

ABSTRACT
Fission yeast phosphate homeostasis entails transcriptional induction of genes encoding phosphate-mobilizing proteins under conditions of phosphate starvation. Transcription factor Pho7, a member of the Zn2Cys6 family of fungal transcription regulators, is the central player in the starvation response. The DNA binding sites in the promoters of phosphate-responsive genes have not been defined, nor have any structure-function relationships been established for the Pho7 protein. Here we narrow this knowledge gap by (i) delineating an autonomous DNA-binding domain (DBD) within Pho7 that includes the Zn2Cys6 module, (ii) deploying recombinant Pho7 DBD in DNase I footprinting and electrophoretic mobility shift assays (EMSAs) to map the Pho7 recognition sites in the promoters of the phosphate-regulated pho1 and tgp1 genes to a 12-nucleotide sequence motif [5′-TCG(G/C)(A/T)xxTTxAA], (iii) independently identifying the same motif as a Pho7 recognition element via in silico analysis of available genome-wide ChIP-seq data, (iv) affirming that mutations in the two Pho7 recognition sites in the pho1 promoter efface pho1 expression in vivo, and (v) establishing that the zinc-binding cysteines and a pair of conserved arginines in the DBD are essential for Pho7 activity in vivo.
KEYWORDS: DNA binding, fission yeast, phosphate homeostasis, transcriptional regulation
IMPORTANCE
Fungi respond to phosphate starvation by inducing the transcription of a set of phosphate acquisition genes that comprise a phosphate regulon. Pho7, a member of the Zn2Cys6 family of fungal transcription regulators, is the central player in the phosphate starvation response in fission yeast. The present study identifies a 12-nucleotide Pho7 DNA binding motif [5′-TCG(G/C)(A/T)xxTTxAA] in the promoters of phosphate-regulated genes, pinpoints DNA and protein features important for Pho7 binding to DNA, and correlates them with Pho7-dependent gene expression in vivo. The results highlight distinctive properties of Pho7 vis-a-vis other fungal zinc binuclear cluster transcription factors as well as the divergent cast of transcription factors deployed for phosphate homeostasis in fission yeast versus budding yeast.
INTRODUCTION
Phosphate homeostasis in the fission yeast Schizosaccharomyces pombe is achieved by regulating the transcription of genes encoding three proteins involved in extracellular phosphate mobilization and uptake: a cell surface acid phosphatase, Pho1; an inorganic phosphate transporter, Pho84; and a glycerophosphate transporter, Tgp1 (1). Expression of the genes coding for these proteins is repressed during growth in phosphate-rich medium and induced during phosphate starvation. Induction of the phosphate-regulated genes during starvation depends on transcription factor Pho7 (1–3), a member of the zinc binuclear cluster family of fungal DNA-binding transcription regulators (4).
System-wide microarray analyses of gene expression in pho7+ and pho7Δ strains and chromatin immunoprecipitation sequencing (ChIP-seq) analyses of DNA occupancy by Pho7-TAP in phosphate-replete and phosphate-starved cells affirmed the role of Pho7 in the phosphate starvation response, while highlighting an additional function in driving expression of multiple stress response genes independent of phosphate status (1).
We have shown that a 283-nucleotide (nt) segment of DNA 5′ of the pho1 transcription start site suffices to drive expression of Pho1 acid phosphatase activity in vivo. Because the 283-nt pho1 promoter generated no acid phosphatase activity in a pho7Δ strain, this DNA segment must include the requisite Pho7 binding site(s) (5). Genome-wide ChIP-seq analysis revealed a peak of Pho7-TAP occupancy from positions −230 to −120 upstream of the pho1 transcription start site (1). However, the DNA element recognized by Pho7 remains unknown, and prior attempts to identify a DNA binding motif based on genome-wide Pho7 ChIP-seq data were unsuccessful (1).
In the present study, we define the DNA site recognized by Pho7, via two independent approaches: (i) a revisited in silico analysis of the Pho7 ChIP-seq data and (ii) biochemical footprinting of the Pho7 DNA-binding domain (DBD) on the pho1 and tgp1 gene promoters. We identify DNA features important for Pho7 binding and correlate them with Pho7-dependent gene expression. Our results provide new insights into the fission yeast phosphate response and illuminate distinctive properties of Pho7 vis-a-vis other fungal zinc binuclear cluster transcription factors.
RESULTS
The zinc-binding cysteines are essential for Pho7 activity in vivo.
The 738-amino-acid (aa) S. pombe Pho7 protein is most closely related to its 683-aa homolog from Schizosaccharomyces octosporus. Alignment of the two polypeptides highlights 291 positions of side-chain identity/similarity across their full lengths, with the highest conservation being in the central Zn2Cys6 domain (aa 282 to 339 in Pho7 [shaded gray in Fig. 1A]) and the C-terminal segment (aa 629 to 722 in Pho7). To gauge the role of the putative zinc-binding cysteines in Pho7 function, we replaced the chromosomal pho7+ gene with a pho7-TAP::hygMX cassette (encoding a C-terminal TAP-tagged Pho7 polypeptide) in which pho7 was either wild type (WT) or had double-alanine mutations of three serial cysteine pairs (C292A-C295A, C302A-C308A, and C311A-C318A). Spot testing for growth on YES (yeast extract with supplements) agar medium showed that the pho7Δ strain grew slower than wild type at 20, 25, and 30°C (as gauged by colony size) and was extremely sick at higher (34 and 37°C) and lower (18°C) temperatures (Fig. 1B). All three of the double-cysteine mutations phenocopied the growth defect of the pho7Δ strain, suggesting that zinc binding by Pho7 is essential for Pho7 function in sustaining growth at extremes of temperature.
FIG 1 .

Features of Pho7 essential for its activity in vivo. (A) The amino acid sequence of S. pombe Pho7 (Spo) is aligned to the sequence of the homologous protein from S. octosporus (Soc). Gaps in the alignment are denoted by dashes. Positions of side-chain identity/similarity are denoted by dots. The putative Zn2Cys6 DNA-binding domain is highlighted in the gray box. Conserved pairs of cysteines and arginines in the DBD that were targeted for alanine substitution are indicated by |. The margins of the N-terminal deletions are indicated by arrowheads. (B) S. pombe strains deleted of pho7 (pho7Δ) or bearing the pho7 alleles as indicated were spot tested for growth at the temperatures specified. (C) Similarity of the Zn2Cys6 DNA-binding domains of S. cerevisiae Gal4 and S. pombe Pho7. The secondary structure elements of Gal4 are indicated above the amino acid sequence, with α-helices depicted as horizontal cylinders. The signature zinc-binding cysteines are indicated by |. Conserved arginines that in Gal4 contact the DNA phosphodiester backbone are shown in white font on a black background. Other positions of side-chain identity/similarity are denoted by dots. The Gal4 lysine that contacts adjacent guanine nucleobases in the DNA target site is indicated by a triangle. (D) Western blots of whole-cell extracts prepared from S. pombe strains with the indicated pho7 alleles. The blots were probed with antibodies recognizing the TAP tag or the Spt5 protein. (E) Acid phosphatase activity of cells bearing the indicated pho7-TAP alleles assayed 5 h after transfer of logarithmically growing cells to medium lacking phosphate.
