

Abstract
Self-assembly of amyloid-β (Aβ) peptides in human brain tissue leads to neurodegeneration in Alzheimer's disease (AD). Amyloid fibrils, whose structures have been extensively characterized by solid state nuclear magnetic resonance (ssNMR) and other methods, are the thermodynamic end point of Aβ self-assembly. Oligomeric and protofibrillar assemblies, whose structures are less well understood, are also observed as intermediates in the assembly process in vitro and have been implicated as important neurotoxic species in AD. We report experiments in which the structural evolution of 40-residue Aβ (Aβ40) is monitored by ssNMR measurements on frozen solutions prepared at four successive stages of the self-assembly process. Measurements on transient intermediates are enabled by ssNMR signal enhancements from dynamic nuclear polarization (DNP) at temperatures below 30 K. DNP-enhanced ssNMR data reveal a monotonic increase in conformational order from an initial state comprised primarily of monomers and small oligomers in solution at high pH, to larger oligomers near neutral pH, to metastable protofibrils, and finally to fibrils. Surprisingly, the predominant molecular conformation, indicated by 13C NMR chemical shifts and by sidechain contacts between F19 and L34 residues, is qualitatively similar at all stages. However, the in-register parallel β-sheet supramolecular structure, indicated by intermolecular 13C spin polarization transfers, does not develop before the fibril stage. This work represents the first application of DNP-enhanced ssNMR to the characterization of peptide or protein self-assembly intermediates.
Graphical abstract
Introduction
The detailed molecular basis for Alzheimer's disease (AD) is still largely unknown. The amyloid cascade hypothesis relates disease onset and progression to a dysregulation of amyloid-β (Aβ) peptide production and clearance that leads to Aβ self-assembly in brain tissue1. Amyloid fibrils, consisting primarily of 40- and 42-residue Aβ peptides (Aβ40 and Aβ42), are the thermodynamic endpoint of the Aβ self-assembly process and comprise the major component of self-assembled Aβ in the brain tissue of AD patients. However, the total quantity of fibrillar Aβ in brain tissue does not correlate strongly with the progression of neurodegeneration, as indicated by studies of human brain extracts2 and transgenic mice3. Polymorphism of Aβ fibrils4 may be one factor that weakens this correlation, as several types of experiments indicate that different polymorphs can have different neurodegenerative effects5. Alternatively, nonfibrillar Aβ assemblies may be important neurotoxic species, as suggested by a variety of observations in humans and mouse models2,6. Nonfibrillar assemblies include oligomers of various sizes and shapes7, as well as protofibrils, which are metastable, fibril-like assemblies with greater curvature and shorter typical lengths8. Oligomers produced by aggregation of synthetic Aβ peptides in vitro show higher toxicity in cell cultures than an equivalent mass of fibrils7d,9. Given the diversity of neurotoxicity mechanisms that have been proposed for Aβ assemblies in AD, including membrane disruption10, interactions with specific cell-surface receptors11, oxidative damage associated with binding of metal ions12, stimulation of destructive inflammation13, and disruption of vasculature14, it is conceivable that both fibrillar and nonfibrillar species contribute significantly to neurodegeneration, possibly through different mechanisms. Thus, characterization of Aβ structures at all stages of self-assembly is an important goal.
Detailed structural models for Aβ fibrils have been developed from a variety of experimental measurements, especially solid state nuclear magnetic resonance (ssNMR) 5a,15. In these models, molecules with approximately U-shaped conformations stack in an in-register parallel manner, forming parallel cross-β structures4,16. Structurally distinct, self-propagating fibril polymorphs5b differ in the detailed conformations at certain sites, the number of cross-β units within the minimal fibril structure, the contacts between cross-β units, and other features. For example, salt bridge interactions between oppositely charged D23 and K28 sidechains have been observed by ssNMR in some Aβ40 fibril polymorphs5a,15a, but not in other polymorphs15b.
A detailed molecular structural model for protofibrils formed by the Asp23-to-Asn “Iowa mutant” (D23N-Aβ40) has been developed from ssNMR data17, showing that U-shaped D23N-Aβ40 molecules stack in an antiparallel manner to form a double-layered, antiparallel cross-β structure in the protofibrils. In contrast, D23N-Aβ40 fibrils contain double-layered, parallel cross-β structural motifs15f.
Various other oligomeric and protofibril Aβ assemblies, prepared under a variety of conditions, have been partially characterized by ssNMR. These include approximately spherical Aβ40 and Aβ42 assemblies that form in aqueous buffers7c,7d,18, Aβ42 oligomers prepared by dialysis of a detergent solution7b, Aβ40 protofibrils stabilized by interaction with the B10AP antibody19, small disc-like Aβ42 oligomers20, and Aβ40 oligomers formed in the presence of epigallocatechin-3-gallate (EGCG) 21. In addition, oligomers formed by a GroES-ubiquitin-Aβ42 fusion protein have been studied by electron paramagnetic resonance with spin labeling22. A structural model for protofibrils formed by an internally crosslinked, double cysteine mutant of Aβ42 has also been developed from ssNMR data23. The consensus of these studies is that Aβ conformations in oligomers and protofibrils are similar to Aβ conformations in fibrils, although the supramolecular organization may be rather different.
To date, most ssNMR studies of Aβ self-assembly intermediates have been performed on samples that were concentrated by lyophilization after oligomer formation7b-7d,17c,18a,19-20,23. Lyophilization has been required because Aβ concentrations in oligomer preparations in vitro are typically on the order of 1 mM or less. With typical ssNMR sample volumes in the 10-100 μl range, the quantity of Aβ in the oligomer solution is then on the order of 100 nmole or less. Although ssNMR measurements can be performed successfully on frozen solutions of peptides and proteins24, higher concentrations and/or larger sample volumes are needed for adequate signal-to-noise, even with measurement times of several days or more. Lyophilization allows greater quantities of Aβ assemblies to be packed into smaller volumes, greatly accelerating data acquisition.
It is well established that lyophilization does not perturb the molecular structures of amyloid fibrils significantly, as NMR chemical shifts of lyophilized and lyophilized/rehydrated fibril samples are the same as those of non-lyophilized, “as grown” samples25. For certain Aβ oligomers and protofibrils, evidence has been presented that lyophilization also does not perturb molecular structures significantly7c,7d,17c,18a. However, in the case of smaller Aβ assemblies and assemblies that are not metastable, lyophilization may produce significant changes in molecular conformations, intermolecular interactions, and assembly sizes.
In the experiments described below, we use dynamic nuclear polarization (DNP) to enhance the sensitivity of ssNMR measurements on frozen solutions of Aβ assemblies, thereby avoiding the need for lyophilization and permitting measurements on transient species. DNP is an effect in which microwave irradiation of electron spin transitions leads to large enhancements of nuclear spin polarizations of nuclei, and hence large enhancements of ssNMR signals26. In recent ssNMR studies, DNP has been applied to membrane proteins27, amyloid fibrils28, and viral DNA29. To our knowledge, the experiments described below represent the first application of DNP in ssNMR studies of transient or metastable species with biological or biophysical relevance.
We describe experiments on four successive stages of Aβ40 self-assembly: (1) A freshly-prepared Aβ40 solution at pH 12, where the peptide is primarily monomeric; (2) the same solution shortly after adjustment to pH 7.5, where the peptide exists as a mixture of monomers and oligomers of various sizes and morphologies; (3) the same solution after incubation at pH 7.5 for at least 4 h, at which time metastable Aβ40 protofibrils are the predominant species; (4) Aβ40 fibrils that develop from protofibrils during repetitive sonication/incubation cycles. Our ssNMR data indicate a progressive reduction in conformational disorder as self-assembly proceeds through these four stages. The predominant molecular conformation, indicated by 13C chemical shifts, is qualitatively similar at all stages. Long-range tertiary contacts between sidechains of F19 and L34, which are known to be a characteristic feature of many Aβ405a,15a,15b and Aβ4220 fibril polymorphs, are also detectable at all stages. Surprisingly, these contacts are most pronounced at the protofibril stage. We find that parallel intermolecular alignment in Aβ40 assemblies does not develop until the fibril stage.
Methods
Peptide synthesis
Aβ40 was synthesized on a 0.15 mmol scale by fluorenylmethoxycarbonyl (FMOC) chemistry on a Tribute TPS-110 automated peptide synthesizer (Protein Technologies), with activation by O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HCTU) and N,N-diisopropylethylamine (DIPEA) and FMOC-Wang resins (0.27 mEq/g substitution). Isotopic labeling patterns of Aβ40 samples for ssNMR measurements are summarized in Table 1. Valine residues were deuterated to reduce the intrinsic 1H spin-lattice relaxation rates within Aβ40 assemblies and thereby increase DNP signal enhancements. To improve yields, pseudoproline dipeptides were used at D7-S8 and G25-S26. Isotopically labeled residues were double-coupled (2 h coupling with labeled amino acid in four-fold excess, followed by 1 h coupling with unlabeled amino acid in ten-fold excess). The crude product was purified by reverse-phase high performance liquid chromatography, using a water/acetonitrile gradient with 1% acetic acid and a C3 preparative column at room temperature. Purity is estimated to be greater than 95% by mass spectrometry. For samples II-IV, the synthesis was performed up to the 13C-labeled residue (i.e., A21, A30, or G33), then half of the resin was removed from the reaction vessel, the carbonyl-labeled amino acid was coupled in the reaction vessel for 2 h, the uncoupled resin was returned to the reaction vessel, the aliphatic-labeled amino acid was coupled for 2 h, and unlabeled amino acid was coupled for 1 h. The ratio of carbonyl-labeled to aliphatic-labeled peptides in the final product was determined from one-dimensional NMR spectra in dimethyl sulfoxide (DMSO) solution.
Table 1.
Isotopic labeling patterns of peptide samples for ssNMR measurements. All Aβ40 samples were also perdeuterated at V12, V18, V24, V36, and V39.
| peptide sample | labeling of component A | labeling of component B | A:B ratio |
|---|---|---|---|
| Aβ40 I | U-15N,13C at F19, L34, G38; 13CO at A30 | U-15N,13C at F19, L34, G38; 13Cβat A30 | 50:50 |
| Aβ40 II | 13CO at A21 | 13Cβat A21 | 28:72 |
| Aβ40 III | 13CO at A30 | 13Cβat A30 | 56:44 |
| Aβ40 IV | 13CO at G33 | 13Cαat G33 | 40:60 |
| Aβ11-25 | 13CO at F20; 13Cβ at A21; 15N at L17 | -- | -- |
Aβ40 self-assembly protocols
To produce the four types of Aβ40 assemblies examined in this work, purified, lyophilized peptide was first disaggregated by dissolution in hexafluoroisopropanol (HFIP) to a concentration of 2 mg/ml, incubation at room temperature for 0.5-1.0 h, and lyophilization again. HFIP-treated peptide was then dissolved at 2.5 mM concentration in 20 mM NaOH, pH ≈ 12. Negatively-stained transmission electron microscope (TEM) images of this high pH sample, on grids that were prepared within 2 min of peptide dissolution, showed a nearly clear TEM grid surface, with a low coverage of amorphous aggregates that may develop during drying of the grid (Fig. 1A). The pH was then lowered by addition of 250 mM phosphate (pH 7.4) in a 1:4 ratio, bringing the final buffer concentration to 50 mM. A small quantity of concentrated HCl was added to adjust the final pH to 7.5. TEM images of this neutral pH sample, on grids that were prepared within 2 min of pH adjustment, showed a high density of irregularly shaped assemblies (Fig. 1B). After quiescent incubation at 24° C for 4-5 hours, a high density of relatively short and curved “protofibrils” was observed by TEM (Fig. 1C). These structures were stable for at least several weeks in quiescent solutions at pH 7.5. After sonication of the protofibril solution (Branson S-250A sonifier, 10% duty cycle, lowest power setting, 50 pulses) and incubation at 24° C for 12 h, TEM images showed a mixture of protofibrils and thicker, longer, and less curved fibrils. After 3-4 repetitions of the sonication/incubation procedure, conversion to mature fibrils was complete (Fig. 1D).
