


Abstract
A series of analogues based on serine as lead structure were designed, and their agonist activities were evaluated at recombinant NMDA receptor subtypes (GluN1/2A–D) using two-electrode voltage-clamp (TEVC) electrophysiology. Pronounced variation in subunit-selectivity, potency, and agonist efficacy was observed in a manner that was dependent on the GluN2 subunit in the NMDA receptor. In particular, compounds 15a and 16a are potent GluN2C-specific superagonists at the GluN1 subunit with agonist efficacies of 398% and 308% compared to glycine. This study demonstrates that subunit-selectivity among glycine site NMDA receptor agonists can be achieved and suggests that glycine-site agonists can be developed as pharmacological tool compounds to study GluN2C-specific effects in NMDA receptor-mediated neurotransmission.
Keywords: Ionotropic glutamate receptor, NMDA, superagonist, d-serine, d-cycloserine, subtype selectivity
Graphical abstract
(S)-Glutamate (Glu, 1, Chart 1), the major excitatory neurotransmitter in the central nervous system, activates a heterogeneous population of receptors comprised of G protein-coupled metabotropic receptors as well as ionotropic Glu receptors (iGluRs).1,2 These receptors are crucial for normal brain functions,3 and dysregulation of iGluRs has been linked to a number of neurological and psychiatric disorders.2,4
Chart 1. Chemical Structures of Glu and Agonists for the Glycine Binding Site of NMDA Receptorsa.

aGly (2) and d-Ser (3) are endogenous agonists, and d-cycloserine (4) as well as vinyl glycines (5) are natural products with agonist properties.1 Moreover, Urwyler et al. reported a series of glycine site agonists based on the general structure 6.7
The iGluRs are ligand-gated ion channels2 that can be divided into three major classes based on sequence identity and the selective activation by the agonists N-methyl-d-aspartic acid (NMDA), (S)-2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)-propionic acid (AMPA) and kainate (KA). NMDA receptors are tetrameric assemblies of subunits5,6 and are assembled from GluN1, GluN2A–D, and/or GluN3A,B subunits. While GluN2 subunits recognize Glu, the GluN1 and GluN3 subunits recognize the endogenous coagonists glycine (Gly, 2) and d-serine (d-Ser, 3). The distinct distribution of GluN2A–D subunits in different brain regions and at the cellular level,3 suggests different physiological roles of these subunits in normal brain functions and disease. During the past several decades, a great number of iGluR ligands have been developed, but few of them are specific for a single subtype. For the glycine site in the GluN1 subunit of NMDA receptors, a large number of antagonists exists, but relatively few full and partial agonists, such as compounds 4–6 (Chart 1), have been reported.1,7–9
In recent years, a large number of structures of isolated iGluR agonist binding domains (ABDs) have disclosed important information on the molecular basis for orthosteric ligand recognition, and the mechanisms underlying activation, desensitization, and allosteric modulation.10–14 These studies also show that structural differences exist in the dimer interface between ABDs of GluN1 and the different GluN2A–D subunits.12,13 Here, we have aimed at exploiting these structural differences to develop agonists capable of differentiating between the glycine binding site of GluN1 in a GluN2 subunit-dependent manner. The glycine site agonists developed in our study display pronounced variation in activity among GluN1/2A–D NMDA receptor subtypes. These findings expand the pharmacology of glycine site agonists and highlight more opportunities for the development of subunit-selective NMDA receptor ligands than previously anticipated.
RESULTS AND DISCUSSION
Inspection of the GluN1 ABD structure with bound d-Ser (PDB: 1PB810) overlaid with the GluN1/GluN2 ABD dimer structure with Gly and Glu (PDB: 5I5712) shows a cavity of water molecules protruding from the hydroxy group of d-Ser (Figure 1). The structure suggests that ligand substituents may be accommodated in this cavity and that glycine-site agonists can extend into this cavity toward the GluN1–GluN2 ABD dimer interface where structural variation among GluN2 subunits exist.
Figure 1.

