

Summary
Macrophages fulfill most of their microbicidal duties in their phagosomes following uptake of microbes. However, some microbes, such as Mycobacterium tuberculosis, have evolved mechanisms to subvert the normal maturation process of their phagocytic compartment to limit the hostility of this environment. The experimental analysis of this process, and its subsequent impact on bacterial fitness is technically-demanding and has required the development of a broad range of readouts to correlate function and outcome. In this chapter we detail two technically divergent platforms to measure the environment within the phagosomal compartment that contains Mtb in the short term, and more long-term readouts of bacterial fitness and Mtb’s reaction to host-derived stresses. The readouts are all fluorescence-based and are adaptable to measurement by a range of platforms, including spectrofluorometry, confocal microscopy and flow cytometry.
Keywords: Macrophage, Mycobacterium tuberculosis, phagosome, phagocyte
1. Introduction
It has been known for many years that the success of pathogenic Mycobacterium spp. is linked to their ability to block the fusion of the phagosomes in which they reside with the lysosomal network of their host cell (1–3). Early studies used electron microscopy to document this behavior and only since the mid-90s have more physiological readouts been exploited to characterize the nature of the Mtb-containing phagosomes. In 1994, we used fluorescein to measure the pH of the phagosomes containing Mycobacterium avium and found them to equilibrate to pH 6.2–6.4 (4). This analysis was repeated for Mycobacterium tuberculosis in 2004 (5). Examination of the trafficking of a range of ligands such as transferrin and cholera toxin B, and the relative distribution of several host cell proteins such as the transferrin receptor, the early endosomal marker EEA1 and the pro-enzyme form of the lysosomal hydrolase cathepsin D led to the conclusion that the Mtb-containing phagosome behaved like an early endosome (6–10). The vacuole was highly dynamic and fused readily with other early endosomal compartments. Measurement of the intravacuolar pH was a useful indicator of phagosome maturation and, by inference, the relative “fitness” of the bacterium. Activation of the macrophage with inflammatory cytokines such as interferon-γ (IFN-γ) overcomes this blockage and delivers the bacilli to compartments with a reduced pH of 5.2 (10, 11). Finally, mutant bacteria defective in the modulation of their phagosome showed reduced survival inside murine macrophages (5).
Despite its usefulness there are distinct limitations to the pH assay. It is achieved through the labeling of reactive amino groups on the bacterial surface with the succinimidyl ester form of carboxyfluorescein. This label comes off the bacterium within hours of uptake by macrophages and is therefore only useful for analysis within the first few hours of infection in tissue culture models. Over the years we have been developing alternative readouts of the intracellular environment experienced by Mtb, and its impact on bacterial fitness. Transcriptional profiling of the response of Mtb following internalization by the host macrophage has allowed us to identify specific environmental cues detected by Mtb that induce defined regulons (12–15). pH and chloride concentration are interdependent parameters that vary during phagosome maturation – as the pH drops the Cl− ion concentration increases (12, 15). Mtb exhibits an overlapping transcriptional response to both stimuli. Promoter elements that are responsive to these environmental cues were placed in replicating plasmids upstream of the gfp gene and introduced into Mtb. These early reporter bacterial strains could be induced to express GFP under appropriate environmental conditions in the test tube and showed levels of expression that correlated with the inferred status of their phagosome (12, 15).
More recently, we have developed a new generation of reporter Mtb strains that constitutively express mCherry and express GFP under immune-regulated environmental stresses, such as nitric oxide (hpsX’::GFP, smyc’::mCherry), or that expresses GFP as a fusion partner with the single stranded binding protein (SSB) (SSB-GFP, smyc’::mCherry) (15, 16). This latter construct had been developed previously in Escherichia coli, and used to score bacterial replication (17). Formation of the replisome leads to the bacteria possessing green fluorescent foci during replication of the bacterial chromosome. In Mtb these foci persist for 76%–80% of the division cycle of the bacterium (16). Therefore by scoring the number of red bacilli with green foci, one can determine the relative replication status of a bacterial population. We have used these reporter strains to probe the relative fitness of bacterial populations in in vivo experimental mouse infection under differing immune conditions (16). These reporter strains have the opposite limitation to the CF-SE-labeled Mtb in that some of these readouts are best observed in the context of longer-term infection, and maintaining the health of host cells in tissue culture for an appropriate duration is extremely challenging. These reporter strains are thus much more suited to in vivo infection models lasting several weeks.
These assays, developed over the past 20 years, have the capacity to probe the host environment and its impact on bacterial fitness in short-term infections in tissue culture, or in long-term in vivo infections. As mentioned, all these assays have caveats or qualifiers and none of the assays are universally applicable. In this chapter we will present the methods behind the assays and highlight their application under differing experimental conditions.
