

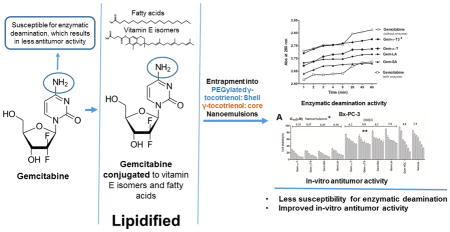
Abstract
Gemcitabine is the first line therapy for pancreatic cancer. It is, however, extensively metabolized to the inactive form by deamination enzymatic reaction. Conjugation of gemcitabine with fatty acids on its 4-amino group was found to protect it from deamination deactivation reaction. The objective of the present study was to test the in-vitro anticancer activity of gemcitabine conjugated to the γ-tocotrienol isomer of vitamin E against pancreatic tumor cells. This objective was based on reported studies in which it was demonstrated that free tocotrienol isomers of vitamin E can potentiate the anticancer activity of gemcitabine. To accomplish this objective, a full synthesis scheme for gemcitabine conjugation to fatty acids (stearic and linoleic) and the tocopherol and tocotrienol isomers of vitamin E (α-T and γ-T3) was presented. The conjugates were characterized by 1H-NMR and mass spectrometry analysis and tested for their susceptibility to deamination. Also discussed is the impact of entrapping the conjugates into nanoemulsions on the physiochemical properties of the delivery system and the in vitro anticancer activity of gemcitabine against Bx-PC-3 and PNAC-1 pancreatic cancer cells. In-vitro enzymatic deamination study showed that the γ-T3 conjugate of gemcitabine was least affected by deamination deactivation reaction when compared with the free and conjugated gemcitabine in solution. Furthermore, in-vitro cytotoxicity study demonstrated that entrapment of gemcitabine-lipid conjugates into nanoemulsions significantly enhanced their anticancer activity when compared to the free drug. It was concluded that conjugation to the γ-T3 isomer is a viable option for gemcitabine delivery and is worthy of further investigation.
Keywords: Gemcitabine, vitamin E, tocopherols, tocotrienols, lipid-conjugation, synthesis, 1H-NMR characterization, enzymatic deamination, nanoemulsions, in-vitro anticancer activity
Graphical Abstract
1. Introduction
Gemcitabine is the first line treatment for pancreatic cancer [1]. Tumor cells, however, often develop resistance to gemcitabine therapy by different mechanisms [2, 3], one of which is by rapid deamination of the free amino group on the pyrimidine ring structure of gemcitabine upon its entry into the cells [4]. Because of its rapid deamination, gemcitabine has also been reported to have a very short half-life (8–17 min) [5]. Consequently, in order to achieve therapeutic drug levels, gemcitabine is administered as a 30-min intravenous infusion at a dose of 1000 mg/m2 [5]. To overcome deamination, a prodrug strategy has been explored whereby gemcitabine is conjugated with long chain fatty acids [6]. This strategy was shown to improve gemcitabine metabolic stability and in vivo cytotoxic activity [5]. Eli Lilly, for example, patented the synthesis of saturated and monounsaturated C18 and C20 long-chain 4-(N)-acylderivatives and 5′-esters of gemcitabine that showed better cytotoxic activity than the parent compound [5]. In addition to utilizing saturated fatty acids, there has been an interest in testing the antitumor activity of gemcitabine conjugates with polyunsaturated fatty acids (PUFA), including linoleic acid [7, 8].
It was also shown that lipid conjugation improves the lipid solubility of gemcitabine and thereby facilitating its entrapment into lipid nanoparticles leading to a further improvement in its anticancer activity [6]. For example, an increase in the survival rate of mice with murine metastatic leukemia (L1210 wt) was observed when gemcitabine was coupled with 1,1′,2-tris-nor-squalenic acid and the resultant 4-(N)-tris-nor-squalenoyl-gemcitabine (SQdFdC) was formulated into nanoparticles [9]. Similarly, 4- (N)-stearoyl-gemcitabine (GemC18), a stearic acid amide derivative of gemcitabine, was shown to have an improved antitumor activity in mouse models as compared to gemcitabine alone when the conjugate was entrapped into solid lipid nanoparticles [6].