The central Zn2Cys6 domain of Pho7 is homologous to the N-terminal Zn2Cys6 DNA-binding domain (DBD) of Saccharomyces cerevisiae Gal4, a prototypal fungal zinc cluster transcription factor. The DBD consists of a folded zinc finger module of two α-helices that recognizes the target DNA sequence, a linker that contacts the DNA phosphate backbone, and a helical coiled-coil module that mediates dimerization of the DBD (6, 7) (Fig. 1C). It is noteworthy that the Lys18 side chain in Gal4 that makes hydrogen bonds to the adjacent guanine nucleobases in the major groove of the CGG target sequence (indicated by the open triangle in Fig. 1C) is not conserved in Pho7 (where the corresponding residue is Asn299). However, the Gal4 Arg46 and Arg51 side chains (located in the linker and at the beginning of the dimerization α-helix, respectively) that contact target site DNA phosphates are conserved in Pho7 as Arg326 and Arg331 (highlighted in white font on a black background in Fig. 1C). We found that a strain with an R326A-R331A double-alanine mutation in pho7 grew better than the pho7Δ null strain at high and low temperatures, although not as well as the wild type (Fig. 1B), indicating that the loss of putative interactions of these conserved arginines with DNA elicits a hypomorphic phenotype during vegetative growth.
Protein segments flanking the DBD of fungal zinc cluster proteins are involved in transcription activation and responses to regulatory inputs. Guided by the alignment of Pho7 with its S. octosporus homolog, we deleted either the N-terminal 183 or 271 aa and replaced pho7+ with the pho7(184–738)-TAP and pho7(272–738)-TAP alleles. Both truncation strains grew like the wild type at 18 to 30°C; the truncation strains grew better than the pho7Δ mutant at 34 and 37°C, albeit with colony sizes slightly smaller than wild type (Fig. 1B).
Whole-cell extracts of the pho7-TAP strains were subjected to SDS-PAGE and Western blotting with anti-TAP antibody (Fig. 1D). Western blotting with antibody to S. pombe Spt5 was used as a loading control. An immunoreactive ~130-kDa polypeptide corresponding to Pho7-TAP was detected in extracts from the pho7-TAP strains but not in extracts from pho7Δ cells. TAP-reactive Pho7 polypeptides of serially smaller molecular masses were detected in extracts of the two truncation strains (Fig. 1D).
The pho7-TAP strains were tested for responsiveness to phosphate starvation, by assay of cell surface Pho1 acid phosphatase activity 5 h after transfer of the cells from rich medium to synthetic medium lacking phosphate. The C292A-C295A, C302A-C308A, C311A-C318A, and R326A-R331A strains manifested no Pho1 induction during phosphate starvation (Fig. 1E). Thus, disruption of the zinc-binding cluster or loss of the pair of putative DNA-binding arginines eliminates Pho7 function in phosphate homeostasis. The extents of Pho1 induction by the pho7(184–738) and pho7(272–738) truncation strains were 22% and 11% of that of the wild type, respectively (Fig. 1E).
In silico identification of a candidate Pho7 recognition element.
The available genome-wide Pho7-TAP ChIP-Seq data from phosphate-replete and phosphate-starved cells (1) (GEO accession no. GSE39498) were processed for Pho7-TAP peak localization above background, and the peak sequences were screened for enriched motifs in MEME (8) as described in Materials and Methods. The separate analyses of the phosphate-starved and phosphate-replete data sets yielded virtually identical candidate motifs, with E values of 1.6 × 10−15 and 9.6 × 10−14, respectively (Fig. 2). The consensus motif for the two data sets is a dodecamer, 5′-TCG(G/C)AxTTTxAA. Occurrences of this motif within individual Pho7-TAP ChIP peaks as determined in FIMO (9) and the identities of S. pombe genes within 1 kb of the peaks are compiled in Tables S1 and S2, respectively, in the supplemental material.
FIG 2 .
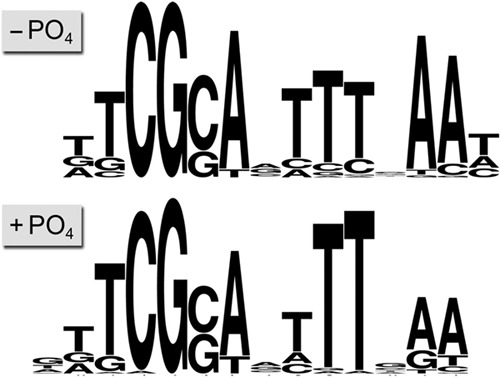
In silico identification of a candidate Pho7 recognition element. Motifs were identified via analysis of previously reported Pho7-TAP ChIP-seq data (1) from phosphate-replete and phosphate-starved cells as described in the text.
Individual motif occurrences within the sequences of the Pho7 ChIP peaks, identified by FIMO (9), are listed (cutoff, motif P value of ≤0.001) on separate pages for phosphate-starved (−Pi) and phosphate-replete (+Pi) conditions. The motifs that correlate with the Pho7 DBD DNase I footprints in the pho1 and tgp1 promoters are highlighted in yellow. Download TABLE S1, XLSX file, 0.1 MB (47.1KB, xlsx) .
Copyright © 2017 Schwer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Annotated genes located within 1 kb downstream of the Pho7 ChIP peaks (group 1) and those that have a Pho7 ChIP peak within the annotated gene (group 2) are listed on separate pages for phosphate-starved (−Pi) and phosphate-replete (+Pi) conditions. Download TABLE S2, XLSX file, 0.1 MB (119.9KB, xlsx) .
Copyright © 2017 Schwer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Recombinant Pho7 Zn2Cys6 DBD binds and footprints the pho1 promoter.
Pho7(279–368), which spans the conserved Zn2Cys6 domain, was produced in Escherichia coli as a His10-Smt3 fusion protein and isolated from a soluble bacterial extract by nickel affinity chromatography. The His10-Smt3 tag was removed with the Smt3-specific protease Ulp1, and the native Pho7(279–368) protein (henceforth referred to as Pho7 DBD) was separated from the tag by a second round of nickel affinity chromatography and purified further by gel filtration. SDS-PAGE analysis revealed a predominant polypeptide that migrated at ~17 kDa relative to size standards (Fig. 3C), although the calculated size of the recombinant polypeptide is 10 kDa. An initial DNase I footprinting experiment was performed using a 300-bp 5′ 32P-labeled DNA fragment comprising 249 nt upstream of the pho1 transcription start site and extending 51 nt into the transcription unit. The DNA was end labeled on the top DNA strand. Limited DNase I digestion in the absence and presence of increasing levels of Pho7 DBD revealed two segments of protection (denoted by brackets in Fig. 3B) separated by an unprotected spacer segment. Analysis in parallel with a 5′ 32P-labeled primer extension sequencing ladder (Fig. 3B) demarcated the two regions of DNase I protection as −188 to −173 and −159 to −142 upstream of the pho1 transcription start site (Fig. 3D). We proceeded to footprint the bottom DNA strand using a 160-bp DNA fragment from nt −249 to −90 upstream of the pho1 transcription start site. Here we again observed two segments of DNase I protection (Fig. 3A, denoted by brackets). Reference to a 5′ 32P-labeled primer extension sequencing ladder (Fig. 3A) assigned the two bottom strand DNase I footprints as −192 to −174 and −158 to −142 upstream of the pho1 transcription start site, respectively (Fig. 3D). Both of the Pho7 footprint sites include a 12-nt motif, 5′-TCG(G/C)(A/T)xxTTxAA (Fig. 3D), that resembles the candidate Pho7 recognition element identified via our in silico analysis (Fig. 2).