2. Materials
2.1. Cells, Reagents, and Buffers
Macrophages: Bone marrow–derived murine macrophages (BMMØ) are our cells of choice for these assays (see Note 1). BMMØ are derived from the bone marrow extracted from the femur, tibia, and ilium of euthanized mice and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 1 mM sodium pyruvate, and 10% L-cell conditioned media (BMMØ media).
If we have to use a cell line we use J774 cells (available from the American Type Culture Collection, Rockville, MD), which are maintained in DMEM supplemented with 10% FBS and 2 mM L-glutamine + 1 mM sodium pyruvate.
Mycobacterium tuberculosis is maintained in liquid culture in Middlebrook 7H9 medium (BD, Franklin Lakes, NJ).
Cover slips: Clean 0.13 mm × 12.5 mm × 25 mm cover glass (see Note 2). Sterilize by autoclaving.
Cuvette buffer: Tissue culture tested phosphate-buffered saline (PBS) pH 7.2 adjusted to contain 1 mM CaCl2, 2.7 mM KCl, 0.5 mM MgCl2, 5 mM dextrose, 10 mM HEPES, and 0.1 % cold water teleost gelatin (see Note 3).
Binding dish: A microbiological petri dish containing a square piece of Parafilm adhered to the dish by a couple of drops of water.
The amine reactive fluorescent reagent: 5-(and-6)-carboxyfluorescein, succinimidyl ester (mixed isomers) (CF-SE) (Life Technologies Corporation). Dissolve in high quality anhydrous dimethyl sulfoxide (DMSO) at 5 mg/ml before use. Stock solutions can be aliquoted and stored at −20°C. Protect reagents from light and moisture.
Coupling buffer: 0.1 M sodium borate in ddH2O, adjusted to pH 8.0 with 10 M NaOH. Filter sterilize through 0.22-μm filter.
Tween 80 detergent.
Reference pH buffers (pH 4.5–5.5): 0.15 M potassium acetate. Adjust pH to 4.5, 5.0, and 5.5 with 10 M NaOH.
Reference pH buffers (pH 6–7.5): 0.1 M piperazine-N,N′–bis(2-ethanesulfonate (PIPES), 0.1 M KCl. Adjust pH to 6.0, 6.5, 7.0 and 7.5 with 5 M HCl.
Nigericin (InvivoGen, San Diego, CA).
Reporter strains of Mtb were generated by transformation of the parental strain with pCherry3 replicating plasmid that expresses mCherry under regulation of the smyc promoter (18), together with the reporter construct (15, 16). In this Chapter we use Mtb transformed with plasmids containing hspX’::GFP, smyc’::mCherry to report on immune-mediated stress due to iNOS expression at sites of infection, and SSB-GFP, smyc’::mCherry encoding a fusion protein of the single stranded binding protein SSB with GFP to indicate the relative replication rates of Mtb under differing immune pressures (15, 16). Additional details regarding these bacterial strains is provided in Note 4.
Alexa Fluor 647 conjugated phalloidin, Alexa Fluor 514 conjugated goat anti-rabbit IgG and DAPI (Life Technologies Corporation). Stored in DMSO at −20°C in aliquots of 10 mg/ml
Rabbit anti-murine iNOS antibody (BD Transduction Laboratories, San Jose, CA).
Mounting medium containing anti-fade.
High vacuum grease (Dow Corning, Auburn, MI).
2.2 Instruments
For pH measurements we utilized a temperature controlled-spectrofluorometer with variable excitation and emission monochromators. The authors use the QMSE4 model spectrofluorometer from Photon Technologies International (Lawrenceville, NJ) equipped with a thermostat-controlled four-chambered turret for simultaneous measurement of four experimental variables. The QMSE4 is interfaced with a PC-compatible computer and is managed by Felix32 software (Photon Technologies International).
For confocal microscopy we used a Leica SP5 spectral confocal laser-scanning system with an inverted microscope (Leica Microsystems GmbH, Germany). Leica Application Suite Advanced Fluorescence (LAS-AF 2.6) was utilized for image acquisition. Volocity image analysis software (Perkin Elmer Life Sciences, USA) was used for image analysis and quantification of fluorescence signals.
Data are finally analyzed and displayed using standard mathematical software such as Microsoft Excel® or MATLAB®.
