

Abstract
Purpose
Abnormal activation of the NF-κB pathway induces a more aggressive phenotype of cutaneous melanoma. Understanding the mechanisms involved in melanoma NF-κB activation may identify novel targets for this pathway. KPC1, an E3 ubiquitin ligase, is a regulator of NF-κB pathway. The objective of this study was to investigate the mechanisms regulating KPC1 expression and its clinical impact in melanoma.
Experimental Design
The clinical impact of KPC1 expression and its epigenetic regulation were assessed in large cohorts of clinically well-annotated melanoma tissues (tissue micro-arrays; n=137, JWCI cohort; n=40) and The Cancer Genome Atlas database (TCGA cohort, n=370). Using melanoma cell lines, we investigated the functional interactions between KPC1 and NF-κB, and the epigenetic regulations of KPC1, including DNA methylation and microRNA expression.
Results
We verified that KPC1 suppresses melanoma proliferation by processing NF-κB1 p105 into p50, thereby modulating NF-κB-target gene expression. Concordantly, KPC1 expression was down-regulated in AJCC stage IV melanoma compared to early stages (stage I/II p=0.013, stage III p=0.004), and low KPC1 expression was significantly associated with poor overall survival in stage IV melanoma (n=137, Hazard Ratio 1.810, p=0.006). Furthermore, our data showed that high miR-155-5p expression, which is controlled by DNA methylation at its promoter region (TCGA; Pearson’s r −0.455, p<0.001), is significantly associated with KPC1 down-regulation (JWCI; p=0.028, TCGA; p=0.003).
Conclusions
This study revealed novel epigenetic regulation of KPC1 associated with NF-κB pathway activation, promoting metastatic melanoma progression. These findings suggest the potential utility of KPC1 and its epigenetic regulation as theranostic targets.
Keywords: melanoma proliferation, KPC1, NF-κB1, miR-155-5p, miR methylation
Introduction
Cutaneous melanoma is a highly aggressive cancer in patients with metastatic disease (1,2). Despite recent advances in molecular targeted therapies and immunotherapies, the long-term survival of these patients remains poor, particularly in metastatic melanoma (3–6). One of the mechanisms that switch non-aggressive melanoma to an aggressive phenotype is dysregulation in the nuclear factor-κB (NF-κB) pathway (7–11). NF-κB is a key transcription factor regulating the expression of multiple genes involved in various cellular functions, such as immune or inflammatory responses, cell proliferation, differentiation, and progression of multiple tumor types (7,8,12–14). In melanoma, up-regulation of the NF-κB pathway is correlated with melanoma progression through suppression of the apoptosis and promotion of metastasis (7–11). Furthermore, there are autocrine mechanisms promoting constitutive activation of the NF-κB pathway in melanoma cells (7,8). However, the mechanisms that control abnormal activation of NF-κB pathway in cutaneous melanoma remain poorly understood.
One of the most important steps in NF-κB regulation, particularly NF-κB1, is the processing of precursor NF-κB1 p105 into mature p50 (13,15). We recently identified that, following p105 phosphorylation by IKKβ, ubiquitination mediated by KPC1 (Kip1 ubiquitination-promoting complex subunit 1, gene name; RNF123 (Ring finger protein 123), an E3 ubiquitin ligase) leads to proteasomal processing of p105 into p50, which resulted in downstream regulation of the NF-κB pathway (15). Although p105 ubiquitinated by β-Transducin Repeat Containing Protein (βTrCP) E3 ubiquitin ligase is completely degraded via the 26S proteasome (16), ubiquitination by KPC1 leads to proteasomal processing of p105, which results in accumulation of p50 and regulation of NF-κB pathway (15). KPC1 has a suppressive effect on cell proliferation through NF-κB pathway regulation (15).
We hypothesized that KPC1 is involved in abnormal NF-κB pathway activation in cutaneous melanoma progression. In this study, we demonstrated that KPC1 induces NF-κB1 p105 processing into p50, which modulates NF-κB-target gene expression and exerts a suppressive effect on melanoma cell proliferation. The clinical impact of this mechanism is evinced by the significant association of KPC1 down-regulation with poor prognosis in American Joint Committee on Cancer (AJCC) stage IV melanoma. We further investigated epigenetic mechanisms that regulate KPC1 expression in melanoma. In particular, we examined the mechanistic role of miR-155-5p, whose expression is controlled by DNA methylation at its promoter region, targeting KPC1 mRNA to down-regulate KPC1 expression. Overall, this study revealed epigenetic regulation of KPC1 associated with abnormal NF-κB pathway activation, suggesting KPC1 and its epigenetic regulation’s potential as theranostic targets in cutaneous melanoma.
Materials and Methods
Institutional approval and informed consent
This study followed the principles in the Declaration of Helsinki. All human samples and clinical information for this study were obtained according to the protocol guidelines approved by the Saint John’s Health Center (SJHC) / John Wayne Cancer Institute (JWCI) Western Institutional Review Board. Informed consent was obtained from all participants.
Melanoma cell lines
Twenty seven established cutaneous melanoma cell lines were used in this study. Twenty three early-passage (<20) melanoma lines were established from AJCC stage III and IV melanoma patients who received elective surgery at JWCI and were authenticated with autologous peripheral blood leukocytes. Stage I/II melanoma lines (WC00060, WC00080, WC00081 and WC00062) were obtained from Coriell Institute (Camden, NJ) and have been tested by short tandem repeat DNA profiles. Cells were cultured in a humidified chamber with 5% CO2 at 37°C in either RPMI-1640 (Corning, Corning, NY) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gemini Bio-Products, Sacramento, CA) and 1% penicillin-streptomycin, or MCDB-153 (Sigma-Aldrich, St. Louis, MO) / Leibovitz’s L-15 (Life Technologies, Carlsbad, CA) supplemented with 2% FBS, Insulin (5 μg/ml, Sigma-Aldrich), and 1% penicillin-streptomycin (9,17). All the cell line experiments were completed within 12 passages or <4 months from thawing.
Cell viability, colony-formation, and 3D spheroid formation assays
Cells (2.5 × 103) were cultured in a 96-well plate (Thermo Fisher Scientific, Waltham, MA), and the number of viable cells was assessed every 24 hours using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI) according to the manufacturers’ instructions. A soft agar colony-formation assay was performed as follows: 1.5 ml phosphate-buffered saline (PBS; Life Technologies) containing 0.7% low melting point agarose (Lonza, Allendale, NJ) was poured into 6-well dishes (Thermo Fisher Scientific), and the layer was covered with 5.0 × 103 cells suspended in 3 ml medium containing 0.35% low melting point agarose and 10% heat-inactivated FBS (Gemini Bio-Products). After two weeks incubation, colonies were stained with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (5 mg/ml, Sigma-Aldrich) and counted using ImageJ software (http://imagej.nih.gov/ij/). For assessing the 3-dimentional (3D) spheroid formation assay, 2.5 × 103 cells were cultured on a 96-well Corning Spheroid Microplate (Corning) for up to 9 days, and images were obtained every 48 hours (18). The number of viable cells in spheroids at days 7 or 9 was quantified using the CellTiter-Glo Luminescent Cell Viability Assay (Promega).
Cell migration and invasion assays
Cell migration and invasion was quantified using transwell migration chambers and BD BioCoat Matrigel Invasion Chambers (BD Biosciences, Franklin Lakes, NJ) according to manufacturer’s instructions. Cells (2.5 or 5 × 104 cells) were incubated for 24 hours in the 24-well chambers. Cells that had migrated were fixed with 100% methanol and stained with 2% crystal violet solution. The membranes were mounted on a slide and observed under a Nikon Eclipse Ti microscope and NIS elements software (Nikon, Melville, NY). The number of cells from four randomly selected fields was evaluated for each membrane (19).
Transfection of KPC1
To establish stable KPC1-overexpressing clones, cells (5 × 105 in 60 mm dishes (Corning)) with low KPC1 expression (IM-0223 and MH-0331) were transfected with Myc-tagged KPC1 vector (KPC1; OriGene, Rockville, MD) using jetPRIME (Polyplus transfection, New York, NY) and were selected using Geneticin (500 μg/ml, Life Technologies). Overexpression of KPC1 was validated using western blot (WB) after establishment of stable transfected clones. All experiments were performed within ten passages after the establishment of cell line clones.