(A) Crystal structure of the NMDA receptor agonist binding domain (ABD) heterodimer (PDB: 5I5712) consisting of GluN1 (yellow) with Gly and GluN2A (orange) with Glu. The cavity extending from the glycine binding site into the ABD dimer interface is shown in cyan (see Supporting Information). (B) Detailed view of the GluN1/2A ABD dimer structure aligned with the structure of the GluN1 ABD (pink) with bound d-Ser (PDB: 1PB810). The hydroxy group of d-Ser points toward the subunit dimer interface into the cavity.
For example, V783 in GluN2A is a nonconserved residue among GluN2A–D subunits (phenylalanine in GluN2B and leucine in GluN2C–D) and has been exploited for the development of GluN2A-selective negative and positive allosteric modulators.12,13,15 This residue is in contact with F754 in GluN1 (Figure 1B), and site-directed mutagenesis of GluN2A V783 and GluN1 F754 residues profoundly affects glycine potency, despite their location >10 Å away from the glycine ligand.13
We hypothesize that agonists with substituents extending toward the GluN1–GluN2 ABD dimer interface can engage in GluN2-specific interactions, thereby enabling variation in activity among GluN1/GluN2 NMDA receptor subtypes.16,17 To evaluate this hypothesis, we designed and synthesized a series of amino acids carrying bulky substituents and characterized them by two-electrode voltage-clamp (TEVC) electrophysiology using Xenopus oocytes expressing recombinant NMDA receptor subtypes. We generated concentration–response data for the compounds in the continuous presence of a saturating concentration of Glu (100–300 µM) at the four GluN1/GluN2A–D NMDA receptor subtypes.
In the crystal structure of d-Ser in complex with the GluN1 ABD, the hydroxy group of d-Ser is pointing toward the dimer interface between GluN1 and GluN2 (Figure 1B). We therefore used d-Ser as a lead structure initially to obtain d-Ser analogues O-butyl 7, O-benzyl 8a, and O-naphtyl 9a (Chart 2).
Chart 2. Chemical Structures of d-Ser Derivatives as Well as Amide and Thioether Containing Analogues.

Compounds 7–9 and 10–12 were synthesized (Scheme S1 in the Supporting Information) from Boc-protected Ser or cysteine by deprotonation with NaH or K2CO3, alkylation with the respective alkyl halides, followed by Boc-deprotection under acidic conditions. Compounds 13–16 were synthesized by reacting Boc-protected amino alanine with the respective acid chlorides followed by Boc-deprotection under acidic conditions.
Consistent with our rationale, compounds 7, 8a, and 9a displayed variation in agonist efficacy among GluN1/2A–D subtypes (Table 1 and Figure 2). Compound 7 was a partial agonist with low agonist efficacy at GluN1/2A (36% compared to glycine), GluN1/2B (61%), and GluN1/2D (69%), and higher agonist efficacy at GluN1/C (91%). Compound 8a had a different pharmacological profile and was a partial agonist at GluN1/2A (53%), GluN1/2B (54%), and GluN1/2D (88%), but a full agonist at GluN1/2C (108%). Compound 9a showed no activity at GluN1/2A and GluN1/2B, but where partial agonists at GluN1/2C (61%) and GluN1/2D (55%). The l-forms 8b and 9b did not show agonist activity at any NMDA receptor subtype.
Table 1.