3. Methods
3.1 Measurement of the pH of Mtb-containing phagosomes (all manipulations conducted under BSL3 conditions)
Mycobacterium tuberculosis is a human pathogen and must be handled under Biosafety Level 3 containment by trained personnel. Such facilities have to be maintained under registered institutional safety and employee health monitoring programs. Some investigators use non-pathogenic Mycobacterium spp., such as Mycobacterium smegmatis, as a surrogate but the relevance of data generated with non-pathogenic organisms is open to obvious concerns regarding validity.
3.1.1. Fluorescent labeling of Mtb
A mid-log phase culture of Mtb Erdman is harvested by centrifugation at 2,000 g for 12 min at 4°C. The medium discarded into a bleach container and the bacteria washed 2X with PBS supplemented with 0.1% Tween 80. The bacteria are resuspended in Coupling buffer with 0.1% Tween 80 to a concentration of 5 × 108/ml. 5 μl of CF-SE stock solution is added to the bacterial suspension and the tube is covered with aluminum foil and incubated on a nutator mixer for 20 min at room temperature.
Following incubation the bacteria are harvested by centrifugation in an tabletop microcentrifuge with sealed rotor. The bacteria are washed 3X with Cuvette buffer, adjusted to 2.5 × 108/ml in Cuvette buffer and stored in the dark on ice while preparing the spectrofluorometer.
3.1.2. Macrophage monolayer preparation and handling
Fully differentiated BMMØ monolayers are grown to confluency in untreated petri dishes. Growth media is removed and replaced with cold PBS pH 7.2 (without Ca2+ and Mg2+) and incubated at 4°C for 10 min to facilitate BMMØ detachment from the plastic. BMMØ are then gently dislodged with a rubber policeman and harvested by centrifugation at 230 g at 4°C for 10 min.
Sterile, clean 12.5 × 25 mm cover slips are placed in a sterile quadrant petri dish (2 per quadrant) using fine-point forceps that have been dipped in 70% ethanol and flamed (see Note 5).
BMMØ are gently resuspended in 1 ml BMMØ media, counted using a hemocytometer, and diluted in BMMØ media to a density of ~1.25 × 106 macrophages/ml.
10 ml of BMMØ suspension is added to the petri dish and incubated at 37°C for 24 hrs to allow a monolayer to establish on the cover slips. Care should be exercised to prevent excessive movement of the cover slips in the petri dish. The BMMØ monolayer–covered cover slips (subsequently referred to as monolayers) are then ready for infection.
3.1.3. Spectrofluorometer Setup and Operation
The spectrofluorometer should be set up according to manufacturer’s directions such that optimal measurements can be taken using the desired wavelengths (see Note 6).
Clean quartz cuvettes containing Cuvette buffer are inserted into a thermostat-controlled sample holder and warmed to 37°C prior to the loading of the monolayers (see Note 7).
Using fine-point forceps, a monolayer-covered cover slip is grasped at one end (see Note 8) and dipped 4 times into a 50-ml tube containing Cuvette buffer to remove BMMØ growth media and loosely adhered cells. The cover slip is then placed in the cuvette with a vertical orientation (length of the cover slip parallel to the long axis of the cuvette), on the diagonal (width of the cover slip at 45 degrees to the short axes of the cuvette) and with the cellular side facing the emission slit (Fig. 1A).
At this point, the background measurements of each monolayer are acquired for the wavelengths required.
At the conclusion of the background determination, the cover slips are carefully removed from the cuvettes using forceps and placed with the cellular side up on the Parafilm in the binding dish inside a biosafety cabinet.
90 μl of CF-SE-labeled Mtb suspension is carefully laid over each monolayer. The bacterial suspension is incubated with the cell monolayer at room temperature for 5 min (see Note 9).
The cover slips are then dipped 10 times in Cuvette buffer at room temperature to remove unbound bacteria and are placed in the cuvettes with the same orientation. The cuvettes are sealed with caps, removed from the biosafety cabinet and placed in the heated cuvette holder in the spectrofluorometer chamber.
Emission at 520 nm is recorded with excitation alternating between 450 and 490 nm (see Note 10). Typically an integration time of 1 s per data point is optimal. Data are routinely collected for a 3 hr time period to follow the acidification of the Mtb-containing phagosomes.
Fig. 1. Measuring phagosomal pH using a spectrofluorometer.