FIG 3 .

Recombinant Pho7 Zn2Cys6 DBD binds and footprints the pho1 promoter. (A and B) DNase I footprint analyses of the bottom strand (A) and top strand (B) of the pho1 promoter are shown. Binding reaction mixtures contained 0.75 pmol of 5′ 32P-labeled DNA and either no added Pho7 DBD (lanes –) or increasing amounts of Pho7 DBD (13, 17, 22, 33, and 66 ng in panel A or 20, 27, 40, and 80 ng in panel B). The DNase I digestion products were analyzed by denaturing PAGE in parallel with a series of DNA-directed primer extension reactions (using a 32P-labeled primer identical to the labeled 5′ end of the footprinted DNA) that contained mixtures of standard and chain-terminating nucleotides. (The chain terminator is specified above the lanes.) The margins of the footprints are indicated by brackets. (C) SDS-PAGE analysis of recombinant Pho7 DBD. The Coomassie blue-stained gel is shown. The positions and sizes (in kilodaltons) of marker polypeptides are indicated on the left. (D) Nucleotide sequence of the top and bottom strands of the pho1 promoter from −196 to −136 upstream of the transcription start site. The DNase I footprints on each strand are denoted by magenta brackets. A conserved dodecamer sequence within the footprints is highlighted in gold. (E) Gel shift assay on Pho7 binding to the pho1 promoter. Reaction mixtures (10 µl) containing 1 pmol of a 160-nt 32P-labeled DNA fragment, 0.34 µg poly(dI-dC), and either no protein (–) or increasing amounts of Pho7 DBD (8.3, 17, 33, or 66 ng) were incubated for 10 min at room temperature and then analyzed by native PAGE. An autoradiograph of the gel is shown. The arrowheads on the right indicate positions of DNA-protein complexes.
We also assessed binding of Pho7 DBD to the pho1 promoter by electrophoretic mobility shift assay (EMSA) using the 160-bp 5′ 32P-labeled DNA probe comprising nt −249 to −90 upstream of the pho1 transcription start site. Pho7 DBD formed a single discrete DNA-protein complex at lower Pho7 DBD concentrations that was converted to a more slowly migrating complex as the Pho7 DBD concentration was increased (Fig. 3E). The evolution of the DNA-protein complexes as a function of protein concentration is indicative of the sequential (noncooperative) mode of Pho7 binding to the two sites footprinted in the pho1 promoter. If binding had been highly cooperative, we would have expected to see accumulation of the two-site-occupancy DNA-protein complexes at protein concentrations at which there was a significant fraction of residual unbound DNA.
To query whether the Pho7 DBD requires tandem recognition sites or binds independently to each of the target sites in the pho1 promoter, we performed EMSAs with a 5′ 32P-labeled 73-bp DNA probe spanning both sites and with 40- and 43-bp DNA probes corresponding to only site 1 and site 2, respectively (Fig. 4). The results showed that Pho7 binds sequentially to two sites in the 73-bp DNA to give rise to two discrete shifted complexes and binds independently to site 1 and site 2 DNAs to form a single shifted complex in each case (Fig. 4). Quantification of the yield of the Pho7-DNA complex as a function of input protein indicated that Pho7 had 2-fold-higher affinity for site 2 than for site 1.
FIG 4 .

Pho7 DBD binds independently to two target sites in the pho1 promoter. The sequences of three pho1 promoter DNAs containing both Pho7 binding sites or individual Pho7 binding sites are shown at the bottom. The consensus motifs are shaded in gray, and the 5′ 32P labels on the top strands are indicated by large dots. Reaction mixtures (10 µl) containing 1 pmol 32P-labeled DNA, 0.34 µg poly(dI-dC), and either no protein (–) or increasing amounts of Pho7 DBD (8.3, 17, 33, or 66 ng) were incubated for 10 min at room temperature. The mixtures were analyzed by native PAGE. An autoradiograph of the gel is shown.
Effect of Pho7 site mutations in the pho1 promoter on Pho1 expression in vivo.
A plasmid-based reporter containing 283 nt of promoter DNA 5′ of the pho1 transcription start site suffices to drive expression of Pho1 acid phosphatase in fission yeast cells in which the chromosomal pho1+ locus has been deleted (5). To gauge the role of the Pho7 binding sites in pho1 promoter activity, we made a series of promoter mutants in this plasmid in which dinucleotides were deleted, either within the margins of the Pho7 footprints (mutants 1, 2, 4, and 5), between the two Pho7 sites (mutant 3), or between the Pho7 sites and a putative TATA box element, −34TATTTAA−28, preceding the transcription start site (mutants 6 and 7). The deleted dinucleotides are indicated by brackets above the pho1 promoter sequence in Fig. 5. In parallel, we replaced the TATTTAA sequence with CGCCCGG. pho1Δ cells carrying the wild-type and promoter mutant plasmids were assayed for acid phosphatase activity. Mutation of the TATA box sequence effaced Pho1 expression, as did the dinucleotide deletions 1, 2, and 4 within the Pho7 binding sites (Fig. 5). Each of these dinucleotide deletions alters the Pho7 site by shifting the flanking DNA sequences to create two possible mutant versions of the dodecamer (indicated by “or” in Fig. 5), both of which deviate greatly from the Pho7 site consensus. However, deletion 5 within Pho7 site 2 had only a modest effect, reducing Pho1 expression to 47% of the wild-type value. We attribute the residual activity to the fact that one of the frameshifted versions of site 2 mutant 5 (indicated by the arrowhead in Fig. 5) retains many of the consensus nucleotides of the Pho7 dodecamer 5′-TCG(G/C)AxTTTxAA while replacing the triplet of T’s with A’s. The dinucleotide promoter deletions 3, 6, and 7 supported 40, 75, and 55% of wild-type Pho1 expression, respectively. These experiments highlight that both Pho7 sites and the TATA box are critical for pho1 promoter activity in vivo.
FIG 5 .

Effect of Pho7 site mutations in the pho1 promoter on Pho1 expression in vivo. The sequence of the pho1 promoter from −206 to +7 relative to the pho1 transcription start site (denoted by an arrowhead) is shown at the top. Pho7 binding sites 1 and 2 are shaded in gold, and dinucleotides that were deleted in promoter mutations 1 to 7 are marked by blue brackets above the sequence. A putative TATA element (outlined in blue) was replaced by the sequence shown in red. The sequences at right in the bottom panel show the two possibilities of how the dinucleotide deletions alter the Pho7 binding motifs for site 1 and site 2. The arrowhead indicates the more likely scenario of how promoter mutation 5 alters the Pho7 binding site. Reporter plasmids with wild-type and the mutated pho1 promoters were introduced into a strain with the endogenous pho1 gene deleted. Plasmid-containing cells were grown logarithmically in YES medium and assayed for acid phosphatase activity.