Agonist Potencies and Efficacies of Compounds at Recombinant GluN1/2A–D Receptors Measured Using TEVC Electrophysiologya
| GluN1/2A | GluN1/2B | GluN1/2C | GluN1/2D | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|||||
| EC50 (µM) | Rmax (%) rel. to Gly | EC50 (µM) | Rmax (%) rel. to Gly | EC50 (µM) | Rmax (%) rel. to Gly | EC50 (µM) | Rmax (%) rel. to Gly | |
| Gly (2) | 0.95 | 100 | 0.24 | 100 | 0.20 | 100 | 0.09 | 100 |
| d-Ser (3) | 1.0 | 96 | 0.51 | 98 | 0.18 | 119 | 0.15 | 95 |
| 7 | 123 | 36 | 49 | 61 | 38 | 91 | 12 | 69 |
| 8a | 95 | 53 | 40 | 54 | 21 | 108 | 14 | 88 |
| 8b | NR | >300 | >300 | >300 | ||||
| 9a | NR | NR | 45 | 61 | 94 | 55 | ||
| 9b | NR | NR | NR | NR | ||||
| 10a | NR | NR | >300 | >300 | ||||
| 10b | NR | >300 | >300 | >300 | ||||
| 11a | NR | NR | 87 | 169 | >300 | |||
| 11b | NR | NR | >300 | >300 | ||||
| 12a | NR | NR | NR | NR | ||||
| 12b | NR | NR | >300 | >300 | ||||
| 13a | >300 | >300 | 206 | 223 | >300 | |||
| 13b | NR | NR | >300 | >300 | ||||
| 14a | 84 | 46 | 56 | 17 | 38 | 71 | 115 | 98 |
| 14b | >300 | >300 | >300 | >300 | ||||
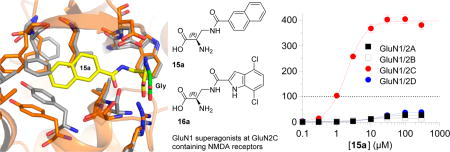
| 15a | 12.3 | 27 | 3.83 | 15 | 1.97 | 398 | 20.1 | 40 |
| 15b | NR | NR | 274 | 148 | >300 | |||
| 16a | 2.56 | 13 | 0.4 | 5 | 0.32 | 308 | 0.3 | 8 |
| 16b | NR | NR | 93 | 27 | NR | |||
The relative maximal currents (Rmax) are the maximal responses to the indicated agonists obtained by fitting the full concentration-response data normalized to the maximal response activated by glycine in the same recording. Where full concentration–response data could not be determined, EC50 is indicated as >300. NR indicates responses <5% at 300 µM of the compound. See Table S1 in the Supporting Information for an extended version with statistical details.
Figure 2.

Mean concentration–response curves for compounds 8a, 15a, and 16a and representative recordings obtained using TEVC electrophysiology with Xenopus oocytes coexpressing GluN1 and GluN2A–D. The curves are normalized to the maximal current response to Gly in the same recording. All EC50 values are listed in Table 1. Error bars (SEM) are small and may be contained within the symbols. The vertical scale bar represents 100 nA and the horizontal scale bar represents 30 s.
These differences spurred us to extent the compound series by exchanging the ether linkage of these compounds with an amide and thioether linkage (Chart 2). Each stereoisomer (a/b) of compounds 10–15 was characterized on the four GluN1/2A–D receptor subtypes.
S-Butyl l- and d-cysteine 10a and 10b did not have noticeable agonist activity, but the S-benzyl substituted l-cysteine (R-form) 11a showed significant superagonistic activity (169% response relative to glycine) at the GluN1/2C receptor subtype (Table 1). Although the potency of 11a was low (87 µM) at GluN1/2C, some selectivity was observed for NMDA receptors containing the GluN2C subunit over the other receptor subtypes (>300 µM). The S-benzylated d-cysteine 11b did not show marked agonist activity below 300 µM, and both enantiomers S-naphtyl 12a and 12b showed little or no activity. The agonist efficacy at GluN1/2C of pentanamido analogue 13a was more than double that of glycine (223%), whereas the enantiomer 13b showed little activity (Table 1). The agonist efficacy of benzamido analogue 14a displayed a broad range of agonist efficacies varying from 17% at GluN1/2B to 98% at GluN1/2D receptors with potencies ranging from 38 to 115 µM, whereas the enantiomer 14b displayed little activity (Table 1). 15a displayed subunit-specific variation in potencies spanning an order of magnitude among NMDA receptor subtypes (Table 1 and Figure 2). Interestingly, 15a is a highly efficacious superagonist at GluN1/2C with maximal currents of 398% relative to glycine, but a low efficacy partial agonist at other subtypes. Racemate 15 was previously characterized in radioligand binding experiments without functional characterization.7
We decided to include both enantiomers of the previously reported racemate 16,7 to establish the subtype profile, since it was reported to be partial agonist with low efficacy (32%) in a membrane binding assay using the channel blocker [3H]MK-801.7 Compound 16a potently (0.32 µM) activated GluN1/GluN2C with maximal currents of 308% relative to glycine (Table 1 and Figure 2), while having a very low agonist efficacy (5–13%) relative to glycine on the other subtypes.