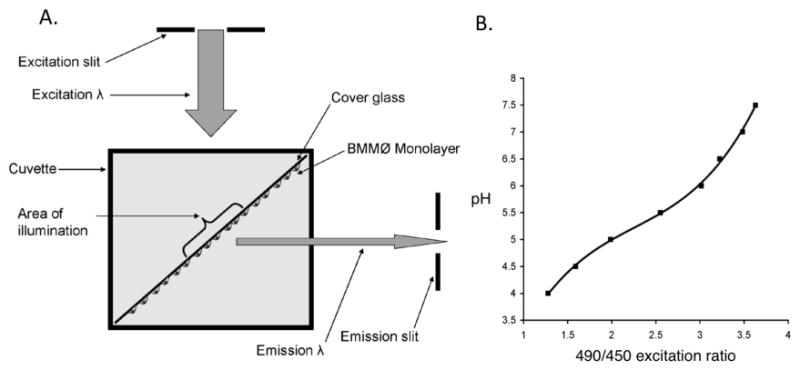
A. Diagram illustrating the orientation of the BMMØ monolayer in the spectrofluorometer. Cover glass is oriented to achieve a 45° incidence with the excitation beam (excitation λ), and with the cells on the side facing the emission slit. B. Excitation ratio versus pH standard curve generated by CF-SE-labeled beads in buffers of known pH (19, 20). The curve of best-fit can be described by the equation y = 0.4233x3 − 2.8693x2 + 7.3391x − 1.5901 (where x=490/450 excitation ratio and y=pH).
3.1.4. Conversion of Excitation Ratio to pH
-
1
At the conclusion of the experiment the cuvettes are removed from the spectrofluorometer and transferred to the biosafety cabinet. The Cuvette buffer is removed and replaced with the pH reference buffers 5.5, 6.0, 6.5 and 7.0 with 10 μM nigericin (see Note 11). The cuvettes are returned to the spectrofluorometer and calibration readings at these defined pHs are acquired for 5 min at 450/520 nm and 490/520 nm.
-
3
Extensive calibration of CF-SE-labeled beads has been conducted and documented in an earlier Chapter in this series (19). The CF-SE-bead 490/450 excitation ratio for each pH standard (minus background) is calculated and plotted against pH. Confirm that the pKa of the bound carboxyfluorescein is close to the pH that is of most interest and that the curve generated is reasonably flat over the pH range required (see Note 11).
-
5
The polynomial equation that best describes the curve is calculated (Fig. 1B). Usually a third or fourth order polynomial is sufficient. This can be done with standard mathematical software such as Microsoft Excel® or MATLAB®.
-
6
This equation is used to convert the real time phagosomal 490/450 excitation ratios into pH units and these values are plotted against time (Fig. 2) (see Note 10).
-
7
Figure 2 shows the pH plots from the phagosomes containing wild-type Mtb in comparison to those containing a mutant defective in rv3378c that was isolated in a genetic screen to identify genetic loci required for successful modulation of phagosome maturation (5). The wild-type bacteria maintain their vacuoles at pH 6.4, whereas the Rv3378c mutant is delivered into lower pH compartments, approximately pH 5.8. This behavior is reflected in the acquisition of lysosomal tracers, Fe2+-dextran (Fig. 2C), and a reduction in the survival and/or growth of the mutants inside their host macrophages (Fig. 2D).
Fig. 2. Measuring the pH of Mtb-containing phagosomes.
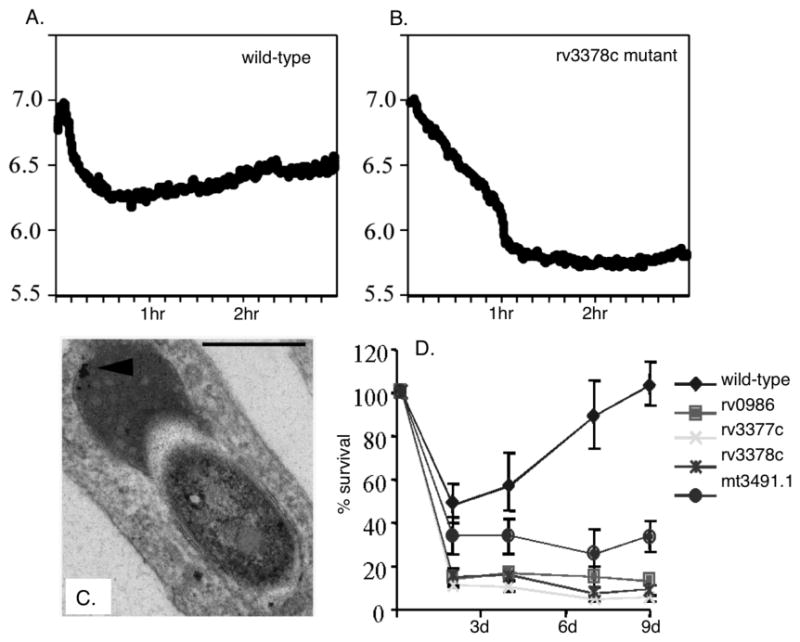
A. Graphs showing the acidification of the phagosomes of BMMØ infected with CF-SE-labeled wild-type Mtb (5). The phagosomes containing wild-type viable bacteria acidify to pH 6.4 within the time frame of the assay (3 hrs). B. In contrast, the phagosomes containing the rv3378c mutant are able to acidify further, down to pH 5.8. C. Electron microscopy examination of macrophages infected with the mutant bacteria demonstrated that these phagosomes exhibited an increased association with lysosomes, defined by their dense cargo, and the presence of an iron dextran tracer (arrowhead). D. The survival kinetics of wild-type Mtb compared to the mutants isolated as defective in the regulation of their phagosome indicates that these mutants are unable to enter into growth phase following uptake by the BMMØ.
