

Abstract
Purpose
Although most human cancers display a single histology, there are unusual cases where two or more distinct tissue types present within a primary tumor. One such example is metaplastic breast carcinoma, a rare but aggressive cancer with a heterogenous histology, including squamous, chondroid, and spindle cells. Metaplastic carcinomas often contain an admixed conventional ductal invasive or in situ mammary carcinoma component, and are typically triple-negative for estrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 (HER-2) amplification/overexpression. An unanswered question is the origin of metaplastic breast cancers. While they may arise independently from their ductal components, their close juxtaposition favors a model that postulates a shared origin, either as two derivatives from the same primary cancer, or one histology as an outgrowth of the other. Understanding the mechanism of development of these tumors may inform clinical decisions.
Experimental Design
We performed exome sequencing for paired metaplastic and adjacent conventional invasive ductal carcinomas in eight patients and created a pipeline to identify somatic variants and predict their functional impact, without having normal tissue. We then determined the genetic relationships between the histologically distinct compartments.
Results
In each case, the tumor components have nearly identical landscapes of somatic mutation, implying that the differing histologies do not derive from genetic clonal divergence.
Conclusions
A shared origin for tumors with differing histologies suggests that epigenetic or noncoding changes may mediate the metaplastic phenotype, and that alternative therapeutic approaches, including epigenetic therapies, may be required for metaplastic breast cancers.
Translational relevance
Metaplastic breast cancers are a rare but treatment refractory subtype of breast cancer. It remains unclear whether these tumors are separate cancers that arise independently from their ductal counterparts, or are genetically related, resulting from dedifferentiation of cells. Knowledge of the origin of metaplastic breast cancers may help to develop therapeutic strategies for this group of recalcitrant cancers. Using whole exome sequencing and new bioinformatics tools, we compared variants between paired breast cancer samples exhibiting invasive ductal carcinoma and metaplastic components. Based on the high degree of shared variants between paired tissue types from eight patients, we conclude that invasive ductal carcinomas with an associated metaplastic component are genetically related, and likely the result of epigenetic changes leading to multiple histologies. These results provide new insights into the origin of metaplastic breast cancers that could have implications for treatment strategies.
Keywords: metaplastic breast cancer, exome sequencing, cancer evolution, cancer histology, cancer genetic variation
Introduction
A primary cancer originates from cells with strongly oncogenic mutations that grow and accumulate additional mutations. At some point, cells from the cancer gain mutations that enable metastasis. A clinically apparent cancer thus harbors thousands of genetic and epigenetic changes, some of which contribute to the cancer phenotype and most of which do not. This complexity is compounded by tumor heterogeneity; expansion of cells with highly compromised DNA repair systems within the tumor creates geographically and genetically distinct populations. Metaplastic breast carcinoma presents an interesting challenge to this paradigm, as it is a histologically unique tumor but is often intertwined with other common histologic subtypes of breast carcinoma.
Metaplastic breast carcinoma is a heterogenous group of primary breast carcinomas that display heterologous differentiation, with or without an associated conventional invasive mammary carcinoma component (1). The heterologous elements may be pure or mixed and include squamous, chondroid, osseous, spindle and pleomorphic differentiation (1-5). Although accounting for less than ∼0.2-5% of all primary breast carcinomas, metaplastic carcinomas present a significant challenge, diagnostically and clinically. Histologically, metaplastic carcinomas of pure mesenchymal differentiation are difficult to distinguish from primary breast sarcomas, phyllodes tumors and metastatic sarcomas, which have differing therapies and prognoses. Metaplastic carcinomas are commonly triple-negative for the estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor 2 receptor (HER-2) (6) and thus do not benefit from targeted anti-hormonal or anti-HER-2 therapy. By molecular profiling, metaplastic carcinomas segregate as basal-like (7, 8) or claudin-low(8-11) phenotypes, though by transcription profiling, they subtype as mesenchymal-like, unstable or mixed type (8). They are more often resistant to neoadjuvant chemotherapy (12), more likely to present with metastatic disease (13-15), and carry a worse overall prognosis (12, 13, 16) than conventional triple-negative carcinomas.
The cell of origin of metaplastic carcinomas and the genomic relationship between the various histologic components of metaplastic carcinomas are unclear. Early studies suggested that the tumors are clonal, using several assays including karyotyping (17), loss of heterozygosity assays (18, 19), TP53 point mutations (20-23) and human androgen receptor (HUMARA) X-chromosome inactivation assays (21, 24, 25). Microarray comparative genomic hybridization and TP53 sequencing and X-chromosome inactivation assessment on six metaplastic carcinomas suggested clonality of the morphologically distinct components in four cases (26), but with some evidence of subclone development.
We propose three models for the evolution of tumors containing cells of mixed histology. Figure 1A depicts a model in which cells from two different primary tumors grow together as a single mass with intertwined histologies. A different scenario is shown in Figure 1B, in which two subclones from a primary tumor independently generate tumors with different histologies, that happen to occupy the same anatomical space. Finally, Figure 1C depicts a model in which a cell from a single subclone of a primary tumor forms a mass with two different histologies, created perhaps from secondary mutations or epigenetic changes. Interestingly, these models can be differentiated by the landscape of mutations shared by the two histologic subtypes. Mutations can be characterized by their expected biological impact. High impact variants include frameshift, stop gain, or splice site variants, which are likely deleterious, and possibly driver mutations. Low impact variants, including silent and evolutionarily tolerated synonymous changes, are often passenger mutations, with little effect on the cell. Modifier variants generally lie in noncoding regions and have uncertain impact. Moderate impact variants carry intermediate functional consequences. We reasoned that tumors that are independently propagated from two subclones of a primary tumor (Figure 1B) may share high-impact variants (as these are more likely to be tissue-specific driver mutations (27)), but their continued and separate evolution will produce low frequency, low-impact variants that will be unique to each lesion. In contrast, two sections from a tumor that has originated from a single cell (Figure 1C) will have a high rate of shared low-impact, low frequency variants, a high proportion of shared copy number and structural variants, and a common subclone structure. Importantly, a shared subclone structure implicates that many cells from a mature tumor adopted a divergent histology.