Mimic micro-RNA (miR) transfection
Cells (2 × 105) with high KPC1 expression (SR-0788 and LP-0024) were transfected with 30 nM of miR-155-5p precursor (Ambion, Austin, TX) or negative control in 6-well dishes using jetPRIME (Polyplus transfection). Transfection was repeated on the cells 48 hours following the initial transfection. RNA and protein extraction were performed 48 hours after the second transfection, and miR-155-5p overexpression was validated using reverse transcriptional-quantitative polymerase chain reaction (RT-qPCR) (20).
siRNA for KPC1 and NF-κB1
Cells (2 × 105) were transfected with 10 nM ON-TARGETplus SMARTpool siRNA (Dharmacon, Lafayette, CO) to down-regulate human KPC1 or NF-κB1 using jetPRIME (Polyplus transfection). The following siRNA were used: KPC1 (GCGCUACUAUUGGGAUGAA, CAACUGGGCCUUCUCUGAA, GCACAUGGCGGACCUCCUA, GGUGAAGCUUCUAGGUAUA), NF-κB1 (GGAGACAUCCUUCCGCAAA, GAUGGGAUCUGCACUGUAA, GAAAUUAGGUCUGGGGAUA, GCAGGAAGGACCUCUAGAA), or Non-targeting Pool (UGGUUUACAUGUCGACUAA, UGGUUUACAUGUUGUGUGA, UGGUUUACAUGUUUUCUGA, UGGUUUACAUGUUUUCCUA). The suppression of gene expression was validated 48 hours after transfection by WB or RT-qPCR.
Cycloheximide chasing assay
IM-0223 or SR-0788 cells (1 × 105) in 12-well dishes were transiently transfected with cDNA coding for human p105 in pFLAG-CMV2 as previously described (15), using jetPRIME (Polyplus transfection). 24 hours after transfection, cycloheximide (50 μg/ml, Sigma-Aldrich) was added, and protein extraction was performed at the hours indicated for WB.
Demethylating agent treatment
Cells with low miR-155-5p expression (JT-1045 and WP-0614) were treated with 2.5 μM of 5-Aza-2′-deoxycytidine (5-Aza-2-dC, Sigma-Aldrich). Medium supplemented with 5-Aza-2-dC was refreshed every 24 hours. Dimethyl sulfoxide (DMSO) (Thermo Fisher Scientific) was used as a non-treated control. RNA extraction was performed after 72 hours of treatment.
RNA isolation
Total RNA isolation from cell lines was performed using Direct-zol RNA MiniPrep kit (Zymo Research, Irvine, CA), according to manufacturer’s instructions. Total RNA for formalin-fixed paraffin-embedded (FFPE) tissues from 40 melanoma patients who underwent surgery at SJHC were isolated by microdissection as previously described (21,22).
RT-qPCR analysis of KPC1 and miR-155-5p
RT-qPCR for mRNA and miRNA was performed as previously described (20,23,24). Quantitative expression of KPC1 was referenced by the expression of human SDHA (Succinate Dehydrogenase Complex, Subunit A). Gene-specific oligonucleotide primers and probe were designed as follows: KPC1, 5′-GTGGGTGTCTCCGATGATGTC-3′ (forward), 5′-CAAGGATGTCCTTCCTCCTCTT-3′ (reverse) and 5′-/56-FAM/TGAATACGC/ZEN/TATGGCTCTGAGGGACACA/3IABkFQ/-3′ (probe); SDHA, 5′-TCAGCATGCAGAAGTCAAT-3′ (forward) and 5′-GAACGTCTTCAGGTGCTTT-3′ (reverse). Quantitative expression of miR-155-5p was referenced by the expression of small nuclear RNA, RNU6. Primers (miR-155-5p and RNU6) were acquired from PerfeCTa miRNA Assays (Quanta Biosciences, Gaithersburg, MD).
Western blot
Protein extraction and WB were performed as previously described (25). The following antibodies (Abs) were used: mouse anti-human KPC1 Ab (1:300, #ab57549; Abcam, Cambridge, MA), rabbit anti-human NF-κB1 p50 Ab (1:200, #sc-114; Santa Cruz Biotechnology, Dallas, TX), mouse anti-Myc-tag Ab (1:1000, #05-724; Millipore, Billerica, MA), mouse anti-Flag Ab (1:1000, #TA50011-100; OriGene), mouse anti-human β-actin Ab (1:10000, #A5441; Sigma-Aldrich), or horseradish peroxidase-conjugated Abs (sheep anti-mouse Ab (1:4000, #NA931; GE Healthcare, Pittsburgh, PA) or donkey anti-rabbit Ab (1:4000, #NA934; GE Healthcare)). Immunoreactive bands were visualized with the SuperSignal West Femto Maximum Sensitivity Substrate (Life Technologies), and the densities of protein bands were quantified using ImageJ software (http://imagej.nih.gov/ij/).
Genomic DNA extraction and detection of methylated MIR155HG
DNA isolation from cells was performed using Quick-gDNA MiniPrep kit (Zymo Research), according to the manufacturer’s instructions. Sodium bisulfate modification was applied to 500ng of extracted genomic DNA using EZ DNA Methylation-Direct kit (Zymo Research). DNA methylation level of MIR155HG gene promoter region was analyzed in bisulfate-modified DNA with a quantitative methylation-specific PCR (MSP) assay (17,26). The methyl-specific primers for MIR155HG gene were designed as follows: 5′-GTCGAGTTCGGGTTTAGC-3′ (forward) and 5′-GCGAAACTAAAATCGACGTAC-3′ (reverse). DNA methylation level was referenced against the amplification of the human ACTB gene promoter region that is free of CpG sites using the following primers: 5′-GTGGTGATGGAGGAGGTTTAGTA-3′ (forward), 5′-ACCAATAAAACCTACTCCTCCCTTA-3′ (reverse) (27). The quantitative amplification of MIR155HG-methylated alleles and ACTB alleles were performed and evaluated using a standard curve of serial dilutions consisting of the universal methylated control DNA as previously described (26).
Protein-protein interactions
Whole cell lysate from IM-0223 cells overexpressing both Flag-p105 and myc-KPC1 was incubated with anti-NF-κB1 p105 Ab (#sc-114; Santa Cruz Biotechnology) or rabbit-IgG control Ab (#NB810-56910; Novus Biologicals, Littleton, CO) and immunoprecipitated using Protein A-Agarose beads (Roche, Indianapolis, IN). Beads were washed twice with RIPA lysis buffer (Santa Cruz Biotechnology) and three times with PBS. The immunoprecipitated proteins were analyzed by WB with anti-KPC1 Ab or anti-p105 Ab.
IHC for tissue microarrays
Paraffin-embedded tissue microarrays (TMA) for AJCC stage IV melanoma established at JWCI included 262 stage IV metastatic tumors with paired stage III tumor tissues, each in duplicate, from 137 patients (28,29). TMA also contained seven normal tissues with limited or no proliferation (adrenal gland, brain, kidney, and liver), in duplicate. TMA were clinically well-annotated with greater than 5 years follow-up (28,29). Immunohistochemistry (IHC) was performed as previously described (28,29), using the following Abs: mouse anti-human KPC1 Ab (1:50 dilution, #ab57549; Abcam), rabbit anti-human NF-κB1 p50 Ab (1:200 dilution, #sc-114; Santa Cruz Biotechnology), or mouse anti-human p27 Ab (1:100 dilution, #610241; BD Biosciences). Photographs were obtained using a Nikon Eclipse Ti microscope and NIS elements software (Nikon). To exclude the influence of pigmentation and background staining, IHC staining without primary Ab was subtracted from the corresponding specimen. Immunostaining was assessed by two independent observers, blinded to the patients’ data. Specimens were scored for KPC1 according to the intensity of cytoplasmic staining (0: none, 1: weak, 2: moderate, 3: strong) and for p50 based on localization of the staining (no staining, cytoplasmic or nucleus staining) as previously described (15). Nucleus p27 staining was scored (0: <5%, 1: 5–50%, 2: >50% positive cells) and classified as low (score 0, 1) or high (score 2).
Immunofluorescence staining
IM-0223 cells (2 × 104 cells) were incubated in 8-well chamber slides (Thermo Fisher Scientific), and stained for immunofluorescence as previously described (28), using the following Abs: mouse anti-human KPC1 Ab (1:50 dilution, #ab57549; Abcam), rabbit anti-human NF-κB1 p50 Ab (1:50 dilution, #sc-114; Santa Cruz Biotechnology), Alexa Fluor 488-conjugated goat anti-mouse IgG (2.5 μg/ml, #115-545-003, Jackson ImmunoResearch, West Grove, PA) or Cy3-conjugated goat anti-rabbit IgG (2.5 μg/ml, #111-165-003, Jackson ImmunoResearch). The slides were stained with 4′,6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific) in mounting medium. Images were obtained using a Nikon Eclipse Ti microscope and NIS elements software (Nikon).