Pho7 binding site in the tgp1 promoter.
We employed reverse transcriptase primer extension analysis to locate the 5′ end of the tgp1 mRNA. A 32P-labeled DNA primer complementary to nt 2 to 23 of the of the tgp1 open reading frame (ORF) was annealed to RNA isolated from phosphate-starved yeast cells and then subjected to reverse transcription (RT). The RT primer extension product was analyzed in parallel with a chain-terminated sequencing ladder generated by DNA polymerase extension of the same 32P-labeled primer annealed to a DNA template containing the tgp1 locus (not shown). A single 5′ end was thereby located 42 nt upstream of the start codon of the tgp1 open reading frame (ORF) (Fig. 6A).
FIG 6 .

Plasmid reporter of tgp1 promoter function. The DNA sequence from positions −13 to +51 relative to the tgp1 transcription start site (indicated by the arrow) is shown. The tgp1 ATG translation start site is underlined. (B) In the tgp1-pho1 reporter plasmids, the pho1 ORF was fused immediately downstream of a fragment of genomic DNA encompassing the tgp1 transcription start site plus 42 bp of downstream sequences and 871 bp (−871) or 122 bp (−122) of upstream tgp1 sequences. (C) Acid phosphatase activity of S. pombe pho1Δ strains bearing the indicated tgp1-pho1 reporter plasmids during logarithmic growth in YES medium. The strains were either pho7+ or pho7Δ as specified. A dinucleotide deletion (*) in the Pho7 binding site, indicated by a bracket in the sequence at right, was introduced in the context of the −871 tgp1-pho1 reporter (−871*).
To interrogate the tgp1 promoter, we constructed a plasmid reporter in which a genomic DNA segment containing nt −871 to +42 of the tgp1 transcription unit (with +1 being the mRNA start site) was fused to the pho1 ORF (Fig. 6B). Because this plasmid generated vigorous acid phosphatase activity when introduced into a pho1Δ strain (Fig. 6C), we surmised that the 871-nt segment upstream of the transcription start site embraces a tgp1 promoter and potential regulatory elements. Acid phosphatase activity driven by the tgp1 promoter reporter plasmid was reduced by 85% in a pho7Δ strain background (Fig. 6C), signifying that the 871-nt segment contains a site or sites that are bound by the Pho7 transcription factor.
To locate such sites, a series of overlapping 32P-labeled DNA fragments spanning the genomic region from nt −871 to −15 preceding the transcription start site of the tgp1 gene was prepared and tested by EMSA for binding to Pho7 DBD. Whereas there was no protein-DNA complex formed in DNA fragments in the region from −871 to −219, the 223-nt DNA fragment from −237 to −15 was bound by Pho7 DBD to form a single protein-DNA complex (Fig. 7A). Pho7 DBD also formed a single protein-DNA complex on a 129-nt DNA fragment from −237 to −109 (Fig. 7A). This DNA segment was used in DNase I footprinting experiments shown in Fig. 7B. A single region of protection from DNase I cleavage spanned nt −193 to −178 on the top strand and nt −194 to −180 on the bottom strand. The footprint is denoted by brackets in Fig. 7B, and it embraces a 12-mer sequence (5′-TCGGACATTCAA) that is identical at 10 of 12 positions to Pho7 recognition site 2 in the pho1 promoter.
FIG 7 .

Pho7 binding site in the tgp1 promoter. (A) EMSAs using 32P-labeled DNA fragments embracing the indicated sequences of the tgp1 locus upstream of the tgp1 transcription start site (defined as +1). The nucleotide margins of the DNA segments are indicated at the top. Reaction mixtures (10 µl) containing 0.6 pmol labeled DNA, 340 ng poly(dI-dC), and 0, 33, or 66 ng of Pho7 DBD were incubated for 10 min at room temperature. The mixtures were analyzed by native PAGE. An autoradiograph of the gel is shown. The asterisks above the gel denote tgp1 promoter segments that are bound by Pho7; the arrowheads at right mark the positions of the DNA-protein complexes. (B) DNase I footprinting analyses of the top strand (left panel) and bottom strand (right panel) of the tgp1 promoter are shown along with the respective sequencing ladders. Binding reaction mixtures contained 1 pmol of 5′ 32P-labeled DNA and either no added Pho7 DBD (lanes –) or increasing amounts of Pho7 DBD (12.5, 25, or 50 ng). The margins of the footprints are indicated by brackets. The tgp1 sequence from −216 to −148 upstream of the tgp1 transcription start site is shown at the bottom. The Pho7 binding motif is shown in white font on a black background. The brackets denote the DNase I footprints on the top and bottom strands.
With this information in hand, we truncated the −871 tgp1 promoter reporter plasmid to position −122, thereby deleting the Pho7 site. Pho1 acid phosphatase activity expressed by the truncated promoter was reduced to 17% of the level driven by the −871 promoter (Fig. 6C), thereby phenocopying the effect of pho7Δ on the longer promoter. Indeed, the residual acid phosphatase expression driven by the −122 tgp1 promoter was maintained in a pho7Δ background (Fig. 6C). To query whether the effect of promoter truncation is caused by loss of the Pho7 site, we deleted the GG dinucleotide from the Pho7 site in the context of the −871 tgp1 promoter; this dinucleotide deletion (−871*) reduced acid phosphatase expression by 85%, mimicking the effects of pho7Δ and the promoter truncation (Fig. 6C). We conclude that the Pho7 binding site identified by EMSA and footprinting is essential for Pho7-dependent tgp1 promoter activity.
Effect of nucleotide substitutions on Pho7 binding to its DNA target.
The three Pho7 binding sites mapped biochemically in the pho1 and tgp1 promoters agree with the consensus 5′-TCG(G/C)AxxTTxAA site identified in silico. To understand the role of nucleobase sequence in Pho7 recognition, we prepared a series of 5′ 32P-labeled 24-bp DNA duplexes based on the footprinted Pho7 site in the tgp1 promoter that corresponded to the native wild-type dodecamer DNA sequence 5′-TCGGACATTCAA or had 2-base substitutions (underlined) as follows: 5′-TGCGACATTCAA (Mut1), 5′-TCGGACAAACAA (Mut2), or 5′-TCGGACATTCTT (Mut3). The 24-mers were tested by EMSA for binding to Pho7 DBD (Fig. 8A). DNA binding was effaced by Mut1 that changed the CG dinucleotide at positions 2 and 3 of the dodecamer element to GC. The loss of DNA binding in vitro by altering the CG dinucleotide is consistent with the loss of pho1 expression in vivo when the CG (site 1) or the GG (site 2) dinucleotide is altered in the site 1 or site 2 dodecamers of the pho1 promoter (Fig. 5). In contrast, the Mut2 and Mut3 dinucleotide changes reduced but did not eliminate Pho7 DBD affinity for the tgp1 promoter site: i.e., 27 ng of input Pho7 DBD sufficed to bind 81% of the wild-type 24-mer DNA but only 45% and 23% of the Mut2 and Mut3 DNAs, respectively (Fig. 8A). It is instructive that the ~2-fold decrease in Pho7 binding in vitro elicited by the TT-to-AA change in the dodecamer correlates with the 2-fold decrease in pho1 expression when the same TT-to-AA change is created in site 2 of the pho1 promoter (Fig. 5).