Figure 1.
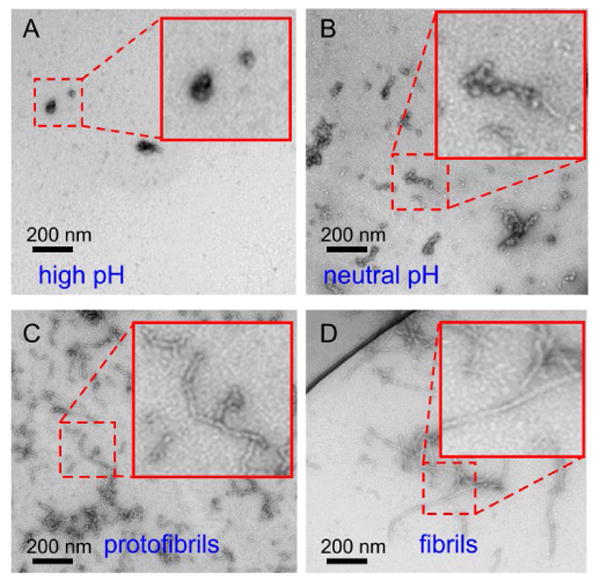
Negatively stained TEM images of Aβ40 at four stages of self-assembly. (A) “High pH” sample, after initial preparation of an Aβ40 solution at pH 12. (B) “Neutral pH” sample, shortly after adjustment to pH 7.5. (C) “Protofibril” sample, after 4 h incubation at pH 7.5. (D) “Fibril” sample, after four rounds of sonication and incubation. Insets are expanded views of the indicated regions.
Thioflavin T (ThT) fluorescence measurements on Aβ40 solutions (Fig. S1) showed a lag period of 1.0 h after adjustment to pH 7.5, followed by an increase in fluorescence that corresponds to the development of the protofibrillar assemblies seen by TEM. The fluorescence level reached a plateau after approximately 10 h, at which time the fluorescence level was about 40% of the level observed after conversion of protofibrils to mature fibrils by sonication/incubation. Thus, it appears that nonfibrillar Aβ40 assemblies in our experiments are not ThT-active, while protofibrils exhibit lower ThT fluorescence than fibrils.
In addition, Aβ40 fibrils were prepared by seeded growth from pre-formed fibrils with the three-fold-symmetric structure reported by Paravastu et al.15b Seeds were prepared by sonication of a solution of pre-formed fibrils. A 6 mM solution of Aβ40 in DMSO was diluted into 10 mM sodium phosphate buffer, pH 7.4, to create a 100 μM Aβ40 solution. Seeds were added in a 1:19 molar ratio (by Aβ40 monomers), and the mixture was gently vortexed. After 1 h incubation, a 10% aliquot of the mixture was withdrawn, sonicated, and returned to the mixture. After overnight incubation, abundant fibrils were visible by TEM.
Biophysical characterization of Aβ40 assemblies
TEM images were obtained with an FEI Morgagni microscope, operating at 80 kV. Grids were glow-discharged carbon films, supported by lacey carbon on 300 mesh copper. Samples were diluted ten-fold in their own solvents (20 mM NaOH or 10 mM phosphate) before application of a 5 μl drop to the grid surface. After 1 min adsorption, grids were blotted with filter paper, rinsed twice with water, stained for 30 s with 5 μl of 3% uranyl acetate, blotted, and dried in air.
Experiments with the photoinduced cross-linking of unmodified proteins (PICUP) technique30 were performed by adding 10 μl of 1 mM tris(2,2′-bipyridyl)dichlororuthenium (Ru(II)) and 10 μl of 20 mM ammonium persulfate (APS) to 2.0 μl samples of Aβ40 assemblies. A 2.0 mM solution of the 35-residue villin headpiece subdomain protein (HP35) in 50 mM phosphate buffer, pH 7.4, was used as a non-aggregating control31. The mixtures were then irradiated at 24° C for periods ranging from 0.5 s to 5 min, using the output of a 150 watt incandescent fiber optic lamp (Dolan-Jenner model 180) and an electronically timed shutter (Melles-Griot model IES 3). Cross-linking reactions were then quenched immediately by adding 10 μl of 1 M dithiothreitol. Cross-linked products were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). For SDS-PAGE, 4 μl of 20 μM cross-linked Aβ40 or HP35 was dissolved in NuPAGE LDS buffer (Life Technologies) and loaded onto a 10% NuPAGE Bis-Tris gel (Life Technologies). Gels were run in MES buffer for 40-45 min with 80-100 mA current. Silver staining was performed with a SilverXpress kit (Life Technologies). Gel images were analyzed with ImageJ software32.
Dynamic light scattering (DLS) measurements at 658 nm wavelength were carried out with a DynaPro NanoStar (Wyatt Technology) instrument on undiluted samples (50 μl in Eppendorf UVettes) equilibrated at 25° C. Light scattering autocorrelation functions were recorded for 60 s and averaged over 10 repetitions.
Sedimentation velocity experiments were conducted at 20.0° C on a Beckman Coulter ProteomeLab XL-I analytical ultracentrifuge. High pH samples were studied at rotor speeds of 50,000 and 60,000 rpm. Neutral pH and protofibril samples were studied at 20,000, 40,000, 45,000 and 50,000 rpm. Samples were loaded into two-channel centerpiece cells, allowed to equilibrate at 20.0° C for 2 h and then analyzed at the indicated speeds. Concentrated samples of Aβ40 were loaded into 3 mm path-length cells (100 μl), whereas diluted samples were loaded into 12 mm path-length cells (400 μl). Data were collected using both absorbance (280 nm) and Rayleigh interference optical detection systems and analyzed in SEDFIT 14.4f 33 in terms of a continuous c(s) distribution of Lamm equation solutions with a resolution of 0.05 – 0.10 S, depending on the extent of aggregation. Regularization was carried out using the method of maximum entropy with a confidence level of 0.68. The partial specific volume of Aβ40 was calculated in SEDNTERP (http://sednterp.unh.edu/) based on the amino acid composition. The buffer density and viscosity were also calculated based on their composition and sedimentation coefficients were corrected to standard conditions of 20° C in water (s20,w).
Thioflavin T (ThT) fluorescence measurements were performed at room temperature with a StellarNet BLACK-Comet-TEC fiber optic spectrofluorometer. For each time point, an aliquot of the Aβ40 sample was diluted by a factor of 100 with 10 mM sodium phosphate buffer (pH 7.4) containing 100 μM ThT. Fluorescence intensity at 490 nm was measured immediately after dilution, with excitation at 423 nm.
Circular dichroism (CD) spectra were recorded with a JASCO J-715 spectropolarimeter, using 0.01 mm path length quartz cuvettes. Acquisition of CD spectra began immediately after sample preparation. Three spectra of each sample were recorded in succession, with approximately 11 min acquisition time per spectrum, to confirm that the spectra did not change on the time scale of the CD measurements.
Size exclusion chromatography (SEC) experiments were performed with a BioRAD NGC chromatography system. Aβ40 samples (50 μl injected volume) were separated at 0.5 ml/min on a Superose 6 10/300 GL column (GE Healthcare), with a total column volume of Vc= 24 ml. Prior to separation, the column was equilibrated with either 10 mM sodium phosphate buffer (pH 7.4) or 20 mM NaOH (with or without 150 mM NaCl). To check reproducibility, several runs were carried out for each condition, each followed by flushing with 3-4 Vc to remove any residual Aβ40 from the column. Molecular weight calibration runs were performed with a HMW Calibration Kit (GE Healthcare).
DNP and ssNMR measurements
DNP-enhanced ssNMR experiments were carried out at 9.4 T (400.9 MHz 1H NMR frequency, 264.0 GHz microwave frequency) using the low-temperature ssNMR and DNP instrumentation described previously26d,34. In brief, the home-built magic-angle spinning (MAS) ssNMR probe used elongated MAS rotors with 4 mm outer diameter and 80 μl sample volume. Nitrogen gas was used for MAS drive and bearings, while the sample volume near the center of the rotor was cooled with helium from a liquid helium transfer line connected to the MAS module of the ssNMR probe. Experiments were performed at sample temperatures of 25 ± 2 K and at 6.7 kHz MAS frequency. Sample temperatures were determined from 79Br spin-lattice relaxation rates of KBr powder, contained in capillary tubes within the MAS rotors35. 1H radio-frequency (rf) fields for decoupling were 80-85 kHz. 1H-13C cross-polarization used 1H rf fields of 54 kHz and 13C rf fields of 47 kHz. 1H-15N cross-polarization used 1H rf fields of 50 kHz and 15N rf fields of 43 kHz. 15N-13C cross-polarization used 15N rf fields of 24 kHz and 13C rf fields of 31 kHz. An extended interaction oscillator (Communications and Power Industries) was used as the microwave source, with continuous wave output power of approximately 1.4 W. Microwaves were circularly polarized with a quasi-optical interferometer (Thomas Keating, Ltd.) and transmitted to the sample through a corrugated waveguide within the ssNMR probe.
NMR data were collected with a Bruker Avance III spectrometer console and processed with nmrPipe36, Bruker Topspin, and Sparky (available from https://www.cgl.ucsf.edu/home/sparky) software. Two-dimensional (2D) 13C-13C ssNMR spectra were acquired with either 13C-13C spin diffusion or rf-assisted diffusion/dipolar-assisted rotational resonance37 (RAD/DARR) during the mixing period between t1 and t2 periods. 2D 15N-13C ssNMR spectra were acquired with 5 ms 15N-13C band-selective cross-polarization periods and (for protofibril and fibril samples) 23.8 ms 13C-13C spin diffusion periods between t1 and t2 Recycle delays were approximately 5 s for high pH and neutral pH samples and 10 s for protofibril and fibril samples, corresponding to approximately 1.3 times the characteristic build-up times for 1H spin polarizations under DNP. Total measurement times were 1.5-5 h for 2D 13C-13C spectra and 4.5-10 h for 2D 15N-13C spectra. In 2D measurements on high pH and neutral pH samples, the number of scans per t1 point decreased with increasing t138. Measurements of intermolecular 13C polarization transfers were performed as described below, with total measurement times of 2-4 h for each sample.
