

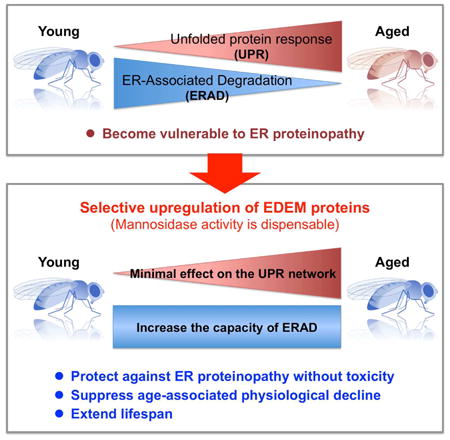
Summary
The unfolded protein response (UPR), which protects cells against accumulation of misfolded proteins in the endoplasmic reticulum (ER), is induced in several age-associated degenerative diseases. However, sustained UPR activation has negative effects on cellular functions and may worsen disease symptoms. It remains unknown whether and how UPR components can be utilized to counteract chronic ER proteinopathies. We found that promotion of ER-associated degradation (ERAD) through upregulation of ERAD-enhancing α-mannosidase-like proteins (EDEMs) protected against chronic ER proteinopathy without inducing toxicity in a Drosophila model. ERAD activity in the brain decreased with aging, and upregulation of EDEMs suppressed age-dependent behavioral decline and extended lifespan without affecting the UPR gene expression network. Intriguingly, EDEM mannosidase activity was dispensable for these protective effects. Therefore, upregulation of EDEM function in the ERAD protects against ER proteinopathy in vivo, and thus represents a potential therapeutic target for chronic diseases.
Keywords: endoplasmic reticulum (ER) stress, unfolded protein response (UPR), ER-associated degradation (ERAD), protein misfolding, neurodegeneration, aging, Drosophila
Graphical abstract
Introduction
Nearly one-third of newly synthesized proteins are secreted or membrane proteins initially targeted to the endoplasmic reticulum (ER) (Ghaemmaghami et al., 2003). In the ER, stringent quality-control systems ensure that only correctly folded proteins are sent to their final destinations. Destabilizing mutations, stress conditions, or metabolic challenges increase the risk of protein misfolding (Hartl and Hayer-Hartl, 2009), and terminally misfolded proteins must be eliminated from the ER. Accumulation of misfolded ER proteins is harmful to cells and causes several degenerative diseases (Guerriero and Brodsky, 2012).
The unfolded protein response (UPR) evolved as a defense mechanism against accumulation of defective proteins in the ER (Guerriero and Brodsky, 2012). Upon protein overload in the ER, UPR signaling via the ATF6, PERK, and IRE1/XBP1 pathways is activated to repress translation, increase ER capacity, and remove excess and misfolded proteins via ER-associated degradation (ERAD) (Kimata and Kohno, 2011). Among the branches of the UPR, the IRE1/XBP1 pathway is the most highly conserved, and plays a major role in initiation of ERAD by inducing ERAD-related genes (Mori, 2009).
ERAD is regulated by a multiprotein complex containing proteins involved in the recognition, delivery, retrotranslocation, and ubiquitination of misfolded ER proteins and their subsequent proteosomal degradation (Araki and Nagata, 2011). Several ER chaperones and α-mannosidase-like proteins participate in recognition of misfolded polypeptides and their delivery to the dislocon machinery (Bernasconi and Molinari, 2011). The HRD1 dislocon complex, comprising the HRD1 E3 ubiquitin ligase, SEL1L, DER1, and several other accessory proteins, is responsible for retrotranslocation and ubiquitination of misfolded proteins and their subsequent delivery to proteasomes for degradation (Bernasconi and Molinari, 2011).
The UPR is activated in several chronic degenerative diseases, presumably to protect against ER stress occurring during pathogenesis (Guerriero and Brodsky, 2012; Kim et al., 2008; Wang and Kaufman, 2012). Modest UPR induction over the short term is beneficial in cellular and animal models of neurodegenerative diseases (Sado et al., 2009; Valenzuela et al., 2012). However, sustained activation of the UPR can induce apoptosis (Kim et al., 2008; Lin et al., 2007; Marciniak et al., 2004; Tabas and Ron, 2011). Moreover, overactivation of the IRE1/XBP1 pathway causes uncontrolled and inappropriate degradation of mRNAs and proteins localized to the ER (Denic et al., 2006; Hollien and Weissman, 2006), negatively affecting cellular functions (Allagnat et al., 2010; Wang and Kaufman, 2012). Thus, the canonical UPR may not be prepared to protect cells against sustained ER stress, and it remains unclear whether and how UPR components can be utilized to prevent or treat diseases caused by chronic ER stress (Walter and Ron, 2011).
Using a transgenic Drosophila model, we searched for genes that could counteract chronic ER proteinopathy in vivo. We found that selective upregulation of Drosophila orthologs of ERAD-enhancing α-mannosidase-like proteins (EDEMs) (Hosokawa et al., 2001), a family of mannosidases that create a glycan-based quality-control signal for ERAD (Ninagawa et al., 2015) and also participate in degradation of non-glycoprotein substrates (Shenkman et al., 2013), promoted ERAD without inducing toxicity, and also protected against neurodegeneration caused by chronic ER proteinopathy in brain. ERAD activity in fly brains decreased with aging, but upregulation of Drosophila EDEMs (dEDEMs) suppressed age-associated behavioral decline without altering the UPR gene expression network. Notably, EDEM mannosidase activity was dispensable for these protective effects. These results suggest that selective upregulation of EDEM function in the ERAD is sufficient to increase cellular resistance to ER stress. Thus, EDEM proteins are candidate therapeutic targets for age-associated diseases caused by chronic ER proteinopathy.
Results
A Drosophila model of chronic ER proteinopathy in the brain
As a Drosophila model of chronic ER proteinopathy, we utilized transgenic Drosophila expressing aggregation-prone, neurotoxic, amyloid-β42 peptides (Aβ42) in the ER of central nervous system neurons (Iijima et al., 2008; Iijima et al., 2004). To direct Aβ42 expression in the ER, human Aβ42 peptides were directly fused to the signal sequence of the rat preproenkephalin peptide, and neuron-specific expression of Aβ42 was achieved using the GAL4/UAS system with the pan-neuronal elav-GAL4 driver (Brand and Perrimon, 1993). Western blot and immunoprecipitation/mass spectrometry analyses showed that Aβ42 peptides are expressed from this construct in fly neurons (Figure 1A) (Iijima et al., 2004). In addition, immunoelectron microscopy (immuno-EM) confirmed that the expressed Aβ42 peptides were localized to the ER (Figure 1B) (Iijima et al., 2008).
Figure 1. Chronic activation of the Ire1/Xbp1 pathway reduces Aβ42 levels, but causes age-dependent behavioral deficits, in a Drosophila model of chronic ER proteinopathy.
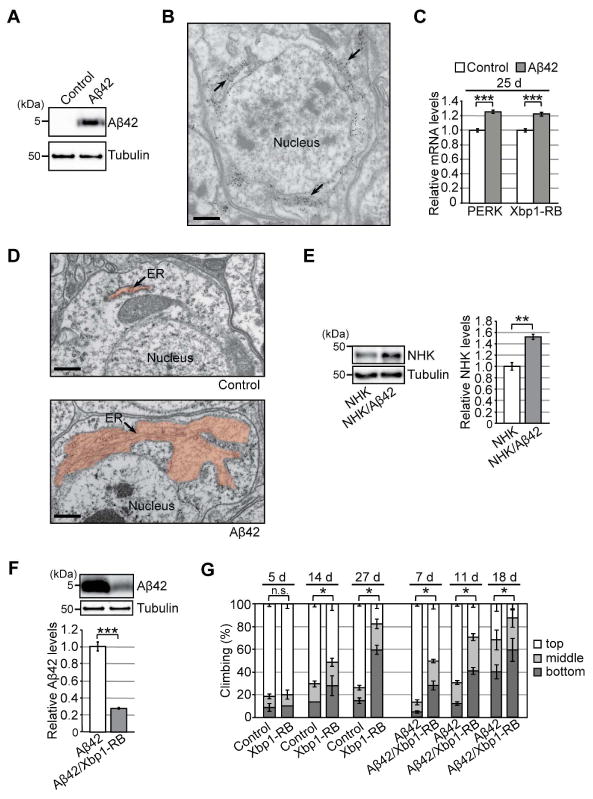
(A) Expression of Aβ42 peptides in fly brains. Heads from flies carrying the elav-GAL4 driver alone (Control) or the elav-GAL4 driver and UAS-Aβ42 (Aβ42) were subjected to western blotting using an anti-Aβ antibody. Tubulin was used as the loading control. (B) Immuno-EM analysis of Aβ42 distribution in fly brain neurons. Gold particles were detected in the ER. Scale bar: 0.5 μm. (C) mRNA levels of PERK and Xbp1-RB were increased in Aβ42 fly brains, as determined by qRT-PCR. Mean ± SEM, n = 4, ***p < 0.001 by Student's t-test. (D) Abnormal enlargement of the ER was observed in neurons expressing Aβ42 (indicated by arrow). Scale bars: 0.5 μm. (E) Levels of exogenously expressed null Hong Kong α1-antitrypsin (NHK) protein were elevated in Aβ42 fly brains. Mean ± SEM, n = 3–4, **p < 0.01 by Student's t-test. (F) Overexpression of Xbp1-RB reduced Aβ42 levels in fly brains. Mean ± SEM, n = 4, ***p < 0.001 by Student's t-test. (G) Overexpression of Xbp1-RB in neurons caused age-dependent locomotor deficits and worsened behavioral deficits in Aβ42 flies. Average percentages of flies that climbed to the top (white) or middle (light gray), or stayed at the bottom (dark gray), of the vials. Ages (d, days after eclosion) are indicated on the top of the graph. Percentages of flies that stayed at the bottom were subjected to statistical analyses. Mean ± SD, n = 5, *p < 0.05 by Mann–Whitney U test. n.s.: not significant. See also Figure S1 and Table S5.