Although numerous studies have been reported on the conjugation of gemcitabine with fatty acids, conjugation of gemcitabine with the γ-tocotrienol (γ-T3) and α-tocoperol (α-T) isomers of vitamin E has not been reported. Vitamin E refers to a family of four tocopherol (T) and four tocotrienol (T3) isomers [10]. Unlike T, T3 isomers possess potent anticancer activity [11] and were found to sensitizes pancreatic tumor cells to gemcitabine by suppressing NF-κB–mediated inflammatory pathways [12]. Therefore, the first objective of the current study was to synthesize and characterize gemcitabine conjugates with the γ-T3 and α-T isomers of vitamin E. As a control, gemcitabine conjugates with stearic and lineoleic acid were also synthesized via a conjugation reaction procedure using the reactive acyl chloride derivatives of the lipids. The currently available methods for the synthesis of gemcitabine-lipid conjugates use slower catalyst-based chemical reactions [3, 4] to facilitate conjugating the carboxylic group of the fatty acid to the amino group of gemcitabine [3, 7].
Since the entrapment of lipid conjugates into nanoparticles is expected to enhance their anticancer activity, the second objective of the current study was to entrap the gemcitabine-vitamin E conjugates into nanoemulsions and to examine their in-vitro anticancer activity against Bx-PC-3 and PANC-1 pancreatic cancer cells.
2. Materials and methods
2.1. Materials
Gemcitabine HCL was obtained from LC Laboratories (Woburn, MA). α-T and γ-T3 isomers were extracted from Tocotrol™ L50P, a Tocotrienol-rich fraction from palm oil (Fuji Health Science, Inc., Burlington, NJ). Stearoyl chloride, linoleoyl chloride, Di-tert-butyl dicarbonate (Boc2O), and dioxane were from TCI America Inc. (Portland, OR). 4-Dimethylaminopyridine (DMAP), Dimethylformamide (DMF), dichloromethane (DCM), trifluoroacetic acid (TFA), and pyridine were obtained from EMD Chemical Inc, (Gibbstown, NJ). Triethylamine, succinic anhydride, and oxalyl chloride were from Alfa Aesar (Ward Hill, MA). Ethyl acetate (EtOAc) was from Pharmco-AAPER (Brookfield, CT). Potassium hydroxide (KOH) was from BDH, VWR International LLC (West Chester, PA). Sodium sulfate (Na2SO4) was from AMRESCO LLC (Solon, OH). All chemicals and solvents were of reagent grade or higher and were used as supplied without further modification.
2.2. Synthesis of gemcitabine-lipid conjugates
Before conjugating the gemcitabine with vitamin E and fatty acids, the hydroxy groups of the gemcitabine were first protected by tert-butyloxycarbonyl groups (BOC) (Fig. 1). BOCylation of gemcitabine was carried out via a procedure adapted from Lansakara et al. [3] as follows; 200 mg of gemcitabine HCl in 13.3 mL of 1 N potassium hydroxide (KOH) was cooled to 4°C. To this solution, di-tert-butyl dicarbonate (Boc2O, 1.5 g) in anhydrous dioxane (13.3 mL) was added over 10 min. The reaction mixture was stirred at 22°C for 1 h, followed by an extraction with ethyl acetate (EtOAc) twice. The combined organic layers were washed with brine, dried over anhydrous sodium sulfate (Na2SO4), filtered and the solvent was removed by rotovap. As estimated by TLC, this step yielded a 50:50 mixture of gemcitabine BOCylated on just one hydroxy group and gemcitabine BOCylated on the two free hydroxy groups. Therefore, the above procedure of adding Boc2O was repeated to complete the reaction to give gemcitabine protected on both hydroxy groups. The crude product was purified by column chromatography (DCM: acetone, 1:1). The residue was dried by rotovap and N2 to yield ~ 235 mg Bocylated gemcitabine (Fig. 1). For gemcitabine-vitamin E conjugates synthesis, α-T and γ-T3 succinate derivatives were first synthesized (Figs. 2 and 3). The reaction procedure was adapted from Constantinou et al. [13] as follows; a mixture of 200 mg of γ-T3 or α-T3 in pyridine was stirred at 20°C. A catalytic quantity (approximately 20 mg) of 4-Dimethylaminopyridine (DMAP) and 400 