MI, USA; 21Center for Human Genetics, Marshfield Clinic Research Foundation, Marshfield, WI, USA; 22Scheie Eye Institute, Department of Ophthalmology, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA; 23Department of Ophthalmology, Erasmus Medical Center, Rotterdam, The Netherlands; 24Department of Epidemiology, Erasmus Medical Center, Rotterdam, The Netherlands; 25School of Medicine, Menzies Research Institute Tasmania, University of Tasmania, Hobart, Tasmania, Australia; 26Department of Ophthalmology, Wilmer Eye Institute - Johns Hopkins University School of Medicine, Baltimore, MD, USA; 27Department of Neuroscience - Johns Hopkins University School of Medicine, Baltimore, MD, USA; 28University Hospital of Cologne, Department of Ophthalmology, Cologne, Germany; 29Department of Ophthalmology, University of California San Diego and VA San Diego Health System, La Jolla, CA, USA; 30Division of Epidemiology and Clinical Applications, Clinical Trials Branch, National Eye Institute, National Institutes of Health, Bethesda, MD, USA; 31Department of Ophthalmology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA; 32Department of Ophthalmology, Hadassah Hebrew University Medical Center, Jerusalem. Israel; 33UCL Institute of Ophthalmology, University College London, London, UK; 34Moorfields Eye Hospital, London, UK; 35Centre for Ophthalmology and Visual Science, Lions Eye Institute, University of Western Australia, Perth, Western Australia, Australia; 36Department of Population and Quantitative Health Sciences, Case Western Reserve University School of Medicine, Cleveland, OH, USA; 37John P. Hussman Institute for Human Genomics, Miller School of Medicine, University of Miami, Miami, FL, USA; 38Department of Ophthalmology, Flinders Medical Centre, Flinders University, Adelaide, South Australia, Australia; 39Clinical and Experimental Sciences, Faculty of Medicine, University of Southampton, UK; 40Department of Ophthalmology, The University of Alabama at Birmingham, Birmingham, AL, USA; 41Department of Ophthalmology and Visual Sciences, University of Utah, Salt Lake City, UT, USA 42Department of Ophthalmology, Donders Institute for Brain, Cognition, and Behaviour, Radboud University Medical Center, Nijmegen, The Netherlands; 43CEA – IG – Centre National de Génotypage, France; 44Department of Human Genetics, Radboud University Medical Centre, Nijmegen, The Netherlands; 45School of Clinical Sciences University of Edinburgh, Scotland, UK; 46Center for Inherited Disease Research (CIDR) Institute of Genetic Medicine Johns Hopkins University School of Medicine, Baltimore, MD, USA; 47Department of Medicine (Biomedical Genetics), Boston University Schools of Medicine and Public Health, Boston, MA, USA; 48Department of Ophthalmology, Boston University Schools of Medicine and Public Health, Boston, MA, USA; 49Department of Neurology, Boston University Schools of Medicine and Public Health, Boston, MA, USA; 50Department of Epidemiology, Boston University Schools of Medicine and Public Health, Boston, MA, USA; 51Department of Biostatistics, Boston University Schools of Medicine and Public Health, Boston, MA, USA; 52Centre for Vision Research, Department of Ophthalmology and Westmead Millennium Institute for Medical Research, University of Sydney, Sydney, Australia; 53Department of Ophthalmology, David Geffen School of Medicine—UCLA, Stein Eye Institute, Los Angeles, CA, USA; 54Department of Human Genetics, David Geffen School of Medicine—UCLA, Los Angeles, CA, USA; 55Cole Eye Institute, Cleveland Clinic, Cleveland OH, USA; 56Institute for Computational Biology, Case Western Reserve University School of Medicine, Cleveland, OH, USA; 57Department of Ophthalmology, Duke University Medical Center, Durham NC, USA; 58Department of Medicine, Duke University Medical Center, Durham NC, USA; 59Duke Molecular Physiology Institute, Duke University Medical Center, Durham NC, USA; 60MRC Human Genetics Unit, Institute of Genetics and Molecular Medicine, University of Edinburgh, Scotland, UK; 61Center for Human Genetics Research, Vanderbilt University Medical Center, Nashville, TN, USA; 62University of Bonn - Department of Ophthalmology – Bonn, Germany; 63Department of Epidemiology, Harvard School of Public Health, Boston, MA, USA; 64Department of Nutrition, Harvard School of Public Health, Boston, MA, USA; 65Department of Biostatistics, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA, USA; 66Center for Human Disease Modeling, Duke University, Durham, NC, USA; 67Department of Cell Biology, Duke University, Durham, NC, USA; 68Department of Pediatrics, Duke University, Durham, NC, USA; 69Department of Ophthalmology, Royal Perth Hospital, Perth, Western Australia, Australia; 70Department of Medical Genetics, Cambridge Institute for Medical Research, University of Cambridge, Cambridge, UK; 71Retina Service, Massachusetts Eye and Ear, Department of Ophthalmology Harvard Medical School, Boston, MA, USA; 72Department of Ophthalmology and Visual Sciences, University of Wisconsin, Madison, WI, USA; 73Casey Eye Institute, Oregon Health & Science University, Portland OR, USA; 74Bascom Palmer Eye Institute, University of Miami Miller School of Medicine, Naples, FL, USA; 75Department of Biostatistics and Epidemiology, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA; 76Department of Ophthalmology, University of California