FIG 8 .

Effect of nucleotide substitutions and deletion of flanking nucleotides on Pho7 binding to its DNA target. EMSAs were performed using the indicated DNA duplexes encompassing the Pho7 binding site in the tgp1 promoter. Reaction mixtures (10 µl) containing 32P-labeled DNAs (1 pmol), 340 ng poly(dI-dC), and Pho7 DBD as specified were incubated for 10 min at room temperature and then analyzed by native PAGE. Autoradiographs of the gels are shown. The sequences of the DNAs are indicated below the gels; the Pho7 binding motif is shaded gray, and the 5′ 32P labels on the top strand are indicated by large dots. (A) Effect of nucleotide changes. The two-nucleotide substitutions in Mut1, Mut2, and Mut3 are denoted in white font on a black background. Reaction mixtures contained 0, 27, or 55 ng Pho7 DBD as indicated. (B) Effect of deleting flanking nucleotides. Reaction mixtures contained either no added Pho7 DBD (lanes –) or increasing amounts of Pho7 DBD (8.3, 17, 33, or 66 ng from left to right in the titration series).
To gauge whether DNA flanking the dodecamer plays a role in Pho7 binding, we tested in parallel the 24-bp DNA duplex from the tgp1 promoter and two shorter versions, a 20-bp duplex and a 16-bp duplex, in which 2 or 4 nt were deleted on both sides of the central dodecamer. The Pho7 DBD bound with similar affinity to all three DNAs (Fig. 8B).
DISCUSSION
The 738-aa Pho7 protein is the key transcriptional activator underlying fission yeast phosphate homeostasis. Here, we purified a recombinant Pho7 DNA-binding domain (DBD) that includes the Zn2Cys6 module and used it in DNase I footprinting experiments to map the Pho7 recognition sites in the promoters of the pho1 and tgp1 genes to a specific 12-nt motif, 5′-TCG(G/C)(A/T)xxTTxAA. We independently identified the same motif as a Pho7 recognition element via in silico analysis of available genome-wide ChIP-seq data (1). The pho1 promoter contains two Pho7 recognition sites, in direct repeat orientation and separated by a 20-nt spacer. EMSAs showed that the Pho7 DBD binds independently, and apparently noncooperatively, to these two sites in the pho1 promoter. Two-nucleotide deletions within each of the Pho7 recognition sites sharply reduced Pho1 expression in vivo, signifying that both Pho7 sites are needed to activate pho1 transcription. (The spacing between Pho7 sites 1 and 2 might also be relevant for pho1 promoter activity, insofar as a dinucleotide deletion between the sites reduced Pho1 expression by half. A systematic interrogation of the effects of contracting and expanding the spacer length is a subject for future studies.) In contrast, the tgp1 promoter appears to contain only a single Pho7 binding site that is important for Pho7-dependent tgp1 promoter activity.
The Pho7 DBD resembles the DBDs of other zinc cluster fungal transcription factors with respect to the presence and spacing of its six conserved cysteines. We find that the three pairs of zinc-coordinating cysteines are essential for Pho7 function in vivo, as gauged by two criteria: complementation of the conditional growth defects of a pho7Δ strain and the ability to mount a Pho1 inductive response to phosphate starvation. We surmise that DNA binding by Pho7 is necessary for its biological activity. The Pho7 DBD also has a pair of arginines that are conserved in Gal4, where they contact the phosphate backbone of the Gal4 DNA recognition site. Mutation of this arginine pair in Pho7 eliminated Pho1 expression during phosphate starvation, again consistent with DNA binding being crucial for Pho7 activity during the starvation response.
In many of the zinc cluster fungal transcription factors, the DBD is located at the very N terminus of the polypeptide (4). This is not the case in Pho7, where the DBD is preceded by a 270-aa segment that is weakly conserved in the Pho7-like protein of S. octosporus (Fig. 1A). A truncated Pho7 lacking this N domain complemented the cold-sensitive growth defect of the pho7Δ mutant, but only partially complemented the temperature-sensitive (ts) growth phenotype. With respect to phosphate homeostasis, deletion of the N domain sharply reduced Pho1 production in response to phosphate starvation. It is noteworthy that eight sites of Pho7 phosphorylation have been annotated within the N domain: S230, S244, S247, S249, S253, S257, T260, and T261 (http://www.pombase.org). Five of these phosphorylation sites are conserved in the S. octosporus N domain.
The Pho7 DNA recognition site 5′-TCG(G/C)(A/T)xxTTxAA differs from those of other well-studied fungal Zn2Cys6 proteins (e.g., Gal4, Leu3, Hap1, Put3, and Ppr1), which typically recognize pairs of CGG triplets that are arranged as inverted, direct, or everted repeats (4, 6, 10–13). The fungal Zn2Cys6 proteins that bind to such DNA elements typically do so as homodimers in which the zinc-binding module confers DNA sequence recognition and a downstream α-helix forms a coiled-coil dimer interface. The pho1 promoter site 2 and tgp1 promoter site that are recognized by Pho7 contain a single CGG triplet, mutation of which to GCG effaces target recognition. The lack of internal symmetry in the Pho7 DNA binding site raises the prospects that (i) it might have a unique binding mode in which the component promoters of a homodimer recognize different nucleotide sequence motifs or (ii) it might bind to DNA as a monomer, a la the Aspergillus transcription factor AlcR (14). Further insights into Pho7 specificity will hinge on cocrystallizing the DBD bound to target DNA and educing structure-activity relations via comprehensive mutagenesis of the DBD.
Finally, it is worth highlighting that whereas both fission and budding yeasts respond to phosphate starvation by inducing the transcription of phosphate acquisition genes (a phosphate regulon), they rely on quite different classes of DNA-binding transcription factors to achieve this response (2). As affirmed here, the Pho7 protein that drives phosphate homeostasis in S. pombe is a member of the fungal Zn2Cys6 transcription factor family. In contrast, transcription of the phosphate regulon in phosphate-starved S. cerevisiae depends on cooperative action of two distinct transcription factors: Pho4 and Pho2 (15–17). Pho2 is a member of the homeodomain family. Pho4 belongs to the basic helix-loop-helix (bHLH) family of transcription factors, and its activity is regulated negatively/positively by phosphorylation/dephosphorylation under phosphate-replete/phosphate-starved conditions (18). The crystal structure of the Pho4 bHLH domain homodimer bound to its 17-bp high-affinity DNA target site revealed a network of amino acid contacts with the nucleobases of the central 7-bp 5′-CACGTGG element (19). There is no similarity in the DNA sequences recognized by S. pombe Pho7 and S. cerevisiae Pho4. Pho4-like bHLH transcription factors drive the phosphate starvation response in the fungal pathogens Candida glabrata and Cryptococus neoformans (20, 21). The evolutionary paths that led to the extreme divergence in the choice of transcription factors that govern phosphate homeostasis in fission versus budding yeast are presently obscure.