Luminescent reporter gene transfections and luciferase assay
2×104 cells (SR-0788) in a 96-well plate were co-transfected with 30 nM of miR-155-5p precursor (Ambion) and 100 ng of KPC1-Wild, KPC1-Mutant 3′-untranslated region (3′-UTR) GoClone reporter constructs or an empty 3′-UTR vector (Switch Gear Genomics, Menlo Park, CA). After 24 hours incubation, LightSwitch Luciferase Assay Reagent (Switch Gear Genomics) was added to each well, and luciferase signal intensity was assessed by GloMax-Multi Detection System (Promega).
RNA sequencing (RNA-seq)
Total RNA was extracted from melanoma line (IM-0223) stably transfected with either Myc-tagged KPC1 or empty-vector (OriGene). mRNA libraries were constructed from total RNA with high quality (RIN≥8.0) and high purity (OD 260/280 1.8–2.0) scores using the Illumina TruSeq RNA Sample Preparation Kit v2 (Illumina Inc., San Diego, CA) as previously described (24). The libraries were sequenced on the Illumina HiSeq 2500 (Illumina Inc.) in Rapid Mode using 50 bp single-end reads and achieved an average read depth of over 30 million reads per sample at the JWCI Sequencing Center. Base calling and demultiplexing were processed using CASAVA v1.8 (Illumina Inc.), reads were mapped to the GENCODE release 19 reference using STAR version 2.4.2a (30), and read counts were generated using the quantMode GeneCounts option in STAR. The GFOLD version 1.1.4 algorithm was used to detect fold change differences in expression between conditions (31).
Reverse-phase protein array (RPPA)
Protein lysate from melanoma lines (IM-0223 and MH-0331) stably transfected with either Myc-tagged KPC1 or empty-vector (OriGene) was extracted as previously described (32), and RPPA analysis was performed by the CCSG-supported RPPA Core Facility at the University of Texas MD Anderson Cancer Center (32). A total of 232 authenticated Abs for total protein expression and 62 Abs for protein phosphorylation were analyzed in this study. A list of the Abs can be accessed from https://www.mdanderson.org/education-and-research/resources-for-professionals/scientific-resources/core-facilities-and-services/functional-proteomics-rppa-core/antibody-lists-protocols/functional-proteomics-reverse-phase-protein-array-core-facility-antibody-lists-and-protocols.html (Ab set 107). Heat maps and Volcano plots were generated from load-corrected log2 data using MultiExperiment Viewer (MeV) v4.7.1. Differences in protein expression between groups were determined using student’s t test with a two-sided p-value < 0.05.
Bioinformatics analysis
Online computational tool, miRANDA (http://www.microrna.org/microrna/) was used to predict miR that effectively binds to 3′-UTR of KPC1 mRNA based on miRSVR score. The prediction was validated using three other computational tools (TargetScanHuman 7.0 (http://www.targetscan.org/), DIANA-microT CDS (http://diana.imis.athena-innovation.gr/DianaTools/), and miRDB (http://mirdb.org/miRDB/)). The Cancer Genome Atlas (TCGA) data sets in melanoma for DNA methylation, mRNA expression, miR expression, somatic mutation and clinical information were obtained in Nov, 2015 (http://cancergenome.nih.gov/). Of 466 samples, 370 tumor samples from primary tumor, regional lymph node, or distant metastasis were included to analyze the correlation between KPC1 expression and NF-κB-target gene expression (http://www.bu.edu/nf-kb/gene-resources/target-genes/) and the correlation between DNA methylation level and expression of KPC1 or miR-155-5p. 272 samples were available for mutation data analyses. Genomic regions with significant focal amplification or deletion and significant arm-level change were identified using TCGA data (n=367, http://www.firebrowse.org, doi:10.7908/C1445KXQ).
Data access
DNA methylation data for the Infinium HumanMethylation450K (HM450K) platform obtained in our previous study (26) and gene expression data for RNA-seq can be accessed from Gene Ontology Omnibus (GEO) under the accession number GSE44661 and GSE79111, respectively.
Statistical analysis
Continuous variables were assessed by using Student’s t test or Wilcoxon rank-sum test and categorical variables were assessed by using χ2 test, Fisher’s exact test, or Cochran-Armitage trend test. The correlation between DNA methylation and gene expression levels was analyzed using the Pearson’s r correlation coefficient for TCGA cohorts (n=370) and ten melanoma lines as previously described (26). Overall survival (OS) was analyzed based on the time from diagnosis with stage IV melanoma using the Kaplan-Meier method and log-rank test, and multivariate analysis was performed by the Cox proportional hazard model. All statistical analyses were performed with JMP, version 11.0 (SAS Institute Inc., Cary, NC), and a two-sided p-value < 0.05 was regarded as statistically significant.
Results
KPC1 overexpression suppresses melanoma cell proliferation
To evaluate the involvement of KPC1 in melanoma proliferation, cells with low KPC1 expression (IM-0223 and MH-0331) were stably transfected with a cDNA coding Myc-KPC1 (Supplementary Fig. S1A, S1B) and were assessed for proliferation. The overexpression resulted in inhibition of melanoma cell proliferation (IM-0223; p<0.001, Fig. 1A, MH-0331; p<0.001, Fig. 1B). Cell proliferation was also evaluated in a 3D condition using a 3D spheroid formation assay. As expected, KPC1 overexpression significantly reduced spheroid growth in both lines (Supplementary Fig. S1C, S1D). Similarly, the percentage of colony-formation of KPC1-overexpressing cells was lower than that of control cells (Supplementary Fig. S1E, S1F). Despite the effect of KPC1 on cell proliferation, KPC1 overexpression did not affect the ability of cell migration or invasion of either line (data not shown). In concordance with the suppressive role of KPC1 on melanoma cell proliferation, melanoma lines from stage IV (n=8) showed lower KPC1 expression compared to those from stage I/II (n=4) or stage III (n=15) at both the mRNA and protein expression levels (Fig. 1C, Supplementary Fig. S1G, S1H), indicating the involvement of KPC1 in advanced metastatic melanomas. The down-regulation of KPC1 in advanced melanomas was also validated in a clinically well-annotated JWCI cohort of melanoma specimens representing different clinical stages (JWCI, n=40); AJCC stage IV tissues (n=19) showed significantly lower KPC1 expression compared to stage I/II (n=11, p=0.013) or stage III tissues (n=10, p=0.004) (Fig. 1D). Interestingly, mitotic rate, which represents proliferation of the primary melanoma (1), was negatively correlated with KPC1 mRNA expression in TCGA primary melanoma (Spearman’s rho −0.51, p=0.03, Supplementary Figure 1I). These results suggested a suppressive role of KPC1 on melanoma proliferation, and indicate that KPC1 down-regulation is involved in advanced metastatic melanoma compared to early stages.
Figure 1. KPC1 suppresses melanoma cell proliferation by inducing NF-κB1 p105 processing into p50.