Figure 1. Models of intratumoral heterogeneity.
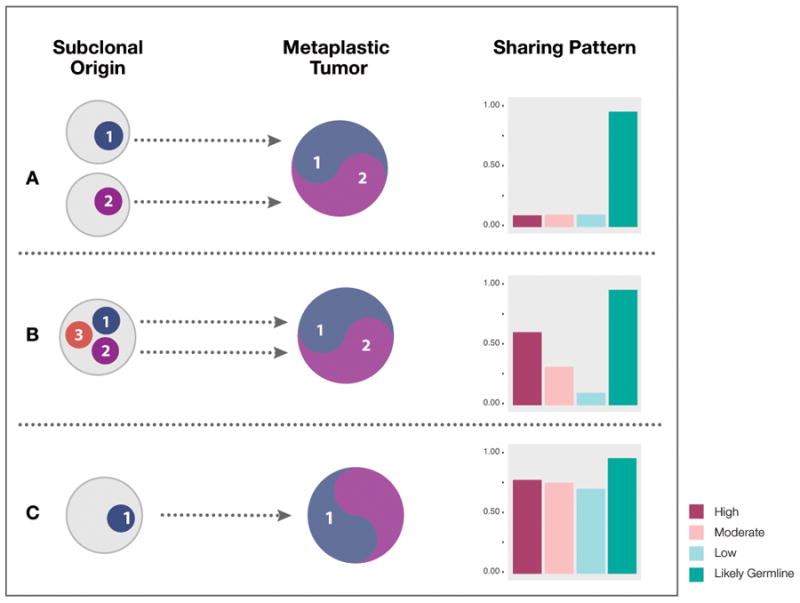
Mechanism A: The metaplastic and conventional ductal carcinoma components may arise from independent primary tumors. This trajectory would result in shared germline variants but few, if any, shared somatic variants in any impact category. Mechanism B: The metaplastic and conventional ductal carcinoma components arise from different subclones in the same primary tumor. This would result in shared high impact alleles (potential driver mutations), but minimal overlap in low impact alleles. Mechanism C: The metaplastic and conventional ductal carcinoma components appear as different phenotypes but share a genotype, as they are derived from a single clone. In this model, the ductal and metaplastic cells share both the high impact and low impact alleles, as well as copy number variation structure and subclone architecture.
In this study, we analyzed whole exome sequencing data from the metaplastic and conventional invasive ductal components of eight primary breast metaplastic carcinomas, and compared the types and frequencies of genetic variants in the two histological subtypes to determine clonality. Without sequencing data from normal tissue, we could not unequivocally determine whether a variant was somatic; therefore we filtered all variants against public databases, to assign somatic versus germline status to each variant in a tumor. In all eight cases examined, we evaluated single nucleotide variants (population SNPs as well as somatic changes), indels, copy number variation, and subclone structure, in the metaplastic and associated invasive components of the tumors. For all eight cases, we found a similar proportion of shared variants in all impact categories and at very similar allele frequencies, a high proportion of shared copy number variations and structural variants, and similar subclone structure. Our work suggests that the invasive ductal and metaplastic components are not separate cancers; rather, our study suggests that diverse histological appearances are manifested by single cancers.
Materials and methods
Case selection of metaplastic breast carcinoma
These studies were conducted in accordance with the U.S. Common Rule, with written consent of participants, and the Institutional Review Board of the Johns Hopkins Medical Institutions reviewed and approved of this study. We searched the pathology archives database for cases of surgically resected primary metaplastic breast carcinomas over the time period of 1995-2013. Selection criteria included the diagnostic terms: “metaplastic breast carcinoma,” “ductal carcinoma with metaplastic features,” “sarcomatoid carcinoma” and “ductal carcinoma with sarcomatoid features.” Patients with a completed course of neoadjuvant therapy prior to resection were excluded; one patient with a partial abbreviated course of neoadjuvant therapy but progressive disease was included. Slides were available for review on 33 patients’ tumors. A board certified pathologist with breast pathology expertise (A.C.M.) verified the histologic diagnosis of all cases.
Of these 33 tumors, 9 potential cases contained both conventional in situ or invasive carcinoma and metaplastic components that were selected for microdissection for DNA extraction, on the basis of geographically distinct regions of tumor of >3×3mm. Of these, 8 cases had sufficient quantity DNA extracted from the formalin fixed, paraffin embedded blocks. We recorded clinicopathologic features from the chart review and review of all slides, including patient age, gender, race, the conventional and metaplastic carcinoma components, tumor grade, tumor ER, PR and HER-2 status, tumor Ki67 proliferation index, tumor size, tumor stage at diagnosis, and clinical outcome.