3.2 Use of reporter Mtb strains to probe bacterial fitness in vivo
3.2.1. Preparation of reporter Mtb strains
Reporter strains of Mtb, generated as detailed previously (see Note 4), are grown to mid-log (OD600 ~ 0.6; this corresponds to ~ 108 bacteria/ml) in Middlebrook 7H9 medium (BD, Franklin Lakes, NJ) with 50 μg/ml hygromycin B to ensure maintenance of the replicating plasmids An equal volume of 20% glycerol is added to the bacterial culture and mixed (final glycerol concentration = 10%). This suspension is aliquoted in 1 ml volumes into 1.5 ml screw cap cryovials and stored at −80°C until required. Prior to initial use, an aliquot of each strain is thawed and serial dilutions plated on Middlebrook 7H10 agar (BD) with 50 μg/ml hygromycin B, and colony-forming units (CFUs) counted after 3–4 weeks to provide a definitive quantification of viable bacteria in each frozen stock.
At the time of challenge, an aliquot of reporter bacteria for each strain required is thawed, passed 5–8x through a tuberculin syringe, and diluted in PBS containing 0.05% Tween 80 to obtain a final concentration of 1,000 CFUs in 25 μl. Serial dilutions of the suspension are also plated on Middlebrook 7H10 agar with 50 μg/ml hygromycin B to verify the inoculum numbers.
3.2.2. Infection of mice with the bacterial reporter strains
We use intranasal challenge as our route of infection. This method has significant advantages as it is inexpensive and easy to use, and generates the infected tissue we require for analysis. However it is important to bear in mind the limitations of this method for modeling infections, for the reason discussed in Note 13.
6–8 week old C57BL/6J female mice (Jackson Laboratories, Bar Harbor, ME) are used routinely for infection studies, see also Note 14. Mice are placed individually in an induction chamber and exposed to 2% isoflurane delivered by a Harvard Systems anesthesia system (Harvard Biosciences, Inc., Holliston, MA).
During this step the mouse is monitored visually, and when lightly anesthetized the mouse is removed, held with its head inclined and 25 μl total of bacterial suspension is carefully delivered down both nares. The mouse is placed in a cage and monitored for its recovery from anesthesia.
At various time intervals post-challenge mice are sacrificed for analysis. Mice are euthanized by CO2 inhalation.
To obtain colony-forming unit counts, the left lung lobe and the accessory lobe of the right lung are manually homogenized in a Whirl-Pak tissue homogenizer bag (Nasco, Fort Atkinson, WI) with 0.5 ml PBS containing 0.05% Tween 80. The homogenate is removed into a clean screw-cap tube, and the residual contents in the homogenizer bag rinsed with an additional 0.5 ml PBS containing 0.05% Tween 80. This is then added to the tube containing the initial 0.5 ml homogenate (total = 1 ml homogenate). The homogenate is serial diluted and plated on Middlebrook 7H10 Agar +/− 50 μg/ml hygromycin B.
For image analysis of the infected tissue, the rest of the right lung lobes are fixed in 4–5 ml of 4% paraformaldehyde in PBS in a 15 ml conical tube, for immunofluorescence microscopy and histological examination. The tubes are surface decontaminated and removed from the BSL-3 facility, and the samples left in fixative overnight at room temperature, protected from light, in a sealed container, prior to immunofluorescence staining.
Thick sections of the fixed lung lobe tissue (~ 0.5 mm) are cut by hand with a razor (see Note 15), then placed in a 1.5 ml microcentrifuge tube and blocked and permeabilized by incubating for 1 hour at room temperature in 500 μl PBS + 3% BSA + 0.1% Triton X-100 (blocking buffer) (see Note 16).