To evaluate if the compounds without agonist activity at any receptor subtype (e.g., 10a and 12a) or on a specific subtype (e.g., 11a) can bind the glycine site and serve as competitive antagonists, we generated concentration–inhibition data for 10a, 11a, and 12a using TEVC recordings. The NMDA receptors were activated by 3 µM glycine plus 300 µM glutamate in the absence and presence of increasing compound concentrations. None of the compounds produced marked inhibition of any NMDA receptor subtype (GluN1/2A–D), demonstrating that these ligands are not high-affinity competitive antagonists at the glycine binding site (Figure S1).
We have identified GluN1 glycine site agonists with pronounced GluN2-dependent activity by designing ligands with large substituents predicted to interact with residues in the vicinity of the ABD dimer interface between GluN1 and GluN2 subunits. To support this prediction, we performed ligand-docking followed by molecular dynamics simulations using the crystal structure of the GluN1/2A ABD heterodimer (see the Supporting Information for procedures).
Briefly, compound 15a was docked into the glycine binding site of GluN1 to obtain two initial high scoring poses (Figure S2) and four molecular dynamics simulations (300 ns each) were performed using these two poses (two simulations for each pose). In all four simulations, compound 15a quickly adopted the same stable binding mode, where the amide nitrogen of 15a can form a hydrogen bond with the side chain carboxylate of GluN1 D732 and the carbonyl of 15a can from hydrogen bonds with the side chain hydroxyl of S688 and the backbone nitrogen of V686. Importantly, the naphtyl group of 15a protrudes into the cavity toward the ABD dimer interface and accommodation of the naphtyl group requires some rearrangement of residues lining this cavity. Specifically, considerable rearrangement of GluN1 F753, F754, and Y692 is required for 15a binding, and the position of GluN2A V783 is shifted compared to glycine bound structure (Figure 3). For comparison, four molecular dynamics simulations (300 ns each) were performed with Gly bound in the GluN1 agonist binding site. Gly dissociated from the GluN1 agonist binding site in one of the four simulations, but with the exception of this simulation, binding of compound 15a did not result in noticeable differences in the global conformation of the GluN1 ABD compared to binding of Gly (Figure S3). Overall, the molecular modeling therefore supports that larger ligand substituents may be accommodated in the cavity toward the GluN1–GluN2 ABD dimer interface. More work is needed to determine the mechanism by which the highly efficacious GluN2C-selective superagonists 15a and 16a may engage in GluN2-specific interactions, presumably at the ABD dimer interface, thereby enabling variation in activity among GluN1/GluN2 NMDA receptor subtypes.
Figure 3.

Ligand-docking and molecular dynamics simulations with 15a in the GluN1/2A ABD heterodimer structure. (A) Representative stable binding mode of 15a adopted in four molecular dynamics simulations using two high scoring poses from ligand-docking into the structure of the GluN1/2A ABD dimer (see the Supporting Information for additional figures and details on the molecular modeling). (B) Overlay of the structure from molecular dynamics simulations with bound 15a (protein in orange; 15a in yellow) and the GluN1/2A ABD crystal structure with bound Gly (protein in gray; Gly in green).
We have presented the design and synthesis of a series of Ser analogues and evaluated whether they can discriminate between glycine binding sites in NMDA receptor complexes with different GluN2 subunits. We predicted that activity might vary if substituents were located in the vicinity of the dimer interface between GluN1 and GluN2 ABDs. Consistent with this hypothesis, d-Ser analogues 7a and 8a show variation in agonist efficacy at GluN1 depending on the coexpressed GluN2 subunit. We extended the series by exchanging the ether linkage with amide and thioether linkers. These analogues also show varying activity. Surprisingly, 11a and 13a showed extensive potentiation (2-fold) of maximal current at GluN1/2C relative to the current induced by Gly. Thus, both compounds are superagonists at the glycine site of the GluN1/2C receptor. This observation was even more pronounced with 15a and 16a showing remarkable enhancements of agonist efficacy (398% and 308%) compared to the endogenous agonist glycine. These levels of superagonistic activity are unprecedented among all NMDA receptor agonists described to date.1,8,16,17 The substituents on 15a and 16a are quite bulky and molecular modeling support that steric effects at GluN1 F754 could be involved in the GluN2-specific agonist activity (Figure 3).