Genomic DNA extraction
We extracted DNA from formalin-fixed, paraffin-embedded tissue. After hematoxylin and eosin (H&E)-stained slide review, and tumor tissue selection, we manually microdissected the corresponding tissue from five unstained, 5-μm-thick tissue sections using Pinpoint reagents according the manufacturer's protocol (ZymoResearch, Orange, CA). We purified DNA from the sample using QIAmp DNA kit (Qiagen, Valencia, CA) and quantified it by spectophotometry.
Whole exome sequencing
DNA samples were submitted to SeqWright for Next Generation Sequencing on an Illumina HiSeq after whole exome capture using the Agilent SureSelect 51Mb kit. Average read depth was over 60× for most samples, ranging from 14× to over 100× (see supplemental table 1 for details).
Alignment and variant calling
The analysis pipeline is depicted in Supplemental figure S1. We aligned paired end whole exome sequencing reads to the human reference genome (GRCh38) using BWA mem (28), with default parameters. We then optimized the alignments for MuTect2. We used sambamba (29) to sort and index the alignments, Picard MarkDuplicates (http://broadinstitute.github.io/picard) to identify duplicate artifacts, and GATK (version 3.6) BaseRecalibrator with knownSites set to dbSNP build 147 (30), to account for systemic base quality errors. We used GATK MuTect 2 (31), a cancer-specific variant caller with high sensitivity and specificity, for variant calling, comparing both samples in each pair to the reference genome (as both are tumor samples), with the common dbSNP build 147 database and and COSMIC coding mutations database version 77 (cancer.sanger.ac.uk) (32). In the absence of matched normals, we selected the ExAC (33) variant calls (ExAC.r0.3.1.sites.vep.hg38.vcf) as our panel of normals; the ExAC database contains genotypes of 60,706 individuals who are not known to have cancer. We used Picard LiftoverVcf to convert the ExAC database file to GRCh38 using chain file hg19toHg38.over.chain. Criteria for variant selection included 20 supporting reads for each locus in both MC and IDC components, a base call quality greater than 30 (phred scale, 99.9% confidence), and a 10% allele frequency for the alternate allele. Command lines: MuTect2 parameters: GenomeAnalysisTK.jar --analysis_type MuTect2 --reference_sequence [GRCh38] --input_file:tumor [recalibrated and sorted bam file] --normal_panel [PON from ExAC] --dbsnp [common dbSNP build 147] --cosmic [COSMIC version 77]. BaseRecalibrator parameters: GenomeAnalysisTK.jar --analysis_type BaseRecalibrator --reference_sequence [GRCh38] --input_file [sorted bam file] --knownSites [common dbSNP build 147] > [recalibrated data table]. PrintReads parameters: GenomeAnalysisTK.jar --analysis_type PrintReads --reference_sequence [GRCh38] --input_file [sorted bam file] --BQSR [recalibrated data table] > recalibrated BAM file)
We used SnpEff (34) to annotate single nucleotide variants and indels, only considering canonical genes present in the GRCh38 genome build, and annotating variants existing in dbSNP build 147 (30) or in the Catalog of Somatic Mutations in Cancer (COSMIC) database version 77 (cancer.sanger.ac.uk) (32). We assessed functional outcomes of variants with SnpEff (34), and we separately analyzed and visualized high, moderate and low impact variants.
All variant calls are available in vcf files (Supplemental Data 1-16).
Copy Number Variant Calling
We determined copy number variations using CNVkit (35), run separately on each sample with the following parameters: cnvkit.py batch [recalibrated and sorted bam file] —normal --targets [exome regions bed file] --fasta [GRCh38] --split --annotate [ftp://hgdownload.cse.ucsc.edu/goldenPath//hg38/database/refFlat.txt.gz] --access [cnvkit-master/data/access-10kb.hg38.bed] --output-reference [SAMPLE].cnn. We plotted the metaplastic carcinoma component and conventional invasive ductal carcinoma copy number calls across the genome, for each patient, using the OmicCircos R package (36).
Subclone Analysis
We performed subclone analysis using the SciClone (37) R Package. With copy number variant (CNV) segment calls from CNVkit and inferred somatic variants from our MuTect2 pipeline. SciClone parameters: minimumDepth=50, maximumClusters = 10, copyNumberMargins = 0.25 (only consider variants with a diploid CN 2.0 +/- 0.25 regions) .
Correlations
We measured the relatedness of single nucleotide variant (SNV) and CNV profiles of any two samples with Pearson correlation coefficients using R (cor.test). We generated the genetic similarity score between MC and IDC variants in all categories, by calculating #shared variants / (# shared variants + (# unique variants / 2).
Results
The clinicopathologic characteristics are illustrated in Figure 2a, with representative paired ductal and metaplastic components in Figures 2b-d. The epithelial components of the tumors were Elston grade III, invasive ductal carcinomas (Figure 2b,d). Characteristics of the metaplastic components are detailed in Figure 2a. All tumors were negative for ER, and HER2 was amplified in one tumor. Two patients were germline BRCA2 mutation carriers, and this was confirmed by the whole exome sequencing.
Figure 2. Clinical and pathologic characteristics.