For DNP-enhanced ssNMR measurements, Aβ40 samples in the four stages of self-assembly described above were prepared in 80% D2O/20% H2O solutions, as solvent deuteration is known to increase DNP signal enhancements and reduce DNP build-up times39. For high pH and neutral pH samples, 13C-depleted, perdeuterated glycerol was then added to achieve 60% (by mass) glycerol concentration, along with an aliquot of the triradical dopant DOTOPA-3OH-Methoxy34 in perdeuterated DMSO to achieve a 6.6 mM dopant concentration. Immediately after mixing, samples were loaded into MAS rotors and frozen by immersion in liquid nitrogen. Protofibril and fibril samples were pelleted by centrifugation at 430,000 X g for 2.0 h (Beckman TLA 100.2 rotor). Supernatants were removed and glycerol and triradical dopant were added. After a second centrifugation for 2 h, pellets were loaded into MAS rotors and frozen. Final dopant concentrations in protofibril and fibril samples were approximately 1 mM. Final high pH and neutral pH samples for ssNMR contained 0.3 mg of Aβ40. Final protofibril and fibril samples contained roughly 5 mg of Aβ40.
Results
Biophysical characterization of Aβ40 assemblies
Fig. 1 shows negatively-stained TEM images of Aβ40 at the four stages of self-assembly described above. From TEM images alone, one can not determine the abundance of aggregated Aβ40 species relative to Aβ40 monomers, especially in the early and intermediate stages of assembly. Four independent techniques were therefore used to characterize the distributions of Aβ40 species. First, PICUP experiments were performed, as previously introduced by Teplow and coworkers for studies of Aβ self-assembly30a,30b. The light exposure time and concentrations of cross-linking reagents were optimized to produce cross-linked oligomer bands in SDS-PAGE that differentiated among various stages of self-assembly (Fig. S2). Results in Figs. 2A and 2B with 0.5 s exposure indicate negligible oligomer formation in the high pH sample, as the intensities of SDS-PAGE bands for cross-linked Aβ40 trimers and tetramers in the high pH sample are no greater than the corresponding bands for the highly soluble protein HP35 that was used as a non-aggregating control. In contrast, neutral pH and protofibril samples show larger trimer and tetramer bands. The dimer bands for HP35 and high pH Aβ40 are attributable to cross-linking of monomers that collide in solution within the lifetime of free radical species generated by the PICUP technique.
Figure 2.
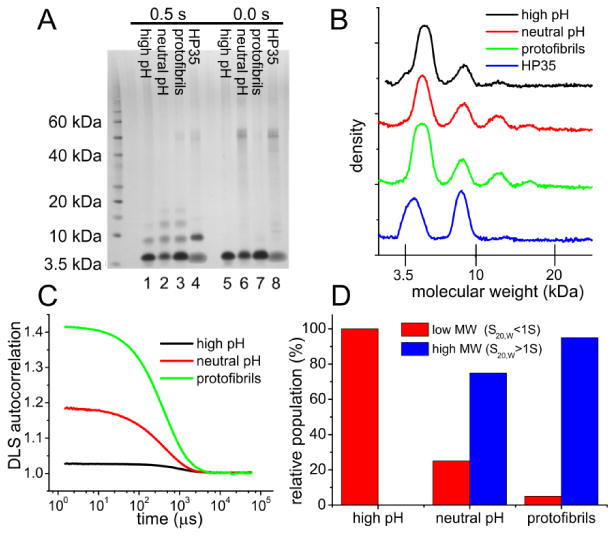
Biophysical characterization of Aβ40 assemblies. (A) SDS-PAGE gel of Aβ40 assemblies after PICUP cross-linking with 0.5 s and 0.0 s light exposure times. A 2 mM solution of the 4.06 kDa protein HP35 in 50 mM phosphate buffer at pH 7.4 is used as a non-aggregating control. (B) Stain density profiles from lanes 1-4 of the SDS-PAGE gel. (C) DLS autocorrelation functions, with the amplitudes before 10 μs indicating the relative concentrations of large aggregates. (D) Populations of small (sedimentation coefficient S20,w< 1 S) and large (S20,w > 1 S) species determined by sedimentation velocity measurements. Here S20,wrepresents the sedimentation coefficient normalized to the standard conditions of 20° C and pure water solvent.
Second, DLS data in Fig. 2C indicate a near absence of large Aβ40 assemblies in the high pH sample, and a higher concentration of large assemblies in the protofibril sample than in the neutral pH sample. DLS measurements were performed within 2 min of initial preparation of the high pH sample, and within 2 min of pH adjustment for the neutral pH sample. In these measurements, large assemblies are defined to be those for which the DLS autocorrelation time is greater than 100 μs, corresponding roughly to particles with hydrodynamic radii greater than 10 nm. (Note that DLS measurements also indicated a monomeric state for HP35 at the 2 mM concentration used in the PICUP experiments.)
Third, sedimentation velocity measurements were performed on high pH, neutral pH, and protofibril samples. Results are summarized in Fig. 2D. For each sample, absorption and interference data sets were analyzed in terms of a continuous distribution c(s) of sedimenting species33. Data for the high pH sample at 2.5 mM Aβ40 concentration (Fig. S3A) indicate a single species with sedimentation coefficient s20,w= 0.37 S and estimated molar mass of 2.3 kDa. Due to the high peptide concentration, repulsive non-ideality reduces the sedimentation coefficient and the apparent molar mass relative to the expected values for Aβ40 monomers. Data for the neutral pH sample at 2.0 mM Aβ40 concentration (Fig. S3B) indicate a distribution of aggregates, accounting for approximately 75% of the loading signal, with sedimentation coefficients broadly distributed around 11 S and estimated molar masses around 0.4 MDa. The remaining material is comprised of slowly sedimenting species near 0.6 S with an apparent molar mass of 4.7 kDa, close to the expected value of 4.3 kDa for monomeric Aβ40. Data for the protofibril sample at 2.0 mM Aβ40 concentration (Fig. S3C) indicate that approximately 95% of the material consists of rapidly sedimenting species, with a broad distribution of sedimentation coefficients around 18 S and apparent molar masses of roughly 10 MDa. For comparison, a single protofibril with a molecular structure similar to that described by Qiang et al.17c for D23N-Aβ40 and with a length of 200 nm would have a molar mass of 1.8 MDa, suggesting that the major species observed in sedimentation velocity experiments are bundles of protofibrils.
For neutral pH and protofibril samples, sedimentation velocity experiments were also performed on diluted samples (see Fig. S2D and S2E). The resulting data indicate dissolution of the large assemblies during these experiments, producing larger fractions of monomeric Aβ40 after dilution. From these data, it appears that the quasi-equilibrium solubilities of Aβ40 assemblies in the neutral pH and protofibril samples are greater than 10 μM, consistent with low thermodynamic stability relative to mature fibrils, for which the quasi-equilibrium solubilities are typically less than 1 μM40.
Based on the TEM, PICUP, DLS, and sedimentation velocity measurements, we conclude that our high pH Aβ40 samples are primarily (>90%) monomeric and that our neutral pH samples are primarily (roughly 75%) oligomeric, with oligomerization numbers ranging from about 10 to more than 100. Our protofibril samples are primarily (>90%) protofibrillar assemblies.
High pH, neutral pH, and protofibril samples were also analyzed by SEC (Fig. S4), but results from SEC are deemed unreliable due to interactions of Aβ40 with the SEC column material and dissolution of Aβ40 assemblies during chromatographic elution. For the high pH sample, a single chromatographic peak was observed when the SEC column was equilibrated with 20 mM NaOH, but at an apparent molecular weight of approximately 150 kDa. This observation is attributable to interaction of negatively charged Aβ40 molecules with the negatively charged agarose matrix of the column, which causes the protein to elute at an earlier volume41. Addition of 150 mM NaCl to the running buffer shifted the peak to a later value, corresponding to an apparent molecular weight of approximately 20 kDa. The discrepancy from the monomer value of 4.3 kDa may be attributable in part to the non-globular structure of monomeric Aβ4042. SEC data for the neutral pH sample also showed primarily a single peak, at an elution volume similar to that observed for the high pH sample. It should be noted that the width of this peak corresponds to a volume of roughly 1 ml, whereas the injected sample volume was 50 μl. Dilution of the neutral pH sample during the SEC measurement may then reduce the apparent concentration of larger assemblies. SEC data for protofibrils show a broad distribution of assembly sizes, including a large fraction of material with apparent molecular weight greater than 1 MDa.
Conformational evolution of Aβ40 probed by DNP-enhanced ssNMR
Structures of Aβ40 assemblies at the four stages described above were examined by ssNMR measurements on frozen solutions. As in previous DNP-enhanced ssNMR studies of biopolymers27-29, glycerol and paramagnetic dopants were added to produce glassy solutions amenable to DNP by the cross-effect mechanism26c,26e. In experiments on high pH and neutral pH samples, glycerol and dopants were added immediately before loading the solutions into MAS rotors and immersing the rotors in liquid nitrogen. In experiments on protofibrils and fibrils, samples were pelleted by ultracentrifugation after addition of glycerol and paramagnetic dopants before these samples were frozen. CD measurements, shown in Fig. 3, suggest that addition of glycerol may stabilize β-strand-like conformations of Aβ40 in high pH and neutral pH samples. The metastability of protofibrils8a,17c and stability of fibrils implies that structural changes during ultracentrifugation were negligible. Samples were subsequently kept below 100 K at all times.
Figure 3.
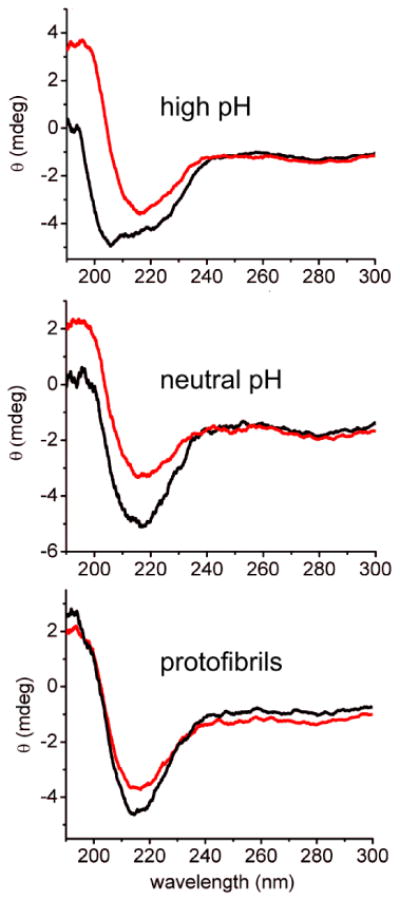
Circular dichroism spectra of high pH, neutral pH, and protofibril samples at 2.0 mM Aβ40 concentration (black spectra) and after mixing with an equal volume of glycerol (red spectra)
Fig. 4 compares one-dimensional, cross-polarized 13C NMR spectra of high pH, neutral pH, protofibril, and fibril samples in the frozen glycerol/water solutions with and without microwave irradiation, at sample temperatures near 25 K and with 6.7 kHz MAS. DNP signal enhancement factors in these spectra are in the 18-80 range. Without these DNP enhancements, the ssNMR measurements described below would have been prohibitively time-consuming.