Although R-isomers generally are more active than S-isomers, it is notable that the cysteine analogue 11a is the active enantiomer since this l-form has opposite geometry. This could mean that substituents are pointing in opposite directions in the GluN1 glycine binding site if the amino acid (including the α-carbon) moiety has an overlapping binding mode. However, the selectivity profile of l-isomers 11a, 15b, and 16b suggests that the substituents point in the same direction toward the ABD dimer interface by overlaying of the amino acid moieties, but not the α-carbon, as can be observed in the cocrystal structures of d- and l-Glu in the GluN2D ABD.18
d-Cycloserine (4) has been intensively studied as a GluN1 glycine site agonist with intriguing neuroactive properties. Administration of d-cycloserine can enhance extinction of fear in rodents and humans, and d-cycloserine has been considered as a potential therapeutic agent in several psychiatric disorders.19,20 Until now, d-cycloserine was the only described glycine site ligand with agonist efficacy that is highly dependent on the glutamate-binding GluN2 subunits and the only described superagonist at GluN1/2C receptors.21,17 Our identification of compounds 15a and 16a as GluN2C-selective superagonists provides new opportunities for in vivo biological and behavioral studies that have previously relied on d-cycloserine for NMDA receptor modulation.
In conclusion, this study demonstrates large variation in potency and agonist efficacy of various Ser analogues at the glycine sites in NMDA receptors, in a GluN2 dependent manner. Exchanging the ether linker in Ser analoges with thioether and amide linkers yielded a series of compounds that displayed large variation in agonist efficacy ranging from very partial agonism (5%) to superagonism (398%) in a GluN2-specific manner. Thus, the results of this study suggest that differential potency, improved agonist efficacy, and subtype-selectivity at the glycine sites in NMDA receptors can be achieved by synthesizing novel ligands designed to exploit structural differences in the NMDA receptor ABDs.
METHODS
Chemistry
Representative experimental procedures for compound 8a, 15a, and 16a. For further experimental details, see the Supporting Information.
O-Benzyl-d-Ser (8a)
To a dispersion of 60% NaH (204 mg, 5.1 mmol, 2.1 equiv) in anhydrous DMF (2.0 mL) at 0 °C was slowly added N-Boc-d-Ser (500 mg, 2.43 mmol) dissolved in anhydrous DMF (4 mL). The mixture was stirred for 1 h at room temperature, cooled to 0 °C, and then benzyl bromide (0.29 mL, 416 mg, 2.43 mmol, 1 equiv) was added. The mixture was allowed to warm to room temperature and stirred for 4 h. The reaction was quenched with sat. NH4Cl (1 mL) and the mixture was partioned between Et2O (20 mL) and icecold 0.1 M HCl (10 mL). The aqueous phase was extracted with Et2O (3 × 50 mL), dried (MgSO4), concentrated, and purified by column chromatography (eluent 0–100% EtOAc in hexane, 1% CH3CO2H) to afford 470 mg (66% yield). For deprotection, the product was dissolved in dioxane (2 mL), HCl in dioxane (4 M, 1 mL) was added dropwise and the reaction stirred overnight. The precipitate was washed with Et2O, dried (MgSO4), and concentrated to afford 178 mg (32% yield) of 8a as white solid. 1H NMR (300 MHz; methanol-d4): δ 7.36−7.29 (m, 5H), 4.66−4.56 (m, 2H), 4.17 (dd, J = 4.8, 3.3 Hz, 1H), 3.88 (dq, J = 11.5, 4.1 Hz, 2H). 13C NMR (75 MHz; methanol-d4): δ 169.7, 138.4, 129.3, 128.91, 128.89, 74.4, 68.2, 54.4. MS calcd for C10H13NO3H+ [M + H]+: 196.1; found: 196.1. Mp: 193.3–194.3 °C.