MATERIALS AND METHODS
Allelic exchange at the pho7 locus.
We constructed a series of pKS-based plasmids carrying pho7-TAP integration cassettes marked with hygMX by PCR amplification using genomic DNA as a template and oligonucleotides that introduced restriction sites for cloning. The cassettes consisted of the following elements, proceeding from 5′ to 3′: (i) a 515-bp segment of genomic DNA 5′ of the pho7+ start codon; (ii) an open reading frame encoding wild-type Pho7, or mutated versions thereof, fused to a 560-bp ORF encoding the TAP tag (22); (iii) a 302-bp segment of genomic DNA 3′ of the pho7+ stop codon; (iv) an hygMX gene (1.69 kbp) conferring resistance to hygromycin; and (v) a 728-bp segment of genomic DNA from nt +2517 to +3245 downstream of the pho7+ start codon. The integration cassettes were excised from the pKS plasmids and transformed into a haploid S. pombe strain. Correct insertions were verified by Southern blotting and sequencing of PCR-amplified DNA segments to ascertain the presence of the desired allele. To gauge the effect of these mutations on vegetative growth, cultures of S. pombe strains containing the indicated pho7-TAP allele were grown in liquid medium until the A600 reached 0.6 to 0.9. The cultures were adjusted to a final A600 of 0.1, and aliquots (3 µl) of serial 5-fold dilutions were spotted on YES agar plates, which were then incubated at 18, 20, 25, 30, 34, and 37°C.
Western blotting.
S. pombe pho7Δ and pho7-TAP strains were grown in YES medium at 30°C until the A600 reached 0.6 to 0.8. Aliquots (10 A600 units) of cells were collected by centrifugation and lysed in 20% trichloroacetic acid. Total acid-insoluble protein was recovered by centrifugation; the pellets were washed with ethanol and resuspended in 1 M Tris-HCl (pH 8.0). Aliquots of the samples, adjusted to contain the same total protein content based on the A280 of the extracts, were adjusted to 2% SDS and 0.1 M dithiothreitol (DTT) and then analyzed by electrophoresis through 8% polyacrylamide gels containing 0.1% SDS. The gel contents were then transferred to a 0.2-µm-pore polyvinylidene difluoride (PVDF) membrane (Bio-Rad). The membranes were probed by Western blotting with either rabbit polyclonal antibody recognizing the TAP tag (Thermo Fisher) or affinity-purified rabbit polyclonal anti-Spt5 antibody (23). Immune complexes were visualized using horseradish peroxidase-linked anti-rabbit IgG and an ECL (enhanced chemiluminescence) Western detection system (Amersham, GE Healthcare).
In silico search for a candidate Pho7 recognition motif.
Pho7 ChIP-seq data sets were accessed from GEO data sets (accession no. GSE39498) (1). The FASTQ files were mapped to the S. pombe genome (ASM294v2.28) using Bowtie-2 2.2.9 (24). The resulting SAM files were converted to BAM files, and all duplicate reads were eliminated using SamTools (25). Pho7 peaks were determined by HOMER (Hypergeometric Optimization of Motif EnRichment) (26). For peak determination, ChIP-seq of Pho7-TAP under phosphate-starved or phosphate-rich conditions was compared to a similarly treated mock immunoprecipitation (IP). Conditions for peak filtering required putative peaks to have normalized sequence read tags 4.5-fold higher than the mock IP. Peak filtering based on local signal required putative peaks to have normalized sequence read tags 3-fold higher than in the surrounding 10-kb region. The sequence within each peak was extracted using a python script (27) and saved as a FASTA file for either Pho7-TAP ChIP in phosphate-replete or phosphate-starved cells. Motif discovery was conducted by MEME (Multiple Em for Motif Elicitation) (8) using the FASTA files. Motifs were predicted within the central 100-nt window of the peak using a zero order background model, with a defined cutoff for minimum-maximum sequence range for a motif to be from 6 to 14 bases with a possibility of any number of motif repetitions. The predicted motif with the lowest E value from both phosphate-replete and phosphate-starved ChIP data was 5′-TCG(C/G)AxTTTxAA. Individual motif occurrences within the sequence of the peaks (FASTA files), conducted by FIMO (Find Individual Motif Occurrences) (9), are listed in Table S1 (cutoff, motif P value of ≤0.001). The motifs that correlate with the Pho7 DBD DNase I footprints in the pho1 and tgp1 promoters are highlighted in yellow. Annotated genes within 1 kb of the peaks were identified using a python script (27) and are listed in Table S2. The position probability matrices for the motifs predicted by MEME and ChIP peaks identified by HOMER for Pho7 under phosphate-starved or phosphate-rich conditions are given in Table S3 and Table S4, respectively, in the supplemental material.
Position probability matrices for the Pho7 motifs predicted by MEME (8) under phosphate-starved (−Pi) and phosphate-replete (+Pi) conditions. Download TABLE S3, XLSX file, 0.1 MB (49.2KB, xlsx) .
Copyright © 2017 Schwer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The Pho7 ChIP peaks identified by HOMER (26) are listed on separate pages for phosphate-starved (−Pi) and phosphate-replete (+Pi) conditions. Download TABLE S4, XLSX file, 0.1 MB (102.9KB, xlsx) .
Copyright © 2017 Schwer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Pho7 DNA-binding domain.
Plasmid pET28b-His10-Smt3-Pho7(279–368) encodes the central Zn2Cys6 domain fused to an N-terminal His10-Smt3 module under the transcriptional control of a T7 RNA polymerase promoter. The plasmid was transfected into E. coli BL21(DE3) cells. Cultures (2 liters) amplified from single kanamycin-resistant transformants were grown at 37°C in Terrific Broth containing 50 µg/ml kanamycin until the A600 reached 0.8. The cultures were chilled on ice for 1 h and adjusted to 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and then incubated for 20 h at 18°C with constant shaking. All subsequent steps of purification were performed at 4°C. Cells were harvested by centrifugation and resuspended in 35 ml buffer A (50 mM Tris-HCl [pH 7.5], 500 mM NaCl, 20 mM imidazole, 10% glycerol). The cells were lysed by sonication, and the insoluble material was removed by centrifugation at 18,000 rpm for 45 min. Supernatants were mixed for 1 h with 4 ml of Ni-nitrilotriacetic acid (NTA) resin (Qiagen) that had been equilibrated with buffer A. The resin was recovered by centrifugation and resuspended in 50 ml buffer A. The washed resin was centrifuged again, resuspended in 50 ml buffer A, and then poured into a column. The bound material was step eluted with buffer A containing 300 mM imidazole. The polypeptide compositions of the flowthrough and eluate fractions were monitored by SDS-PAGE. The 300 mM imidazole eluate fractions containing His10-Smt3-Pho7(279–368) were supplemented with Smt3-specific protease Ulp1 and then dialyzed overnight against 2 liters buffer B (50 mM Tris-HCl [pH 7.5], 200 mM NaCl, 10% glycerol). The dialysates were mixed for 1 h with 2 ml of Ni-NTA resin that had been equilibrated with buffer B. Tag-free Pho7(279–368) was recovered in the flowthrough fractions and then subjected to gel filtration through a Superdex-200 column (GE Healthcare) equilibrated in a mixture of 50 mM Tris-HCl (pH 7.5), 200 to 250 mM NaCl, and 10% glycerol. Peak fractions were pooled, concentrated by centrifugal ultrafiltration, and then stored at −80°C at a concentration of 10 mg/ml. Protein concentration was determined as follows. Aliquots of Pho7 DBD and known amounts of bovine serum albumin (BSA) standard were serially diluted and adjusted to 2% SDS and 0.1 M DTT and then resolved by electrophoresis through a 15% polyacrylamide gel containing 0.1% SDS. The gel contents were stained with Coomassie brilliant blue R-250 followed by destaining in a solution containing 22.9% ethanol and 8.4% acetic acid. The gel was scanned, and the staining intensity of the Pho7 DBD and BSA polypeptides was quantified using ImageJ software. A BSA standard curve was generated by plotting intensity as a function of input BSA protein (micrograms); the concentration of Pho7 DBD was calculated by interpolation to the BSA standard curve.