Figure 4.
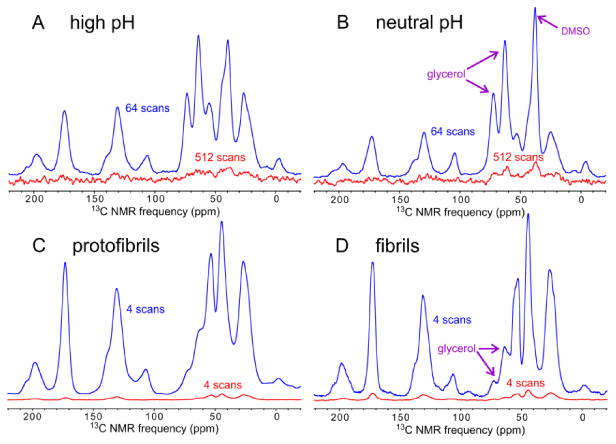
DNP enhancements of ssNMR signals from Aβ40 assemblies in frozen, triradical-doped glycerol/water solutions. (A-D) One-dimensional, cross-polarized 13C spectra of high pH, neutral pH, fibril, protofibril, and high pH samples, respectively, prepared with isotopic labeling pattern I in Table 1. Spectra were obtained at 100.8 MHz 13C NMR frequency, 25 K sample temperatures, and 6.7 kHz MAS frequency. Spectra with (blue) and without (red) microwave irradiation at 264.0 GHz are plotted on the same vertical scale in each panel and with the indicated number of scans for each spectrum. DNP enhancement factors, defined as the ratio of signal amplitudes with and without microwave irradiation, are 80, 60, 32, and 18 in panels A-D, respectively. Recycle delays are 5.6 s, 5.0 s, 25.0 s, and 30.0 s.
DNP enhancement factors for protofibril and fibril samples are lower than those for high pH and neutral pH samples. We attribute this observation in part to binding of triradical dopants to the protofibrils and fibrils, producing high local concentrations of dopants that render DNP less effective. As discussed above, nominal dopant concentrations in protofibril and fibril samples were deliberately lower than in high pH and neutral pH samples. Higher dopant concentrations were found to produce lower enhancement factors for protofibrils and fibrils. Enhancement factors for protofibrils and fibrils are also reduced by the higher density of Aβ40 assemblies in these samples, which reduces the local 1H spin-lattice relaxation times.
Fig. 5 shows 2D 13C-13C NMR spectra of Aβ40 samples with isotopic labeling pattern I (see Table 1), obtained with 23.8 ms spin diffusion mixing periods. 13C NMR lines for uniformly 15N,13C-labeled residues F19 and L34 are quite broad in high pH and neutral pH samples (4.4-7.4 ppm full width at half maximum, FWHM), are somewhat sharper in protofibrils (3.0-5.2 ppm FWHM), and are sharpest in fibrils (2.4-3.2 ppm FWHM). This progression (see Fig. 6) indicates an increasing degree of conformational order. Although 13C ssNMR linewidths in the 2.4-3.2 ppm range are still large compared with linewidths in ssNMR spectra of well-structured, hydrated proteins near room temperature, 13C ssNMR linewidths of well-structured peptides and proteins in frozen glycerol/water solutions are commonly 2 ppm or greater24,26d. 13C ssNMR linewidths in Fig. 5 are not determined by transverse spin relaxation rates (see Table S1).
Figure 5.
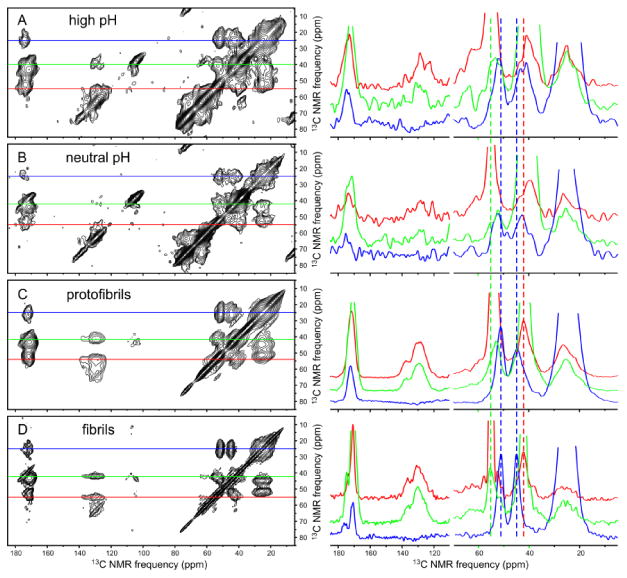
DNP-enhanced 2D 13C ssNMR spectra of Aβ40 assemblies with isotopic labeling pattern I. (A) High pH sample, acquired with 5.6 s recycle delay, 2.7 ms maximum t1 period, total measurement time of 5.0 h, processed with 150 Hz Gaussian apodization in both dimensions. (B) Neutral pH sample, acquired with 5.7 s recycle delay, 2.7 ms maximum t1 period, total measurement time of 5.0 h, and 150 Hz Gaussian apodization. (C) Protofibril sample, acquired with 10.0 s recycle delay, 3.9 ms maximum t1 period, total measurement time of 1.5 h, and 50 Hz Gaussian apodization. (D) Fibril sample, acquired with 10.2 s recycle delay, 3.9 ms maximum t1 period, total measurement time of 1.7 h, and 50 Hz Gaussian apodization. 1D slices at 13C chemical shifts of L34 Cγ (blue), F19 Cβ (green), and F19 Cα (red) are shown to the right of each 2D spectrum. Vertical dashed lines indicate peak positions in the fibril spectrum. Samples were in frozen glycerol/water at 25 K with triradical dopants. Spectra were acquired with 6.7 kHz MAS and 23.8 ms spin diffusion mixing periods between t1 and t2 periods. Contour levels increase by successive factors of 1.3.
Figure 6.
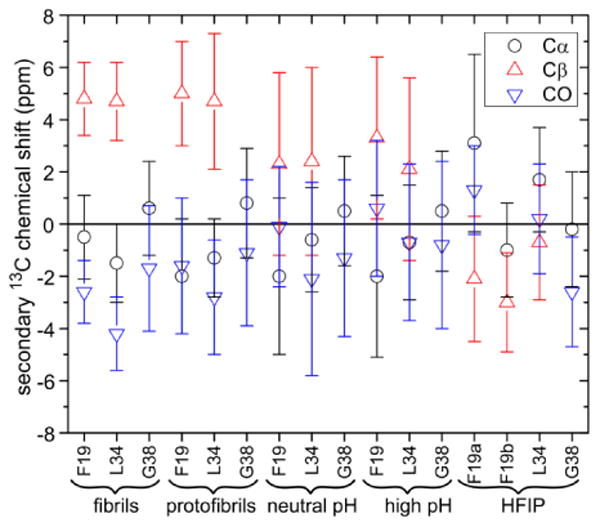
Secondary chemical shifts for carbonyl, Cα, and Cβ sites in F19, L34, and G38, relative to random coil shifts reported by Wishart et al.43 Error bars indicate the full widths at half maxima measured from crosspeaks in 2D ssNMR spectra of the Aβ40 samples. In addition to data from DNP-enhanced spectra of high pH, neutral pH, protofibril, and fibril samples in frozen solutions, data are shown for an Aβ40 powder prepared by lyophilization of an HFIP solution.
Average 13Cα and 13Cβ chemical shifts (i.e., chemical shifts at crosspeak maxima) for L34 are similar in protofibrils and fibrils (vertical blue dashed lines in Fig. 5). For F19, 13Cα chemical shifts of protofibrils and fibrils are significantly different (green dashed line), but 13Cβ chemical shifts are similar (red dashed line). 13C chemical shifts for F19, L34, and G38 are summarized in Fig. 6. 13Cαand 13CO secondary shifts (i.e., differences from random coil values43) are negative and 13Cβ secondary shifts are positive for both F19 and L34 in fibrils and protofibrils, indicating β-strand-like conformations at these residues44. 13CO secondary shifts for G38 are negative, also indicating a β-strand-like conformation. Average 13Cα secondary shifts for G38 in fibrils and protofibrils are similar but can not be interpreted in terms of conformational preference, as 13Cα secondary shifts for glycine residues in proteins do not correlate strongly with secondary structure.
Average 13C secondary shifts in neutral pH and high pH samples exhibit the same patterns (except for 13CO of F19), indicating a predominance of extended, β-strand-like conformations despite the greater conformational disorder in these samples. For comparison, 13C secondary shifts in a 2D 13C-13C NMR spectrum of a lyophilized Aβ40 powder after HFIP treatment, recorded at room temperature, are quite different and are consistent with predominant α-helical conformations at F19 and L34 (Fig. S5).
DNP-enhanced 2D 15N-13C NMR spectra of high pH, neutral pH, protofibril, and fibril samples were also acquired (Fig. S6). The reduced disorder in Aβ40 fibrils relative to the other samples is also apparent in these spectra.
Additional information about the Aβ40 conformation comes from the 2D 13C-13C NMR spectra in Fig. 7, which were obtained with 2.1 s RAD mixing periods. The spectrum of fibrils (Fig. 7D) shows a strong crosspeak between L34 methyl signals (near 25 ppm) and F19 aromatic signals (near 130 ppm), indicating proximity of L34 and F19 sidechains. This crosspeak is expected in the fibril spectrum, since previously reported structural models for Aβ40 fibrils include F19-L34 contacts within double-layered, parallel cross-β motifs, as depicted in Fig. 8A. Surprisingly, F19-L34 crosspeaks are also observed in 2D RAD spectra of high pH and neutral pH samples (Figs. 7A and 7B), suggesting that F19-L34 contacts are present even before structurally ordered assemblies develop. The F19-L34 crosspeak in the 2D RAD spectrum of protofibrils (Fig. 7C) is even more intense than in the spectrum of fibrils, suggesting a shorter distance between F19 and L34 sidechains in the protofibrils and providing an initial indication that the protofibril structure may be significantly different from the fibril structure (see below). F19-L34 crosspeak volumes, normalized to intra-residue L34 crosspeaks in the same spectra, are quantified in Fig. 8B.
Figure 7.