(R)-3-(2-Naphthamido)-2-aminopropanoic Acid (15a)
To Boc-d-3-aminopropionic acid (500 mg, 2.4 mmol) in THF (5 mL) was added DIPEA (2.1 mL, 12.2 mmol, 5 equiv) and 2-naphthoyl chloride (460 mg, 2.4 mmol, 1 equiv). The reaction was stirred overnight, and then 1 M HCl was added (until pH = 2–3). The mixture was extracted with EtOAc (2 × 20 mL), washed with brine (2 × 10 mL), dried (MgSO4), and evaporated. Purification by column chromatography (eluent 30% EtOAc in heptane, 2% CH3CO2H) afforded 320 mg (37% yield) as colorless oil. For deprotection, the product was stirred in 2 M HCl in Et2O (7 mL, 20 equiv) at room temperature for 2.5 h. The precipitate was then washed with Et2O and dried to afford 135 mg (68% yield) of 15a as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 13.89 (s, 1H), 8.99 (t, J = 5.8 Hz, 1H), 8.53 (s, 1H), 8.48 (brs, 3H), 8.03−7.94 (m, 4H), 7.70−7.53 (m, 2H), 4.14 (t, J = 5.8 Hz, 1H), 3.94−3.71 (m, 2H). 13C NMR (151 MHz, DMSO) δ 169.1, 166.9, 134.2, 132.0, 131.0, 128.8, 128.0, 127.8, 127.73 127.6, 126.8, 124.3, 52.4, 39.4. MS calcd for C14H14N2O3H+ [M + H]+: 259.1; found: 259.1. Mp: 226.3–232.4 °C.
(R)-2-Amino-3-(4,7-dichloro-1H-indole-2-carboxamido)-propanoic Acid (16a)
To 4,7-dichloro-1H-indole-2-carboxylic acid (0.3 g, 1.3 mmol) in THF (5 mL) was added 2 M oxalyl chloride in CH2Cl2 (1.3 mL, 2.6 mmol, 2 equiv) and a drop of DMF. The reaction was stirred at room temperature for 1 h and then concentrated. The crude material was redissolved in THF (5 mL), and then Boc-aminopropanoic acid (0.27 g, 1.3 mmol, 1 equiv) and DIPEA (1.1 mL, 6.5 mmol, 5 equiv) were added. The mixture was stirred at room temperature for 1 h, then diluted with H2O (10 mL) and 1 M HCl (2 mL, to pH = 2–3), and extracted with EtOAc (2 × 20 mL). The combined organic phases were washed with brine (2 × 10 mL), dried (MgSO4), and concentrated. For deprotection, the purified compound was dissolved in 2 M HCl in Et2O and stirred for 2.5 h. The final product was obtained as a white solid (yield: 9%, 3 steps for 16a). 1H NMR (600 MHz, DMSO-d6) δ 13.99 (brs, 1H), 12.22 (s, 1H), 9.04 (t, J = 5.8 Hz, 1H), 8.42 (brs, 3H), 7.32 (d, J = 8.1 Hz, 1H), 7.31 (d, J = 1.8 Hz, 1H), 7.18 (d, J = 8.1 Hz, 1H), 4.13−4.09 (m, 1H), 3.84 (dt, J = 14.3, 5.2 Hz, 1H), 3.78−3.72 (m, 1H). 13C NMR (151 MHz, DMSO) δ 169.0, 160.5, 134.0, 133.7, 126.8, 124.4, 124.0, 120.4, 115.8, 104.1, 52.3, 38.9. MS calcd for C12H11Cl2N3O3H+ [M + H]+: 316.0; found: 316.0. Mp: 218.7–224.8 °C.
Pharmacology
DNA Constructs and Expression in Xenopus Oocytes
cDNAs encoding GluN1-1a (Genbank accession number U11418 and U08261), GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), and GluN2D (L31611) were generously provided by Dr. S. Heinemann (Salk Institute, La Jolla, CA), Dr. S. Nakanishi (Osaka Bioscience Institute, Osaka, Japan), and Dr. P. Seeburg (University of Heidelberg). Amino acid residues are numbered based on the full-length polypeptide sequence, including the signal peptide (initiating methionine is 1).