DNA binding by EMSA.
32P-labeled DNA fragments were generated by PCR amplification of pho1 and tgp1 promoter segments using 5′ 32P-labeled forward primers (prepared with [γ-32P]ATP and T4 polynucleotide kinase) and nonlabeled reverse primers. The PCR fragments were purified by electrophoresis through a native 8% polyacrylamide gel in 1× TBE buffer (80 mM Tris-borate, 1.2 mM EDTA), eluted from an excised gel slice, ethanol precipitated, and resuspended in 10 mM Tris-HCl (pH 7.4)–1 mM EDTA at a concentration of 0.6 to 1 µM. Alternatively, 5′ 32P-labeled oligonucleotides containing Pho7 binding sites were separated from [γ-32P]ATP by Sephadex G25 gel filtration and annealed to a 1.5-fold molar excess of a complementary nonlabeled DNA oligonucleotide in a mixture of 10 mM Tris-HCl (pH 7.4), 1 mM EDTA, and 250 mM NaCl by heating for 20 min at 65°C followed by slow cooling to room temperature. The labeled oligonucleotide duplexes were ethanol precipitated, resuspended in 10 mM Tris-HCl (pH 7.4)–1 mM EDTA–10% glycerol, and then purified by electrophoresis through a native 12% polyacrylamide gel. EMSA reaction mixtures (10 µl) containing 50 mM Tris-HCl (pH 7.4), 10% glycerol, 340 ng poly(dI-dC) (Sigma), 0.6 or 1 pmol 32P-labeled DNA (as specified in figure legends), and 2 µl Pho7 DBD (serially diluted in buffer containing 50 mM Tris-HCl [pH 7.5], 250 mM NaCl, 10% glycerol, and 0.1% Triton X-100) were incubated for 10 min at room temperature. The mixtures were analyzed by electrophoresis through a native 6% polyacrylamide gel containing 2.5% (vol/vol) glycerol in 0.25× TBE buffer. 32P-labeled DNAs (free and Pho7 bound) were visualized by autoradiography. Where indicated, the extent of DNA binding to Pho7 (as a percentage of total DNA in the sample) was quantified by scanning the gel with a phosphorimager.
DNase I footprinting.
32P-labeled DNA fragments of the pho1 and tgp1 promoters were generated by PCR amplification as described above, using either a 5′ 32P-labeled forward primer and nonlabeled reverse primer (to label the top strand) or a 5′ 32P-labeled reverse primer and nonlabeled forward primer (to label the bottom strand). Footprinting reaction mixtures (10 µl) containing 50 mM Tris-HCl (pH 7.4), 10% glycerol, and 32P-labeled DNA and Pho7 DBD as specified were incubated for 10 min at room temperature. The mixtures were then adjusted to 2.5 mM MgCl2 and 0.5 mM CaCl and reacted with 0.04 U DNase I (New England Biolabs) for 90 s at room temperature. The DNase I reaction was quenched by adding 200 µl of stop solution (50 mM sodium acetate [pH 5.2], 1 mM EDTA, 0.1% SDS, 30 mg/ml yeast tRNA). The mixture was phenol-chloroform extracted, ethanol precipitated, and resuspended in 90% formamide–50 mM EDTA. The samples were heated at 95°C and then analyzed by urea-PAGE. 32P-labeled DNAs were visualized by autoradiography.
Assay of Pho1 induction during phosphate starvation.
Aliquots of exponentially growing S. pombe cultures in YES (yeast extract with supplements) medium were harvested, the cells were washed in water and adjusted to A600 of ~0.3 in PMG (Pombe glutamate) medium without phosphate. After incubation for 5 h at 30°C, cells were harvested, washed and suspended in water. To quantify Pho1 acid phosphatase activity, reaction mixtures (200 µl) containing 100 mM sodium acetate (pH 4.2), 10 mM p-nitrophenylphosphate, and cells (0.02 or 0.1 A600 unit) were incubated for 5 min at 30°C. The reactions were quenched by adding 1 ml of 1 M sodium carbonate, the cells were removed by centrifugation, and the absorbance of the supernatant at 410 nm was measured. Acid phosphatase activity is expressed in Fig. 1E as the ratio of A410 (p-nitrophenol production) to A600 (cells). Each data point is the average (±standard error of the mean [SEM]) from three phosphatase assays using cells from three independent cultures.
Assay of Pho1 plasmid reporter activity.
To measure acid phosphatase activity using the pho1 (Fig. 5) and tgp1-pho1 (Fig. 6) plasmid reporters, pho1Δ cells, in which the endogenous pho1 gene is deleted (5), were transfected with the kanMX-marked reporter plasmids. Single colonies of G418-resistant transformants (≥20) were pooled, and cultures were grown in YES medium containing G418 (150 µg/ml). Aliquots of exponentially growing cultures were harvested, washed, and suspended in water. Pho1 acid phosphatase activity was quantified as described above. Each data point in the bar graphs in Fig. 5 and 6C is the average (±SEM) from at least three phosphatase assays using cells from at least three independent cultures.
ACKNOWLEDGMENT
This research was supported by NIH grant GM52470 (to S.S. and B.S.).
Footnotes
Citation Schwer B, Sanchez AM, Garg A, Chatterjee D, Shuman S. 2017. Defining the DNA binding site recognized by the fission yeast Zn2Cys6 transcription factor Pho7 and its role in phosphate homeostasis. mBio 8:e01218-17. https://doi.org/10.1128/mBio.01218-17.