For immunohistochemistry (Figure 3), samples are incubated with primary antibodies overnight at 4°C, washed 3 × 5 minutes with blocking buffer at room temperature, then incubated with secondary antibodies for 2 hours at room temperature. After washing 3 × 5 minutes with blocking buffer, samples are mounted with mounting medium containing antifade (see Note 17). All staining and washing steps are carried out on a rocking platform, with the samples protected from light. We routinely stain 2 sections/sample in the same 1.5 ml microcentrifuge tube, in 200 μl volumes of antibody solutions. Wash steps are carried out in 500 μl volumes. All antibodies are diluted in blocking buffer. Rabbit anti-iNOS was used at 1:100, and Alexa Fluor 514 goat anti-rabbit IgG used at 1:200 for secondary detection. Alexa Fluor 647 conjugated phalloidin (1:50) was used for visualization of the actin cytoskeleton and nuclei were visualized with DAPI (1:500).
Samples are imaged with a Leica SP5 spectral confocal microscope (see Note 18), and z-stacks reconstructed into 3D using Volocity software. We routinely image a depth of 10 μm, with 0.5 μm steps.
For quantification of hspX’::GFP signal (Fig. 3), the volume of each bacterium was measured via the mCherry channel, with the corresponding sum of the GFP signal for that bacterium simultaneously acquired, to obtain GFP/μm3 values. Settings for the GFP channel are maintained throughout the imaging of samples within each experimental set to allow comparison of values (see Note 19).
For quantification of SSB-GFP signal (Fig. 4), bacteria with or without SSB-GFP foci are scored manually by inspection of 3D reconstructed and extended focus images in Volocity. At least 100 bacteria, taken across multiple images, should be quantified for each variable under experimentation. Statistical differences between data sets are determined by a non-parametric Mann-Whitney test.
Fig. 3. Measuring the induction of hspX’-driven expression of GFP.
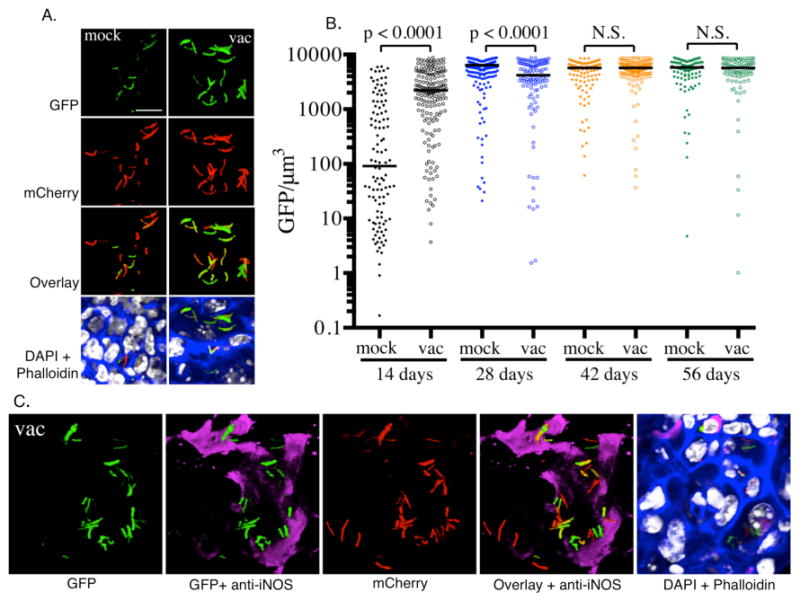
A. Illustrates the levels of expression of hspX’ promoter dependent GFP at 14 days post-infection in naïve mice (mock) and mice vaccinated with heat-killed Mtb (vac), as reported previously (16). At 14 days the vaccinated mice have a robust immune response, while this response is not fully developed in the naïve mice. B. Images are acquired using the Leica LAS software and are quantified by Volocity. The bacterial volume is defined by the mCherry signal, and the total GFP signal intensity is measured for each bacterial volume. Each dot on the scatter plot represents a single bacterium or a cluster of bacteria that cannot be separated. The average level of expression of GFP at 14 days is markedly lower in naïve mice than in vaccinated mice. By 28 days, high levels of induction of GFP expression is observed in both naïve and vaccinated groups. The horizontal bars represent the median value for each group and p-values were generated using a Mann-Whitney statistical test. C. The hspX promoter is regulated by the dosR regulon, which is activated by hypoxia and nitric oxide. Nitric oxide is generated by the inducible nitric oxide synthase (iNOS), which is expressed in mouse macrophages in the presence of IFN-γ and TNF-α. Probing the murine lung tissue with an anti-iNOS antibody reveals the presence of the host enzyme in regions that contain GFP-expressing Mtb.
Fig. 4. Using a SSB-GFP fusion protein to assess bacterial replication in tissue.