Expression of Aβ42 in the ER at very high levels induced chronic ER stress. Of the three branches of the mammalian UPR, the PERK and IRE1/XBP1 pathways are well conserved in Drosophila (Mori, 2009). In fly, the Ire1-dependent splicing of Xbp1 mRNA generates an active Xbp1 isoform, Xbp1-RB. The mRNA levels of Xbp1-RB and PERK were significantly upregulated in Aβ42 fly brains (Figure 1C), indicating activation of canonical UPR pathways. In addition, EM revealed abnormal enlargement of the ER in Aβ42 fly brain neurons (Figure 1D). Moreover, the level of exogenously expressed null Hong Kong α1-antitrypsin (NHK), a well-known ERAD substrate, was significantly elevated in Aβ42 fly brains (Figure 1E) with no corresponding increase in mRNA level (Figure S1), suggesting that the capacity and efficiency of ERAD were compromised in Aβ42 fly brains. Importantly, these Aβ42 flies developed age-dependent sequential progression of memory loss, locomotor defects, and neurodegeneration (Iijima et al., 2008; Iijima et al., 2004). These results suggest that our Aβ42 flies can be used as a genetic model of chronic ER proteinopathy in vivo.
Chronic activation of the IRE1/Xbp1 pathway by overexpression of Xbp1-RB reduces the Aβ42 level in the ER but causes age-dependent behavioral deficits
Short-term modest activation of IRE1/XBP1 exerts beneficial effects in several neurodegenerative disease models (Sado et al., 2009; Valenzuela et al., 2012). Because this pathway was activated in our model (Figure 1C), we examined the effects of its upregulation on the Aβ42 level in the ER. As expected from its role in induction of ERAD, activation of Ire1/Xbp1 signaling by neuronal overexpression of Xbp1-RB significantly reduced Aβ42 levels in brain (Figure 1F).
However, long-term activation of the Ire1/Xbp1 pathway in neurons was detrimental. Chronic overexpression of Xbp1-RB caused age-dependent locomotor defects (Figure 1G, Control vs. Xbp1-RB) and worsened behavioral deficits in Aβ42 flies (Figure 1G, Aβ42 vs. Aβ42/Xbp1-RB), suggesting that chronic activation of the canonical Ire1/Xbp1 pathway in fly brain neurons reduces levels of misfolding-prone protein in the ER, but has other consequences that are deleterious.
Neuronal knockdown of Sin3A repressor complex suppresses chronic ER proteinopathy in Aβ42 fly brains
To identify a way to protect against chronic ER proteinopathy in vivo, we performed a genetic modifier screen in Aβ42 flies to identify genes that reduce both the levels of Aβ42 in the ER and the associated toxicity. Using age-dependent locomotor defects and premature death as readouts, we performed a small-scale genetic modifier screen including 113 genes selected from various biological processes (Table S1). The screen revealed that loss of one copy (i.e., heterozygous mutation) of Sin3A (Sin3ALOF), a component of an evolutionarily conserved transcriptional co-repressor complex (Grzenda et al., 2009), suppressed Aβ42-induced locomotor defects (Figure 2A, Aβ42 vs. Aβ42/Sin3ALOF). In the absence of Aβ42 expression, Sin3ALOF did not affect locomotor function (Figure 2A, Control vs. Sin3ALOF).
Figure 2. Neuronal knockdown of Sin3A protects against chronic ER proteinopathy in Aβ42 fly brains.
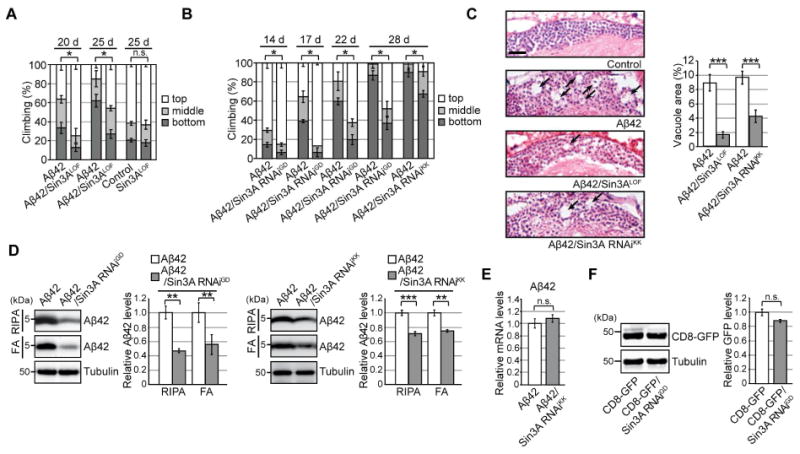
(A–B) Heterozygous loss-of-function mutation (Sin3ALOF) or RNAi-mediated knockdown (Sin3A RNAiGD and Sin3A RNAiKK) of Sin3A suppressed Aβ42-induced locomotor defects. Mean ± SD, n = 5, *p < 0.05 by Mann–Whitney U test. n.s.: not significant. (C) Sin3ALOF or Sin3A RNAi suppressed Aβ42-induced neurodegeneration. Percentages of vacuole areas in fly brain cortices are shown (indicated by arrows). Scale bar: 10 μm. Mean ± SEM, n = 7–12 hemispheres, ***p < 0.001 by Student's t-test. (D) RNAi-mediated knockdown of Sin3A in neurons reduced Aβ42 levels in fly brains. Detergent-soluble (RIPA) and -insoluble (FA) fractions from heads were analyzed by western blotting with anti-Aβ antibody. Mean ± SEM, n = 3–4. **p < 0.01 and ***p < 0.001 by Student's t-test. (E) Neuronal knockdown of Sin3A did not alter Aβ42 mRNA levels in fly brains, as determined by qRT-PCR. Mean ± SEM, n = 3–4, n.s.: not significant (Student's t-test). (F) Neuronal knockdown of Sin3A did not reduce levels of CD8-GFP protein. Mean ± SEM, n = 3, n.s.: not significant (Student's t-test). See also Figure S2 and Table S5.
To determine whether Sin3A knockdown in neurons protects against Aβ42 toxicity, we used two independent transgenic lines expressing RNAi constructs targeting different regions of Sin3A (Sin3A RNAiGD and Sin3A RNAiKK). Both RNAi transgenes significantly reduced Sin3A mRNA levels in neurons (Figure S2A) and suppressed Aβ42-induced locomotor defects (Figure 2B, Aβ42 vs. Aβ42/Sin3A RNAi). In the absence of Aβ42, neuronal knockdown of Sin3A did not improve locomotor functions (Figure S2B and C, Control vs. Sin3A RNAi). Neuronal expression of a control RNAi targeting firefly luciferase (Luc RNAi) did not suppress locomotor defects in Aβ42 flies, indicating a specific effect of Sin3A knockdown (Figure S2D).
Brain vacuolation, often observed following neurodegeneration in Drosophila, is a morphological hallmark of neuronal deterioration (Kretzschmar et al., 1997). For example, expression of Aβ42 in the ER causes late-onset, progressive neurodegeneration resulting in the appearance of vacuoles in the brain (Iijima et al., 2008; Iijima et al., 2004). Loss of one copy of Sin3A or neuronal knockdown of Sin3A significantly suppressed Aβ42-induced neurodegeneration (Figure 2C, Aβ42 vs. Aβ42/Sin3ALOF or Aβ42/Sin3A RNAi). By contrast, neuronal expression of control RNAi (Luc RNAi) did not affect Aβ42-induced neurodegeneration (Figure S2E).
Previous studies using Drosophila models of Huntington's disease and Parkinson's disease show that mutant forms of huntingtin or α-synuclein translocate to the nucleus and cause significant reductions in global histone acetylation levels. In these models, Sin3A loss of function confers protective effects by restoring histone acetylation levels to normal levels (Kontopoulos et al., 2006; Steffan et al., 2001). By contrast, in our model, Aβ42 did not translocate to the nucleus (Figure 1B) or reduce global histone acetylation levels (Figure S2F), suggesting that the protective mechanisms of Sin3A knockdown in Aβ42 flies differ from those in Huntington's and Parkinson's disease models.
Aβ42 levels were reduced in Sin3A-knockdown fly brains (Figure 2D and S2G). Aβ42 peptides can be sequentially extracted from brains in detergent-soluble fractions (RIPA) and detergent-insoluble/70% formic acid (FA) soluble fractions, with most aggregated forms of Aβ42 accumulating in the FA fractions. Loss of one copy of Sin3A, or RNAi-mediated knockdown of Sin3A, significantly reduced Aβ42 levels in both fractions (Figure 2D and S2G). Similar effects were observed in eyes when the pan-retinal GMR-GAL4 driver was used to express Aβ42 and Sin3A RNAi (Figure S2H); by contrast, control RNAi did not affect Aβ42 levels (Figure S2I). Neuronal knockdown of Sin3A did not reduce Aβ42 mRNA levels (Figure 2E), suggesting that lower levels of Aβ42 in the Sin3A-knockdown background were not due to reduction in transgene transcription. Moreover, neuronal knockdown of Sin3A did not affect protein levels of CD8-GFP, a control membrane protein (Figure 2F), suggesting that Sin3A knockdown does not disrupt translation or cause global reductions in the levels of exogenous proteins in the ER.