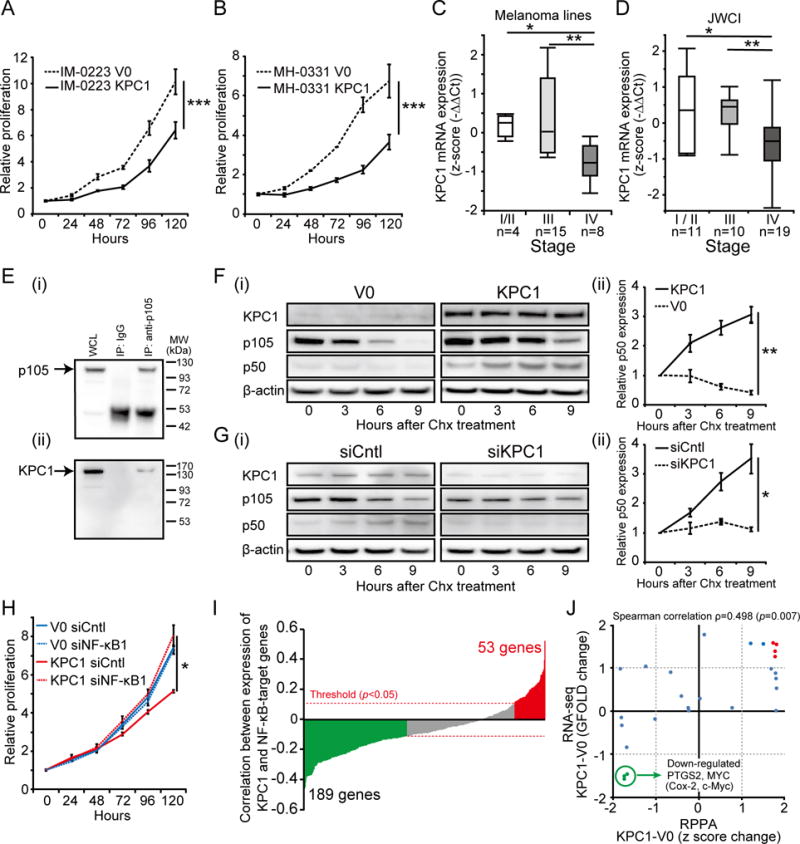
(A & B) IM-0223 or MH-0331 were stably transfected with empty vector (V0) or cDNA coding Myc-KPC1 (KPC1). Melanoma cell proliferation after the transfection was assessed. KPC1 overexpression inhibited melanoma cell proliferation in (A) IM-0223 (B) MH-0331 compared to control at 120 hours (t-test, *** p<0.001). (C) Box plot of KPC1 expression in melanoma lines from AJCC stage I/II (n=4), stage III (n=15), and stage IV (n=8) assessed by RT-qPCR (Wilcoxon-test, * p<0.05, ** p<0.01). (D) KPC expression was analyzed in melanoma patients’ tissues. Boxplot of KPC1 expression in melanoma FFPE samples from AJCC stage I/II (n=11), stage III (n=10), and stage IV (n=19) assessed by RT-qPCR (JWCI cohort, n=40) (Wilcoxon-test, * p<0.05, ** p<0.01). (E) Co-immunoprecipitation of KPC1 and p105. Immunoprecipitation (IP) was performed using anti-p105 or control IgG Abs for IM-0223 cell lysate overexpressing both KPC1 and p105. p105 was detected using anti-p105 Ab (i), and KPC1 was detected using anti-KPC1 Ab (ii) by WB. Whole cell lysate (WCL) was used as a positive control. (F) (i) Cycloheximide chasing assay was performed in IM-0223. Cycloheximide (Chx, 50 μg/ml) was added after transfection of cDNA coding for human p105 and incubated for the indicated time before protein extraction. WB was performed using anti-KPC1 Ab, anti-Flag Ab (to detect exogenous p105 and p50), or anti-β-actin Ab. (ii) p50 expression relative to 0 hour was quantified. p50 expression was higher in KPC1-overexpressing cells compared to control cells at 9 hours (t-test, ** p<0.01). (G) (i) SR-0788 was transfected with control si-RNA (siCntl) or siRNA for KPC1 (siKPC1), and cycloheximide chasing assay was performed. WB was performed using anti-KPC1 Ab, anti-Flag Ab, or anti-β-actin Ab. (ii) p50 expression relative to 0 hour was quantified. p50 expression was lower in cells with KPC1 suppression compared to control cells at 9 hours (t-test, * p<0.05). (H) IM-0223 cells (V0 and KPC1) were transfected with control siRNA (siCntl) or siRNA for NF-κB1 (siNF-κB1). p105 knock-down promoted proliferation in KPC1-overexpressing cells at 120 hours (t-test, * p<0.05). (I) Correlation (Spearman’s rank correlation rho) between KPC1 expression and expression of NF-κB-target genes (413 genes) from TCGA cohort (n=370) was analyzed. Bar plots showing the negative correlation for 189 genes (green bars, p<0.05) and positive correlation for 53 genes (red bars, p<0.05)). Red dotted lines indicate the statistical significant threshold (p=0.05) for correlation analysis. (J) RPPA and RNA-seq were performed for IM-0223 (V0 and KPC1) to demonstrate the downstream regulation of NF-κB-target genes. Scatter plots showing protein expression change (RPPA) and RNA expression change (RNA-seq) from 28 proteins / genes that are targeted by NF-κB pathway. Targets were considered to be up-regulated (red dots) when z score change for RPPA (p<0.05) and GFOLD change for RNA-seq were both more than one, and down-regulated (green dots) when less than -1. Error bars represent means ± standard deviation (SD) from replicates (n=3). WB images were cropped for clarity and focus on relevant bands.
KPC1 induces processing of NF-κB1 p105 into p50 in melanoma
We recently demonstrated that KPC1 interacts with NF-κB1 p105 in human embryonic kidney cells (15). This interaction results in an increase in p50 levels, a negative regulator of the NF-κB pathway (15). To evaluate potential interactions between p105 and KPC1 in melanoma cells, we performed co-immunoprecipitation using cell lysate from IM-0223 cells overexpressing Flag-p105 and Myc-KPC1 (Fig. 1E). p105 and KPC1 were co-immunoprecipitated, demonstrating that KPC1 directly interacts with p105 in melanoma cells. To evaluate the involvement of KPC1 in the processing of p105 into p50, cycloheximide chasing assays with transient Flag-p105 transfection were performed on IM-0223 cells (Fig. 1F). KPC1 overexpression induced significantly higher levels of p50 produced from exogenous Flag-p105 (p=0.005), but not in control cells (V0), demonstrating that KPC1 is involved in processing into p50. Despite lower accumulation of p50 observed in IM-0223 V0, p105 diminished strongly, presumably due to other factors such as proteasomal degradation of p105 through ubiquitination by βTrCP ubiquitin ligase (16). In concordance with exogenous p105 processing, endogenous p50 expression was also higher in KPC1-overexpressing cells compared to control cells (p=0.013, Supplementary Fig. S2A). Conversely, KPC1 suppression in melanoma cells with high KPC1 expression (SR-0788) attenuated the processing of exogenous Flag-p105 into p50 (p=0.026, Fig. 1G). We further evaluated p50 expression and cellular location in KPC1-overexpressing IM-0223 cells using immunofluorescence staining (Supplementary Fig. S2B). In accordance with the functional interaction between KPC1 and p105 observed in melanoma cells, control cells demonstrated weak p50 expression in the nucleus, while KPC1-overexpressing cells presented increased p50 staining in both the nucleus and cytoplasm. To discern whether the suppressive effect of KPC1 overexpression on proliferation is not due to some non-specific effects, we evaluated the effect of NF-κB1 p105 knock-down on cell proliferation using IM-0223. NF-κB1 p105 knock-down abrogated the suppressive effect of KPC1 overexpression on melanoma cell proliferation (IM-0223, p=0.011, Fig. 1H, Supplementary Fig. S2C), indicating that the effect of KPC1 overexpression is specific to the increase of p50. These results strongly suggested the suppressive effect of KPC1 on melanoma cell proliferation is due to its regulatory activity on processing p105 into p50.
KPC1 regulates the expression of NF-κB-target genes
To investigate the regulation of KPC1 on NF-κB pathway, we assessed correlation between KPC1 expression and expression of 413 genes recognized as direct targets for the NF-κB transcription factor (http://www.bu.edu/nf-kb/gene-resources/target-genes/) using melanoma TCGA cohort (n=370). Compatible with the role of KPC1 as a potential negative regulator of the NF-κB pathway (15), 189 target genes were negatively correlated with KPC1 expression, whereas 53 were positively correlated (Fig. 1I, p<0.05). To further investigate the downstream mechanisms controlled by KPC1, we performed RNA-seq and RPPA for IM-0223 cells overexpressing KPC1 and evaluated associated changes in mRNA expression and protein levels of NF-κB-target genes (Fig. 1J). 28 NF-κB-target genes were screened both on RNA-seq and RPPA, and seven genes were significantly affected at mRNA and protein levels after KPC1 overexpression. Interestingly, these changes included significant down-regulation of the genes PTGS2 (Cox-2) and MYC (c-Myc) (IM-0223, p<0.05, Fig. 1J, Supplementary Fig. S2D (i)), known tumor promoters (33–36), and significant down-regulation of Cox-2 and c-Myc was also validated in MH-0331 from RPPA analysis (p<0.05, Supplementary Fig. S2D (ii)).