mg of succinic anhydride were added to the reaction mixture. The reaction mixture was stirred for 20 h. The mixture was then poured into 100 mL of water and extracted with ethyl acetate (EtOAc) twice. The combined organic layers were washed with 5% HCL solution to remove the pyridine and finally washed for the second time with saturated sodium chloride (brine) solution. The solution was dried over anhydrous Na2SO4, and finally concentrated in vacuo to afford α-T succinate as a white crystal powder and γ-T3 as brownish yellow syrup. After the succinate formation, the carboxylic acid groups of the succinates derivatives were activated to acyl chloride derivative as follows (Figs. 2 and 3) [14]; a mixture of α-T succinate or γ-T3 succinate (~ 150 mg) and oxalyl chloride (0.15 ml) in dichloromethane (DCM) was stirred at room temperature. A catalytic quantity of DMF (50 μL) was added, and the mixture was stirred for 2 h. The solvent and oxalyl chloride were then removed under reduced pressure to afford the succinate chloride derivatives of α-T and γ-T3 isomers. For gemcitabine-fatty acids synthesis, the chloride derivatives of the stearic and linoleic acids were used (Figs. 4 and 5). The Bocylated gemcitabine (Fig. 1) was then conjugated with the acyl chloride derivatives of α-T and γ-T3 (Figs. 2 and 3), and with stearic and linoleic acids (Fig. 4 and 5) as the following; equimolar amounts of bocylated gemcitabine and acyl chloride derivatives of vitamin E isomers or fatty acids in DCM were stirred at room temperature. Approximately 50 μL of triethylamine (Et3N) was then added to the reaction mixtures and stirred for 7 h. The reaction residues were purified by column chromatography (EtOAc: hexane, 3:7). The Boc groups of gemcitabine-lipid conjugates were removed by adding trifluoroacetic acid (TFA) to the residues of the conjugates in DCM. The mixtures were stirred for 1 h at room temperature. Excess TFA was removed by co-distillation with DCM 4 times. The crude samples were chromatographed on silica gel (DCM: ethanol, 94:6) and finally concentrated in vacuo to afford off-white viscous Gem-α-T, orange viscous Gem-γ-T3, and off-white powders of Gem-SA and Gem-LA conjugates.
Fig. 1.

Synthesis scheme of the Bocylated gemcitabine
Fig. 2.

Synthesis scheme of the gemcitabine-α-tocopherol (Gem-α-T) conjugate
Fig. 3.

Synthesis scheme of the gemcitabine-γ-tocotrienol (Gem-γ-T3) conjugate
Fig. 4.

Synthesis scheme of the gemcitabine-stearic acid (Gem-SA) conjugate
Fig. 5.

Synthesis scheme of the gemcitabine- linoleic acid (Gem-LA) conjugate
2.3. 1H-NMR analysis of gemcitabine-lipid conjugates
Proton-NMR studies were carried out to confirm the formation of vitamin E and fatty acid conjugates of gemcitabine. Samples of each conjugate were prepared in CDCL3. JEOL Eclipse NMR spectrometer (JEOL USA, Inc., MA) operating at 400 MHz and 20°C was used for analysis. Delta™ NMR Data Processing Software (JEOL USA, Inc., MA) was used for data collection and spectral processing with chemical shifts reported in ppm (d). One-dimensional spectra were collected with 64 scans of 16 K points over 20 ppm and a recycle delay of 5s.
2.4. Mass spectrometry (MS) analysis of the gemcitabine-lipid conjugates
MS analysis was carried out using a JEOL AccuTOF™ time-of-flight mass spectrometer (JEOL Ltd., Tokyo, Japan) equipped with orthogonal spray electrospray ionization (ESI) ion source. Samples were dissolved in HPLC grade methanol, and 50 μL was injected through Rheodyne 6-port valve injector. The mass spectrometer was operated in positive-ion mode (ESI + ve) with needle voltage (2000 V). The atmospheric pressure interface potentials were set to the following values: orifice 1 = 55 V, ring lens voltage = 5 V and orifice 2 = 6 V. The detector voltage was set to 1900 V. Orifice 1 temperature was adjusted to 80°C with dissolving temperature at 250°C. Nebulizing and desolvation gas (N2) were adjusted to 2 and 5 L/min flow rate, respectively.