Neuronal Sin3A knockdown increases expression of Drosophila EDEMs (dEDEMs), and overexpression of dEDEMs reduces Aβ42 levels in brain
Because Sin3A functions as an organizer of a histone deacetylase complex that epigenetically represses specific sets of genes, neuronal knockdown of Sin3A may induce expression of genes whose protein products decrease Aβ42 levels and associated toxicity in fly brains. To test this idea, we selected candidate genes involved in ER quality control (Araki and Nagata, 2011), as well as fly homologs of the mammalian Aβ-degrading enzymes (summarized in Table S2). We then systematically screened for genes that fit three criteria: first, mRNA levels should be increased by neuronal knockdown of Sin3A; second, overexpression of the protein should reduce Aβ42 levels in fly brains; and third, overexpression of the protein should suppress age-dependent behavioral deficits and neurodegeneration in Aβ42 flies. This approach identified two Drosophila homologs of ERAD-enhancing α-mannosidase-like proteins (dEDEMs), dEDEM1 and dEDEM2, the only Drosophila homologs of human EDEM proteins (Kang and Ryoo, 2009) (Figure 3, and also see Figure S3A–D for a subset of screening results).
Figure 3. Neuronal knockdown of Sin3A increases mRNA levels of Drosophila EDEMs (dEDEMs), and upregulation of these factors is sufficient to reduce Aβ42 levels in brain.
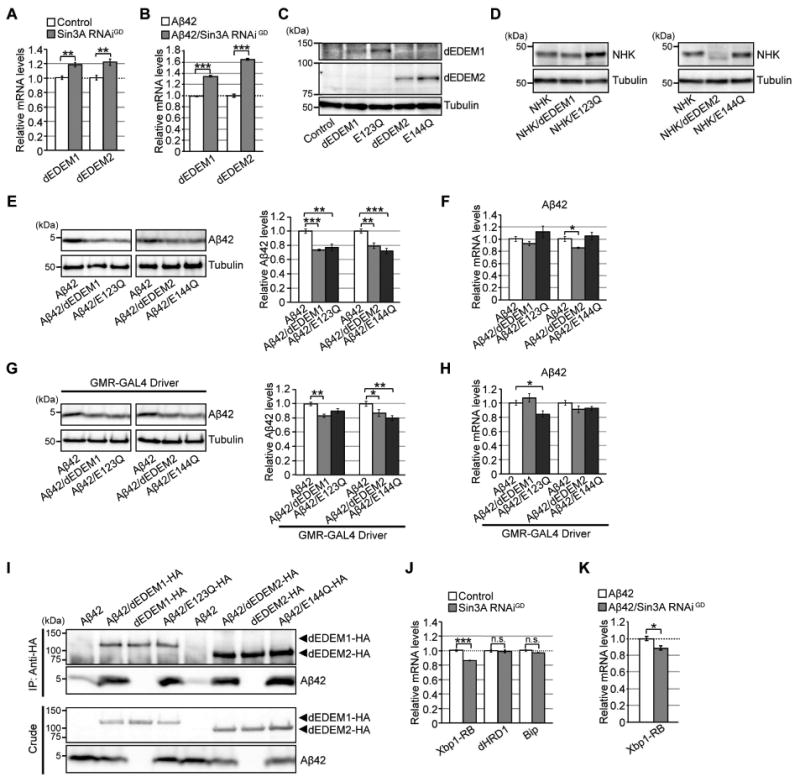
(A–B) Neuronal knockdown of Sin3A increased mRNA levels of dEDEM1 or dEDEM2 in both Control and Aβ42 fly brains. Mean ± SEM, n = 4, **p < 0.01 and ***p < 0.001 by Student's t-test. (C) Expression of dEDEM proteins in transgenic fly brains. Heads of flies expressing dEDEMs were analyzed by western blotting with anti-HA antibody. (D) Mannosidase activities of dEDEMs were critical for degradation of misfolded glycoprotein NHK in fly brains. Heads of flies expressing NHK alone or co-expressing NHK and dEDEMs were analyzed by western blotting. (E, G) Overexpression of dEDEMs reduced Aβ42 levels in neurons (E) and eyes (G). Heads of flies expressing Aβ42 alone or co-expressing Aβ42 and dEDEMs were analyzed by western blotting with anti-Aβ antibody. Mean ± SEM, n = 3–4, *p < 0.05, **p < 0.01, and ***p < 0.001 by Student's t-test. (F, H) Overexpression of dEDEMs had minimal effects on Aβ42 mRNA levels in neurons (F) and eyes (H), as determined by qRT-PCR. Mean ± SEM, n = 4, *p < 0.05 by Student's t-test. (I) dEDEMs co-immunoprecipitated with Aβ42 peptides. Drosophila S2 cells were transiently transfected with HA epitope-tagged dEDEMs-HA, Aβ42 and dEDEMs-HA, or Aβ42 alone. Cell lysates were subjected to immunoprecipitation with anti-HA antibody, followed by western blotting with anti-Aβ or anti-HA antibody. Top two panels show immunoprecipitates, and bottom two panels show crude lysates. (J–K) Neuronal knockdown of Sin3A did not increase mRNA levels of Xbp1-RB, dHRD1, or BiP in control or Aβ42 fly brains, as determined by qRT-PCR. Mean ± SEM, n = 4, *p < 0.05 and ***p < 0.001. n.s.: not significant (Student's t-test). See also Figure S3 and Tables S2 and S5.
Quantitative RT-PCR (qRT-PCR) analyses revealed that neuronal knockdown of Sin3A significantly increased dEDEM1 and dEDEM2 mRNA levels in both control and Aβ42 brains (Figure 3A and B). According to DIOPT (DRSC Integrative Ortholog Prediction Tool), human EDEM2 is the best ortholog of dEDEM1 (DIOPT score 9, 55% identity and 70% similarity in primary amino acid sequence), whereas human EDEM3 is the best ortholog of dEDEM2 (DIOPT score 9, 50% identity and 69% similarity). Human EDEM1 exhibits moderate similarity to both dEDEM1 (DIOPT score 3, 43% identity and 58% similarity) and dEDEM2 (DIOPT score 3, 44% identity and 61% similarity). Mammalian EDEM proteins belong to the glycosylhydrolase 47 family (Class I α-mannosidases) including ER α1,2-mannosidase I and Golgi α1,2-mannosidases, and this mannosidase homology domain is conserved in dEDEM1 and dEDEM2.
To determine whether overexpression of dEDEMs can reduce Aβ42 levels and associated toxicity in Aβ42 flies, we established new transgenic fly lines that express C-terminally HA-tagged dEDEM1 and dEDEM2 with their intact N-terminal signal sequences (for details, see STAR Methods). To further examine the role of dEDEM mannosidase activity, we also established two lines expressing catalytically inactive versions of dEDEM1 (E123Q) and dEDEM2 (E144Q), which harbor mutations within the EF hand motif essential for α1,2-mannosidase activity (Ninagawa et al., 2014).
Western blotting confirmed that all four transgenic proteins (dEDEM1, E123Q, dEDEM2, and E144Q) were detected at the expected sizes in fly heads (Figure 3C). We then characterized their effects on degradation of NHK, whose degradation involves mannose trimming by mammalian EDEMs. Overexpression of wild-type dEDEM2 significantly reduced steady-state levels of NHK proteins, whereas the effect of dEDEM1 was modest (Figure 3D). By contrast, overexpression of both E123Q and E144Q significantly increased steady-state levels of NHK proteins (Figure 3D). Thus, dEDEM mannosidase activity is involved in NHK degradation in fly neurons, and dEDEM2 overexpression is sufficient for degradation of NHK.
Overexpression of either dEDEM1 or dEDEM2 in neurons reduced Aβ42 levels in brain (Figure 3E), with minimal effects on Aβ42 mRNA levels (Figure 3F), suggesting that dEDEM1- or dEDEM2-mediated reductions in Aβ42 levels were not due to reductions in transcription. Moreover, overexpression of E123Q and E144Q also significantly reduced Aβ42 levels, confirming that dEDEM mannosidase activity is not required for degradation of Aβ42, a non-glycoprotein substrate (Figure 3E). Similar effects were observed in the eye when the pan-retinal GMR-GAL4 driver was used to express Aβ42 and dEDEM (Figure 3G and H). Overexpression of dEDEM1 or dEDEM2 did not reduce the levels of the cytoplasmic microtubule-associated protein tau (Figure S3E), suggesting that the effects of dEDEM1 or dEDEM2 are specific to misfolding-prone ER proteins.
In mammals, EDEMs can bind to non-glycoprotein substrates (Shenkman et al., 2013). Co-immunoprecipitation assays with Aβ42 and dEDEM1, E123Q, dEDEM2, or E144Q in Drosophila S2 cells revealed that all four proteins formed a complex with Aβ42 peptides expressed in the ER (Figure 3I). These results suggest that the overexpressed dEDEMs serve a chaperone-like function, i.e., they formed a complex with Aβ42, independent of their mannosidase activity, to promote its degradation.
Expression of many ERAD components is induced by activation of the IRE1/XBP1 pathway (Mori, 2009). However, neuronal knockdown of Sin3A decreased mRNA levels of Xbp-1RB in both control and Aβ42 fly brains (Figure 3J and K). In addition, mRNA levels of other ERAD components downstream of the Ire1/Xbp1 pathway, dHRD1 and the ER chaperone BiP, were not significantly elevated in Sin3A-knockdown flies (Figure 3J), suggesting that Sin3A knockdown increases EDEM levels without activating the canonical UPR. To determine whether Sin3A directly regulates EDEM expression in flies, we used genome-wide ChIP-Seq data regarding Sin3A-binding sites from the modEncode database (http://www.modencode.org/) (Celniker et al., 2009). This analysis revealed that Sin3A directly binds the dEDEM2 promoter region, suggesting that it can suppress expression of dEDEM2.