KPC1 expression is regulated by miR-155-5p
We then investigated the potential mechanisms that regulate KPC1 expression in melanoma. Analysis of amplification or deletion in the genomic region 3p21, where the RNF123 gene is located, showed no significant changes in the TCGA melanoma cohort (data not shown). After stratification of cutaneous melanoma into four subtypes characterized by genomic mutation pattern (mutation in BRAF, RAS (N/H/KRAS), NF1 and Triple-wild-type) (37), mutation status was not associated with KPC1 mRNA expression in the TCGA cohort (n=272, Supplementary Fig. S3A–D), suggesting KPC1 expression is independent of this genomic classification or these known melanoma driver gene mutation. Additionally, DNA methylation level of 17 CpG sites (Chr3:49,723,947–49,727,474) associated with the RNF123 gene including the promoter region was analyzed using the HM450K platform in metastatic melanoma lines (n=10) and melanoma tissue specimens (TCGA cohort, n=370). We identified no significant correlation (melanoma lines) or weak correlation (TCGA, Pearson’s r −0.189) between KPC1 mRNA expression and DNA methylation levels of each 17 CpG sites (Supplementary Fig. S3E), indicating that KPC1 expression is independent of the DNA methylation level of KPC1 promoter region. Therefore, to further investigate other potential epigenetic mechanisms affecting KPC1 expression, we evaluated the role of specific miR in KPC1 mRNA stability. Initially, putative miRs that bind to 3′-UTR on KPC1 mature mRNA were assessed using miRANDA. This computational tool predicted five different miRs that potentially targets 3′-UTR of KPC1 mRNA, and miR-155-5p presented strongest score. Effective binding of miR-155-5p on KPC1 mRNA was validated in other different computational tools, TargetScan, DIANA TOOL, and miRDB, and importantly, miR-155-5p was the only miR commonly predicted to target KPC1 mRNA by all these tools (Fig. 2A). To verify whether miR-155-5p regulates KPC1 expression, melanoma lines with high KPC1 expression (SR-0788 and LP-0024) were transfected with pre-miR-155-5p (miR-155-5p) or miR control (miR-Cntl). miR-155-5p overexpression induced reduction of KPC1 expression at both mRNA and protein expression levels (Fig. 2B, Supplementary Fig. S3F). Importantly, in accordance with the regulation of NF-κB p50 by KPC1, miR-155-5p overexpression reduced NF-κB p50 expression (SR-0788, Supplementary Fig. S3G). Furthermore, KPC1 overexpression on melanoma cells overexpressing miR-155-5p abrogated the decrease of NF-κB p50 expression, indicating that miR-155-5p overexpression in melanoma cells results in down-regulation of KPC1 and affects NF-κB p50 expression. Contrarily, miR-155-5p overexpression in melanoma cells with low KPC1 expression (IM-0221) induced KPC1 reductions, however did not affect the amount of NF-κB p50 (Supplementary Fig. S3H), suggesting less involvement of KPC1 on NF-κB p50 regulation in this context. To demonstrate direct binding of miR-155-5p to the KPC1 3′-UTR, a luciferase reporter activity assay was performed. SR-0788 cells were co-transfected with miR precursor and KPC1-Wild 3′-UTR (WT) or KPC1-Mutant 3′-UTR (Mutant) on a RenSP vector (Fig. 2C, 2D). miR-155-5p suppressed luciferase reporter activity by targeting the KPC1-Wild 3′-UTR, but did not affect the activity of the construct containing the KPC1-Mutant 3′-UTR. The luciferase reporter activity assay demonstrated that miR-155-5p directly and specifically targets the 3′-UTR of KPC1 mRNA.
Figure 2. Epigenetic regulatory mechanism of KPC1 expression.
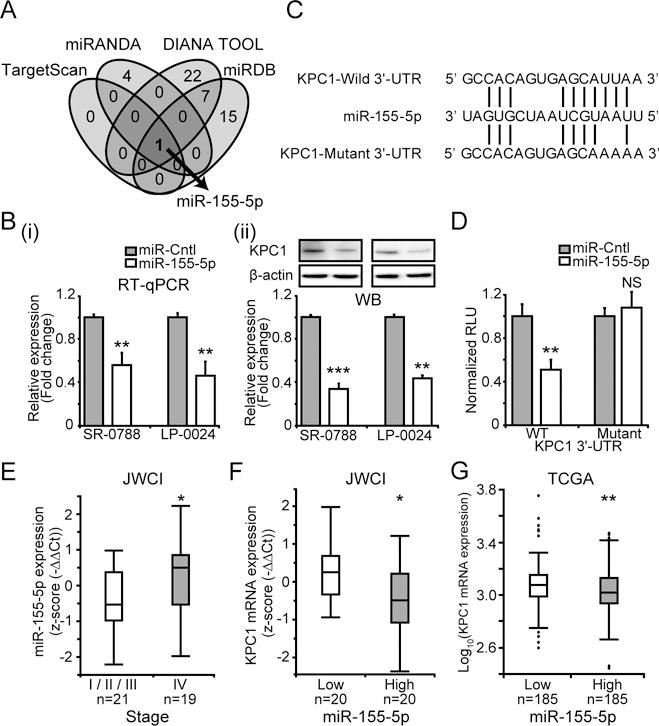
(A) Venn diagram showing putative miRs that target 3′-UTR of KPC1 mRNA predicted by different computational tools (TargetScan, miRANDA, DIANA TOOL, and miRDB). (B) SR-0788 and LP-0024 were transfected with pre-miR-155-5p (miR-155-5p) or miR control (miR-Cntl). KPC1 expression was quantified using (i) RT-qPCR (ii) WB after miR-155-5p transfection (t-test, ** p<0.01, *** p<0.001). (C) miR-155-5p sequence aligned with human KPC1-Wild 3′-UTR (WT) and KPC1-Mutant 3′-UTR (Mutant) sequences. (D) A luciferase reporter activity assay to determine miR-155-5p targets 3′-UTR of KPC1 using human KPC1-Wild 3′-UTR (WT) and KPC1-Mutant 3′-UTR (Mutant) sequences on RenSP vector (t-test, NS p≥0.05, ** p<0.01). (E) Boxplot of miR-155-5p expression in JWCI cohort assessed by RT-qPCR (n=40, Wilcoxon-test, * p<0.05). (F) Boxplot of KPC1 expression in the patients with miR-155-5p low (n=20) or high (n=20) expression (classified based on median of miR-155-5p expression) from JWCI cohort (n=40, Wilcoxon-test, * p<0.05). (G) Boxplot showing KPC1 expression in patients with miR-155-5p low (n=185) and high (n=185) expression (classified based on median of miR-155-5p expression) from TCGA cohort (t-test, ** p<0.01). Error bars represent means ± SD from replicates (n=3). WB images were cropped for clarity and focus on relevant bands.
To further validate the regulation of KPC1 expression by miR-155-5p in melanoma tissue, we analyzed miR-155-5p expression and KPC1 expression in the above melanoma tissue JWCI cohort (n=40). AJCC stage IV tissues exhibited significantly higher expression of miR-155-5p compared to those from stage I to III (JWCI, p=0.019, Fig. 2E). Importantly, higher miR-155-5p expression was associated with lower KPC1 expression in both JWCI (p=0.028, Fig. 2F) and TCGA (p=0.003, Fig. 2G) cohorts, supporting that up-regulation of miR-155-5p leads to down-regulation of KPC1 in melanoma. Overall, these results strongly suggested that miR-155-5p regulates KPC1 expression in cutaneous melanoma.
miR-155-5p expression is regulated by miR promoter DNA methylation
To further investigate the mechanisms that regulate miR-155-5p expression in melanoma, we assessed DNA methylation level of seven CpG sites including the MIR155HG gene promoter regions (Chr21:26,934,197–26,934,885). Remarkably, in both melanoma lines (n=10) and melanoma tissue specimens (TCGA cohort, n=370), there was a strong negative correlation between miR-155-5p expression and DNA methylation level of its promoter region (TCGA; Pearson’s r −0.455, p<0.001, melanoma lines; Pearson’s r −0.908, p<0.001, Fig. 3A). To confirm the role of DNA methylation in the regulation of miR-155-5p expression, melanoma lines with low miR-155-5p expression were treated with DNA methyltransferases inhibitor, 5-Aza-2-dC, which significantly enhanced miR-155-5p expression in cell lines (JT-1045; p=0.005, WP-0614; p=0.004, Fig. 3B). Furthermore, MSP analysis revealed that 5-Aza-2-dC treatment significantly reduced the DNA methylation level of the MIR155HG gene promoter region (JT-1045, p=0.008, Fig. 3C). We further validated the regulation of miR-155-5p expression by DNA methylation level of its gene promoter region in the TCGA cohort (n=370). Tissues with lower DNA methylation exhibited significantly higher miR-155-5p expression compared to tissues with higher DNA methylation (Fig. 3D). Importantly, in concordance with the regulatory role of miR-155-5p on KPC1 expression, lower DNA methylation of the miR was associated with lower KPC1 expression (Fig. 3E). These results confirmed that miR-155-5p is regulated by DNA methylation of its CpG island spanning the promoter region in melanoma cell lines and clinical tumor specimens.