2.5. Preparation and characterization of gemcitabine nanoemulsions
Gemcitabine-lipid conjugates were loaded into a nanoemulsion delivery platform composed of γ-T3 as an oil core and γ-T-mPEG 2000 as an emulsifier. γ-T-mPEG 2000 was synthesized in our lab as described previously [15]. Nanoemulsions were prepared by the solvent evaporation method as follows; free γ-T3 (10 mg), gemcitabine-Lipid conjugates equivalent to 1.6 mg gemcitabine HCL, and γ-T-mPEG 2000 (10 mg) were solubilized in DCM. The mixture was then vortexed until a homogeneous phase was obtained. The organic solvent was removed under a stream of nitrogen at 60°C to form a thin film at the bottom of the vials. The dried film was then hydrated with 10 mL of preheated distilled water under vortexing to form translucent dispersions. Dispersions were then sonicated for 5 min using an ultrasonic homogenizer at 60% of its maximum capacity (Model 150VT, Biologics, Inc., Manassas, VA). The mean particle size of the nanoemulsions was measured by photon correlation spectroscopy (PCS) using a NicompTM380 ZLS submicron particle size analyzer (Particle Sizing System, Port Richey, FL) at 25°C and 90° lasers light scattering. Before analysis, samples were diluted with deionized water to avoid side scattering and to achieve a scattering intensity of 300 kHz. The number-weighted mean diameter of the particles was calculated based on Stokes–Einstein law by curve fitting of the correlation function. Zeta-potential of the samples in deionized water were measured using the same instrument under the zeta mode.
2.6. In vitro deamination enzymatic assay of gemcitabine-lipid conjugates
The susceptibility of the gemcitabine-lipid conjugates to deamination was evaluated using Human Cytidine deaminase (CDA) ELISA kit (BioVisiosn Inc., Milpitas, CA). The CDA enzymatic solution was diluted with 50 mM Tris-HCL buffer (pH 7.5) to make an enzymatic solution with a concentration of 0.25 μg/50μL. Solutions of free gemcitabine HCL (100 μM) or gemcitabine-lipid conjugates (solubilized in DMSO, 100 μM equivalent to gemcitabine HCL) were prepared in 50 mM Tris-HCL (pH 7.5). The solutions of the free gemcitabine or gemcitabine-lipid conjugates (150 μL) were pipetted into a 96-well white plate. The enzymatic solution (50 μL) was then added to each well, and the plate was placed on a shaker for 2 min. The absorbance of each well was measured at 280 nm for 60 min at 37°C using a Synergy 2 Multi-Mode BioTek plate reader (BioTek Instruments, Inc. Winooski, VT).
2.7. In vitro cytotoxicity of gemcitabine-lipid conjugates
The cytotoxicity of nanoemulsions loaded with the gemcitabine-lipid conjugates was evaluated against BxPC-3 and PANC-1 pancreatic cancer cells. Cells were obtained from ATCC™ (Manassas, VA). PANC-1 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM), whereas BxPC-3 cells were maintained RPMI-1640 Medium. All media were supplemented with 10% fetal bovine serum, 1% insulin, and 1% penicillin–streptomycin, all from Invitrogen Inc. (Carlsbad, CA). Cells at a density of 5000 cells/well (50 μL) were seeded into 96-well plates and incubated at 37°C with 5% CO2. After overnight incubation, 50 μL of fresh medium containing the treatment was added to the cells. After 72 h of incubation, the medium was replaced with 20 μL of CellTiter-Blue reagent (Promega Corporation, Madison, WI). The plates were allowed to incubate at 37°C for 1 h. Fluorescence was measured using a BioTek Synergy HT multimode microplate reader (BioTek, Winooski, VT). Cell viability was calculated as the percentage of cells remaining viable compared to the untreated cells.
2.8. Statistical analysis
The statistical significance in the in-vitro deamination enzymatic assay and the cytotoxicity data of the gemcitabine-lipid conjugates alone or in nanoemulsions was analyzed by ANOVA with Tukey post-test calculation. A difference of P value < 0.05 was considered to be statistically significant.
3. Results and Discussions
3.1. Synthesis and characterization of the gemcitabine-lipid conjugates
In this study, conjugation of gemcitabine with vitamin E isomers and fatty acids was carried out using the acyl chloride derivatives of the lipids. Methods that are currently available for the conjugation of gemcitabine with lipids use coupling reagents such as 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and 1-Hydroxy-7-azabenzotriazole (HOAt) [3], or protecting groups such as ethylchlorocarbonate [4, 7], which require either long reaction time (~40 h) or extremely low reaction temperature conditions (−15 °C). These requirements make the reaction procedures unfavorable. On the other hand, chloride derivatives of lipids required a maximum of 5 h to complete the reaction.