San Francisco Medical School, San Francisco, CA, USA; 77Department of Ophthalmology, Weill Cornell Medical College, New York, NY, USA; 78Department of Ophthalmology, Seoul National University Bundang Hospital, Seongnam, Republic of Korea; 79University Hospital Southampton, Southampton, UK; 80University Eye Clinic, Ludwig-Maximilians-University Munich, Germany; 81Fondation Ophtalmologique Adolphe de Rothschild, Paris, France; 82Institute of Ophthalmology, University College of London, London, UK; 83Académie des Sciences–Institut de France, Paris, France; 84Center for Translational Medicine, Moran Eye Center, University of Utah School of Medicine, Salt Lake City, UT, USA; 85Division of Preventive Medicine, Brigham & Women's Hospital, Harvard Medical School, Boston, MA, USA; 86Department of Ophthalmology, Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK; 87Centre for Experimental Medicine, Queen's University, Belfast, UK; 88Department of Ophthalmology, NYU School of Medicine, New York, NY, USA; 89Hôpital Intercommunal de Créteil, Hôpital Henri Mondor - Université Paris Est Créteil, France; 90Molecular Medicine Research Center, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Sichuan, China; 91Department of Ophthalmology, University of Thessaly, School of Medicine, Larissa, Greece; 92Eye Centre Southwest, Stuttgart, Germany; 93Department of Human Genetics, Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA, USA; 94Sichuan Provincial Key Laboratory for Human Disease Gene Study, Hospital of the University of Electronic Science and Technology of China and Sichuan Provincial People's Hospital, Chengdu, China; 95Sichuan Translational Medicine Hospital, Chinese Academy of Sciences, Chengdu, China; 96Department of Molecular Biology and Genetics, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 97Institute of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 98Institue de la Vision, Université Pierre et Marie Curie, Paris, France; 99Quantitative Biomedical Research Center, Department of Clinical Science, University of Texas Southwestern Medical Center, Dallas, TX, USA; 100Center for the Genetics of Host Defense, University of Texas Southwestern Medical Center, Dallas, TX, USA.
References
- 1. Jager RD, Mieler WF, Miller JW. . Age-related macular degeneration. N Engl J Med. 2008; 358: 2606– 2617. [DOI] [PubMed] [Google Scholar]
- 2. Fritsche LG, Igl W, Bailey JN,et al. . A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016; 48; 134– 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kenney MC, Chwa M, Atilano SR,et al. . Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3 and EFEMP1 genes: implications for age-related macular degeneration. PLoS One. 2013; 8: e54339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Canter JA, Olson LM, Spencer K,et al. . Mitochondrial DNA polymorphism A4917G is independently associated with age-related macular degeneration. PLoS One. 2008; 3: e2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Udar N, Atilano SR, Memarzadeh M,et al. . Mitochondrial DNA haplogroups associated with age-related macular degeneration. Invest Ophthalmol Vis Sci. 2009; 50: 2966– 2974. [DOI] [PubMed] [Google Scholar]
- 6. SanGiovanni JP, Arking DE, Iyengar SK,et al. . Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS One. 2009; 4: e5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mueller EE, Schaier E, Brunner SM,et al. . Mitochondrial haplogroups and control region polymorphisms in age-related macular degeneration: a case-control study. PLoS One. 2012; 7: e30874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miceli MV, Jazwinski SM. . Nuclear gene expression changes due to mitochondrial dysfunction in ARPE-19 cells: implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2005; 46: 1765– 1773. [DOI] [PubMed] [Google Scholar]
- 9. Fritsche LG, Chen W, Schu M,et al. . Seven new loci associated with age-related macular degeneration. Nat Genet. 2013; 45: 433– 439.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Purcell S, Neale B, Todd-Brown K,et al. . PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81: 559– 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kraft P, Yen YC, Stram DO, Morrison J, Gauderman WJ. . Exploiting gene-environment interaction to detect genetic associations. Hum Hered. 2007; 63: 111– 119. [DOI] [PubMed] [Google Scholar]
- 12. R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. Available at: http://www.R-project.org/. [Google Scholar]
- 13. Chongsuvivatwong V. . Analysis of epidemiological data using R and Epicalc. Thailand: Epidemiology Unit, Prince of Songkla University; 2008. [Google Scholar]
- 14. Imboden M, Kumar A, Curjuric I,et al. . Modification of the association between PM10 and lung function decline by cadherin 13 polymorphisms in the SAPALDIA cohort: a genome-wide interaction analysis. Environ Health Perspect. 2015; 123: 72– 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stranger BE, Stahl EA, Raj T. . Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011; 187: 367– 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gauderman WJ, Morrison JM. QUANTO. 1.2.4: A computer program for power and sample size calculations for genetic-epidemiology studies. Available at http://biostats.usc.edu/software on August 28, 2016. University of Southern California (USC), Los Angeles, California; 2009.