Figure 3. Regulatory mechanism of miR-155-5p expression in melanoma.
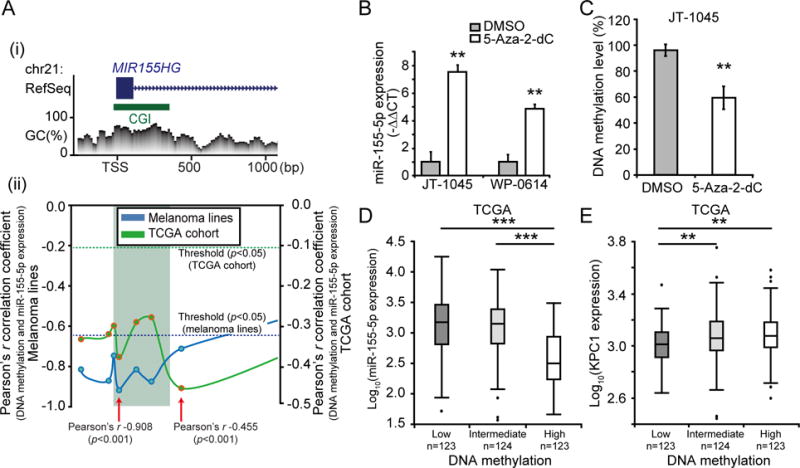
(A) Promoter DNA methylation level of MIR155HG gene were analyzed to investigate the regulatory mechanism of miR-155-5p expression. (i) MIR155HG gene structure [based on RefSeq Feb. 2009 (GRCh37/hg19) assembly] and CpG Context at promoter region. Blue box represents exon and green box CGI (CpG island). (ii) Correlation analysis between DNA methylation and miR-155-5p expression levels from ten melanoma lines and TCGA cohort (n=370). The correlations were calculated using the Pearson’s r correlation coefficient for each CpG sites. Each point represents one CpG site and solid lines indicate the variation of correlation at the promoter region of MIR155HG gene. Dotted lines indicate the statistical significant threshold (p=0.05) for correlation analysis. CpG sites which demonstrated the strongest negative correlation are indicated by red arrows with its Pearson’s r in the figure. (B) JT-1045 and WP-0614 were treated with medium supplemented with 5-Aza-2-dC or control (DMSO), and miR-155-5p expression was quantified using RT-qPCR (t-test, ** p<0.01). (C) Boxplot showing DNA methylation level of miR-155-5p promoter region quantified by MSP from JT-1045 treated with 5-Aza-2-dC or control (DMSO) (t-test, ** p<0.01). (D & E) Association between miR-155-5p promoter DNA methylation level (Chr21:26,934,456, cg23433889) and miR-155-5p or KPC1 expression were analyzed for TCGA cohort (n=370). Boxplot showing (D) miR-155-5p expression or (E) KPC1 expression in patients with low (n=123), intermediate (n=124) and high (n=123) DNA methylation level (classified based on tertile of DNA methylation level) (t-test, ** p<0.01, *** p<0.001). Error bars represent means ± SD from replicates (n=3).
Low KPC1 expression is associated with poor prognosis
To further investigate the clinical relevance of KPC1 and p50 in melanoma tissues, we performed IHC using tissue microarrays (TMA) (n=262 from 137 stage IV patients, Fig. 4A, Supplementary Fig. S4) (28,29). Initially, stage IV melanoma tissues demonstrated lower KPC1 expression compared to normal tissues (n=7) (KPC1 IHC score 3, 19.1% vs 57.1%, respectively, Fisher’s exact text, p=0.032). Supporting our previous observation in the functional interaction between KPC1 and p50, we observed a strong positive association between KPC1 and cytoplasmic p50 staining (n=262, χ2 test, p<0.0001, Fig. 4B). Indeed, there was also a positive association between KPC1 and nucleus p50 staining (Fisher’s exact test, p=0.016, Fig. 4B). KPC1 also ubiquitinates p27, which results in p27 degradation by the 26S proteasome (38); however, there was no association between KPC1 and p27 (χ2 test, p=0.18, Fig. 4B). Finally, we evaluated the association between KPC1 expression and clinically relevant features (n=137). While there was no significant association between KPC1 expression and known clinicopathological variables of stage IV melanoma patients (Supplementary Table S1), overall survival (OS) analysis revealed that KPC1 low expression group (n=44) had significantly shorter OS compared to KPC1 high expression group (n=93, log rank test, p=0.004, Fig. 4C). Importantly, in multivariate analysis, independent factors associated with poor OS were low KPC1 expression (Hazard Ratio 1.810, 95% Confidence Interval (CI) 1.196–2.702, p=0.006, Table 1) and advance M substages (p=0.035).
Figure 4. KPC1, p50 and p27 expression in melanoma patients’ tissues.
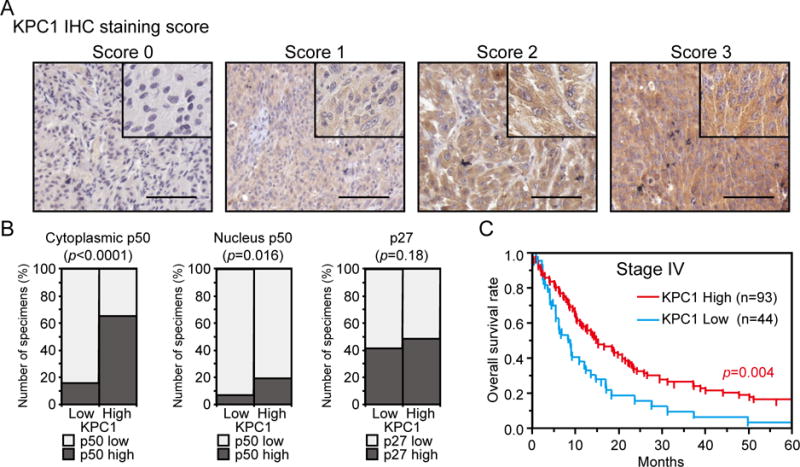
KPC1, NF-κB1 p50, and p27 expression were analyzed in melanoma patients’ tissues. (A) Representative images of AJCC stage IV melanoma TMA samples immunostained using anti-KPC1 Ab are shown. Staining intensity was evaluated from 0 (negative) to 3 (strong). The magnifications of the low-power and high-power images are ×100 and ×200, respectively. Scale bar = 100 μm. (B) Association between KPC1 expression and cytoplasmic p50, nucleus p50 or p27 expression in stage IV melanoma TMA samples (n=262). (C) Kaplan-Meier curves showing OS for KPC1 high (Score 2 and 3) or low (Score 0, 1) expression patients from stage IV melanoma TMA (n=137). Significance of log rank is shown.
Table 1.