Before conjugation of the vitamin E isomers and the fatty acids to gemcitabine, the free hydroxy groups on gemcitabine were protected by BOC groups (Fig. 1). Gemcitabine HCL in KOH solution was mixed with Boc2O and stirred at room temperature for one hour. This step was repeated to ensure that both hydroxy groups were protected, which was confirmed by 1H-NMR. For vitamin E isomer conjugation, the α-T and γ-T3 isomers were first converted to succinate derivatives using 4-dimethylaminopyridine (DAMP) as a catalyst (Figs 2 and 3). The succinate formation was confirmed by 1H-NMR with ~95% yield. Dimethylformamide (DMF)-assisted reaction was employed on the succinate derivatives with oxalyl chloride to form the acyl chloride derivatives of α-T and γ-T3 isomers. The acyl chloride derivatives were also confirmed by 1H-NMR. The acyl chloride derivatives of fatty acids were provided by their suppliers. Acyl chloride derivatives of vitamin E (Figs. 2 and 3) and fatty acids (Fig. 4 and 5) were then reacted at room temperature with BOCylated gemcitabine in DMC and triethylamine (Et3N) to afford the gemcitabine-lipid conjugates with ~94% yield. Fig. 6 shows the 1H-NMR and MS spectra of the vitamin E and fatty acids conjugates of gemcitabine. The conjugation of gemcitabine to the individual lipids was confirmed by the formation of an amide bond, which resulted in downfield shifts of succinate protons of vitamin E isomers from 2.77 to 2.91 ppm and the α-carbon’s protons of the fatty acids from 2.55 to 2.75 ppm (f annotation in Fig. 6A, B, C, and D). The aromatic protons of gemcitabine in the conjugates were located at 7.95 ppm and 7.25 ppm (a annotation in Fig. 6A, B, C, and D). The protons of the sugar moiety of gemcitabine were located at 6.2–6.34 ppm, 4.35 ppm, 3.85–4.1 ppm, and 3.6–3.65 ppm (b, c, d, & e annotation in Fig. 6A, B, C, and D). Carbon chain signals of vitamin E isomers were located at 0.8–1.8 ppm (h annotation in Fig. 6A and B), and carbon chain’ protons of fatty acids were located at 0.8–2.3 ppm (g annotation in Fig. 6C and D). The synthesis of gemcitabine-lipid conjugates was further confirmed by time-of-flight mass spectrometry. The average molecular weights of Gem-α-T, Gem-γ-T3, Gem-SA, and Gem-LA were observed with peaks at m/z of 686.3, 673.3, 586.215 and 586.218, respectively (Na+ adducts), which were in agreement with the expected MWs of the conjugates (Fig. 6A, B, C, and D).
Fig. 6.

1H-NMR in CDCl3 and ESI mass spectra of gemcitabine-lipid conjugates; (A) Gemcitabine-α-tocopherol (Gem-α-T), (B) Gemcitabine-γ-tocotrienol (Gem-γ-T3), (C) Gemcitabine-stearic acid (Gem-SA) and (D) Gemcitabine-linoleic acid (Gem-LA)
3.2. Characterization of nanoemulsions loaded with gemcitabine-lipid conjugates
Entrapment of the conjugates into a delivery vehicle is necessary since the lipid conjugates of gemcitabine are water insoluble. Increasing evidence has pointed out that entrapment of gemcitabine into nanosystems enhances its anticancer activity, both in-vivo and in-vitro [3, 6, 16]. Therefore, in the current study gemcitabine conjugates were entrapped into a nanoemulsion platform using γ-T3-PEG conjugates that were previously synthesized by our group [15]. The size and zeta potential of the nanoemulsion were recorded and are shown in Fig. 7. Gem-α-T nanoemulsion had the largest particle (~250 nm) probably due to the unfavorable packing of the more lipophilic α-T content of the conjugate into the less lipophilic γ-T3 oil core of the nanoemulsions. Gem-LA nanoemulsions particles had the smallest size of approximately 150 nm, whereas Gem-γ-T3 and Gem-SA nanoemulsion particles were comparable in size (~200 nm). All nanoemulsions had sufficient zeta potential (Fig. 7). Higher zeta potential often indicates favorable stability for the nanosystem [17]. The PEG corona of the shell is also expected to impart stability to the particles. The long-term stability of the nanoemulsions, however, was not evaluated and was beyond the scope of the present investigation.
Fig. 7.