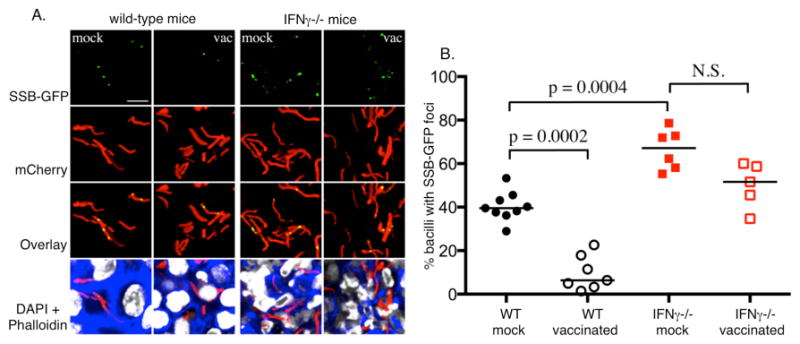
Naïve (mock) and vaccinated (vac) wild-type and IFN-γ-deficient mice were challenged with Mtb expressing mCherry and SSB-GFP, as reported previously (16). The mice were sacrificed at 28 days and the lung tissue analyzed by confocal microscopy. A. Shows confocal images from the infected lung tissue demonstrating the presence of green foci amongst some of the bacteria in the section. The presence or absence of GFP foci within the bacteria was scored manually. B. A scatter plot of the % of GFP foci-positive bacteria in each mouse in each group. The dots each represent a single mouse. The horizontal bars represent the median value for each group and p-values were obtained with a Mann-Whitney statistical test. The graph indicates that the number of SSB-GFP foci-positive bacteria was inversely proportional to the robustness of the immune response. This suggests that replication was best controlled in vaccinated mice, and least controlled in mice deficient in the macrophage-activating cytokine IFN-γ.
Acknowledgments
ST and DGR are supported by the following awards from the National Institutes of Health, USA; AI114952 (ST) and AI067027, AI118582 and HL055936 (DGR). RMY is supported by awards from the Canadian Institutes of Health Research and Natural Sciences and the Engineering Research Council of Canada.
Footnotes
Primary BMMØ are generally preferred for their enhanced phagocytic proficiency and adhesion.
0.13 × 12.5 × 25 mm cover glass is not commercially available. Cover glass can be custom ordered from ProSciTech (Thuringowa, Qld, Australia). Alternatively, 25 × 25 mm cover glass is available from Fisher Scientific (Pittsburgh, PA, USA) and can be cut in half by diamond pencil in house.
Gelatin is used as an alternative to FBS for spectrofluometric assays as it has low autofluorescence with excitation wavelengths above 450 nm. If assays are expected to take greater than 6 hours, FBS is preferred for sustained macrophage viability.
Reporter Mtb strains are generated using the replicating plasmid pCherry3, which encodes mCherry under regulation of the constitutive promoter smyc. We use replicating plasmids because they ensure high levels of expression of fluorescent protein. While integrating plasmids might represent a more stable solution the loss in signal is often too great. Expression of these fluorescent protein constructs can come at some loss to fitness so it is important to assess the maintenance of the plasmid by replica plating bacteria from infected mice on agar plates with and without hygromycin B selection (50 μg/ml) and confirming that they still express GFP under appropriate conditions.
Arrange cover slips so as not to overlap, taking care not to overcrowd them in the petri dish as cover slips can move after monolayers have been established and damage to BMMØ can occur. Alternatively, cover slips can be separated from each other using partitioned petri dishes or 6 well plates.
Some general considerations are: the focusing of illumination on the sample, the addition of long-pass and short-pass filters, and the adjustment of excitation and emission slit widths to maximize signal to noise ratio and to minimize photobleaching.
Cuvette buffer should be of a similar temperature to the cuvettes at addition. This prevents bubble formation that can create unwanted scatter of light.
Cover slips should only be grasped by forceps at the uppermost edge to avoid damaging the area of the monolayer that is illuminated in the spectrofluorometer.
The Mtb suspension has an approximate multiplicity of infection (MOI) of 25:1. This is at vast excess and is required to guarantee binding of adequate numbers of CF-SE-labeled bacteria to generate sufficient signal return to ensure the accuracy of the readings. In actuality, under these conditions, we observe around 5 bacilli bound per macrophage following washing of the coverslips.
Carboxyfluorescein has two wavelengths of maximal excitation that lead to fluorescence emission at 520 nm. Excitation at 450 nm induces a lower signal but the signal is pH insensitive and acts as an internal standard. In contrast, excitation at 490 nm results in a stronger fluorescent signal at 520 nm but this signal is pH sensitive and is quenched by the protonation of the dye at lower pH.