REFERENCES
- 1.Carter-O’Connell I, Peel MT, Wykoff DD, O’Shea EK. 2012. Genome-wide characterization of the phosphate starvation response in Schizosaccharomyces pombe. BMC Genomics 13:697. doi: 10.1186/1471-2164-13-697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henry TC, Power JE, Kerwin CL, Mohammed A, Weissman JS, Cameron DM, Wykoff DD. 2011. Systematic screen of Schizosaccharomyces pombe deletion collection uncovers parallel evolution of the phosphate signal pathways in yeasts. Eukaryot Cell 10:198–206. doi: 10.1128/EC.00216-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwer B, Sanchez AM, Shuman S. 2015. RNA polymerase II CTD phospho-sites Ser5 and Ser7 govern phosphate homeostasis in fission yeast. RNA 21:1770–1780. doi: 10.1261/rna.052555.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacPherson S, Larochelle M, Turcotte B. 2006. A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol Mol Biol Rev 70:583–604. doi: 10.1128/MMBR.00015-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chatterjee D, Sanchez AM, Goldgur Y, Shuman S, Schwer B. 2016. Transcription of lncRNAprt, clustered prt RNA sites for Mmi1 binding, and RNA polymerase II CTD phospho-sites govern the repression of pho1 gene expression under phosphate-replete conditions in fission yeast. RNA 22:1011–1025. doi: 10.1261/rna.056515.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marmorstein R, Carey M, Ptashne M, Harrison SC. 1992. DNA recognition by GAL4: structure of a protein-DNA complex. Nature 356:408–414. doi: 10.1038/356408a0. [DOI] [PubMed] [Google Scholar]
- 7.Hong M, Fitzgerald MX, Harper S, Luo C, Speicher DW, Marmorstein R. 2008. Structural basis for dimerization in DNA recognition by Gal4. Structure 16:1019–1026. doi: 10.1016/j.str.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2:28–36. [PubMed] [Google Scholar]
- 9.Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marmorstein R, Harrison SC. 1994. Crystal structure of a PPR1-DNA complex: DNA recognition by proteins containing a Zn2Cys6 binuclear cluster. Genes Dev 8:2504–2512. doi: 10.1101/gad.8.20.2504. [DOI] [PubMed] [Google Scholar]
- 11.Swaminathan K, Flynn P, Reece RJ, Marmorstein R. 1997. Crystal structure of a PUT3-DNA complex reveals a novel mechanism for DNA recognition by a protein containing a Zn2Cys6 binuclear cluster. Nat Struct Biol 4:751–759. doi: 10.1038/nsb0997-751. [DOI] [PubMed] [Google Scholar]
- 12.King DA, Zhang L, Guarente L, Marmorstein R. 1999. Structure of a HAP1-DNA complex reveals dramatically asymmetric DNA binding by a homodimeric protein. Nat Struct Biol 6:64–71. doi: 10.1038/4940. [DOI] [PubMed] [Google Scholar]
- 13.Fitzgerald MX, Rojas JR, Kim JM, Kohlhaw GB, Marmorstein R. 2006. Structure of a Leu3-DNA complex: recognition of everted CGG half-sites by a Zn2Cys6 binuclear cluster protein. Structure 14:725–735. doi: 10.1016/j.str.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 14.Cahuzac B, Cerdan R, Felenbok B, Guittet E. 2001. The solution structure of an AlcR-DNA complex sheds light onto the unique tight and monomeric DNA binding of a Zn2Cys6 protein. Structure 9:827–836. doi: 10.1016/S0969-2126(01)00640-2. [DOI] [PubMed] [Google Scholar]
- 15.Barbarić S, Münsterkötter M, Svaren J, Hörz W. 1996. The homeobox protein Pho2 and the basic-helix loop-helix protein Pho4 bind DNA cooperatively at the yeast PHO5 promoter. Nucleic Acids Res 24:4479–4486. doi: 10.1093/nar/24.22.4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbaric S, Münsterkötter M, Goding C, Hörz W. 1998. Cooperative Pho2-Pho4 interactions at the PHO5 promoter are critical for binding of Pho4 to UASp1 and for efficient transactivation by Pho4 at UASp2. Mol Cell Biol 18:2629–2639. doi: 10.1128/MCB.18.5.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou X, O’Shea EK. 2011. Integrated approaches reveal determinants of genome-wide binding and function of the transcription factor Pho4. Mol Cell 42:826–836. doi: 10.1016/j.molcel.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komeili A, O’Shea EK. 1999. Roles of phosphorylation sites in regulating activity of the transcription factor Pho4. Science 284:977–980. doi: 10.1126/science.284.5416.977. [DOI] [PubMed] [Google Scholar]
- 19.Shimizu T, Toumoto A, Ihara K, Shimizu M, Kyogoku Y, Ogawa N, Oshima Y, Hakoshima T. 1997. Crystal structure of PHO4 bHLH domain-DNA complex: flanking base recognition. EMBO J 16:4689–4697. doi: 10.1093/emboj/16.15.4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He BZ, Zhou X, O’Shea EK. 2017. Evolution of reduced co-activator dependence led to target expansion of a starvation response pathway. Elife 6:e25157. doi: 10.7554/eLife.25157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lev S, Kaufman-Francis K, Desmarini D, Juillard PG, Li C, Stifter SA, Feng CG, Sorrell TC, Grau GE, Bahn YS, Djordjevic JT. 2017. Pho4 is essential for dissemination of Cryptococcus neoformans to the host brain by promoting phosphate uptake and growth at alkaline pH. mSphere 2:e00381-16. doi: 10.1128/mSphere.00381-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 23.Schwer B, Schneider S, Pei Y, Aronova A, Shuman S. 2009. Characterization of the Schizosaccharomyces pombe Spt5-Spt4 complex. RNA 15:1241–1250. doi: 10.1261/rna.1572709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The sequence alignment/map format and SAM tools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garg A. 2015. Transcriptional programs in the fission yeast cell cycle. PhD thesis SUNY, Stony Brook, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Individual motif occurrences within the sequences of the Pho7 ChIP peaks, identified by FIMO (9), are listed (cutoff, motif P value of ≤0.001) on separate pages for phosphate-starved (−Pi) and phosphate-replete (+Pi) conditions. The motifs that correlate with the Pho7 DBD DNase I footprints in the pho1 and tgp1 promoters are highlighted in yellow. Download TABLE S1, XLSX file, 0.1 MB (47.1KB, xlsx) .
Copyright © 2017 Schwer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Annotated genes located within 1 kb downstream of the Pho7 ChIP peaks (group 1) and those that have a Pho7 ChIP peak within the annotated gene (group 2) are listed on separate pages for phosphate-starved (−Pi) and phosphate-replete (+Pi) conditions. Download TABLE S2, XLSX file, 0.1 MB (119.9KB, xlsx) .
Copyright © 2017 Schwer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Position probability matrices for the Pho7 motifs predicted by MEME (8) under phosphate-starved (−Pi) and phosphate-replete (+Pi) conditions. Download TABLE S3, XLSX file, 0.1 MB (49.2KB, xlsx) .
Copyright © 2017 Schwer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The Pho7 ChIP peaks identified by HOMER (26) are listed on separate pages for phosphate-starved (−Pi) and phosphate-replete (+Pi) conditions. Download TABLE S4, XLSX file, 0.1 MB (102.9KB, xlsx) .
Copyright © 2017 Schwer et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.