Size and zeta potential of the nanoemulsions loaded with the gemcitabine-lipid conjugates in water. Each value represents the mean ± SD for triplicate samples
3.3. In vitro deamination enzymatic assay of the gemcitabine-lipid conjugates
It has been reported that gemcitabine is rapidly and extensively deaminated by cytidine deaminase (CDA) in the blood, liver, kidney and other tissues to the inactive metabolite 2′,2′-fluorodeoxyuridine (dFdU), and excreted in the urine [4, 18]. Elevated CDA activity in some patients was shown to cause the resistance of leukemic cells to deoxycytidine analogs [18]. Other studies showed that overexpression of CDA confers resistance to gemcitabine in cell culture as well [19]. Several approaches have therefore been attempted to improve gemcitabine metabolic stability, and consequently, it’s in vivo cytotoxic activity. In one such approach, lipid conjugation to gemcitabine was found to reduce its enzymatic deamination after administration [5]. Eli Lilly, for example, patented monounsaturated C18 and C20 long-chain 4-amide derivatives of gemcitabine, which showed better cytotoxic activity than the parent compound [4]. Unsaturated C-18 4-amide derivatives of gemcitabine were also reported [4, 7]. Since this is the first report on the conjugation of gemcitabine to the α-T and γ-T3 isomers of vitamin E, it was necessary to examine their susceptibility to the deaminase enzymatic activity, which was carried out using in-vitro CDA ELISA kit. As shown in Fig. 8, a significant reduction in the absorbance intensity of gemcitabine (P value < 0.05) was observed when gemcitabine was added in the free form to the CDA enzyme solution, which indicated a fast deamination reaction as was previously observed [14]. On the other hand, gemcitabine-lipid conjugates showed higher absorption intensity compared to the free gemcitabine, which indicates that the free gemcitabine is more susceptible for enzymatic deamination. Gem-γ-T3 was least affected by enzymatic deamination as it showed comparable absorbance intensity to the free gemcitabine HCL when tested without the enzyme. This was followed by Gem-α-T and Gem-LA. Gem-SA was the most vulnerable conjugate for enzymatic degradation as its absorbance intensity was close to the free gemcitabine HCL solution with the enzyme (Fig. 8). The results from the in-vitro enzymatic deamination study demonstrated that γ-T3 conjugate of gemcitabine significantly (P-value < 0.05) enhanced the stability of gemcitabine against deamination when compared to the other conjugates.
Fig. 8.

In-vitro enzymatic deamination activity of the gemcitabine-lipid conjugates. Human recombinant cytidine deaminase (CDA) was used in the current study. 100 μM of free gemcitabine HCL or equivalents from gemcitabine-lipid conjugates were incubated with 0.25 μg of CDA at 37°C and the absorbance was recorded at λ = 220 nm for 60 min. the decrease in the absorbance intensity after CDA addition indicated that deamination reaction had taken place [12]. * P value < 0.05: indicates that Gem-γ-T3 was significantly less affected by deamination than the free gemcitabine HCL
3.4. In vitro cytotoxicity of nanoemulsions loaded with the gemcitabine-lipid conjugates
In the current study, the anticancer activity of gemcitabine-lipid conjugates was evaluated against Bx-PC-3 and PANC-1 pancreatic cancer cells (Fig. 9). Bx-PC-3 and PNAC-1 pancreatic cell lines were selected because they were derived from different parts of the pancreas from a patient with pancreatic adenocarcinoma. Bx-PC-3 was from the body of the pancreas with no evidence of metastasis whereas PANC-1 was derived from the head of the pancreas, with evidence of metastasis in peripancreatic lymph node [20]. PANC-1 is considered a more resistant cell line to gemcitabine chemotherapy than BxPC-3 cells [21]. This was evident in the current study where the free gemcitabine was more active on the Bx-PC-3 cell line than the PANC-1 cells. Gemictabine conjugates in DMSO had comparable anticancer activity with the free gemcitabine against both cell lines, with the exception of the γ-T3 conjugate that showed significantly higher anticancer activity than the other treatments (P value < 0.05). As demonstrated by the deaminase experiment, gemcitabine conjugated to the γ-T3 isomer was the least affected by enzymatic deamination (Fig. 9), which might explain the observed higher anticancer activity of this conjugate. These results demonstrated that the γ-T3 isomer of vitamin E holds greater promise for gemcitabine therapy than the other lipid conjugates. Entrapment of gemcitabine conjugates into nanoemulsions significantly enhanced their anticancer activity against both cell lines. This observation was in agreement with numerous studies that showed enhanced uptake and activity of drugs when entrapped into nanoparticles [6, 16, 22]. It is also probable that the enhancement in activity could be partially attributed to the free γ-T3 oily core and the γ-T3-PEG corona, both of which were found in previous studies to exert anticancer activity against pancreatic tumor cells [15]. Furthermore, it was reported that γ-T3 sensitizes tumor cells to gemcitabine chemotherapy [12]. No differences, nonetheless, were observed between the nanoemulsions as a function of conjugate types, with the exception of the linoleic acid conjugate, which was the least active against both cell lines.