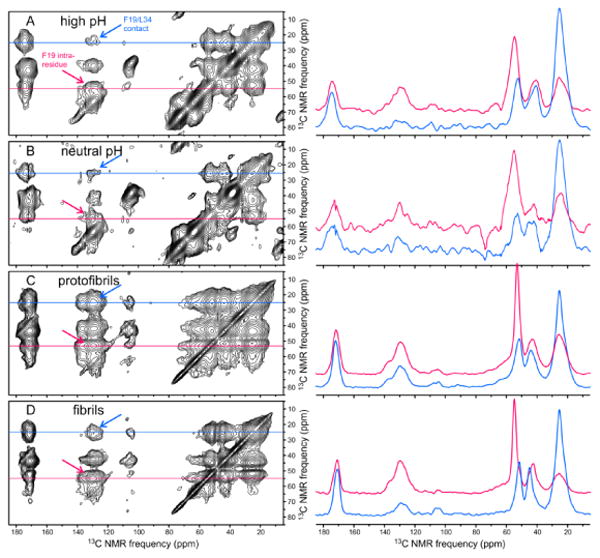
DNP-enhanced 2D 13C ssNMR spectra of Aβ40 assemblies, obtained with 2.1 s RAD mixing periods. (A-D) Spectra of high pH, neutral pH, protofibril, and fibril samples, with 1D slices at 13C chemical shifts of L34 Cγ(red) and F19 Cα (blue) shown to the right of each 2D spectrum. Samples were prepared with isotopic labeling pattern I. Sample temperatures, MAS frequencies, maximum t1 periods, and recycle delays were as in Figure 4. Total measurement times were 5.0 h, 5.0 h, 1.7 h, and 3.0 h, respectively. Gaussian apodizations were 250 Hz, 250 Hz, 150 Hz, and 150 Hz, respectively. Blue arrows indicate inter-residue crosspeaks between F19 aromatic and L34 aliphatic signals. Red arrows indicate crosspeaks between F19 aromatic and F19 Cα signals.
Figure 8.
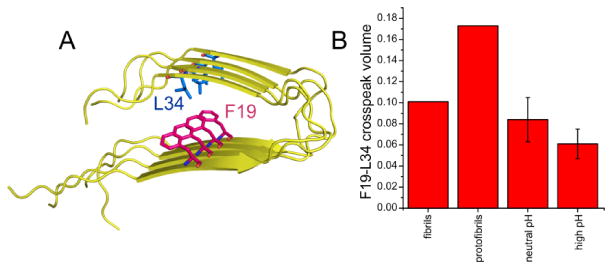
(A) Cartoon representation of a cross-β structural motif in Aβ40 fibrils, adapted from PDB file 2LMP, showing the proximity of F19 and L34 sidechains in the core of the double-layered parallel β-sheet structure that is known to exist in Aβ40 fibrils. (B) Inter-residue F19-L34 crosspeak volumes from 2D 13C ssNMR spectra in Fig. 5, normalized to the sum of intra-residue L34 CO-Cγ, Cα-Cγ, and Cβ-Cγ crosspeak volumes in the same spectra. Error bars for neutral pH and high pH data represent uncertainty due to the root-mean-squared noise in the experimental spectra. Uncertainties for fibril and protofibril data are at least five times smaller.
Liquid state NMR studies of full-length Aβ peptides in aqueous solution near neutral pH and at 4-10° C have shown that these peptides are largely unstructured in the unassembled state, 42 with partial population of β-strand conformations in certain segments, including segments that contain F19 and L34.42b In our experiments, the primarily monomeric state is prepared at pH 12 and higher Aβ40 concentrations, and glycerol is added prior to freezing. The freezing process occurs within several seconds, allowing conformational changes until the solvent approaches its glass transition temperature. These differences in sample preparation may produce differences in conformational distributions observed by liquid state NMR and ssNMR.
Supramolecular structure in Aβ40 protofibrils and fibrils
Aβ40 fibrils have been shown to contain cross-β structures comprised of in-register parallel β-sheets, meaning that identical residues of neighboring molecules align with one another within the β-sheets (through intermolecular hydrogen bonds between backbone amide and carbonyl groups of residue k in one β-strand and carbonyl and amide groups of residues k-1 and k+1 in a neighboring β-strand). Less is known about supramolecular structures in protofibrils. A double-layered antiparallel β-sheet structure has been identified in protofibrils formed by D23N-Aβ40 under the specific conditions described by Qiang et al.17c In these D23N-Aβ40 protofibrils, residues 17-21 form an antiparallel β-sheet with inter-strand hydrogen bonds between residue 19+k and residue 19-k, while residues 30-36 form an antiparallel β-sheet with inter-strand hydrogen bonds between residue 33+k and 33-k. It is not known whether the same antiparallel supramolecular structure exists in wild-type Aβ40. As one alternative, it has been suggested that Aβ protofibrils may contain β-sheet structures comprised of β-hairpins, in which the β-sheets would involve both intermolecular and intramolecular hydrogen bonds among two separate β-strand segments in the Aβ sequence23,45. It should be emphasized that no intramolecular hydrogen bonds are present in existing ssNMR-based models for wild-type or mutant Aβ fibrils or D23N-Aβ40 protofibrils5a,15a-15c,15e,15f,17c,46, and that these models do not contain true β-hairpins.
The rf pulse sequence in Fig. 9A was used to measure intermolecular 13C spin polarization transfers between labeled carbonyl and aliphatic sites, which depend on intermolecular 13C-13C distances and hence on supramolecular structure. In this pulse sequence, longitudinal 13C polarization was first created by 1H-13C cross-polarization, a 13C “flip-back” pulse, and a period τz for dephasing of transverse polarization. A weak Gaussian-shaped π pulse at the carbonyl 13C NMR frequency was then either present or absent on successive free-induction decay (FID) acquisitions, effectively allowing carbonyl 13C polarization to be prepared selectively by taking the difference between successive FIDs. Carbonyl polarization was then allowed to transfer to other 13C sites during the polarization transfer period τpt. Values of τpt ranged from 0.6 ms to 4.0 s.
Figure 9.
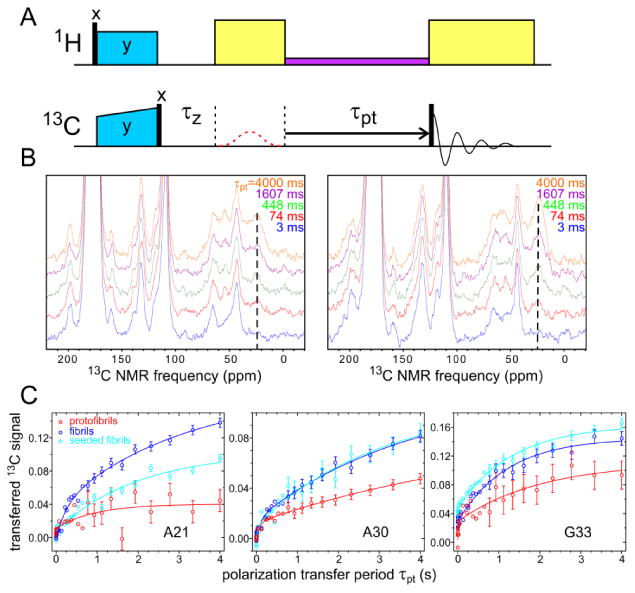
Characterization of supramolecular structures in Aβ40 protofibrils and fibrils by measurements of intermolecular 13C spin polarization transfers. (A) Radio-frequency pulse sequence for polarization transfer experiments, showing 1H-13C cross-polarization (blue period), a 3 ms “z-filter” dephasing period τz, a 0.6 ms Gaussian-shaped frequency-selective π pulse applied to carbonyl 13C sites on alternating signal acquisitions (dashed red line), the incremented polarization transfer period τpt with RAD irradiation (purple), and signal acquisition after a 13C π/2 pulse (black bars). Yellow blocks indicate 1H decoupling. (B) Examples of 13C difference spectra (with and without the frequency-selective π pulse) for Aβ40 protofibrils (left) and fibrils (right) with isotopic labeling pattern III, showing the growth of A30 Cβ signals with increasing τpt (dotted lines). (C) Polarization transfer curves for fibrils and protofibrils with isotopic labeling patterns II, III, and IV, showing the growth of signals from A21 Cβ sites, A30 Cβ sites, and G33 Cα sites due to polarization transfers from A21, A30, and G33 carbonyl sites, respectively. For each curve, Cβor Cα signals in difference spectra, integrated over 7-10 ppm intervals, are normalized to integrated carbonyl signals in the same spectra. Solid lines are least-squares fits with a double exponential function, as described in the text. Seeded fibrils were grown by using sonicated fragments of Aβ40 fibrils with the three-fold symmetric structure described by Paravastu et al.15b as seeds.
Alternative ssNMR methods for characterizing supramolecular structure in Aβ assemblies, such as the dipolar recoupling techniques used in previous studies of selectively labeled amyloid fibrils5a,15b,16a,25a,47, were also considered but were ruled out by the relatively short transverse spin relaxation times in paramagnetically doped frozen solutions (Table S1) and the relatively low MAS frequencies that are achievable at low temperatures. It should also be noted that methyl group rotation is fully quenched near 25 K, so that (unlike the typical situation at temperatures above 100-150 K) methyl 13C labels have even stronger 1H-13C dipole-dipole couplings than methylene 13C labels.
As a test of the pulse sequence in Fig. 9A under low-temperature DNP conditions, measurements were performed on a sample of fibrils formed by a peptide comprised of residues 11-25 of Aβ (Aβ11-25), synthesized with 13C labels at the carbonyl site of F20 and the Cβ site of A21. Aβ11-25 fibrils have been the subject of previous structural studies by ssNMR, which show that F20 and A21 are contained in a β-strand segment25a, implying an intramolecular distance between carbonyl and Cβ labels of 3.4 ± 0.2 Å. For these measurements, Aβ11-25 fibrils (approximately 5 mg) were suspended in the same glycerol/water solvent as in measurements on Aβ40 assemblies, with 1 mM DOTOPA-3OH-Methoxy. Data in Fig. S7 show a build-up of Cβ NMR signals in difference spectra with increasing τpt on the 425 ms time scale, with a maximum polarization transfer of approximately 40%.
Measurements to probe supramolecular structure in Aβ40 fibrils and protofibrils were performed on samples II-IV (Table 1), in which assemblies were formed from mixtures of molecules with single 13C labels at carbonyl and aliphatic sites. To ensure random mixtures within the assemblies, carbonyl-labeled and aliphatic-labeled molecules were synthesized and purified simultaneously as described in the Methods section. Fig. 9B shows examples of 13C NMR difference spectra for protofibrils and fibrils prepared from Aβ40 sample III. With τpt= 3 ms, the difference spectra show strong carbonyl lines from the 13C-labeled A30 carbonyl site and natural-abundance carbonyl 13C sites (near 177 ppm) as well as weaker signals from glycerol (near 65 ppm) and MAS sidebands of carbonyl and glycerol signals (near 199 ppm, 132 ppm, 110 ppm, and 43 ppm). As τpt increases, signals from 13C-labeled A30 Cβ sites (near 25 ppm) increase, primarily due to intermolecular polarization transfers from 13C-labeled A30 carbonyl sites.