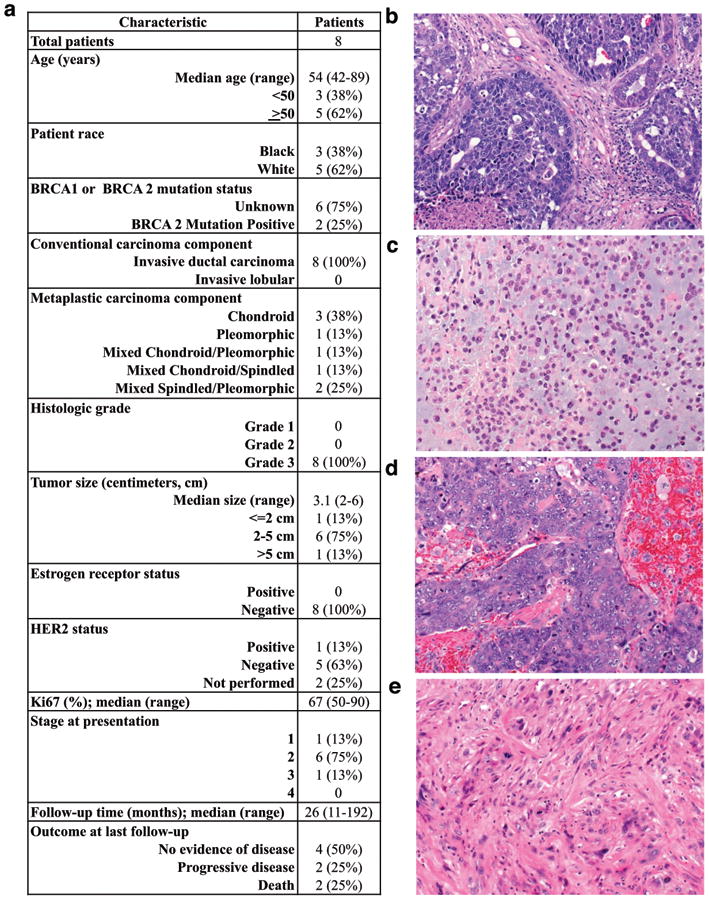
(a) Clinicopathologic characteristics of eight patients with metaplastic breast carcinoma containing both metaplastic and conventional invasive ductal components. (b-c) Paired samples from one patient's tumor (Patient F) contain regions of high grade invasive ductal carcinoma (b) with an adjacent metaplastic component showing chondroid differentiation (c). (d-e) Paired samples from a second patient's tumor (Patient A) contain regions of high grade invasive ductal carcinoma (d) with an adjacent metaplastic component showing spindled and pleomorphic differentiation (e).
We first performed microdissection on formalin fixed, paraffin embedded sections of the tumors for DNA extraction and whole exome sequencing, with alignment of sequencing reads to the human reference genome (GRCh38) using BWA mem (28), followed by copy number variation analysis with CNVkit (35). We used MuTect2 to detect single nucleotide variants (31); this program typically uses a matched normal sample to filter germline variants, but in the absence of normal samples we used the ExaC (33) variant calls as our panel of normals. Finally, we used SciClone (37) to determine subclone populations. We used SnpEff and SnpSift (34) to annotate variants and to categorize changes according to predicted functional consequence (high, moderate and low impact).
To examine large-scale genetic changes, we used CNVkit to determine copy number across each sample. Both the magnitude of copy number variations, as well as the position of the breakpoints, were strongly correlated between metaplastic and ductal components within each patient, but dissimilar among different patients, suggesting that the alterations are not a feature of all tumors of this type, nor a sequencing artifact (Figure 3). Supplemental figure 2 has copy number profiles for all patients.
Figure 3. Copy number variation (CNV) for metaplastic (MC) and invasive ductal carcinoma (IDC) are similar.
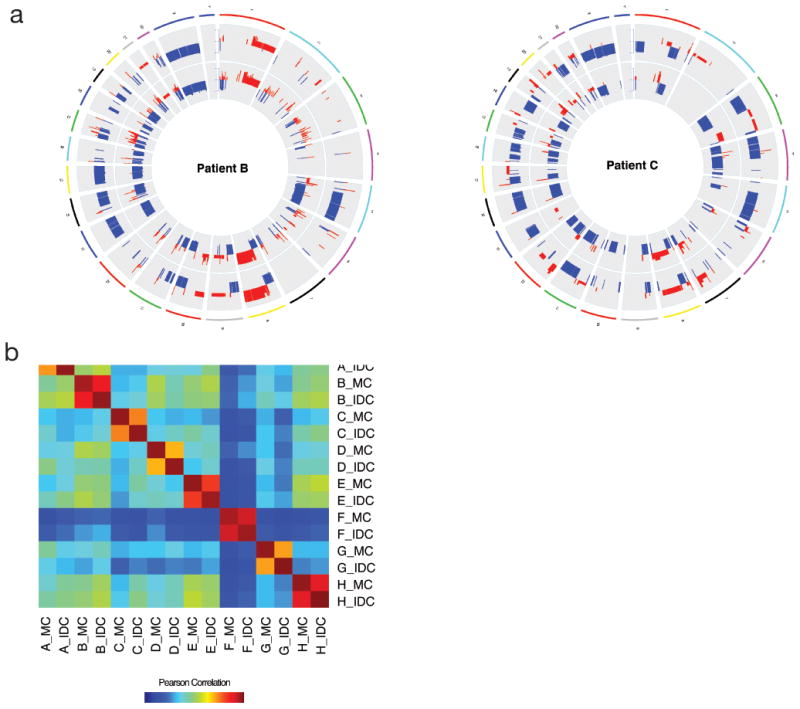
A.CNV plots for patients B and C. Copy number in IDC component is on the outer ring, MC is on the inner ring. B. Pearson correlation coefficients for copy number profiles of all 16 samples sequenced.