Fig. 9.

In-vitro anticancer activity of gemcitabine-lipid conjugates nanoemulsions against pancreatic cancer cell lines; (A) Bx-PC-3 and (B) PANC-1. Cells were treated for 72 hr. Values reported are the mean ± SD for triplicate samples. * P value < 0.05: indicates that gemcitabine-lipid conjugates nanoemulsions had significantly higher anticancer activity than the gemcitabine-lipid conjugates in DMSO against both cell lines. ** P value < 0.05: indicates that the Gem-γ-T3 conjugate in DMSO had significantly higher anticancer activity than the other Gem-lipid conjugates in DMSO against both cell lines
4. Conclusion
Gemcitabine conjugates were successfully synthesized using the acyl chloride derivatives of vitamin E isomers and fatty acids instead of the common coupling reagents that are currently employed in the literature for gemcitabine conjugation, which was confirmed by 1H-NMR and mass spectrometry analysis. When tested for in-vitro enzymatic deamination, conjugates, as expected, were less susceptible to inactivation. Of the treatments, the γ-T3 conjugate was the least susceptible to deamination. Although this observation is not entirely understood, it may explain why the γ-T3 conjugate was the most potent treatment against both Bx-PC-3 and PANC-1 pancreatic cancer cells. Conjugates were also readily entrapped into 150–250 nm nanoemulsions that have been stabilized by γ-T3-mPEG 2000 as an emulsifier while using the free γ-T3 isomer as the core. Our previous studies showed that both γ-T3-mPEG 2000 and free γ-T3 exert anticancer activity, which explains the observed effect of the vehicle on the cells. Entrapment of the conjugates into the nanoemulsions also significantly enhanced their anticancer activity against both cell lines when compared to their effect when treated in DMSO or when compared to the free drug. No differences between the conjugates when entrapped into nanoemulsion, however, were observed in-vitro. It is probable, nonetheless, that the conjugates would perform differently when tested in animal models. In-vivo testing, however, was beyond the scope of the present investigation.
Acknowledgments
This work was supported in part by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103424. Drs. Jennifer L. Carroll and Ana-Maria Dragoi from the Feist-Weiller Cancer Center, Innovative Northwest Louisiana Experimental Therapeutics (INLET) at Louisiana State University Health Sciences Center, Shreveport, Louisiana, USA, are acknowledged for their assistance with the in-vitro cell culture studies. Acknowledgment is also extended to Dr. Robert Cody from JEOL USA, Inc. for his assistance with mass spectrometry analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Company, E.L.a. GEMZAR (gemcitabine for injection) leaflet. 2014 Available from: https://pi.lilly.com/us/gemzar.pdf.