Chronic upregulation of EDEMs in neurons suppresses behavioral deficits and neurodegeneration in Aβ42 flies
Unlike activation of the Ire1/Xbp1 pathway (Figure 1G), upregulation of ERAD through neuronal overexpression of dEDEM1 or dEDEM2 suppressed Aβ42-induced locomotor defects (Figure 4A, Aβ42 vs. Aβ42/dEDEMs) and neurodegeneration (Figure 4B, Aβ42 vs. Aβ42/dEDEMs). Differences in the fold increases in the mRNA levels of Xbp1-RB and the dEDEMs were not responsible for the phenotypes, since the levels of overexpression of the mRNAs of these transgenes were comparable (Figure S4A). Similar protection was observed upon neuronal overexpression of E123Q or E144Q (Figure 4A and B, Aβ42 vs. Aβ42/E123Q or E144Q), suggesting that dEDEM mannosidase activity is dispensable for these effects. We also examined the protective effects of dEDEMs on neurodegeneration using the rough eye phenotype caused by expression of Aβ42 under the control of the pan-retinal GMR-GAL4 driver. Co-expression with dEDEM2, and to a lesser extent with dEDEM1, suppressed this phenotype (Figure 4C, Aβ42 vs. Aβ42/dEDEM2 or dEDEM1). Similar rescue effects were observed in flies expressing catalytically inactive versions of dEDEMs (Figure 4C, Aβ42 vs. Aβ42/E144Q or E123Q), but not in flies co-expressing a control protein, CD8-GFP (Figure 4C, Aβ42 vs. Aβ42/CD8-GFP). The rough eye phenotypes induced by expression of human tau, a cytoplasmic aggregation-prone protein, were not suppressed by co-expression of dEDEMs (Figure S4B), indicating that the protective effects of dEDEMs in Aβ42-expressing flies were specific to neurodegeneration caused by ER proteinopathy.
Figure 4. Overexpression of dEDEMs protects against chronic ER proteinopathy in Aβ42 flies.
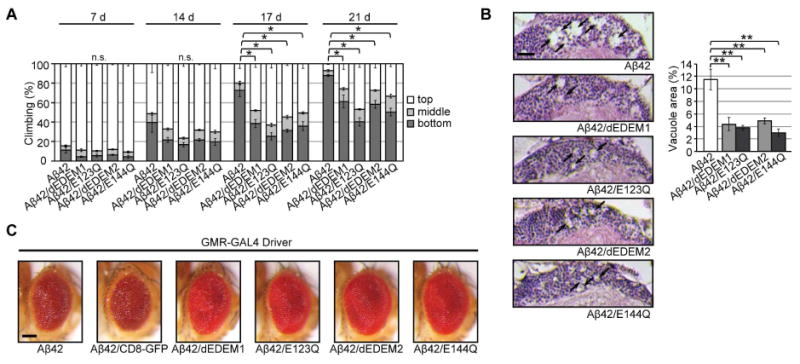
(A) Neuronal overexpression of dEDEMs suppressed locomotor defects in Aβ42 flies. Mean ± SEM, n = 6, *p < 0.05 by Mann–Whitney U test. (B) Neuronal overexpression of dEDEMs suppressed neurodegeneration in Aβ42 flies. Percentages of vacuole areas (indicated by arrows) in fly cortices are shown. Scale bar: 10 μm. Mean ± SEM, n = 7–12 hemispheres, **p < 0.01 by Student's t-test. (C) dEDEM overexpression suppressed retinal degeneration induced by Aβ42. Eyes of flies expressing Aβ42 alone, co-expressing Aβ42 and the control protein CD8-GFP, or co-expressing Aβ42 and dEDEMs under the control of the eye-specific driver GMR-GAL4 are shown. Scale bar: 100 μm. See also Figure S4 and Table S5.
Neither overexpression of ER chaperone BiP or HRD1 E3 ubiquitin ligase nor elevation of proteasome activity protects against chronic ER proteinopathy in Aβ42 flies
Because our results suggested that chaperone-like functions of dEDEMs confer protective effects against ER proteinopathy caused by Aβ42, we asked whether overexpression of BiP, which is also involved in the recognition and delivery step of ERAD, could decrease Aβ42 levels in the ER and associated toxicity. Overexpression of BiP (Figure S5A, 1.5-fold increase) slightly increased Aβ42 levels in fly brains (Figure 5A) and significantly worsened behavioral deficits (Figure 5B). These results suggest that not all ER chaperone proteins can phenocopy the protective effects of dEDEMs.
Figure 5. Neither overexpression of the ER chaperone BiP or HRD1 E3 ubiquitin ligase nor elevation of proteasome activity protects against chronic ER proteinopathy in Aβ42 flies.
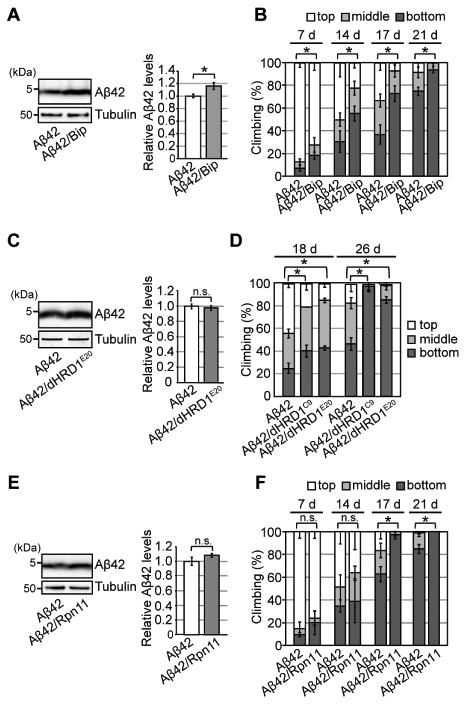
(A) Overexpression of BiP increased Aβ42 levels in fly brains. Heads of flies expressing Aβ42 alone or co-expressing Aβ42 and BiP were analyzed by western blotting with anti-Aβ antibody. Mean ± SEM, n = 4, *p < 0.05 by Student's t-test. (B) Overexpression of BiP in neurons worsened behavioral deficits in Aβ42 flies. Mean ± SD, n = 5, *p < 0.05 by Mann–Whitney U test. n.s.: not significant. (C) Overexpression of dHRD1 did not reduce Aβ42 levels in fly brains. Mean ± SEM, n = 4, n.s.: not significant (Student's t-test). (D) Overexpression of dHRD1 (dHRD1C9 and dHRD1E20) in neurons worsened behavioral deficits in Aβ42 flies. Mean ± SD, n = 5, *p < 0.05 by Mann–Whitney U test. n.s.: not significant. (E) Overexpression of Rpn11 did not reduce Aβ42 levels in fly brains. Mean ± SEM, n = 4, n.s.: not significant (Student's t-test). (F) Overexpression of Rpn11 in neurons worsened behavioral deficits in Aβ42 flies. Mean ± SD, n = 5, *p < 0.05 by Mann–Whitney U test. n.s.: not significant. See also Figure S5 and Table S5.
EDEMs deliver misfolded proteins to the HRD1 E3 ubiquitin ligase complex to promote their degradation (Bernasconi and Molinari, 2011). Hence, we examined whether overexpression of Drosophila HRD1 could reduce Aβ42 levels in the ER and associated toxicity. Overexpression of HRD1 (Figure S5B, 2-fold increase) did not reduce Aβ42 levels (Figure 5C) and worsened behavioral deficits in Aβ42 flies (Figure 5D, Aβ42 vs. Aβ42/dHRD1). Because overexpression of HRD1 alone promotes degradation of not only misfolded proteins, but also normal proteins (Denic et al., 2006; Kang and Ryoo, 2009), chronic overexpression of dHRD1 in neurons may be toxic to flies (Figure S5C).
ERAD eliminates misfolded proteins from the ER via proteasome-mediated degradation (Araki and Nagata, 2011; Guerriero and Brodsky, 2012). Hence, we investigated whether elevation of proteasome activity protects against ER proteinopathy. Overexpression of Rpn11 (Figure S5D, 3.8-fold increase), which increases proteasomal activity in flies (Tonoki et al., 2009), did not reduce Aβ42 levels (Figure 5E) and instead significantly exacerbated the locomotor deficits (Figure 5F). Thus, elevated proteosomal clearance activity also fails to phenocopy the protective effects of dEDEMs, highlighting the unique role of EDEMs in protecting against chronic ER proteinopathy.
Expression levels of UPR-related genes are elevated, whereas ERAD activity is reduced, in aged fly brains
The stability of the proteome is compromised over the course of aging (Ben-Zvi et al., 2009; Walther et al., 2015). Hence, we examined the effects of normal aging on ERAD and UPR activity. In aged fly brains, Xbp-1RB and PERK mRNA levels were elevated (Figure 6A), suggesting that canonical UPR pathways were activated. To detect age-dependent alterations in the UPR gene expression network, we performed genome-wide DNA microarray analyses to compare mRNA expression levels in the brains of young (2-day-old) and aged (25-day-old) fly brains (see STAR Methods). Among 232 genes associated with UPR-related functions (see STAR Methods and Table S3 for gene selection criteria), 77 were upregulated in aged brains (Table S4). Among these 77 genes, 21 genes contained an Xbp1-binding sequence motif in their 5′- or 3′-regulatory regions (Herrmann et al., 2012). qRT-PCR analysis confirmed that the mRNA levels of 16 out of those 21 genes were upregulated in aged brains (Figure 6B). These analyses suggested that the canonical UPR is activated in fly brains during normal aging.
Figure 6. Expression levels of UPR-related genes are elevated, whereas ERAD activity is reduced, in aged fly brains.