Difference spectra for all Aβ40 samples are shown in Fig. S8. Fig. 9C shows the dependences of A21 and A30 Cβ signal areas (for Aβ40 samples II and III) and G33 Cαsignal areas (for Aβ40 sample IV) on τpt. The normalized signal areas are calculated from the expression ameth/(acarb+ ameth), where ameth and acarb are Cβ (or Cα) and carbonyl signal areas in the difference spectra, so that the plots in Fig. 9C represent fractional polarization transfers. Data are shown for protofibrils and fibrils, prepared as described above, and also for fibrils that were grown from pre-formed Aβ40 fibril seeds with the three-fold symmetric molecular structures reported previously by Paravastu et al.15b (cyan symbols in Fig. 9C). In all cases, the data can be fit with double-exponential functions (solid lines in Fig. 9C) of the form S(τpt) = (S1+ S2) – S1 exp(-τpt / τ1) – S2 exp(-τpt / τ2). Values of the best-fit parameters are given in Table 2.
Table 2. Best-fit parameters for polarization transfer data in Figs. 7, S8, and S9.
| peptide sample | assembly type | S1 | S2 | τ1 (ms) | τ2 (ms) |
|---|---|---|---|---|---|
| Aβ40 II | protofibrils | 0.011±0.005 | 0.029±0.008 | 2.6±4.6 | 950±700 |
| fibrils | 0.036±0.014 | 0.140±0.024 | 250±110 | 3100±1600 | |
| seeded fibrils | 0.008±0.003 | 0.095±0.009 | 5.6±6.9 | 2000±440 | |
| Aβ40 III | protofibrils | 0.012±0.001 | 0.074±0.032 | 77±23 | 6200±3800 |
| fibrils | 0.015±0.003 | 0.101±0.018 | 102±41 | 3800±1300 | |
| seeded fibrils | 0.010±0.004 | 0.099±0.022 | 61±59 | 3200±1300 | |
| Aβ40 IV | protofibrils | 0.029±0.004 | 0.082±0.0.018 | 4.4±2.9 | 2000±990 |
| fibrils | 0.029±0.002 | 0.116±0.005 | 0.3±0.2 | 1210±140 | |
| seeded fibrils | 0.048±0.002 | 0.116±0.004 | 0.9±0.2 | 1330±130 | |
| Aβ11-25 | fibrils | 0.014±0.003 | 0.392±0.006 | 0.1±0.7 | 425±20 |
We attribute the rapidly-increasing signal components (parameters S1 and τ1) to intramolecular polarization transfers from carbonyl 13C labels or natural-abundance carbonyl 13C to natural-abundance aliphatic 13C, and from natural-abundance carbonyl 13C to aliphatic 13C labels. We attribute the slowly-increasing signal components (parameters S2 and τ2) to intermolecular polarization transfers from carbonyl 13C labels. For all fibril samples, S2≈ 0.12 ± 0.03. For fibrils prepared from Aβ40 samples II and III, τ2≈ 3 ± 1 s. The similar values of these parameters for different fibril samples indicates similar intermolecular carbonyl-Cβ distances for A21 and A30, as expected in an in-register parallel β-sheet structure. For fibrils prepared from Aβ40 sample IV, τ2≈ 1.2 ± 0.2 s. This smaller value of τ2 is also expected, because the nearest-neighbor intermolecular carbonyl-Cα distance for G33 is expected to be less than the nearest-neighbor intermolecular carbonyl- Cβ distances for A21 and A30. In an ideal in-register parallel β-sheet, these distances would be approximately 4.2 Å for G33 and 4.5 Å for A21 and A30, which would imply a ratio of approximately 1.5 for the polarization transfer rates in the crude approximation that these rates are strictly proportional to R-6, with R being the nearest-neighbor distance between carbonyl and aliphatic 13C labels.
Polarization transfer data for Aβ40 fibrils are also consistent with the results for Aβ11-25 fibrils described above and shown in Fig. S7. Assuming polarization transfer rates to be proportional to R-6, values of τ2 for Aβ40 fibrils should be about 5.4 times (for samples II and III) or 3.6 times (for sample IV) greater than the value of τ2 for Aβ11-25 fibrils. Results in Table 2 are in reasonable agreement with this crude prediction.
The observation that S2 for Aβ11‐25 fibrils is roughly 3-4 times larger than S2 for Aβ40 fibrils is attributable to the larger contribution of natural-abundance 13C to the total carbonyl NMR signal area in difference spectra of Aβ40 fibrils and to the fact that the Aβ40 fibrils are comprised of mixtures of carbonyl-labeled and aliphatic-labeled molecules. Natural-abundance 13C accounts for approximately 50% of the total carbonyl signal area acarb in the Aβ40 fibril samples, but only 17% in the Aβ11-25 fibrils. In addition, only about 50% of carbonyl-labeled Aβ40 molecules have an aliphatic-labeled nearest neighbor.
For all Aβ40 protofibrils, the polarization transfer data in Fig. 9C are fit with smaller and more variable values of S2 (0.02-0.10) and with more variable values of τ2 (0.1-10 s), compared with data for Aβ40 fibrils. The smaller polarization transfer amplitudes for Aβ40 protofibrils indicate longer intermolecular carbonyl-aliphatic distances for the labeled sites in protofibrils than in fibrils. Data for protofibrils prepared from Aβ40 sample II show very little polarization transfer, indicating nearest-neighbor intermolecular carbonyl-Cβ distances for A21 that greatly exceed 5 Å. Thus, these data support the absence of in-register parallel β-sheet structures in wild-type Aβ40 protofibrils.
In the structural model for D23N-Aβ40 protofibrils developed by Qiang et al.17c (Protein Data Bank file 2LNQ), nearest-neighbor intemolecular distances between G33 carbonyl and G33 Cα sites are 5.0 ± 0.2 Å, while nearest-neighbor intermolecular distances between A30 carbonyl and A30 Cβ sites or between A21 carbonyl and A21 Cβ sites are greater than 9 Å.If the same double-layered antiparallel β-sheet structure exists in wild-type Aβ40 fibrils, then one would expect the time scale for polarization transfer in protofibrils prepared from Aβ40 sample IV to be roughly three times greater than in fibrils prepared from Aβ40 sample IV, and the time scale for polarization transfer in protofibrils prepared from Aβ40 samples II and III to be more than 10 times greater than in the corresponding fibrils. Data in Fig. 9 do not agree completely with these expectations, particularly in the case of Aβ40 sample III. Therefore, our data do not prove that wild-type Aβ40 protofibrils have the same supramolecular structure as D23N-Aβ40 protofibrils.
Discussion
Summary of conclusions from DNP-enhanced ssNMR data
Experiments described above provide new information about the structural properties of intermediates in the Aβ40 assembly process. First, it is clear from the 13C NMR linewidths in Figs. 5 and 6 that the degree of conformational order increases progressively as assembly proceeds from the largely monomeric state prepared initially at high pH, to the largely oligomeric state that develops quickly after pH neutralization, to the metastable protofibrillar state that subsequently develops over a period of several hours, and finally to the mature fibrillar state that is produced by several rounds of sonication and incubation. Remarkably, the predominant molecular conformations are similar at all stages. In particular, average 13C secondary chemical shifts for carbonyl, Cα, and Cβ sites of F19, L34, and G38 indicate extended, β-sheet-like conformations in ssNMR spectra of high pH, neutral pH, protofibril, and fibril samples. Absolute values of secondary shifts for carbonyl sites are somewhat reduced in neutral pH and high pH samples, consistent with the absence of ordered β-sheet structures prior to the formation of protofibrils.
In addition, F19-L34 sidechain contacts are evident in the 2D spectra in Fig. 7, indicating that Aβ40 conformations similar to the U-shaped conformation in mature fibrils (Fig. 8A) are highly populated at all stages, even before formation of ordered β-sheets and before formation of oligomers that are large enough to be visible in TEM images. Thus, the predominant molecular conformation can be considered to be independent of the supramolecular structure in Aβ40 assemblies. In principle, disruption of this conformation might simultaneously prevent the formation of oligomers, protofibrils, and fibrils.
Protofibrils form within several hours, but then persist for at least 20 days at room temperature in the absence of sonication or agitation of the Aβ40 solution. Conversion to fibrils is accelerated by periodic sonication. This observation can be explained by the existence of a small population of fibrils that develops spontaneously from monomers or small oligomers, possibly during the same period when the majority of Aβ40 molecules are self-assembling into protofibrils. Sonication breaks the fibrils and protofibrils into shorter fragments, creating more fibril and protofibril ends and thereby accelerating the transfer of Aβ40 molecules from the less thermodynamically stable protofibrils to the more stable fibrils. This transfer presumably occurs by gradual dissolution of the less stable structures and extension of the more stable structures, as discussed by Qiang et al.40a Under the conditions of our experiments, nucleation of protofibril structures is apparently more rapid than nucleation of fibril structures, accounting for the much greater abundance of protofibrils than fibrils prior to sonication.
The metastability of protofibrils suggests that their molecular structures must be significantly different from those of fibrils, so that conversion of protofibrils to fibrils can not occur by internal structural rearrangements. In other words, protofibrils are “off-pathway” intermediates and are not simply defective fibrils. 13C spin polarization transfer data in Fig. 9 indicate a significant difference in supramolecular organization, implying that protofibril-to-fibril conversion requires a rearrangement of the hydrogen bonding patterns within β-sheets, which can not occur at an appreciable rate within an intact assembly. Data in Fig. 9 show that the in-register, parallel β-sheet structure of mature Aβ40 fibrils does not exist within Aβ40 protofibrils. Polarization transfer curves for protofibrils in Fig. 9C are consistent with antiparallel β-sheets, but the precise intermolecular alignment in the protofibrils can not be determined from these data.
It should be noted that the Aβ40 concentration and solvent composition in our experiments are quite different from those in physiological settings, as are many other factors. Therefore, the stages of Aβ40 self-assembly examined in our experiments are not necessarily relevant to the physiological self-assembly process.