- 2.Andersson R, et al. Gemcitabine chemoresistance in pancreatic cancer: molecular mechanisms and potential solutions. Scand J Gastroenterol. 2009;44(7):782–6. doi: 10.1080/00365520902745039. [DOI] [PubMed] [Google Scholar]
- 3.Lansakara PD, Rodriguez BL, Cui Z. Synthesis and in vitro evaluation of novel lipophilic monophosphorylated gemcitabine derivatives and their nanoparticles. Int J Pharm. 2012;429(1–2):123–34. doi: 10.1016/j.ijpharm.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Immordino ML, et al. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J Control Release. 2004;100(3):331–46. doi: 10.1016/j.jconrel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Immordino ML, et al. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. Journal of Controlled Release. 2004;100(3):331–346. doi: 10.1016/j.jconrel.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Sloat BR, et al. In vitro and in vivo anti-tumor activities of a gemcitabine derivative carried by nanoparticles. Int J Pharm. 2011;409(1–2):278–88. doi: 10.1016/j.ijpharm.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tao XM, et al. Enhanced anticancer activity of gemcitabine coupling with conjugated linoleic acid against human breast cancer in vitro and in vivo. Eur J Pharm Biopharm. 2012;82(2):401–9. doi: 10.1016/j.ejpb.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Naguib YW, et al. Synthesis, Characterization, and In Vitro and In Vivo Evaluations of 4-(N)-Docosahexaenoyl 2′, 2′-Difluorodeoxycytidine with Potent and Broad-Spectrum Antitumor Activity. Neoplasia. 2016;18(1):33–48. doi: 10.1016/j.neo.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu S, et al. Lysosomal delivery of a lipophilic gemcitabine prodrug using novel acid-sensitive micelles improved its antitumor activity. Bioconjug Chem. 2012;23(5):966–80. doi: 10.1021/bc2005945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abu-Fayyad A, et al. PEGylated γ-tocotrienol isomer of vitamin E: Synthesis, characterization, in vitro cytotoxicity, and oral bioavailability. Eur J Pharm Biopharm. 2015;96:185–95. doi: 10.1016/j.ejpb.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 11.Aggarwal B, Nesaretnam K. Vitamin E tocotrienols: life beyond tocopherols. Genes Nutr. 2012 Jan;7(1):1. doi: 10.1007/s12263-011-0234-x. Epub 2011 May 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunnumakkara AB, et al. γ-Tocotrienol Inhibits Pancreatic Tumors and Sensitizes Them to Gemcitabine Treatment by Modulating the Inflammatory Microenvironment. Cancer research. 2010;70(21):8695–8705. doi: 10.1158/0008-5472.CAN-10-2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Constantinou C, et al. Induction of DNA Damage and Caspase-Independent Programmed Cell Death by Vitamin E. Nutrition and Cancer. 2012;64(1):136–152. doi: 10.1080/01635581.2012.630167. [DOI] [PubMed] [Google Scholar]
- 14.Cai J, et al. Bone-targeting glycol and NSAIDS ester prodrugs of rhein: synthesis, hydroxyapatite affinity, stability, anti-inflammatory, ulcerogenicity index and pharmacokinetics studies. Eur J Med Chem. 2012;55:409–19. doi: 10.1016/j.ejmech.2012.07.053. [DOI] [PubMed] [Google Scholar]
- 15.Abu-Fayyad A, Nazzal S. Synthesis, characterization, and in-vitro antitumor activity of the polyethylene glycol (350 and 1000) succinate derivatives of the tocopherol and tocotrienol isomers of Vitamin E. Int J Pharm. 2017;519(1–2):145–156. doi: 10.1016/j.ijpharm.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sandoval MA, et al. EGFR-targeted stearoyl gemcitabine nanoparticles show enhanced anti-tumor activity. J Control Release. 2012;157(2):287–96. doi: 10.1016/j.jconrel.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim DM, et al. Identification of an emulsifier and conditions for preparing stable nanoemulsions containing the antioxidant astaxanthin. Int J Cosmet Sci. 2012;34(1):64–73. doi: 10.1111/j.1468-2494.2011.00682.x. [DOI] [PubMed] [Google Scholar]
- 18.Bouffard DY, Laliberté J, Momparler RL. Kinetic studies on 2′,2′-difluorodeoxycytidine (gemcitabine) with purified human deoxycytidine kinase and cytidine deaminase. Biochemical Pharmacology. 1993;45(9):1857–1861. doi: 10.1016/0006-2952(93)90444-2. [DOI] [PubMed] [Google Scholar]
- 19.Neff T, Blau CA. Forced expression of cytidine deaminase confers resistance to cytosine arabinoside and gemcitabine. Exp Hematol. 1996;24(11):1340–6. [PubMed] [Google Scholar]
- 20.Deer EL, et al. Phenotype and Genotype of Pancreatic Cancer Cell Lines. Pancreas. 2010;39(4):425–435. doi: 10.1097/MPA.0b013e3181c15963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Réjiba S, et al. Gemcitabine-Based Chemogene Therapy for Pancreatic Cancer Using Ad-dCK::UMK GDEPT and TS/RR siRNA Strategies. Neoplasia (New York, NY) 2009;11(7):637–650. doi: 10.1593/neo.81686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung WG, et al. Stearoyl gemcitabine nanoparticles overcome resistance related to the over-expression of ribonucleotide reductase subunit M1. J Control Release. 2012;157(1):132–40. doi: 10.1016/j.jconrel.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]