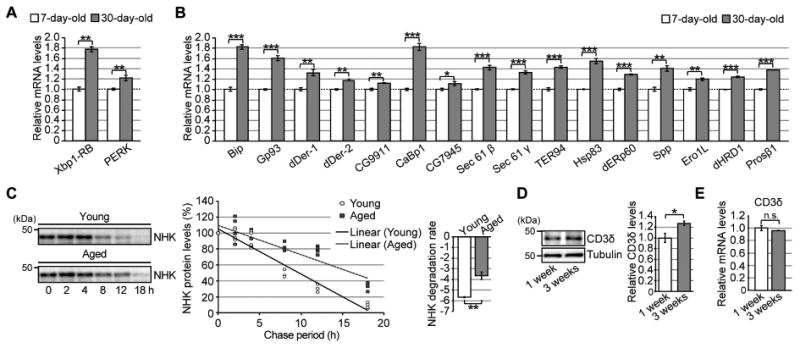
(A) mRNA expression levels of Xbp-1RB and PERK were elevated in aged fly brains. (B) Age-dependent increases in mRNA levels of UPR-related genes downstream of Ire1/Xbp1 in fly brains. For (A–B), mRNA levels in the heads of 7- or 30-day-old flies were analyzed by qRT-PCR. Open bars: 7 days old; filled bars: 30 days old. Mean ± SEM, n = 4–8, *p < 0.05, **p < 0.01, and ***p < 0.001 by Student's t-test. (C) (Left) Degradation of the ERAD substrate NHK was significantly slower in aged fly brains. After induction of NHK expression for 2 days, the decay rate of NHK protein in fly brains was analyzed for up to 18 hours by western blotting. (Right) Degradation rates. Mean ± SEM, n = 4, **p < 0.01 by Student's t-test. (D) Age-dependent increases in an ERAD substrate protein, CD3δ-YFP, in fly brains. Heads of flies expressing CD3δ-YFP at the age of 1 or 3 weeks were analyzed by western blotting. Mean ± SEM, n = 3, *p < 0.05 by Student's t-test. (E) mRNA levels of the CD3δ transgene in fly brains were not altered by age, as determined by qRT-PCR. Mean ± SEM, n = 4, n.s.: not significant (Student's t-test). See also Tables S3–5.
Next, to determine whether ERAD activity was altered over the course of aging, we compared the degradation rate of NHK in the brains of young (7-day-old) and aged (30-day-old) flies. Using a chemically inducible, neuron-specific GeneSwitch GAL4-UAS system (Osterwalder et al., 2001), we transiently induced expression of NHK in fly brains and monitored its decay rate at various time points. The degradation of NHK was significantly slower in aged brain than in young brain (Figure 6C), suggesting that ERAD activity was reduced in the aged flies. To further validate these results with another ERAD substrate protein, we generated transgenic flies expressing CD3δ fused with YFP. When this fusion protein was constitutively expressed, the level of CD3δ-YFP increased in aged fly brains to a level higher than that in young fly brains (Figure 6D), without a corresponding increase in mRNA level (Figure 6E). Thus, ERAD substrate proteins accumulate in fly brains over the course of aging.
Chronic upregulation of dEDEMs protects against age-related decline in locomotor functions and extends lifespan independent of their mannosidase activity
The data presented above demonstrate that the capacity and efficiency of ERAD are reduced under pathological conditions caused by chronic ER stress, and that selective upregulation of Drosophila EDEMs is sufficient to boost ERAD and suppress chronic ER proteinopathy in fly brains (Figures 1–5). In addition, ERAD activity decreased upon aging (Figure 6), suggesting that upregulation of ERAD via EDEMs protects against age-associated changes.
We then characterized the protective effects of chronic upregulation of EDEMs in neurons and found that neuronal expression of dEDEMs improved age-associated decline in locomotor functions. Control flies exhibited a modest decline in climbing ability over the course of aging, and neuronal overexpression of dEDEM1 or dEDEM2 suppressed this phenotype (Figure 7A, Control vs. dEDEMs). Similar protective effects were observed upon neuronal overexpression of E123Q or E144Q (Figure 7A, Control vs. E123Q or E144Q), suggesting that dEDEM mannosidase activity is dispensable for these protective effects. By contrast, neuronal expression of dEDEMs had a modest effect on median lifespan (Control: 47 days, dEDEM1: 50 days, dEDEM2: 50 days); Kaplan–Meier survival analyses with log-rank tests indicated that the effect was significant (Figure 7B). No such protective effects were seen in flies that overexpressed a control protein, CD8-GFP, in the secretory pathway of neurons (Figure S6A and B).
Figure 7. Chronic upregulation of dEDEMs protects against age-related physiological decline with a minimal effect on the UPR gene expression network.
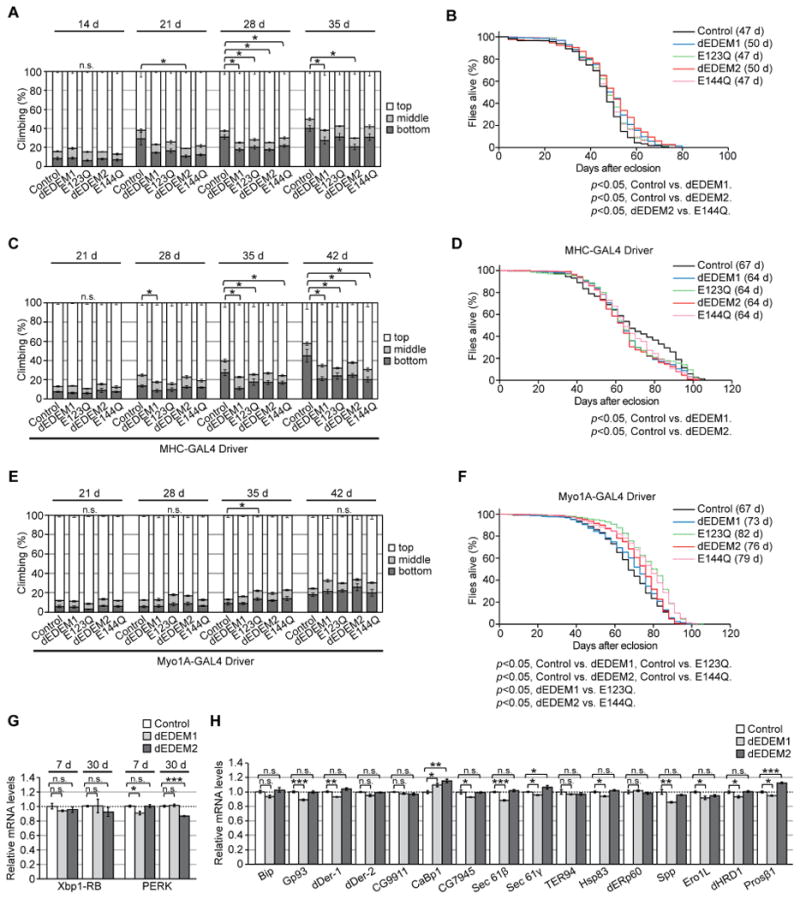
(A) Neuronal overexpression of dEDEMs improved locomotor functions in aged flies. (B) Neuronal overexpression of dEDEMs had a modest effect on median lifespan; p < 0.001 by Kaplan–Meier analyses with log-rank tests (n = 220–233 per group). (C) Overexpression of dEDEMs in muscles improved locomotor functions in aged flies. (D) Overexpression of dEDEMs in muscles had no protective effect on median lifespan; p < 0.001 by Kaplan–Meier survival analyses with log-rank tests (n = 190– 306 per group). (E) Overexpression of dEDEMs in midgut did not affect locomotor functions. (F) Overexpression of dEDEMs in midgut significantly increased median lifespan; p < 0.001 by Kaplan–Meier survival analyses with log-rank tests (n = 493– 636 per group). For (A), (C), and (E), mean ± SEM, n = 4–8 per group, *p < 0.05 by Mann–Whitney U test. n.s.: not significant. (G) Neuronal overexpression of dEDEM1 or 2 did not increase Xbp1-RB or PERK mRNA levels in either young or aged fly brains, as determined by qRT-PCR. (H) Neuronal overexpression of dEDEM1 or 2 did not increase mRNA levels of UPR-related genes in fly brains. Mean ± SEM, n = 4, *p < 0.05, **p < 0.01, and ***p < 0.001. n.s.: not significant (Student's t-test). See also Figure S6 and Table S5.
We next explored the protective effects of dEDEMs in non-neuronal tissues. Chronic overexpression of dEDEM1, dEDEM2, or their catalytically inactive mutants E123Q and E144Q in muscle, under the control of MHC-GAL4, significantly suppressed age-dependent locomotor decline (Figure 7C, Control vs. dEDEM1, E123Q, dEDEM2, or E144Q). These effects were not observed in flies overexpressing CD8-GFP in muscle (Figure S6C). In contrast, expression of dEDEMs in muscle had no protective effect on median lifespan (Control: 67 days, dEDEM1: 64 days, E123Q: 64 days, dEDEM2: 64 days, E144Q: 64 days) (Figure 7D).
Recent studies show that intestinal homeostasis has a significant influence on fly viability (Biteau et al., 2010). Hence, we investigated whether expression of dEDEMs in intestine would exert a protective effect. Expression of dEDEMs in fly midgut muscle using the Myo1A-GAL4 driver did not affect locomotor functions (Figure 7E), but it significantly increased median lifespan (Control: 67 days, dEDEM1: 73 days, E123Q: 82 days, dEDEM2: 76 days, E144Q: 79 days); Kaplan– Meier survival analyses with log-rank tests confirmed that these differences were significant (Figure 7F). Thus, dEDEM mannosidase activity is dispensable for lifespan extension.