Univariate and multivariate analyses of overall survival in stage IV melanoma by Cox proportional hazard model
| Variable | Factor | Number of patients (n=137) | Univariate analysis
|
Multivariate analysis
|
||||
|---|---|---|---|---|---|---|---|---|
| Hazard ratio | 95% CI | p-value | Hazard ratio | 95% CI | p-value | |||
| KPC1 | High (IHC score 2,3) | 93 | 1 | reference | 1 | reference | ||
| Low (IHC score 0,1) | 44 | 1.766 | 1.179–2.605 | 0.006 | 1.810 | 1.196–2.702 | 0.006 | |
|
| ||||||||
| Gender | Female | 40 | 1 | reference | 1 | reference | ||
| Male | 97 | 0.891 | 0.599–1.355 | 0.580 | 0.878 | 0.585–1.347 | 0.544 | |
|
| ||||||||
| Age | 137 | 1.001 | 0.989–1.014 | 0.884 | 1.003 | 0.990–1.016 | 0.652 | |
|
| ||||||||
| M substages | ||||||||
| M1a or M1b | 38 | 1 | reference | 1 | reference | |||
| M1c | 86 | 1.617 | 1.063–2.529 | 0.024 | 1.672 | 1.036–2.749 | 0.035 | |
| Unknown | 13 | 1.631 | 0.604–3.726 | 0.308 | 2.352 | 0.490–17.091 | 0.298 | |
|
| ||||||||
| Number of metastatic organs | ||||||||
| 1 | 73 | 1 | reference | 1 | reference | |||
| ≥2 | 48 | 1.162 | 0.780–1.714 | 0.456 | 0.935 | 0.597–1.461 | 0.769 | |
| Unknown | 16 | 1.242 | 0.544–2.473 | 0.580 | 0.684 | 0.109–2.359 | 0.592 | |
Discussion
In this study, we demonstrated a down-regulation of KPC1 expression in AJCC stage IV melanoma tissue specimens compared to stage I/II or stage III melanomas, indicating the importance of KPC1 during melanoma progression to advanced metastatic disease. Our previous study has shown down-regulation of KPC1 in tumor tissues compared to respective normal tissues in squamous cell carcinoma of head & neck and glioblastoma (15). Consistently, analysis from stage IV melanoma TMA used in this study demonstrated lower KPC1 expression in stage IV melanoma tissues compared to normal tissues. These results suggested the involvement of KPC1 in early primary tumor development; however, this study further emphasizes the importance of KPC1 expression during melanoma progression.
Our study revealed epigenetic mechanisms regulating KPC1 expression in cutaneous melanoma. We demonstrate, for the first time, the detailed mechanisms regulating KPC1 expression in tumor; methylation regulates miR-155-5p expression, and miR-155-5p controls KPC1 expression. The importance of the miR-155-5p-KPC1-NF-κB-axis in melanoma suggested new prospects in melanoma therapy. Abnormal activation of the NF-κB pathway switches non-aggressive melanoma to an aggressive phenotype (7–11), thus, inhibiting NF-κB pathway activation holds therapeutic promise for cutaneous melanoma. Inhibitors that directly target NF-κB pathway, such as IKKβ inhibitors, have been developed; however these drugs also exert NF-κB-independent effects, leading to high risk of side effects and toxicity (12,39–41). Our study widens the potential therapeutic options, regulating KPC1 through targeting the ubiquitin-proteasome system (42) and miR (43–45). We also demonstrated that low KPC1 expression is an independent variable that predicts poor prognosis in melanoma TMA. This suggested the potential of using KPC1 expression as a theranostic target in melanoma.
Besides promoter DNA methylation of miR-155-5p, we investigated other potential mechanisms that regulate miR-155-5p expression in cutaneous melanoma. Genomic region 21q21, where MIR155GH gene is located, was not significantly amplified or deleted in the TCGA melanoma cohort (data not shown). Interestingly, higher miR-155-5p expression was associated with mutation in BRAF (p=0.014, TCGA cohort, n=272, data not shown), one of the major driver gene mutations involved in melanoma progression (37). Although the regulatory mechanisms of miR-155-5p expression with BRAF mutation remains unknown, gene-mutation status is another potential mechanism leading to high miR-155-5p expression and advanced metastatic melanoma.
Our study suggested that KPC1 down-regulates NF-κB-target genes known to promote tumor progression (33–36). The presumable regulatory mechanism involves a KPC1-induced generation of p50, resulting in an increase of p50-p50 homodimers rather than tumor-promoting p65-p50 heterodimers. Because the p50-p50 homodimer lacks transcriptional activity, it is a transcriptional repressor competing with tumor-promoting p65-p50 heterodimers or a modulator of the NF-κB pathway together with other transcriptional modulators such as Bcl-3, p300, or HDAC-1 (46). Although the detailed mechanisms by which excess p50 down-regulates NF-κB-target genes have not been identified, these changes resulted in a suppressive effect on melanoma cell proliferation, as schematically shown in Fig. 5.
Figure 5. Schematic representation of DNA methylation, miR-155-5p, KPC1, and p105 processing into p50 in melanoma.
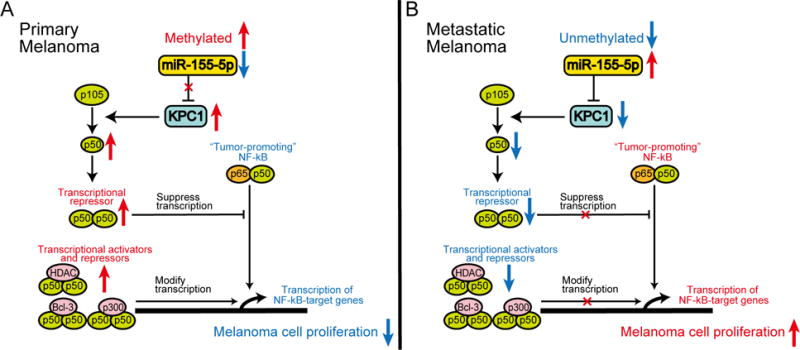
(A) In primary melanoma, where methylation represses miR-155-5p expression and KPC1 is highly expressed, excess p50–p50 homodimers suppress tumor-promoting p65–p50 heterodimers or modify transcription of NF-κB-target genes with other transcriptional modulators, resulting in suppressive effect on melanoma cell proliferation. (B) Contrarily, metastatic melanoma, which has low DNA methylation / high miR-155-5p and low KPC1 expression, lacks excess p50–p50 homodimers, promoting cell proliferation.
In conclusion, this study identified miR-155-5p, which is epigenetically controlled by its promoter methylation, as an epigenetic regulator of KPC1. These interactions promote to down-regulation of KPC1 and abnormal NF-κB pathway activation, leading to highly proliferative melanoma and poor clinical outcomes.
Supplementary Material
Translational relevance of this study.
NF-κB pathway activation promotes a more aggressive phenotype of cutaneous melanoma. Hence, understanding the mechanisms regulating the NF-κB pathway is of critical importance. In this study, we unraveled novel epigenetic mechanisms affecting miR-155-5p and KPC1 expression that lead to an abnormal activation of the NF-κB pathway in melanoma. The importance of the miR-155-5p-KPC1-NF-κB-axis in controlling melanoma proliferation opens new prospects in melanoma therapy, such as targeting the ubiquitin-proteasome system and manipulating micro-RNA. In addition to potential therapies, we demonstrated that KPC1 down-regulation is significantly associated with poor prognosis in advanced melanoma patients, suggesting that assessment of KPC1 expression may contribute to stratification of stage IV melanoma patients. These findings suggest that targeting the miR-155-5p-KPC1-NF-κB-axis holds theranostic promise for melanoma patients.
Acknowledgments
The authors thank Ms. Nousha Javanmardi and Dr. Ian V Hutchinson for their editorial assistance and Dept. of Translational Molecular Medicine Staff from Division of Molecular Oncology (JWCI) for their kind advisory and technical assistance.
Financial support
This work was supported by the National Institutes of Health, National Cancer Institute [R01CA167967 to D.H.], the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (D.H., A.C.), the Leslie and Susan Gonda (Goldschmied) Foundation (D.H.), the Israel Science Foundation (A.C.), and the I-CORE Program of the Planning and Budgeting Committee [Grant1775/12 to A.C.]. The RPPA analysis was supported by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (G.M., M.D.) and the FHCRC/UW Cancer Consortium Cancer Center Support Grant of the National Institutes of Health [P30 CA016672 to G.M.]. The RPPA Core Facility was supported by National Institutes of Health, National Cancer Institute [CA16672 to M.D.].