Turning to single nucleotide variants (SNVs), we compared the proportions of high, moderate and low impact variants, as well as population polymorphisms (here defined as non-pathogenic variants in dbSNP (30) or the panel of normal variants that we constructed), that are shared between the metaplastic and ductal components of each tumor. For any mutation detected in either the invasive ductal or metaplastic component of a patient's cancer, we required that both samples have a minimum of 10 reads at that position, with a Phred score of at least 20. This prevents false positives due to sequencing coverage fluctuations. Figure 4 shows the numbers of shared variants in these categories, for the metaplastic carcinoma and invasive ductal carcinoma components from each patient. Within each patient, the proportions of SNVs shared by the metaplastic component and the conventional ductal component in the different impact categories are the same (Supplemental table 2). As expected, the metaplastic and ductal components of each patient's tumor were highly concordant for the presence and allele frequency of the non-pathogenic dbSNP variants, which are expected to be germline variants and thus present in both components. Interestingly, we did observe the reported RPL39 (A14V) gain-of-function mutation (38) in the metaplastic component only of one patient.
Figure 4. Variants shared by paired metaplastic (MC) and invasive ductal carcinoma (IDC), represented by one stacked bar per patient.
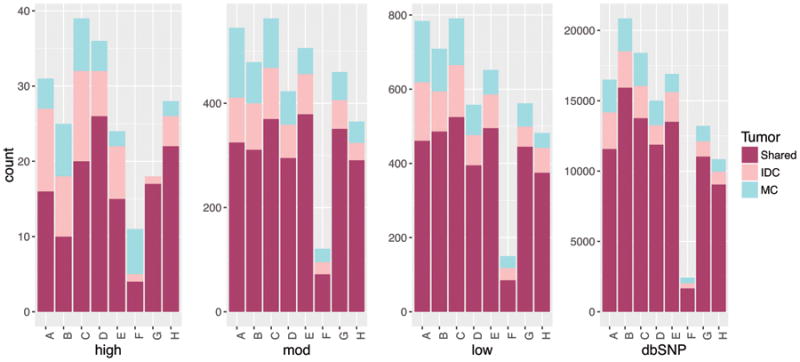
Variants are separated by dbSNP and non-dbSNPs of high, moderate, and low impact. The proportion of variants shared by the specimens is high in the dbSNP category, as expected, and relatively constant across the three stratifications of variant effect.
We also observe that the metaplastic and ductal components of each tumor have strikingly similar allele frequencies for shared high, moderate, and low impact variants, as well as SNPs, even when the comparison is restricted to diploid copy number regions. In Figure 5, we show allele frequencies for shared mutations in low, moderate, and high impact categories, as well as dbSNP variants, for patient B; mutant allele frequencies are nearly identical for high, moderate, and low impact variants. Supplemental Figure 3 shows variant allele frequency scatterplots for all patients.
Figure 5. Allele frequencies are similar for variants in metaplastic (MC) and invasive ductal carcinoma (IDC) samples.
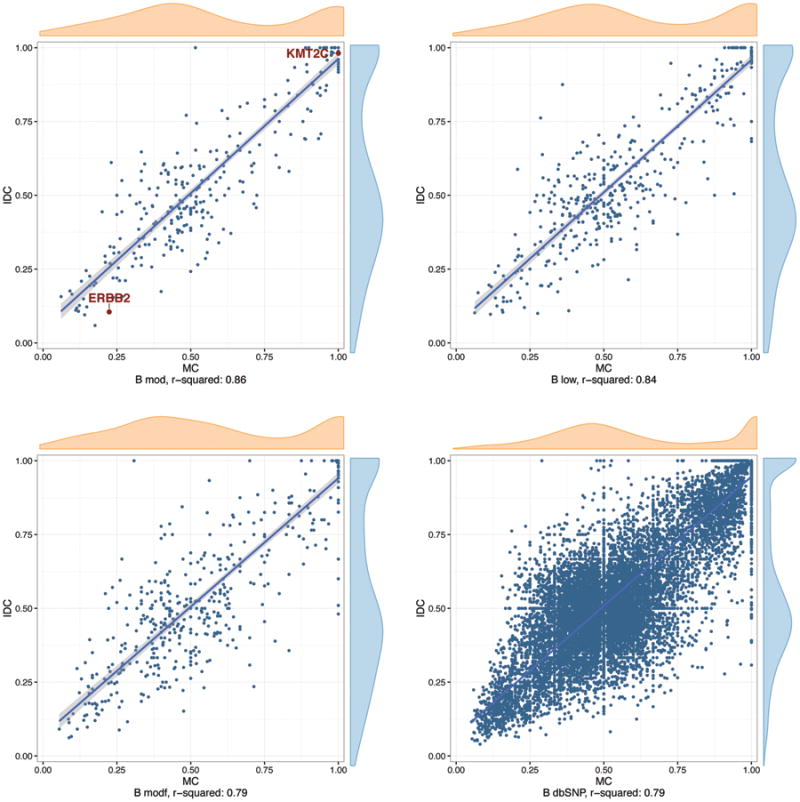
Shown are representative plots from patient B, with canonical mutations highlighted. X and Y axis represent allelic frequencies for MC and IDC components, respectively.
Finally, we examined the subclonal architecture of each tumor. Using SciClone (37), we computationally dissected the genomic clonal architecture of the tumors. Each pair of tumor samples showed substantial overlap of the subclones. The plot in Figure 6a displays the variant frequencies for alleles in each subclone, in the metaplastic carcinoma and invasive ductal carcinoma components for patient B, in which eight shared subclones were identified (represented by different shapes and colors). Each identified subclone, representing a set of cells with very similar allele frequency and copy number variation profiles, is present in roughly the same proportion in each of the two samples. In Figure 6b, we show complementary data, this time restricting the allele frequency plot from Figure 5 to regions that are copy number neutral (diploid) in both the metaplastic carcinoma and invasive ductal carcinoma components. Supplemental Figure 4 shows subclone analysis results for all patients.