For expression in Xenopus laevis oocytes, cDNAs were linearized using restriction enzymes and used as templates to synthesize cRNA using the mMessage mMachine kit (Ambion, Life Technologies, Paisley, UK). Xenopus oocytes were obtained from Rob Weymouth (Xenopus 1, Dexter, MI). The oocytes were injected with cRNAs encoding GluN1 and GluN2 in a 1:2 ratio, and maintained as previously described.22
Two-Electrode Voltage-Clamp Recordings
Two-electrode voltage-clamp (TEVC) recordings were performed on Xenopus oocytes essentially as previously described.19 Oocytes were perfused with extracellular recording solution comprised of 90 mM NaCl, 1 mM KCl, 10 mM HEPES, 0.5 mM BaCl2, and 0.01 mM EDTA (pH 7.4 with NaOH). Current responses were recorded at a holding potential of −40 mV. Compounds were dissolved in extracellular recording solution.
Data were analyzed using GraphPad Prism (GraphPad Software, La Jolla, CA). Agonist concentration–response data for individual oocytes were fitted to the Hill equation as previously described.22
Supplementary Material
Acknowledgments
Funding
This work was supported by the Lundbeck Foundation, the Danish Council for Independent Research - Medical Sciences, the GluTarget Programme of Excellence at University of Copenhagen, University of Montana Research Grant Program, and the National Institute of Health (P20GM103546 and R01NS097536).
ABBREVIATIONS
- ABD
agonist binding domain
- AMPA
(S)-2-amino-3-(3-hydroxy-5-methyl-4-isoxazolyl)propionic acid
- DIPEA
diiso-propylethylamine
- Glu
glutamate
- GluRs
glutamate receptors
- iGluRs
ionotropic glutamate receptors
- KA
kainic acid
- mGluRs
metabotropic glutamate receptors
- NMDA
N-methyl-d-aspartic acid
- TEVC
two-electrode voltage-clamp
Footnotes
ASSOCIATED CONTENT
- Additional details on molecular modeling and synthetic procedures for the preparation of all compounds; additional data and molecular modeling; SEM values (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- 1.Bräuner-Osborne H, Egebjerg J, Nielsen EO, Madsen U, Krogsgaard-Larsen P. Ligands for glutamate receptors: design and therapeutic prospects. J. Med. Chem. 2000;43:2609–2645. doi: 10.1021/jm000007r. [DOI] [PubMed] [Google Scholar]
- 2.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riedel G, Platt B, Micheau J. Glutamate receptor function in learning and memory. Behav. Brain Res. 2003;140:1–47. doi: 10.1016/s0166-4328(02)00272-3. [DOI] [PubMed] [Google Scholar]
- 4.Javitt DC. Glutamate as a therapeutic target in psychiatric disorders. Mol. Psychiatry. 2004;9:984–997. doi: 10.1038/sj.mp.4001551. [DOI] [PubMed] [Google Scholar]
- 5.Lee C-H, Lü W, Michel JC, Goehring A, Du J, Song X, Gouaux E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature. 2014;511:191–197. doi: 10.1038/nature13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urwyler S, Floersheim P, Roy BL, Koller M. Drug design, in vitro pharmacology, and structure-activity relationships of 3-acylamino-2-aminopropionic acid derivatives, a novel class of partial agonists at the glycine site on the N-methyl-D-aspartate (NMDA) receptor complex. J. Med. Chem. 2009;52:5093–5107. doi: 10.1021/jm900363q. [DOI] [PubMed] [Google Scholar]
- 8.Chen PE, Geballe MT, Katz K, Erreger K, Livesey MR, O’Toole KK, Le P, Lee J, Snyder J, Traynelis SF, Wyllie DJA. Modulation of glycine potency in rat recombinant NMDA receptors containing chimeric NR2A/2D subunits expressed in Xenopus laevis oocytes. J. Physiol. 2008;586:227–245. doi: 10.1113/jphysiol.2007.143172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monahan JB, Hood WF, Compton RP, Cordi AA, Williams RM. Identification of a novel structural class of positive modulators of the N-methyl-D-aspartate receptor, with actions mediated through the glycine recognition site. Eur. J. Pharmacol., Mol. Pharmacol. Sect. 1990;189:373–379. doi: 10.1016/0922-4106(90)90034-u. [DOI] [PubMed] [Google Scholar]