Abbreviations
- AJCC
American Joint Committee on Cancer
- CI
confidence interval
- FFPE
formalin-fixed paraffin-embedded
- IHC
immunohistochemistry
- miR
micro-RNA
- MSP
methylation-specific PCR
- NF-κB
nuclear factor-κB
- OS
overall survival
- RNA-seq
RNA sequencing
- RPPA
reverse-phase protein array
- RT-qPCR
reverse transcriptional-quantitative polymerase chain reaction
- SD
standard deviation
- TCGA
The Cancer Genome Atlas
- TMA
tissue microarrays
- WB
western blot
- 3′-UTR
3′-untranslated region
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
References
- 1.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199–206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 5.Griewank KG, Scolyer RA, Thompson JF, Flaherty KT, Schadendorf D, Murali R. Genetic alterations and personalized medicine in melanoma: progress and future prospects. J Natl Cancer Inst. 2014;106:2djt435. doi: 10.1093/jnci/djt435. [DOI] [PubMed] [Google Scholar]
- 6.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–32. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 7.Ueda Y, Richmond A. NF-kappaB activation in melanoma. Pigment Cell Res. 2006;19(2):112–24. doi: 10.1111/j.1600-0749.2006.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amiri KI, Richmond A. Role of nuclear factor-kappa B in melanoma. Cancer Metastasis Rev. 2005;24(2):301–13. doi: 10.1007/s10555-005-1579-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, Hua W, Huang SK, Fan K, Takeshima L, Mao Y, et al. RASSF8 regulates progression of cutaneous melanoma through nuclear factor-kappab. Oncotarget. 2015;6(30):30165–77. doi: 10.18632/oncotarget.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kashani-Sabet M, Shaikh L, Miller JR, 3rd, Nosrati M, Ferreira CM, Debs RJ, et al. NF-kappa B in the vascular progression of melanoma. J Clin Oncol. 2004;22(4):617–23. doi: 10.1200/JCO.2004.06.047. [DOI] [PubMed] [Google Scholar]
- 11.Torabian SZ, de Semir D, Nosrati M, Bagheri S, Dar AA, Fong S, et al. Ribozyme-mediated targeting of IkappaBgamma inhibits melanoma invasion and metastasis. Am J Pathol. 2009;174(3):1009–16. doi: 10.2353/ajpath.2009.080207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. 2012;12(2):121–32. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 13.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2(4):301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 14.DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246(1):379–400. doi: 10.1111/j.1600-065X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- 15.Kravtsova-Ivantsiv Y, Shomer I, Cohen-Kaplan V, Snijder B, Superti-Furga G, Gonen H, et al. KPC1-mediated ubiquitination and proteasomal processing of NF-kappaB1 p105 to p50 restricts tumor growth. Cell. 2015;161(2):333–47. doi: 10.1016/j.cell.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Salmeron A, Janzen J, Soneji Y, Bump N, Kamens J, Allen H, et al. Direct phosphorylation of NF-kappaB1 p105 by the IkappaB kinase complex on serine 927 is essential for signal-induced p105 proteolysis. J Biol Chem. 2001;276(25):22215–22. doi: 10.1074/jbc.M101754200. [DOI] [PubMed] [Google Scholar]
- 17.Tanemura A, Terando AM, Sim MS, van Hoesel AQ, de Maat MF, Morton DL, et al. CpG island methylator phenotype predicts progression of malignant melanoma. Clin Cancer Res. 2009;15(5):1801–7. doi: 10.1158/1078-0432.CCR-08-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vinci M, Gowan S, Boxall F, Patterson L, Zimmermann M, Court W, et al. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012;10:29. doi: 10.1186/1741-7007-10-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amersi FF, Terando AM, Goto Y, Scolyer RA, Thompson JF, Tran AN, et al. Activation of CCR9/CCL25 in cutaneous melanoma mediates preferential metastasis to the small intestine. Clin Cancer Res. 2008;14(3):638–45. doi: 10.1158/1078-0432.CCR-07-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohta K, Hoshino H, Wang J, Ono S, Iida Y, Hata K, et al. MicroRNA-93 activates c-Met/PI3K/Akt pathway activity in hepatocellular carcinoma by directly inhibiting PTEN and CDKN1A. Oncotarget. 2015;6(5):3211–24. doi: 10.18632/oncotarget.3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Maat MF, van de Velde CJ, Umetani N, de Heer P, Putter H, van Hoesel AQ, et al. Epigenetic silencing of cyclooxygenase-2 affects clinical outcome in gastric cancer. J Clin Oncol. 2007;25(31):4887–94. doi: 10.1200/JCO.2006.09.8921. [DOI] [PubMed] [Google Scholar]
- 22.Arigami T, Narita N, Mizuno R, Nguyen L, Ye X, Chung A, et al. B7-h3 ligand expression by primary breast cancer and associated with regional nodal metastasis. Ann Surg. 2010;252(6):1044–51. doi: 10.1097/SLA.0b013e3181f1939d. [DOI] [PubMed] [Google Scholar]
- 23.Hoshimoto S, Shingai T, Morton DL, Kuo C, Faries MB, Chong K, et al. Association between circulating tumor cells and prognosis in patients with stage III melanoma with sentinel lymph node metastasis in a phase III international multicenter trial. J Clin Oncol. 2012;30(31):3819–26. doi: 10.1200/JCO.2011.40.0887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Huang SK, Marzese DM, Hsu SC, Kawas NP, Chong KK, et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J Invest Dermatol. 2015;135(2):532–41. doi: 10.1038/jid.2014.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narita N, Tanemura A, Murali R, Scolyer RA, Huang S, Arigami T, et al. Functional RET G691S polymorphism in cutaneous malignant melanoma. Oncogene. 2009;28(34):3058–68. doi: 10.1038/onc.2009.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marzese DM, Scolyer RA, Huynh JL, Huang SK, Hirose H, Chong KK, et al. Epigenome-wide DNA methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. Hum Mol Genet. 2014;23(1):226–38. doi: 10.1093/hmg/ddt420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menigatti M, Truninger K, Gebbers JO, Marbet U, Marra G, Schar P. Normal colorectal mucosa exhibits sex- and segment-specific susceptibility to DNA methylation at the hMLH1 and MGMT promoters. Oncogene. 2009;28(6):899–909. doi: 10.1038/onc.2008.444. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Chong KK, Nakamura Y, Nguyen L, Huang SK, Kuo C, et al. B7-H3 associated with tumor progression and epigenetic regulatory activity in cutaneous melanoma. J Invest Dermatol. 2013;133(8):2050–8. doi: 10.1038/jid.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goto Y, Ferrone S, Arigami T, Kitago M, Tanemura A, Sunami E, et al. Human high molecular weight-melanoma-associated antigen: utility for detection of metastatic melanoma in sentinel lymph nodes. Clin Cancer Res. 2008;14(11):3401–7. doi: 10.1158/1078-0432.CCR-07-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng J, Meyer CA, Wang Q, Liu JS, Shirley Liu X, Zhang Y. GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics. 2012;28(21):2782–8. doi: 10.1093/bioinformatics/bts515. [DOI] [PubMed] [Google Scholar]
- 32.Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun. 2014;5:3887. doi: 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polsky D, Cordon-Cardo C. Oncogenes in melanoma. Oncogene. 2003;22(20):3087–91. doi: 10.1038/sj.onc.1206449. [DOI] [PubMed] [Google Scholar]
- 34.Elmets CA, Ledet JJ, Athar M. Cyclooxygenases: mediators of UV-induced skin cancer and potential targets for prevention. J Invest Dermatol. 2014;134(10):2497–502. doi: 10.1038/jid.2014.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menter DG, Schilsky RL, DuBois RN. Cyclooxygenase-2 and cancer treatment: understanding the risk should be worth the reward. Clin Cancer Res. 2010;16(5):1384–90. doi: 10.1158/1078-0432.CCR-09-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15(21):6479–83. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161(7):1681–96. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, et al. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol. 2004;6(12):1229–35. doi: 10.1038/ncb1194. [DOI] [PubMed] [Google Scholar]
- 39.Gilmore TD, Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006;25(51):6887–99. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- 40.Shtivelman E, Davies MQ, Hwu P, Yang J, Lotem M, Oren M, et al. Pathways and therapeutic targets in melanoma. Oncotarget. 2014;5(7):1701–52. doi: 10.18632/oncotarget.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Waes C. Nuclear factor-kappaB in development, prevention, and therapy of cancer. Clin Cancer Res. 2007;13(4):1076–82. doi: 10.1158/1078-0432.CCR-06-2221. [DOI] [PubMed] [Google Scholar]
- 42.Micel LN, Tentler JJ, Smith PG, Eckhardt GS. Role of ubiquitin ligases and the proteasome in oncogenesis: novel targets for anticancer therapies. J Clin Oncol. 2013;31(9):1231–8. doi: 10.1200/JCO.2012.44.0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kasinski AL, Slack FJ. Epigenetics and genetics MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer. 2011;11(12):849–64. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berindan-Neagoe I, Calin GA. Molecular pathways: microRNAs, cancer cells, and microenvironment. Clin Cancer Res. 2014;20(24):6247–53. doi: 10.1158/1078-0432.CCR-13-2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nature reviews Drug discovery. 2013;12(11):847–65. doi: 10.1038/nrd4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pereira SG, Oakley F. Nuclear factor-kappaB1: regulation and function. Int J Biochem Cell Biol. 2008;40(8):1425–30. doi: 10.1016/j.biocel.2007.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.