Figure 6. Subclone analysis reveals nearly identical structure in the metaplastic (MC) and conventional invasive ductal carcinoma (IDC) components.
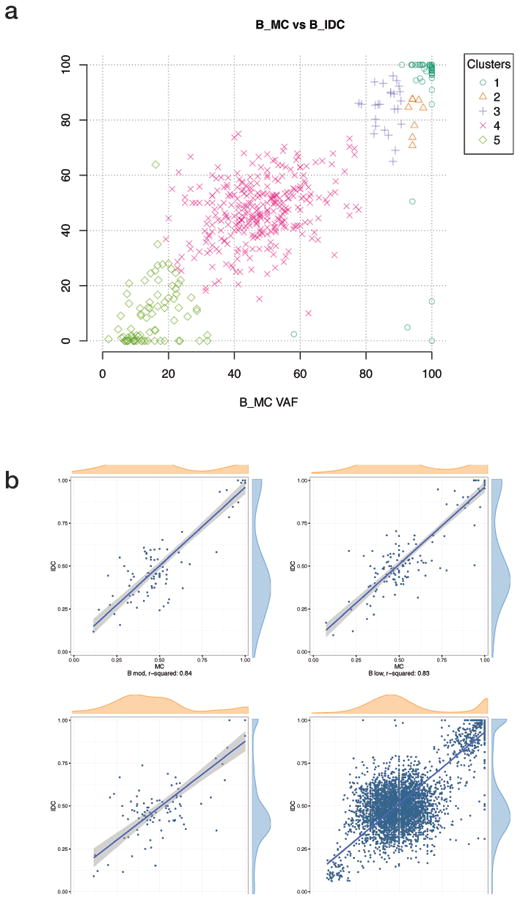
Shown are representative examples from Patient B. (a) Variants shared by the MC and IDC components are assigned to subclones (designated by different colors and shapes) and suggest nearly identical population architecture. (b) Variant allele frequency plots show the same striking shared allele frequencies when restricted to copy number neutral (diploid) regions of the genomes. In this case, skewing of the allele frequencies around 0.5 may reflect either differential normal contamination or an evolutionary process. X and Y axis represent allelic frequencies for MC and IDC components, respectively.
Discussion
Our workflow enables us to determine evolutionary relationships among cancer samples from the same individual when germline genomic information is unavailable. Our assumption, now common in the field, is that high impact variants are present in an initial clone and that further divergence of subclones involves accumulation of low impact variants (27). The high concordance of copy number variation profiles within but not between patients (Figure 3) initially favors the mechanisms postulated in Figure 1B and 1C, as evolution from two separate primary tumors is unlikely to produce the nearly identical copy number changes seen in these samples.
SNV allele frequencies are especially useful. We expect that tumor evolution from two subclones (Figure 1B) will produce cells that share a large proportion of high impact variants but much smaller proportions of low and moderate impact variants, as the different components would accumulate many such variants independently. Evolution from separate primary tumors (Figure 1A) will result in very little sharing across all impact categories, aside from germline dbSNP variants. Our SNV data (Figure 4) are instead consistent with evolution from a single subclone (Figure 1C), as in each patient, the two components share low and moderate impact variants in the same proportion as dbSNP variants.
Simple concordance of the presence and absence of variants, with no further information, is strong evidence in favor of the mechanism depicted in Figure 1C; yet, it could be argued that very recent divergence of a cell from a very homogeneous tumor population would produce similar results. Thus, we examined the frequency of shared variants in each tumor; the highly concordant spectrum of shared allele frequencies is inconsistent with a stepwise model as in Figure 1A or 1B, as mutations, even if gained independently in the evolution of the tumors, would be extremely unlikely to attain the same allele frequencies in each tumor.
The history of a tumor's development is reflected in the landscape of genetic subclones present in the tumor. If the metaplastic and ductal components arose independently from the same seed tumor, they would in time develop different subclones, with different variants and different variant allele frequencies. In fact, the metaplastic and ductal components of each patient's tumor have consistent subclone profiles, suggesting that not only are the two subtypes derived from the same lineage, but they are different phenotypic manifestations of the same population of cells, as in Figure 1C.
Thus, in all eight patients examined, the metaplastic and ductal components of their cancers have the same genotype, and we hypothesize that the differing histology is a phenotype produced by factors other than accelerated clonal evolution. Interestingly, the results also suggest that these tumors, despite the histological suggestion of substantial tumor evolution, do not necessarily have a high mutational load (a feature that could predict a positive response to immune checkpoint inhibitor therapies (39)).
To date, studies of metaplastic breast cancer differ in conclusions on intra-tumor heterogeneity, with some suggesting highly heterogeneous genomic and transcriptomic features and others supporting clonality (40, 41). If, as our study suggests, an intertwined metaplastic tumor is not a separate evolutionary event, the genomic landscape of these tumors will largely be the same as the non-metaplastic component. Interestingly, this implies that the strikingly different histology of these cancers may be driven largely by epigenetic changes, which could have implications for therapy. For example, cancers with identical targetable driver mutations have variable responses depending on the tissue of origin, as demonstrated for BRAF inhibitors and V600-mutation positive cancers (42). Therefore, epigenetic therapies prior to conventional and targeted therapies could provide benefit for metaplastic breast cancers; this is a testable hypothesis for this recalcitrant group of cancers.