Taken together, these behavioral analyses indicate that lifelong activation of ERAD through selective upregulation of dEDEMs in neurons or muscle does not cause obvious toxicity, and instead protects against age-associated physiological decline.
Chronic overexpression of dEDEMs in neurons has a minimal effect on the UPR gene expression network in fly brain
We next investigated whether chronic overexpression of dEDEM1 or dEDEM2 altered the UPR gene expression network in fly brains. Neuronal overexpression of dEDEMs did not increase PERK or Xbp1-RB mRNA levels in young or aged brains (Figure 7G), suggesting that dEDEM overexpression promotes ERAD without activating the canonical UPR. Likewise, chronic overexpression of dEDEMs had little if any effect on expression of UPR-related genes: among 16 genes examined, only two were slightly upregulated (Figure 7H). Thus, upregulation of dEDEMs did not exert a global impact on the canonical UPR network. Therefore, selective upregulation of EDEM function in the ERAD protects against chronic ER proteinopathy in vivo.
Discussion
Our results demonstrate that lifetime upregulation of ERAD through selective induction of individual EDEMs suppresses neurodegeneration caused by chronic ER proteinopathy in vivo. In addition, overexpression of EDEMs suppresses age-dependent behavioral decline and extends lifespan without causing obvious toxicity. Intriguingly, EDEM mannosidase activity is dispensable for these protective effects. Thus, upregulation of EDEM function in ERAD, rather than the entire UPR, represents a promising therapeutic strategy for counteracting age-related chronic ER proteinopathy.
Specific upregulation of EDEM-mediated chaperone-like functions may be an effective strategy for counteracting chronic ER proteinopathy in vivo
UPR activation is observed in neurodegenerative diseases, and short-term and modest induction of the IRE1/XBP1 pathway exerts protective effects in cellular and animal models of neurodegeneration (Sado et al., 2009; Valenzuela et al., 2012). However, the range of activity in which the canonical UPR pathway does not cause detrimental effects is narrow, making it difficult to effectively and safely utilize the UPR to treat chronic disease (Walter and Ron, 2011).
Under our conditions, chronic activation of the Ire1/Xbp1 pathway by overexpression of Xbp1-RB was toxic and caused age-dependent behavioral deficits (Figure 1G). By contrast, overexpression of dEDEMs increased ERAD activity without causing toxicity, thereby preventing chronic ER proteinopathy caused by Aβ42, a non-glycoprotein substrate, in the brain (Figure 4A–C). Moreover, mannosidase activity was dispensable for these protective effects, suggesting that chaperone-like functions of EDEMs may be critical in this context, and providing evidence for the ability of EDEMs to bind to non-glycoproteins and promote their degradation in vivo. However, the protective effects of dEDEMs were not phenocopied by overexpression of BiP or HRD1 E3 ubiquitin ligase, or by elevation of proteasome activity (Figure 5B, D, and F). These results highlight the unique functions of EDEMs in ERAD and suggest that their chaperone-like functions are potential targets for therapies aimed at protecting against chronic ER proteinopathy.
The IRE1/XBP1 pathway also regulates several cellular functions, including Ca2+ homeostasis and lipid metabolism, as well as ERAD initiation. In Drosophila and neuronal cell culture models of Aβ42 toxicity, overexpression of Xbp1 confers neuroprotective effects by reducing accumulation of free calcium in the neurons, independent of Aβ42 degradation via ERAD (Casas-Tinto et al., 2011). We found that a loss of one copy of endogenous Drosophila Xbp1 significantly worsened behavioral deficits in Aβ42 flies (Figure S7A) without altering Aβ42 levels (Figure S7B). Specific activation of both ERAD and ERAD-independent neuroprotective mechanisms downstream of IRE1/XBP1 may additively protect against chronic ER proteinopathy.
Relevance to the pathogenesis of Alzheimer's disease
We overexpressed Alzheimer's disease (AD)-related Aβ peptides in the ER to induce chronic ER proteinopathy in Drosophila brains. However, it may be premature to apply our findings to AD pathogenesis. Although the UPR is activated in AD brains (Reinhardt et al., 2014), and Aβ42 peptides can be generated in or localized to the ER under pathological conditions (LaFerla et al., 2007), the majority of Aβ peptides are produced from amyloid precursor protein (APP) in the late secretory pathway (Gandy, 2005). Thus, ER stress triggered by direct overexpression of Aβ peptides in the ER may not precisely mimic AD pathogenesis. To elucidate the in vivo role of ER stress or EDEMs in metabolism and toxicity of Aβ peptides, it will be necessary to perform studies in mouse models producing Aβ peptides from APP. Because overexpression of APP in the ER may induce ER stress, the use of recently developed knock-in mouse models of Aβ amyloidosis without APP overexpression may be suitable for this purpose (Saito et al., 2014).
The UPR can respond to chronic ER stress, whereas ERAD activity is reduced, in aged fly brains
Aging is a primary risk factor for many protein misfolding diseases, but the underlying molecular mechanisms remain elusive. Age-dependent decline in proteome stability contributes to disease pathogenesis, suggesting that preserving proteostasis would be an effective therapeutic strategy (Brehme et al., 2014; Morimoto and Cuervo, 2014). Activation of IRE-1/XBP-1 signaling in response to ER stressors is lost during aging in C. elegans (Ben-Zvi et al., 2009; Taylor and Dillin, 2013). Moreover, activation of XBP-1 in neurons protects against various ER stressors, and extends lifespan through a cell-nonautonomous mechanism in C. elegans (Taylor and Dillin, 2013).
In Drosophila, the canonical UPR pathways, including PERK and Ire1/Xbp1 signaling, were induced upon ER stress in aged brains (Figure 1). In addition, mRNA levels of many UPR-related genes were significantly elevated during aging (Figure 6A and B), suggesting that the UPR is capable of responding to ER stress in aged fly brains. However, despite activation of the UPR in aged tissues, ERAD activity was reduced (Figure 6C), suggesting that the ERAD machinery may be compromised at a post-transcriptional level. Our results demonstrate that activation of ERAD by selective induction of dEDEMs can prevent age-dependent physiological decline (Figure 7A, C, and F), and that chaperone-like functions of EDEMs are important for these protective effects. Further elucidation of the mechanisms underlying age-dependent reductions in ERAD activity may reveal a strategy for delaying normal aging by counterbalancing alterations in proteostasis.
Modification of intrinsic anti-stress signaling networks represents a novel strategy for preventing aging and age-associated diseases
Multiple anti-stress signaling pathways have evolved to maximize reproduction and survival in the presence of various environmental stressors. During pathogenesis of chronic human diseases, these mechanisms are activated as protective responses. However, canonical stress signaling networks often fail to prevent disease conditions, presumably because they are not optimized for aging or complex diseases. Nonetheless, our data suggest that neurons can counteract chronic diseases if selective components of anti-stress signaling are expressed. Thus, genetic or pharmacological modification of canonical anti-stress signaling pathways represents a novel strategy for counteracting the decline in physiological functions associated with normal aging, as well as age-associated chronic diseases.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Koichi M. Iijima (iijimakm@ncgg.go.jp).
Experimental Model and Subject Details
Drosophila genetics
Flies were maintained in standard cornmeal media at 25°C. cDNA encoding the full length of Drosophila EDEM1 (isoform A∼D) and EDEM2 (isoform A) were cloned by PCR from Drosophila cDNA, and HA tag was fused at the C-terminus, which leave the signal sequence of dEDEM1 or dEDEM2 intact at the N-terminus. These constructs were subcloned into a pUASTattb vector. dEDEM1-E123Q and dEDEM2-E144Q mutants were generated by using site-directed mutagenesis kit (Takara Bio Inc.). Transgenic flies were generated by PhiC31 integrase-mediated transgenesis systems (Best Gene Inc.). Transgenic fly lines carrying UAS-human IDE, UAS-tau and UAS-CD3δ-YFP were established following the standard method and the UAS-Aβ42 and UAS-Luc RNAi transgenic flies were previously described (Iijima et al., 2004; Iijima-Ando et al., 2012). Transgenic fly lines carrying UAS-NHK, UAS-Xbp1-RB, UAS-dHRD1C9, UAS-dHRD1E20 are gifts from Dr. H. D. Ryoo (New York University School of Medicine), MHC-GAL4 is a gift from Dr. F. Demontis (St. Jude Children's Research Hospital), Myo1A-GAL4 is a gift from Dr. B. A. Edgar (Fred Hutchinson Cancer Research Center), UAS-Rpn11 is a gift from Dr. M. Miura (University of Tokyo), UAS-dNEP1 and UAS-dNEP1 RNAi are gifts from Dr. A. Simcox (The Ohio State University), and elav-GeneSwitch is a gift from Dr. H. Keshishian (Yale University). The elav-GAL4, GMR-GAL4, Sin3ALOF (Sin3A08269), UAS-CD8-GFP, UAS-Bip, and Xbp1LOF (Xbp1k13803) were obtained from the Bloomington Stock Center. UAS-Sin3A RNAiGD10808 and UAS-Sin3A RNAiKK105852 were obtained from the Vienna Drosophila RNAi Center. Flies were backcrossed five generations onto a controlled uniform homogeneous genetic background (the Canton-S w1118 (isoCJ1) or w1118 from Vienna Drosophila RNAi center (#60000)) to ensure that the observed phenotypes were not associated with variations in the genetic backgrounds. All experiments were performed using age-matched male flies. Genotypes and ages of all flies used in this study are described in Table S5.