- 17. Kent WJ, Sugnet CW, Furey TS,et al. . The human genome browser at UCSC. Genome Res. 2002; 12: 996– 1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenbloom KR, Sloan CA, Malladi VS,et al. . ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013; 41: D56– D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ward LD, Kellis M. . HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012; 40: D930– D934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Genderen MM, Bijveld MM, Claassen YB,et al. . Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am J Hum Genet. 2009; 85: 730– 736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Z, Sergouniotis PI, Michaelides M,et al. . Recessive mutations of the gene TRPM1 abrogate ON bipolar cell function and cause complete congenital stationary night blindness in humans. Am J Hum Genet. 2009; 85: 711– 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Audo I, Kohl S, Leroy BP,et al. . TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2009; 85: 720– 729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oancea E, Vriens J, Brauchi S, Jun J, Splawski I, Clapham DE. . TRPM1 forms ion channels associated with melanin content in melanocytes. Sci Signal. 2009;2:ra21. [DOI] [PMC free article] [PubMed]
- 24. Ying GS, Maguire MG, Liu C, Antoszyk AN. . Complications of Age-related Macular Degeneration Prevention Trial Research Group. Night vision symptoms and progression of age-related macular degeneration in the Complications of Age-related Macular Degeneration Prevention Trial. Ophthalmology. 2008; 115: 1876– 1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miyata K, Nakayama M, Mizuta S,et al. . Elevated mature macrophage expression of human ABHD2 gene in vulnerable plaque. Biochem Biophys Res Commun. 2008; 365: 207– 213. [DOI] [PubMed] [Google Scholar]
- 26. Dessalces E, Bocquet B, Bourien J,et al. . Early-onset foveal involvement in retinitis punctata albescens with mutations in RLBP1. JAMA Ophthalmol. 2013; 131: 1314– 1323. [DOI] [PubMed] [Google Scholar]
- 27. Hipp S, Zobor G, Glockle N,et al. . Phenotype variations of retinal dystrophies caused by mutations in the RLBP1 gene. Acta Ophthalmol. 2015; 93: e281– e286. [DOI] [PubMed] [Google Scholar]
- 28. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. OMIM #136880, Retinitis punctata albescens. 12/07/2010.
- 29. Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. OMIM: #60747, Newfoundland rod-cone dystrophy; 06/06/ 2016. [Google Scholar]
- 30. Eichers ER, Green JS, Stockton DW,et al. . Newfoundland rod-cone dystrophy, an early-onset retinal dystrophy, is caused by splice-junction mutations in RLBP1. Am J Hum Genet. 2002; 70: 955– 964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Terluk MR, Kapphahn RJ, Soukup LM,et al. . Investigating mitochondria as a target for treating age-related macular degeneration. J Neurosci. 2015; 35: 7304– 7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ohia SE, Opere CA, Leday AM. . Pharmacological consequences of oxidative stress in ocular tissues. Mutat Res. 2005; 579: 22– 36. [DOI] [PubMed] [Google Scholar]
- 33. Jacobson J, Duchen MR. . Interplay between mitochondria and cellular calcium signalling. Mol Cell Biochem. 2004; 256–257: 209– 218. [DOI] [PubMed] [Google Scholar]
- 34. Tilleul J, Richard F, Puche N,et al. . Genetic association study of mitochondrial polymorphisms in neovascular age-related macular degeneration. Mol Vis. 2013; 19: 1132– 1140. [PMC free article] [PubMed] [Google Scholar]
- 35. Morgans CW, Brown RL, Duvoisin RM. . TRPM1: the endpoint of the mGluR6 signal transduction cascade in retinal ON-bipolar cells. Bioessays. 2010; 32: 609– 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Devi S, Kedlaya R, Maddodi N,et al. . Calcium homeostasis in human melanocytes: role of transient receptor potential melastatin 1 (TRPM1) and its regulation by ultraviolet light. Am J Physiol Cell Physiol. 2009; 297: C679– C687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guo H, Carlson JA, Slominski A. . Role of TRPM in melanocytes and melanoma. Exp Dermatol. 2012; 21: 650– 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wen B, Li S, Li H,et al. . Microphthalmia-associated transcription factor regulates the visual cycle genes Rlbp1 and Rdh5 in the retinal pigment epithelium. Sci Rep. 2016; 6: 21208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhiqi S, Soltani MH, Bhat KM,et al. . Human melastatin 1 (TRPM1) is regulated by MITF and produces multiple polypeptide isoforms in melanocytes and melanoma. Melanoma Res. 2004; 14: 509– 516. [DOI] [PubMed] [Google Scholar]
- 40. Miller AJ, Du J, Rowan S, Hershey CL, Widlund HR, Fisher DE. . Transcriptional regulation of the melanoma prognostic marker melastatin (TRPM1) by MITF in melanocytes and melanoma. Cancer Res. 2004; 64: 509– 516. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.