Supplementary Material
Acknowledgments
We would like to thank and acknowledge Dr. Pedram Argani for original diagnosis and review of most cases. This work was supported by the SPORE Career Development Award (A.C.M.), The Avon Foundation (B.H.P.), NIH GM007309 (D.J.Z.), CA168180 (R.L.C.), the QVC and Fashion Footwear Association of New York (R.M.C.), and CA167939 (S.C.). NIH P30 CA006973, the Sandy Garcia Charitable Foundation, the Commonwealth Foundation, the Santa Fe Foundation, the Breast Cancer Research Foundation, the Marcie Ellen Foundation, The Helen Golde Trust, and The Canney Foundation. None of the funding sources influenced the design, interpretation or submission of this manuscript.
Footnotes
Author contributions: A.C.M., B.H.P., R.M.C. conceived of the project
A.C.M. obtained tissues and reviewed histology
K.B., D.J.Z, K.C., K.K.S., S.C., D.C., R.L.C. and C.D.G. performed molecular-biology assays
S.J.W. and B.E.A. designed and performed computational analyses
B.E.A., S.J.W., B.H.P. and A.C.M. interpreted data
Writing and review of the manuscript: All authors
Disclosures: B.H.P. is a paid member of scientific advisory boards for Horizon Discovery, LTD and Loxo Oncology, and has ownership interest in Loxo Oncology. B.H.P. has research contracts with Genomic Health, Inc. and Foundation Medicine, Inc. Under separate licensing agreements between Horizon Discovery, LTD and The Johns Hopkins University, B.H.P. is entitled to a share of royalties received by the University on sales of products. The terms of this arrangement are being managed by the Johns Hopkins University, in accordance with its conflict of interest policies. R.M.C. discloses research funding from Novartis, Genentech, Puma Biotechnology, Merrimack and Clovis. No potential conflicts of interest were disclosed by the other authors. All other authors declare no competing financial interests.
This article contains supporting information online.
References
- 1.Reis-Filho JSLS, Gobbi H, Sneige N. Metaplastic carcinoma. In: Lakhani SREI, schnitt SJ, tan PH, van de vijver MJ, editors. world health organization classification of tumours of the breast. 2012. p. 48. [Google Scholar]
- 2.Wargotz ES, Deos PH, Norris HJ. Metaplastic carcinomas of the breast. II. spindle cell carcinoma. Hum Pathol. 1989;20(8):732–40. doi: 10.1016/0046-8177(89)90065-8. [DOI] [PubMed] [Google Scholar]
- 3.Wargotz ES, Norris HJ. Metaplastic carcinomas of the breast. IV. squamous cell carcinoma of ductal origin. Cancer. 1990;65(2):272–6. doi: 10.1002/1097-0142(19900115)65:2<272::aid-cncr2820650215>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Wargotz ES, Norris HJ. Metaplastic carcinomas of the breast. III. carcinosarcoma. Cancer. 1989;64(7):1490–9. doi: 10.1002/1097-0142(19891001)64:7<1490::aid-cncr2820640722>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 5.Wargotz ES, Norris HJ. Metaplastic carcinomas of the breast. I. matrix-producing carcinoma. Hum Pathol. 1989;20(7):628–35. doi: 10.1016/0046-8177(89)90149-4. [DOI] [PubMed] [Google Scholar]
- 6.Cimino-Mathews A, Verma S, Figueroa-Magalhaes MC, Jeter SC, Zhang Z, Argani P, Stearns V, Connolly RM. A clinicopathologic analysis of 45 patients with metaplastic breast carcinoma. Amer J Clin Pathol. doi: 10.1093/ajcp/aqv097. in press. [DOI] [PubMed] [Google Scholar]
- 7.Weigelt B, Horlings HM, Kreike B, Hayes MM, Hauptmann M, Wessels LF, et al. Refinement of breast cancer classification by molecular characterization of histological special types. J Pathol. 2008;216(2):141–50. doi: 10.1002/path.2407. [DOI] [PubMed] [Google Scholar]
- 8.Weigelt B, Ng CK, Shen R, Popova T, Schizas M, Natrajan R, et al. Metaplastic breast carcinomas display genomic and transcriptomic heterogeneity [corrected] Mod Pathol. 2015;28(3):340–51. doi: 10.1038/modpathol.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009;69(10):4116–24. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lien HC, Hsiao YH, Lin YS, Yao YT, Juan HF, Kuo WH, et al. Molecular signatures of metaplastic carcinoma of the breast by large-scale transcriptional profiling: Identification of genes potentially related to epithelial-mesenchymal transition. Oncogene. 2007;26(57):7859–71. doi: 10.1038/sj.onc.1210593. [DOI] [PubMed] [Google Scholar]
- 12.Hennessy BT, Giordano S, Broglio K, Duan Z, Trent J, Buchholz TA, et al. Biphasic metaplastic sarcomatoid carcinoma of the breast. Ann Oncol. 2006;17(4):605–13. doi: 10.1093/annonc/mdl006. [DOI] [PubMed] [Google Scholar]
- 13.Lai HW, Tseng LM, Chang TW, Kuo YL, Hsieh CM, Chen ST, et al. The prognostic significance of metaplastic carcinoma of the breast (MCB)--a case controlled comparison study with infiltrating ductal carcinoma. Breast. 2013;22(5):968–73. doi: 10.1016/j.breast.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 14.Rayson D, Adjei AA, Suman VJ, Wold LE, Ingle JN. Metaplastic breast cancer: Prognosis and response to systemic therapy. Ann Oncol. 1999;10(4):413–9. doi: 10.1023/a:1008329910362. [DOI] [PubMed] [Google Scholar]