- 10.Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J. 2003;22:2873–2885. doi: 10.1093/emboj/cdg303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inanobe A, Furukawa H, Gouaux E. Mechanism of partial agonist action at the NR1 subunit of NMDA receptors. Neuron. 2005;47:71–84. doi: 10.1016/j.neuron.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 12.Yi F, Mou TC, Dorsett KN, Volkmann RA, Menniti FS, Sprang SR, Hansen KB. Structural Basis for Negative Allosteric Modulation of GluN2A-Containing NMDA Receptors. Neuron. 2016;91:1316–1329. doi: 10.1016/j.neuron.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hackos DH, Lupardus PJ, Grand T, Chen Y, Wang TM, Reynen P, Gustafson A, Wallweber HJA, Volgraf M, Sellers BD, Schwarz JB, Paoletti P, Sheng M, Zhou Q, Hanson JE. Positive allosteric modulators of GluN2A-containing NMDARs with distinct modes of action and impacts on circuit function. Neuron. 2016;89:983–999. doi: 10.1016/j.neuron.2016.01.016. [DOI] [PubMed] [Google Scholar]
- 14.Jespersen A, Tajima N, Fernandez-Cuervo G, Garnier-Amblard EC, Furukawa H. Structural insights into competitive antagonism in NMDA receptors. Neuron. 2014;81:366–378. doi: 10.1016/j.neuron.2013.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volgraf M, Sellers BD, Jiang Y, Wu G, Ly CQ, Villemure E, Pastor RM, Yuen P-W, Lu A, Luo X, Liu M, Zhang S, Sun L, Fu Y, Lupardus PJ, Wallweber HJA, Liederer BM, Deshmukh G, Plise E, Tay S, Reynen P, Herrington J, Gustafson A, Liu Y, Dirksen A, Dietz MGA, Liu Y, Wang T-M, Hanson JE, Hackos D, Scearce-Levie K, Schwarz J. B.Discovery of GluN2A-Selective NMDA Receptor Positive Allosteric Modulators (PAMs): Tuning Deactivation Kinetics via Structure-Based Design. J. Med. Chem. 2016;59:2760–2779. doi: 10.1021/acs.jmedchem.5b02010. [DOI] [PubMed] [Google Scholar]
- 16.Sheinin A, Shavit S, Benveniste M. Subunit specificity and mechanism of action of NMDA partial agonist D-cycloserine. Neuropharmacology. 2001;41:151–158. doi: 10.1016/s0028-3908(01)00073-9. [DOI] [PubMed] [Google Scholar]
- 17.Dravid SM, Burger PB, Prakash A, Geballe MT, Yadav R, Le P, Vellano K, Snyder J, Traynelis SF. Structural Determinants of D-Cycloserine Efficacy at the NR1/NR2C NMDA Receptors. J. Neurosci. 2010;30:2741–2754. doi: 10.1523/JNEUROSCI.5390-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vance KM, Simorowski N, Traynelis SF, Furukawa H. Ligand-specific deactivation time course of GluN1/GluN2D NMDA receptors. Nat. Commun. 2011;2:294. doi: 10.1038/ncomms1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis M, Ressler K, Rothbaum BO, Richardson R. Effects of D-cycloserine on extinction: translation from preclinical to clinical work. Biol. Psychiatry. 2006;60:369–375. doi: 10.1016/j.biopsych.2006.03.084. [DOI] [PubMed] [Google Scholar]
- 20.Ressler KJ, Rothbaum BO, Tannenbaum L, Anderson P, Graap K, Zimand E, Hodges L, Davis M. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch. Gen. Psychiatry. 2004;61:1136–1144. doi: 10.1001/archpsyc.61.11.1136. [DOI] [PubMed] [Google Scholar]
- 21.Sheinin A, Nahum-Levy R, Shavit S, Benveniste M. Specificity of putative partial agonist, 1-aminocyclopropane-carboxylic acid, for rat N-methyl-D-aspartate receptor subunits. Neurosci. Lett. 2002;317:77–80. doi: 10.1016/s0304-3940(01)02430-2. [DOI] [PubMed] [Google Scholar]
- 22.Hansen KB, Tajima N, Risgaard R, Perszyk RE, Jorgensen L, Vance KM, Ogden KK, Clausen RP, Furukawa H, Traynelis SF. Structural determinants of agonist efficacy at the glutamate binding site of N-methyl-D-aspartate receptors. Mol. Pharmacol. 2013;84:114–127. doi: 10.1124/mol.113.085803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.