Comparisons with previous studies of Aβ self-assembly intermediates
The first ssNMR studies of Aβ self-assembly intermediates were reported by Ishii and coworkers7c,7d, who studied large (∼650 kDa), long-lived, spherical Aβ40 assemblies that form at 4° C, pH 7.4, and 100 μM peptide concentration. 13C chemical shifts were found to be quite similar in lyophilized spherical assemblies and in Aβ40 fibrils, consistent with our data for smaller Aβ40 oligomers with shorter lifetimes. Measurements of intermolecular 13C-13C dipole-dipole couplings indicated a predominantly parallel intermolecular alignment in the large spherical assemblies7d, in contrast to our results. TEM images reported by Ishii and coworkers suggest a direct evolution of their spherical assemblies to mature fibrils after more than 50 h of incubation, without the development of a large population of metastable protofibrils and without sonication or agitation of the Aβ40 solution. Thus, it appears that the preference for parallel intermolecular alignment within large, spherical Aβ40 assemblies, under the conditions employed by Ishii and coworkers, may permit an “on-pathway” conversion of the spherical assemblies to fibrillar assemblies by internal structural rearrangements, without the dissolution discussed above. More recently, Ishii and coworkers have reported ssNMR studies of spherical Aβ42 assemblies, called “amylospheroids”, which they also found to contain predominantly parallel intermolecular alignments and molecular conformations similar to the Aβ40 conformation depicted in Fig. 8A.18b
Paravastu and coworkers performed ssNMR studies of small, metastable Aβ42 oligomers, prepared by dialysis from 100 μM peptide solutions that contain SDS7b. Again, 13C chemical shifts indicated similar molecular conformations in Aβ42 oligomers and Aβ42 fibrils, which were prepared separately without SDS. Measurements of intermolecular 13C-13C dipole-dipole couplings indicated the absence of in-register, parallel alignment in the oligomers, although short intermolecular distances between V36 carbonyl sites were detected. Consistent with our results for Aβ40 self-assembly intermediates, differences in β-sheet structure appear to account for the metastability and off-pathway nature of these Aβ42 oligomers.
Smith and coworkers20 performed studies of small Aβ42 oligomers (formed at 200 μM peptide concentration) that were metastable at 4° C, but which converted within 6 h to protofibrils at 37° C, and then to fibrils within 12 days. From a combination of ssNMR, atomic force microscopy, and other measurements, they concluded that these oligomers were pentameric or hexameric discs, in which the Aβ42 conformation was similar to that in Aβ42 fibrils but which lacked β-sheet organization.
Huster and coworkers performed ssNMR studies of Aβ40 protofibrils (formed at 900 μM peptide concentration) that were stabilized by interactions with the antibody B10AP19. Again, their 13C chemical shift data indicate similar conformations in protofibrils and fibrils. Additionally, they observed long-range 13C-13C crosspeak signals between E22 and I31 in 2D ssNMR spectra of B10AP-stabilized protofibrils that are absent from spectra of Aβ40 fibrils, an observation that they interpret as evidence for a significant difference in β-sheet structure.
Reif and coworkers21 performed ssNMR studies of small Aβ40 oligomers (formed at 100 μM peptide concentration) that were stabilized by interactions EGCG, a compound from green tea that is reported to render the oligomers nontoxic. Their data indicate a longer disordered N-terminal tail in EGCG-stabilized oligomers than in Aβ40 fibrils, but otherwise a similar molecular conformation, including the presence of D23-K28 salt bridges in the oligomers.
Madhu and coworkers investigated structural differences between Aβ40 fibrils and small oligomers formed at 25 μM Aβ40 concentration in volatile ammonium acetate buffer18a. 13C chemical shifts were found to be similar in residues 11-21 and 30-40, but significantly different in the N-terminal segment and in residues 23-28. F19-L34 crosspeaks were also observed, as discussed above, in 2D 13C ssNMR spectra of both fibrils and oligomers.
Härd and coworkers have reported ssNMR studies of protofibrils formed by a double cysteine mutant of Aβ42 in which a β-hairpin monomer conformation is enforced by an intramolecular disulfide linkage. From the ssNMR data and computational methods, they propose a structural model in which the protofibrils are stacks of Aβ42 hexamers with an overall β-barrel-like configuration. Although this is an intriguing model, it may not apply to unmodified Aβ protofibrils.
In summary, previous studies have examined Aβ self-assembly intermediates that were prepared according to diverse protocols. In agreement with our data for high pH, neutral pH, and protofibril samples in frozen glycerol/water solutions, the molecular conformations in all intermediates resemble the conformations in Aβ40 and Aβ42 fibrils. Specifically, hydrophobic segments containing residues 17-21 and 30-36 (or somewhat longer segments) adopt extended, β-strand-like conformations. The N-terminal and C-terminal extended segments are separated by a bend or loop, allowing them to interact with one another through sidechain-sidechain contacts (or possibly hydrogen bonds, in the case of self-assembly intermediates with β-hairpin conformations). However, it appears that most self-assembly intermediates do not contain the in-register, parallel β-sheet structure that has been found in all mature Aβ40 and Aβ42 fibrils to date.
Experiments described above are distinct from previous studies in that we have characterized structural changes in a series of successive stages of the Aβ40 self-assembly process, rather than focusing on a single species, we have trapped transient species in frozen solution, and we have used DNP to enhance the sensitivity of the ssNMR measurements, thereby avoiding the need for lyophilization.
Prospects for future studies
DNP-enhanced ssNMR measurements described above represent the first application of DNP to the structural characterization of transient or metastable species that are of biological relevance. Compared with earlier experiments on a protein folding intermediate without DNP24c, the quantity of labeled peptide in each measurement was reduced by a factor of approximately 15 and the time required to acquire 2D 13C ssNMR spectra was reduced by factors of 10-30, depending on linewidths. Thus, ssNMR experiments on transient states in a variety of processes become possible that were previously precluded by limitations on available protein quantities, solubility, and throughput.
Procedures for pH adjustment, mixing, and freezing employed in our experiments are slow, implying that self-assembly proceeds for at least several minutes before the neutral pH samples are frozen for ssNMR measurements. In future studies, we plan to examine earlier stages of self-assembly following a rapid switch from solvent conditions under which Aβ peptides are primarily monomeric to conditions that favor aggregation. Such studies will utilize rapid mixing and rapid freezing methods to trap intermediates on time scales ranging from milliseconds to seconds. With a similar approach, DNP-enhanced ssNMR studies of intermediates in protein folding, ligand binding, membrane insertion, and enzymatic processes are readily envisioned.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health. AP was supported by a postdoctoral research fellowship from the Human Frontier Science Program.
Footnotes
Supporting Information. Figs. S1-S8 and Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Glenner GG, Wong CW. Biochem Biophys Res Commun. 1984;120:885. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]; (b) Hardy JA, Higgins GA. Science. 1992;256:184. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]; (c) Dickson DW. J Neuropathol Exp Neurol. 1997;56:321. doi: 10.1097/00005072-199704000-00001. [DOI] [PubMed] [Google Scholar]; (d) Karran E, Mercken M, De Strooper B. Nat Rev Drug Discov. 2011;10:698. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 2.Terry RD, Masliah E, Salmon DP, Butters N, Deteresa R, Hill R, Hansen LA, Katzman R. Ann Neurol. 1991;30:572. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 3.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Proc Natl Acad Sci U S A. 1999;96:3228. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Tycko R, Wickner RB. Accounts Chem Res. 2013;46:1487. doi: 10.1021/ar300282r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tycko R. Prot Sci. 2014;23:1528. doi: 10.1002/pro.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Cell. 2013;154:1257. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Petkova AT, Leapman RD, Guo ZH, Yau WM, Mattson MP, Tycko R. Science. 2005;307:262. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]; (c) Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Science. 2006;313:1781. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]; (d) Watts JC, Condello C, Stohr J, Oehler A, Lee J, DeArmond SJ, Lannfelt L, Ingelsson M, Giles K, Prusiner SB. Proc Natl Acad Sci U S A. 2014;111:10323. doi: 10.1073/pnas.1408900111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Cohen ML, Kim C, Haldiman T, ElHag M, Mehndiratta P, Pichet T, Lissemore F, Shea M, Cohen Y, Chen W, Blevins J, Appleby BS, Surewicz K, Surewicz WK, Sajatovic M, Tatsuoka C, Zhang S, Mayo P, Butkiewicz M, Haines JL, Lerner AJ, Safar JG. Brain. 2015;138:1009. doi: 10.1093/brain/awv006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Neurobiol Aging. 1995;16:285. doi: 10.1016/0197-4580(95)00013-5. [DOI] [PubMed] [Google Scholar]; (b) Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. Nature. 2006;440:352. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]; (c) Noguchi A, Matsumura S, Dezawa M, Tada M, Yanazawa M, Ito A, Akioka M, Kikuchi S, Sato M, Ideno S, Noda M, Fukunari A, Muramatsu S, Itokazu Y, Sato K, Takahashi H, Teplow DB, Nabeshima Y, Kakita A, Imahori K, Hoshi M. J Biol Chem. 2009;284:32895. doi: 10.1074/jbc.M109.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Proc Natl Acad Sci U S A. 1998;95:6448. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Biochemistry. 2003;42:12749. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]; (b) Tay WM, Huang DT, Rosenberry TL, Paravastu AK. J Mol Biol. 2013;425:2494. doi: 10.1016/j.jmb.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chimon S, Ishii Y. J Am Chem Soc. 2005;127:13472. doi: 10.1021/ja054039l. [DOI] [PubMed] [Google Scholar]; (d) Chimon S, Shaibat MA, Jones CR, Calero DC, Aizezi B, Ishii Y. Nat Struct Mol Biol. 2007;14:1157. doi: 10.1038/nsmb1345. [DOI] [PubMed] [Google Scholar]; (e) Hoshi M, Sato M, Matsumoto S, Noguchi A, Yasutake K, Yoshida N, Sato K. Proc Natl Acad Sci U S A. 2003;100:6370. doi: 10.1073/pnas.1237107100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lasagna-Reeves CA, Glabe CG, Kayed R. J Biol Chem. 2011;286:22122. doi: 10.1074/jbc.M111.236257. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ladiwala ARA, Litt J, Kane RS, Aucoin DS, Smith SO, Ranjan S, Davis J, Van Nostrand WE, Tessier PM. J Biol Chem. 2012;287:24765. doi: 10.1074/jbc.M111.329763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Harper JD, Wong SS, Lieber CM, Lansbury PT. Chem Biol. 1997;4:119. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]; (b) Goldsbury C, Kistler J, Aebi U, Arvinte T, Cooper GJS. J Mol Biol. 1999;285:33. doi: 10.1006/jmbi.1998.2299. [DOI] [PubMed] [Google Scholar]; (c) Williams AD, Sega M, Chen ML, Kheterpal I, Geva M, Berthelier V, Kaleta DT, Cook KD, Wetzel R. Proc Natl Acad Sci U S A. 2005;102:7115. doi: 10.1073/pnas.0408582102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kheterpal I, Chen M, Cook KD, Wetzel R. J Mol Biol. 2006;361:785. doi: 10.1016/j.jmb.2006.06.066. [DOI] [PubMed] [Google Scholar]