Method Details
A small-scale genetic screen for modifiers of Aβ42-induced toxicity
We performed a small-scale genetic screen for modifiers of Aβ42-induced locomotor defects and premature death. The candidate genes were selected from several biological processes and screening results as well as mutant alleles and transgenic lines used in the screen, were summarized in Table S1.
Climbing assay
Approximately 25 flies were placed in an empty plastic vial. The vial was then gently tapped to knock all of the flies to the bottom. The numbers of flies in the top, middle, or bottom thirds of the vial were scored after 10 seconds. The percentages of flies that stayed at the bottom were subjected to statistical analyses. Experiments were repeated more than three times, and a representative result was shown.
Life span analysis
Food vials containing 25 flies were placed on their sides at 25°C under conditions of 70% humidity and a 12:12-h light:dark cycle. Food vials were changed every 2-3 days, and the number of dead flies was counted each time. At least four vials for each genotype were prepared. Experiments were repeated more than three times, and a representative result was shown.
RU feeding
Flies carrying elav-GeneSwitch-GAL4 driver and UAS-NHK were fed the food containing 500 μM mifepristone (Tokyo Chemical Industry) or vehicle (ethanol; final concentration 5%) for 2 days for induction of the transgene expression. Then, flies were transferred to the food vials containing standard cornmeal food and were frozen at indicated time points for western blot analyses.
Histological analysis
Heads of male flies were fixed in 4% paraformaldehyde (Electron Microscopy Science) for 24 h at 4°C and embedded in paraffin. Serial sections (6μm thickness) through the entire heads were prepared, stained with hematoxylin and eosin, and examined by bright-field microscopy. Images of the sections that include the brain cortex were captured with Insight 2 CCD Camera (SPOT Imaging Solutions), and vacuole area was measured using Image J (NIH). Sample preparation and data analysis were performed by different person and were scored blind. Images of the external eye structures were captured with OLYMPUS DP26 CCD Camera (OLYMPUS).
Electron microscopy analyses and immune-gold labeling
Decapitated heads with proboscis removed were immersion-fixed overnight in 4% glutaraldehyde and 2% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS) followed by post-fixation in ferrocyanide-reduced osmium tetroxide (1% osmium tetroxide and 1.5% potassium ferrocyanide). After dehydration in a graded alcohol series and infiltration with LR White resin (2 h in 50% LR White in ethanol and 24 h in 100% LR White), the samples were polymerized overnight at 60°C. Thin sections (100 nm) of Kenyon cells and neuropil regions of the mushroom body were collected on nickel grids (100 mesh, Veco-EMS). For immunogold labelling of Aβ42, thin sections were stained with a rabbit antibody against human Aβ42 (Chemicon-Millipore, AB5078P) diluted 1:10 in PBS. Antibody was detected by incubating grids for 1 h in 10 nm colloidal gold conjugated goat anti-rabbit H&L (GE Healthcare) diluted 1:10 in PBS. For TEM, heads were immersion-fixed overnight in 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M sodium cacodylate buffer at 4°C and post-fixed in 1% osmium tetroxide in 0.1 M sodium cacodylate buffer on ice. After washing, samples were stained en bloc with 0.5% aqueous uranyl acetate for 1 hr, dehydrated with ethanol and embedded in Epon. Thin-sections were collected on copper grids. The sections were stained with 2% uranyl acetate in 70% ethanol and Reynolds' lead citrate solution. Electron micrographs were obtained with a VELETA CCD Camera (Olympus Soft Imaging Solutions GMBH) mounted on a JEM-1010 electron microscope (Jeol Ltd.).
Western blotting
Twenty fly heads for each genotype were homogenized in SDS-Tris-Glycine sample buffer, and the same amount of the lysate was loaded to each lane of multiple 10% Tris-Glycine gels and transferred to nitrocellulose membrane. The membranes were blocked with 5% nonfat dry milk (Nestle), blotted with the antibodies described below, incubated with appropriate secondary antibody and developed using ECL Western Blotting Detection Reagents (GE Healthcare) and analyzed by ImageQuant LAS 4000 (GE Healthcare), or imaging with an Odyssey system (LI-COR Biosciences). The membranes were also probed with anti-tubulin, and used as the loading control for other blots in each experiment. Anti-histone H3 (upstate, Millipore), anti-acetyl Histone H3 (upstate, Millipore), anti-tubulin (Sigma-Aldrich), anti-Aβ 6E10 antibody (Signet, Covance), anti-GFP (Invitrogen, Thermo Fisher Scientific), anti-HA (Santa Cruz biotechnology), anti-α1-antitrypsin (Bethyl laboratories), anti-CD3δ (Santa Cruz biotechnology) and anti-tau (Invitrogen, Thermo Fisher Scientific) were purchased. Most of Western blots were quantified by Odessey western blot system or ImageQuant LAS 4000, in which the signal intensity was confirmed to be in the linear range. In the Figure S2F-G, the signals were detected by ECL system and the signal intensity was quantified using ImageJ (NIH). We have developed at least three films with different signal intensity by changing exposure time to make sure that they are before the saturation points. Western blots were repeated with different animals and representative blots are shown.
For sequential extractions of Aβ42, fly heads were homogenized in RIPA buffer (50 mM Tris-HCl, pH 8.0, 0.5% sodium deoxycholate, 1% Triton X-100, 150 mM NaCl) containing 1% SDS. Lysates were centrifuged at 16,000 × g for 20 min, and supernatants were collected (SDS-soluble fraction). SDS-insoluble pellets were further homogenized in 70% formic acid (ACROS Organics) followed by centrifugation at 16,000 × g for 20 min, and the supernatants were collected (formic acid fraction). Formic acid was evaporated by SpeedVac (Savant, Thermo Fisher Scientific), and protein was resuspended in dimethyl sulfoxide (Sigma-Aldrich). Protein extracts were separated on 10–20% Tris-Tricine gels (Invitrogen, Thermo Fisher Scientific) and transferred to nitrocellulose membranes. The membranes were boiled in PBS for 3 min, blocked with 5% nonfat dry milk, blotted with the anti-Aβ 6E10 antibody (Signet, Covance), incubated with appropriate secondary antibody and developed using ECL Western Blotting Detection Reagents or imaging with an Odyssey system.
RNA extraction and quantitative real time PCR analysis
More than thirty flies for each genotype were collected and frozen. Heads were mechanically isolated, and total RNA was extracted using TRIzol Reagent (Invitrogen, Thermo Fisher Scientific) according to the manufacturer's protocol with an additional centrifugation step (16,000 × g for 10 min) to remove cuticle membranes prior to the addition of chloroform. Total RNA was reverse-transcribed using MuLV Reverse Transcriptase (Invitrogen, Thermo Fisher Scientific) or PrimeScript RT-PCR kit (TaKaRa Bio). qRT-PCR was performed using PowerSYBR (Thermo Fisher Scientific) on a 7500 fast real time PCR system (Applied Biosystems, Thermo Fisher Scientific) or a CFX96 real time PCR detection system (Bio-Rad Laboratories). The average threshold cycle value (CT) was calculated from at least three replicates per sample. Expression of genes of interest was standardized relative to rp49 or GAPDH1. Relative expression values were determined by the ΔΔCT method. Primers were designed using Primer-Blast (NIH) as described in Table S6.
Microarray analysis
Microarray analysis was performed in triplicate on independent RNA samples isolated from heads of flies at 2 day-after-eclosion or 25 day-after-eclosion. Total RNA was extracted with TRIzol followed by RNAeasy columns (QIAGEN). Probe labeling, hybridization to Affymetrix GeneChip Drosophila Genome 2.0 arrays and scanning were performed by Thomas Jefferson University Microarray Core facility. Raw data were normalized with RMA (Bolstad et al., 2003; Irizarry et al., 2003) and analyzed with Bioconductor and Limma (Ritchie et al., 2015). The 232 genes that are associated with GO listed in Table S3 (http://flybase.org/) were selected and subjected to further analyses. Seventy-seven genes upregulated in aged flies with false discovery rate of less than 6% are shown in Table S4. Genes whose regulatory region contains a Xbp-1 binding motif were identified by using i-CisTarget (Herrmann et al., 2012).
Cell culture
Drosophila S2 cells were maintained in Schneider's Drosophila media supplemented with 10% FBS in a 5% CO2 incubator. Aβ42 cDNA was described previously (Iijima et al., 2008). dEDEMs-HA constructs in pUASTattb vector were described above in Drosophila genetics section. S2 cells were transfected with actin-GAL4 and UAS plasmids using HilyMax (Dojindo) following the manufacturer's protocol. Cells were collected at 48 h after transfection and subjected to co-immunoprecipitation and western blot analysis.
Co-Immunoprecipitation
Cells were lysed in buffer (1% NP-40, 150 mM NaCl, 50 mM Tris-HCl, pH8.0) with Complete protease inhibitor (Roche) for 30 min on ice and were centrifuged at 16,000 × g for 20 min. The resulting supernatants were incubated with the anti-HA antibody (Santa Cruz biotechnology) at 4°C overnight. The immunocomplex was recovered using protein G-Sepharose beads (GE Healthcare) and subjected to western blot analysis.
Quantification and Statistical Analysis
All experiments were repeated at least three times, and all results were expressed as mean ± SD (for climbing assay) or mean ± SEM. Unpaired Student's t-test, Mann– Whitney U test, or Kaplan-Meier survival analyses with log-rank tests were used to determine statistical significance as indicated in the figure legends. Excel (Microsoft), SigmaPlot 11.0 (Systat software Inc.), JMP (SAS) and R (R Development Core Team, 2010) were used for the statistical analyses. * indicates p <0.05, ** indicates p < 0.01 and *** indicates p < 0.001 throughout the manuscript.
Data and Software Availability
Affymetrix arrays raw data have been deposited in the Gene Expression Omnibus database under accession number GEO: GSE98554.