- 15.Bae SY, Lee SK, Koo MY, Hur SM, Choi MY, Cho DH, et al. The prognoses of metaplastic breast cancer patients compared to those of triple-negative breast cancer patients. Breast Cancer Res Treat. 2011;126(2):471–8. doi: 10.1007/s10549-011-1359-8. [DOI] [PubMed] [Google Scholar]
- 16.Nelson RA, Guye ML, Luu T, Lai LL. Survival outcomes of metaplastic breast cancer patients: Results from a US population-based analysis. Ann Surg Oncol. 2015;22(1):24–31. doi: 10.1245/s10434-014-3890-4. [DOI] [PubMed] [Google Scholar]
- 17.Teixeira MR, Qvist H, Bohler PJ, Pandis N, Heim S. Cytogenetic analysis shows that carcinosarcomas of the breast are of monoclonal origin. Genes Chromosomes Cancer. 1998;22(2):145–51. [PubMed] [Google Scholar]
- 18.Kung FY, Tse GM, Lo KW, Law BK, Chang AR, Chen MH. Metachronous bilateral mammary metaplastic and infiltrating duct carcinomas: A molecular study for clonality. Hum Pathol. 2002;33(6):677–9. doi: 10.1053/hupa.2002.124906. [DOI] [PubMed] [Google Scholar]
- 19.Zhuang Z, Lininger RA, Man YG, Albuquerque A, Merino MJ, Tavassoli FA. Identical clonality of both components of mammary carcinosarcoma with differential loss of heterozygosity. Mod Pathol. 1997;10(4):354–62. [PubMed] [Google Scholar]
- 20.Wang X, Mori I, Tang W, Yang Q, Nakamura M, Nakamura Y, et al. Metaplastic carcinoma of the breast: P53 analysis identified the same point mutation in the three histologic components. Mod Pathol. 2001;14(11):1183–6. doi: 10.1038/modpathol.3880456. [DOI] [PubMed] [Google Scholar]
- 21.Geyer FC, Weigelt B, Natrajan R, Lambros MB, de Biase D, Vatcheva R, et al. Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J Pathol. 2010;220(5):562–73. doi: 10.1002/path.2675. [DOI] [PubMed] [Google Scholar]
- 22.Lien HC, Lin CW, Mao TL, Kuo SH, Hsiao CH, Huang CS. P53 overexpression and mutation in metaplastic carcinoma of the breast: Genetic evidence for a monoclonal origin of both the carcinomatous and the heterogeneous sarcomatous components. J Pathol. 2004;204(2):131–9. doi: 10.1002/path.1624. [DOI] [PubMed] [Google Scholar]
- 23.Lee JS, Kim YB, Min KW. Metaplastic mammary carcinoma with osteoclast-like giant cells: Identical point mutation of p53 gene only identified in both the intraductal and sarcomatous components. Virchows Arch. 2004;444(2):194–7. doi: 10.1007/s00428-003-0953-5. [DOI] [PubMed] [Google Scholar]
- 24.Thompson L, Chang B, Barsky SH. Monoclonal origins of malignant mixed tumors (carcinosarcomas). evidence for a divergent histogenesis. Am J Surg Pathol. 1996;20(3):277–85. doi: 10.1097/00000478-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Wada H, Enomoto T, Tsujimoto M, Nomura T, Murata Y, Shroyer KR. Carcinosarcoma of the breast: Molecular-biological study for analysis of histogenesis. Hum Pathol. 1998;29(11):1324–8. doi: 10.1016/s0046-8177(98)90266-0. [DOI] [PubMed] [Google Scholar]
- 26.Geyer FC, Weigelt B, Natrajan R, Lambros MB, de Biase D, Vatcheva R, et al. Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J Pathol. 2010;220(5):562–73. doi: 10.1002/path.2675. [DOI] [PubMed] [Google Scholar]
- 27.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics. 2015;31(12):2032–4. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213–9. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, et al. COSMIC: Exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):D805–11. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, et al. Using drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front Genet. 2012;3:35. doi: 10.3389/fgene.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Copy number detection and visualization for targeted sequencing using off-target reads. bioRxiv. 2014 doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu Y, Yan C, Hsu CH, Chen QR, Niu K, Komatsoulis GA, et al. OmicCircos: A simple-to-use R package for the circular visualization of multidimensional omics data. Cancer Inform. 2014;13:13–20. doi: 10.4137/CIN.S13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OL, et al. SciClone: Inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol. 2014;10(8):e1003665. doi: 10.1371/journal.pcbi.1003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dave B, Gonzalez DD, Liu ZB, Li X, Wong H, Granados S, et al. Role of RPL39 in metaplastic breast cancer. J Natl Cancer Inst. 2016;109(6) doi: 10.1093/jnci/djw292. Print 2017 Jun. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weigelt B, Ng CK, Shen R, Popova T, Schizas M, Natrajan R, et al. Metaplastic breast carcinomas display genomic and transcriptomic heterogeneity [corrected] Mod Pathol. 2015;28(3):340–51. doi: 10.1038/modpathol.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geyer FC, Weigelt B, Natrajan R, Lambros MB, de Biase D, Vatcheva R, et al. Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J Pathol. 2010;220(5):562–73. doi: 10.1002/path.2675. [DOI] [PubMed] [Google Scholar]
- 42.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–36. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.