- 9.Deshpande A, Mina E, Glabe C, Busciglio J. J Neurosci. 2006;26:6011. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Arispe N, Pollard HB, Rojas E. Proc Natl Acad Sci U S A. 1993;90:10573. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lashuel HA, Lansbury PT. Q Rev Biophys. 2006;39:167. doi: 10.1017/S0033583506004422. [DOI] [PubMed] [Google Scholar]; (c) Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. J Biol Chem. 2005;280:17294. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 11.(a) Yan SD, Chen X, Fu J, Chen M, Zhu HJ, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. Nature. 1996;382:685. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]; (b) Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Nature. 2009;457:1128. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Huang XD, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE, Bush AI. Biochemistry. 1999;38:7609. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]; (b) Barnham KJ, Masters CL, Bush AI. Nat Rev Drug Discov. 2004;3:205. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]; (c) Parthasarathy S, Long F, Miller Y, Xiao YL, McElheny D, Thurber K, Ma BY, Nussinov R, Ishii Y. J Am Chem Soc. 2011;133:3390. doi: 10.1021/ja1072178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Gonzalez-Scarano F, Baltuch G. Annu Rev Neurosci. 1999;22:219. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]; (b) Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Cell. 2010;140:918. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farkas E, Luiten PGM. Prog Neurobiol. 2001;64:575. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 15.(a) Petkova AT, Yau WM, Tycko R. Biochemistry. 2006;45:498. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paravastu AK, Leapman RD, Yau WM, Tycko R. Proc Natl Acad Sci U S A. 2008;105:18349. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bertini I, Gonnelli L, Luchinat C, Mao JF, Nesi A. J Am Chem Soc. 2011;133:16013. doi: 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]; (d) Niu Z, Zhao WJ, Zhang ZF, Xiao FS, Tang XQ, Yang J. Angew Chem-Int Edit. 2014;53:9294. doi: 10.1002/anie.201311106. [DOI] [PubMed] [Google Scholar]; (e) Schutz AK, Vagt T, Huber M, Ovchinnikova OY, Cadalbert R, Wall J, Guntert P, Bockmann A, Glockshuber R, Meier BH. Angew Chem-Int Edit. 2015;54:331. doi: 10.1002/anie.201408598. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Sgourakis NG, Yau WM, Qiang W. Structure. 2015;23:216. doi: 10.1016/j.str.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 16.(a) Benzinger TLS, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, Meredith SC. Proc Natl Acad Sci U S A. 1998;95:13407. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Margittai M, Langen R. Q Rev Biophys. 2008;41:265. doi: 10.1017/S0033583508004733. [DOI] [PubMed] [Google Scholar]
- 17.(a) Tycko R, Sciarretta KL, Orgel J, Meredith SC. Biochemistry. 2009;48:6072. doi: 10.1021/bi9002666. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qiang W, Yau WM, Tycko R. J Am Chem Soc. 2011;133:4018. doi: 10.1021/ja109679q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Qiang W, Yau WM, Luo YQ, Mattson MP, Tycko R. Proc Natl Acad Sci U S A. 2012;109:4443. doi: 10.1073/pnas.1111305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Sarkar B, Mithu VS, Chandra B, Mandal A, Chandrakesan M, Bhowmik D, Madhu PK, Maiti S. Angew Chem-Int Edit. 2014;53:6888. doi: 10.1002/anie.201402636. [DOI] [PubMed] [Google Scholar]; (b) Parthasarathy S, Inoue M, Xiao Y, Matsumara Y, Nabeshima Y, Hoshi M, Ishii Y. J Am Chem Soc. 2015;137:6480. doi: 10.1021/jacs.5b03373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Scheidt HA, Morgado I, Rothemund S, Huster D, Fandrich M. Angew Chem-Int Edit. 2011;50:2837. doi: 10.1002/anie.201007265. [DOI] [PubMed] [Google Scholar]; (b) Scheidt HA, Morgado I, Huster D. J Biol Chem. 2012;287:22822. doi: 10.1074/jbc.M112.367474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO. Nat Struct Mol Biol. 2010;17:561. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lopez del Amo JM, Fink U, Dasari M, Grelle G, Wanker EE, Bieschke J, Reif B. J Mol Biol. 2012;421:517. doi: 10.1016/j.jmb.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 22.Gu L, Liu C, Stroud JC, Ngo S, Jiang L, Guo ZF. J Biol Chem. 2014;289:27300. doi: 10.1074/jbc.M114.569004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lendel C, Bjerring M, Dubnovitsky A, Kelly RT, Filippov A, Antzutkin ON, Nielsen NC, Hard T. Angew Chem-Int Edit. 2014;53:12756. doi: 10.1002/anie.201406357. [DOI] [PubMed] [Google Scholar]
- 24.(a) Havlin RH, Tycko R. Proc Natl Acad Sci U S A. 2005;102:3284. doi: 10.1073/pnas.0406130102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sharpe S, Kessler N, Anglister JA, Yau WM, Tycko R. J Am Chem Soc. 2004;126:4979. doi: 10.1021/ja0392162. [DOI] [PubMed] [Google Scholar]; (c) Hu KN, Yau WM, Tycko R. J Am Chem Soc. 2010;132:24. doi: 10.1021/ja908471n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R. J Mol Biol. 2004;335:247. doi: 10.1016/j.jmb.2003.10.044. [DOI] [PubMed] [Google Scholar]; (b) Tycko R, Hu KN. J Magn Reson. 2010;205:304. doi: 10.1016/j.jmr.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kloepper KD, Hartman KL, Ladror DT, Rienstra CM. J Phys Chem B. 2007;111:13353. doi: 10.1021/jp077036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Barnes AB, De Paepe G, van der Wel PCA, Hu KN, Joo CG, Bajaj VS, Mak-Jurkauskas ML, Sirigiri JR, Herzfeld J, Temkin RJ, Griffin RG. Appl Magn Reson. 2008;34:237. doi: 10.1007/s00723-008-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Thurber KR, Yau WM, Tycko R. J Magn Reson. 2010;204:303. doi: 10.1016/j.jmr.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Thurber KR, Tycko R. J Chem Phys. 2012;137:084508. doi: 10.1063/1.4747449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Thurber KR, Potapov A, Yau WM, Tycko R. J Magn Reson. 2013;226:100. doi: 10.1016/j.jmr.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hu KN, Song C, Yu HH, Swager TM, Griffin RG. J Chem Phys. 2008;128:052302. doi: 10.1063/1.2816783. [DOI] [PubMed] [Google Scholar]
- 27.(a) Bajaj VS, Mak-Jurkauskas ML, Belenky M, Herzfeld J, Griffin RG. Proc Natl Acad Sci U S A. 2009;106:9244. doi: 10.1073/pnas.0900908106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mao JF, Do NN, Scholz F, Reggie L, Mehler M, Lakatos A, Ong YS, Ullrich SJ, Brown LJ, Brown RCD, Becker-Baldus J, Wachtveitl J, Glaubitz C. J Am Chem Soc. 2014;136:17578. doi: 10.1021/ja5097946. [DOI] [PubMed] [Google Scholar]; (c) Smith AN, Caporini MA, Fanucci GE, Long JR. Angew Chem-Int Edit. 2015;54:1542. doi: 10.1002/anie.201410249. [DOI] [PubMed] [Google Scholar]
- 28.(a) Debelouchina GT, Bayro MJ, van der Wel PCA, Caporini MA, Barnes AB, Rosay M, Maas WE, Griffin RG. Phys Chem Chem Phys. 2010;12:5911. doi: 10.1039/c003661g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bayro MJ, Debelouchina GT, Eddy MT, Birkett NR, MacPhee CE, Rosay M, Maas WE, Dobson CM, Griffin RG. J Am Chem Soc. 2011;133:13967. doi: 10.1021/ja203756x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Potapov A, Thurber KR, Yau WM, Tycko R. J Magn Reson. 2012;221:32. doi: 10.1016/j.jmr.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sergeyev IV, Day LA, Goldbourt A, McDermott AE. J Am Chem Soc. 2011;133:20208. doi: 10.1021/ja2043062. [DOI] [PubMed] [Google Scholar]
- 30.(a) Bitan G, Lomakin A, Teplow DB. J Biol Chem. 2001;276:35176. doi: 10.1074/jbc.M102223200. [DOI] [PubMed] [Google Scholar]; (b) Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Proc Natl Acad Sci U S A. 2003;100:330. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu JX, Sharpe S, Ghirlando R, Yau WM, Tycko R. Prot Sci. 2010;19:1877. doi: 10.1002/pro.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKnight CJ, Doering DS, Matsudaira PT, Kim PS. J Mol Biol. 1996;260:126. doi: 10.1006/jmbi.1996.0387. [DOI] [PubMed] [Google Scholar]
- 32.Schneider CA, Rasband WS, Eliceiri KW. Nat Methods. 2012;9:671. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D. Biophys J. 2002;82:1096. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yau WM, Thurber KR, Tycko R. J Magn Reson. 2014;244:98. doi: 10.1016/j.jmr.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thurber KR, Tycko R. J Magn Reson. 2009;196:84. doi: 10.1016/j.jmr.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 37.(a) Morcombe CR, Gaponenko V, Byrd RA, Zilm KW. J Am Chem Soc. 2004;126:7196. doi: 10.1021/ja047919t. [DOI] [PubMed] [Google Scholar]; (b) Takegoshi K, Nakamura S, Terao T. Chem Phys Lett. 2001;344:631. [Google Scholar]
- 38.Qiang W. J Magn Reson. 2011;213:171. doi: 10.1016/j.jmr.2011.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song CS, Hu KN, Joo CG, Swager TM, Griffin RG. J Am Chem Soc. 2006;128:11385. doi: 10.1021/ja061284b. [DOI] [PubMed] [Google Scholar]
- 40.(a) Qiang W, Kelley K, Tycko R. J Am Chem Soc. 2013;135:6860. doi: 10.1021/ja311963f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kodali R, Williams AD, Chemuru S, Wetzel R. J Mol Biol. 2010;401:503. doi: 10.1016/j.jmb.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golovchenko NP, Kataeva IA, Akimenko VK. J Chromatogr. 1992;591:121. doi: 10.1016/0021-9673(92)80229-n. [DOI] [PubMed] [Google Scholar]
- 42.(a) Fawzi NL, Ying JF, Ghirlando R, Torchia DA, Clore GM. Nature. 2011;480:268. doi: 10.1038/nature10577. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Walti MA, Orts J, Vogeli B, Campioni S, Riek R. Chem Bio Chem. 2015;16:659. doi: 10.1002/cbic.201402595. [DOI] [PubMed] [Google Scholar]
- 43.Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. J Biomol NMR. 1995;5:67. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 44.(a) Wishart DS, Sykes BD, Richards FM. J Mol Biol. 1991;222:311. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]; (b) Spera S, Bax A. J Am Chem Soc. 1991;113:5490. [Google Scholar]; (c) Saito H. Magn Reson Chem. 1986;24:835. [Google Scholar]
- 45.Sandberg A, Luheshi LM, Sollvander S, de Barros TP, Macao B, Knowles TPJ, Biverstal H, Lendel C, Ekholm-Petterson F, Dubnovitsky A, Lannfelt L, Dobson CM, Hard T. Proc Natl Acad Sci U S A. 2010;107:15595. doi: 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Doeli H, Schubert D, Riek R. Proc Natl Acad Sci U S A. 2005;102:17342. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lansbury PT, Costa PR, Griffiths JM, Simon EJ, Auger M, Halverson KJ, Kocisko DA, Hendsch ZS, Ashburn TT, Spencer RGS, Tidor B, Griffin RG. Nat Struct Biol. 1995;2:990. doi: 10.1038/nsb1195-990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.