Supplementary Material
Highlights.
Chronic activation of the IRE1/Xbp1 pathway causes behavioral deficits in flies
Upregulation of EDEMs is sufficient to mitigate ER proteinopathy without toxicity
EDEMs protect against age-related physiological declines without inducing the UPR
The mannosidase activity of EDEMs is dispensable for these protective effects
Acknowledgments
We thank F. Demontis, B.A. Edgar, H. Keshishian, M. Miura, H. D. Ryoo, A. Simcox, and the Bloomington Stock Center and the Vienna Drosophila RNAi center for fly stocks and cDNAs. We thank A. Gatt, L. Granger, C. Shenton, L. Zhao, Y. Ohtake, A. Oishi, S. Chikamatsu, and Y. Ojio for technical assistance. We thank M. E. Fortini and J. Chin for comments on the manuscript. This work was supported by grants from the Gilbert Foundation/American Federation for Aging Research (to K.M.I.) and the National Institutes of Health (R01AG032279-A and U01AG046170-01, to K.M.I. and K.A.); The Research Fund for Longevity Sciences (25-27 and 28-26) from the National Center for Geriatrics and Gerontology, Japan (to K.M.I.); the Takeda Science Foundation, Japan (to K.M.I.); and Grants-in-aid for Scientific Research (26870921 to M.S. and 16K08637 to K.M.I.). The authors declare no competing financial interests.
Footnotes
Author Contributions: Conceptualization, M.S. and K.M.I.; investigation, M.S., A.M.O., S.H., Y.S., N.F., E.S., K.A., and K.M.I.; writing (original draft), M.S., K.A., and K.M.I.; writing (review & editing), M.S., K.A., and K.M.I.; supervision, M.S., K.A., and K.M.I.; funding acquisition, M.S., K.A., and K.M.I
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allagnat F, Christulia F, Ortis F, Pirot P, Lortz S, Lenzen S, Eizirik DL, Cardozo AK. Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia. 2010;53:1120–1130. doi: 10.1007/s00125-010-1699-7. [DOI] [PubMed] [Google Scholar]
- Araki K, Nagata K. Protein folding and quality control in the ER. Cold Spring Harbor perspectives in biology. 2011;3:a007526. doi: 10.1101/cshperspect.a007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A. 2009;106:14914–14919. doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernasconi R, Molinari M. ERAD and ERAD tuning: disposal of cargo and of ERAD regulators from the mammalian ER. Curr Opin Cell Biol. 2011;23:176–183. doi: 10.1016/j.ceb.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biteau B, Karpac J, Supoyo S, Degennaro M, Lehmann R, Jasper H. Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 2010;6:e1001159. doi: 10.1371/journal.pgen.1001159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3312–3317. doi: 10.1073/pnas.0611511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell reports. 2014;9:1135–1150. doi: 10.1016/j.celrep.2014.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez-Velazquez M, Rincon-Limas DE, Fernandez-Funez P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum Mol Genet. 2011;20:2144–2160. doi: 10.1093/hmg/ddr100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celniker SE, Dillon LA, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, et al. Unlocking the secrets of the genome. Nature. 2009;459:927–930. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denic V, Quan EM, Weissman JS. A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell. 2006;126:349–359. doi: 10.1016/j.cell.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Dubnau J, Chiang AS, Grady L, Barditch J, Gossweiler S, McNeil J, Smith P, Buldoc F, Scott R, Certa U, et al. The staufen/pumilio pathway is involved in Drosophila long-term memory. Current biology: CB. 2003;13:286–296. doi: 10.1016/s0960-9822(03)00064-2. [DOI] [PubMed] [Google Scholar]
- Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–1129. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Grzenda A, Lomberk G, Zhang JS, Urrutia R. Sin3: master scaffold and transcriptional corepressor. Biochim Biophys Acta. 2009;1789:443–450. doi: 10.1016/j.bbagrm.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Brodsky JL. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiological reviews. 2012;92:537–576. doi: 10.1152/physrev.00027.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nature structural & molecular biology. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- Herrmann C, Van de Sande B, Potier D, Aerts S. i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 2012;40:e114. doi: 10.1093/nar/gks543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Wada I, Hasegawa K, Yorihuzi T, Tremblay LO, Herscovics A, Nagata K. A novel ER alpha-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001;2:415–422. doi: 10.1093/embo-reports/kve084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Chiang HC, Hearn SA, Hakker I, Gatt A, Shenton C, Granger L, Leung A, Iijima-Ando K, Zhong Y. Abeta42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS One. 2008;3:e1703. doi: 10.1371/journal.pone.0001703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:6623–6628. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima-Ando K, Sekiya M, Maruko-Otake A, Ohtake Y, Suzuki E, Lu B, Iijima KM. Loss of axonal mitochondria promotes tau-mediated neurodegeneration and Alzheimer's disease-related tau phosphorylation via PAR-1. PLoS Genet. 2012;8:e1002918. doi: 10.1371/journal.pgen.1002918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2010. Retrieved from http://www.R-project.org. [Google Scholar]
- Ito K, Awano W, Suzuki K, Hiromi Y, Yamamoto D. The Drosophila mushroom body is a quadruple structure of clonal units each of which contains a virtually identical set of neurones and glial cells. Development. 1997;124:761–771. doi: 10.1242/dev.124.4.761. [DOI] [PubMed] [Google Scholar]
- Jacobsen TL, Cain D, Paul L, Justiniano S, Alli A, Mullins JS, Wang CP, Butchar JP, Simcox A. Functional analysis of genes differentially expressed in the Drosophila wing disc: role of transcripts enriched in the wing region. Genetics. 2006;174:1973–1982. doi: 10.1534/genetics.106.056788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, Edgar BA. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell. 2009;137:1343–1355. doi: 10.1016/j.cell.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MJ, Ryoo HD. Suppression of retinal degeneration in Drosophila by stimulation of ER-associated degradation. Proc Natl Acad Sci U S A. 2009;106:17043–17048. doi: 10.1073/pnas.0905566106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nature reviews Drug discovery. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Kohno K. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr Opin Cell Biol. 2011;23:135–142. doi: 10.1016/j.ceb.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- Kretzschmar D, Hasan G, Sharma S, Heisenberg M, Benzer S. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J Neurosci. 1997;17:7425–7432. doi: 10.1523/JNEUROSCI.17-19-07425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. Journal of biochemistry. 2009;146:743–750. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- Morimoto RI, Cuervo AM. Proteostasis and the aging proteome in health and disease. The journals of gerontology Series A, Biological sciences and medical sciences. 2014;69(Suppl 1):S33–38. doi: 10.1093/gerona/glu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninagawa S, Okada T, Sumitomo Y, Horimoto S, Sugimoto T, Ishikawa T, Takeda S, Yamamoto T, Suzuki T, Kamiya Y, et al. Forcible destruction of severely misfolded mammalian glycoproteins by the non-glycoprotein ERAD pathway. J Cell Biol. 2015;211:775–784. doi: 10.1083/jcb.201504109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninagawa S, Okada T, Sumitomo Y, Kamiya Y, Kato K, Horimoto S, Ishikawa T, Takeda S, Sakuma T, Yamamoto T, et al. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J Cell Biol. 2014;206:347–356. doi: 10.1083/jcb.201404075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder T, Yoon KS, White BH, Keshishian H. A conditional tissue-specific transgene expression system using inducible GAL4. Proc Natl Acad Sci U S A. 2001;98:12596–12601. doi: 10.1073/pnas.221303298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt S, Schuck F, Grosgen S, Riemenschneider M, Hartmann T, Postina R, Grimm M, Endres K. Unfolded protein response signaling by transcription factor XBP-1 regulates ADAM10 and is affected in Alzheimer's disease. FASEB J. 2014;28:978–997. doi: 10.1096/fj.13-234864. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo HD, Domingos PM, Kang MJ, Steller H. Unfolded protein response in a Drosophila model for retinal degeneration. The EMBO journal. 2007;26:242–252. doi: 10.1038/sj.emboj.7601477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sado M, Yamasaki Y, Iwanaga T, Onaka Y, Ibuki T, Nishihara S, Mizuguchi H, Momota H, Kishibuchi R, Hashimoto T, et al. Protective effect against Parkinson's disease-related insults through the activation of XBP1. Brain Res. 2009;1257:16–24. doi: 10.1016/j.brainres.2008.11.104. [DOI] [PubMed] [Google Scholar]
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer's disease. Nat Neurosci. 2014;17:661–663. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- Schuster CM, Davis GW, Fetter RD, Goodman CS. Genetic dissection of structural and functional components of synaptic plasticity. I. Fasciclin II controls synaptic stabilization and growth. Neuron. 1996;17:641–654. doi: 10.1016/s0896-6273(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Shenkman M, Groisman B, Ron E, Avezov E, Hendershot LM, Lederkremer GZ. A shared endoplasmic reticulum-associated degradation pathway involving the EDEM1 protein for glycosylated and nonglycosylated proteins. J Biol Chem. 2013;288:2167–2178. doi: 10.1074/jbc.M112.438275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, Dillin A. XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell. 2013;153:1435–1447. doi: 10.1016/j.cell.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonoki A, Kuranaga E, Tomioka T, Hamazaki J, Murata S, Tanaka K, Miura M. Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol Cell Biol. 2009;29:1095–1106. doi: 10.1128/MCB.01227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela V, Collyer E, Armentano D, Parsons GB, Court FA, Hetz C. Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell death & disease. 2012;3:e272. doi: 10.1038/cddis.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M, Mann M, et al. Widespread Proteome Remodeling and Aggregation in Aging C. elegans. Cell. 2015;161:919–932. doi: 10.1016/j.cell.2015.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.