Standard curves are generated using phagocytosed Mtb on BMMØ monolayers using the ionophore nigericin (10 μM) in K+-containing buffers of known pH. This eliminates the possibility of modification of the fluorochrome’s pKa by the intracellular environment. For convenience we routinely generate standard curves using a suspension of CF-SE-labeled Mtb. In each experimental setup however, it should be determined that these curves are equivalent to those generated with Mtb-containing monolayers treated with nigericin. Standard pH curves should be generated at the conclusion of every phagosomal pH experiment. Subtle changes to components of the experiment such as degree of CF-SE labeling, slit width and PMT voltage, can have profound effects on the relationship between excitation ratio and actual pH.
The pKa of free carboxyfluorescein-SE is 6.4. We have found that when bound to proteins or to the surface of Mtb, the pKa is shifted to ~5.5 making it particularly useful in the generation of phagosomal pH profiles. However, should the pKa of the CF-SE conjugates be inappropriately high thus rendering measurement of lower pH values inaccurate, then Oregon green-SE should be used instead of carboxyfluorescein-SE.
Intranasal infection has significant advantages for in vivo Mtb infection experiments in its convenience and low cost. However, it is important to recognize that this delivery method is relatively non-physiological compared to an aerosol delivery, because Mtb infection in nature is initiated by the inhalation of extremely small numbers of bacilli into the deep lung.
In addition to wild-type mice we have also examined immune-compromised C57BL/6J mice that were defective in expression of IFN-γ, and immune-enhanced mice that were vaccinated with heat-killed Mtb 4 weeks prior to challenge (16).
We do not process the samples with cryostat or paraffin-based sectioning, thus minimizing manipulation of the tissue, and enabling thick section imaging as desired. It is important to obtain tissue sections that are even, to allow proper mounting of the sample after staining. To do this, we make an initial cut through the middle of the lung lobe, then subsequently slice from this cut face to obtain the tissue sections.
If no primary antibody stain is being done, this first 1 hour incubation step can be skipped, and the sections placed directly into the solution of secondary antibodies (e.g. phalloidin and DAPI) in blocking buffer for the 2 hour incubation at room temperature.
To ensure that the sample is not compressed on mounting, we use inert vacuum grease to make a “chamber” in which the sample sits. Placing the vacuum grease in a 10 ml syringe allows for easy dispensing. The coverslip is then gently and evenly pressed down such that the sample is held in place on the slide, without being unduly compressed. Use of a mounting medium that does not harden allows for re-mounting of the sample as needed. An alternative to the use of vacuum grease is to use SecureSeal™ imaging spacers (Electron Microscopy Sciences, Hatfield, PA).
Use of a spectral confocal system is particularly helpful for 5-color imaging, to enable proper separation of the various fluorophores used – in particular here, for the separation of GFP versus Alexa Fluor 514 versus mCherry signal. We also use sequential scans, to further minimize signal bleed-through.
For quantification of hspX’::GFP signal, it is also important that saturation in the GFP channel be minimized, to allow the greatest dynamic range and accurate measurements. It is thus best to first test settings on both samples with the lowest and highest signals, to obtain parameters that will be acceptable across all samples to be compared, so that the same settings can be used throughout the experiment and valid comparisons made.
References
- 1.Draper P, Hart PD, Young MR. Effects of anionic inhibitors of phagosome-lysosome fusion in cultured macrophages when the ingested organism is Mycobacterium lepraemurium. Infect Immun. 1979;24:558–561. doi: 10.1128/iai.24.2.558-561.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hart PD, Armstrong JA. Strain virulence and the lysosomal response in macrophages infected with Mycobacterium tuberculosis. Infect Immun. 1974;10:742–746. doi: 10.1128/iai.10.4.742-746.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hart PD, Young MR. Ammonium chloride, an inhibitor of phagosome-lysosome fusion in macrophages, concurrently induces phagosome-endosome fusion, and opens a novel pathway: studies of a pathogenic mycobacterium and a nonpathogenic yeast. J Exp Med. 1991;174:881–889. doi: 10.1084/jem.174.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, Haddix PL, Collins HL, Fok AK, Allen RD, Gluck SL, Heuser J, Russell DG. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science. 1994;263:678–681. doi: 10.1126/science.8303277. [DOI] [PubMed] [Google Scholar]
- 5.Pethe K, Swenson DL, Alonso S, Anderson J, Wang C, Russell DG. Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc Natl Acad Sci U S A. 2004;101:13642–13647. doi: 10.1073/pnas.0401657101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clemens DL, Horwitz MA. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J Exp Med. 1995;181:257–270. doi: 10.1084/jem.181.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clemens DL, Horwitz MA. The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J Exp Med. 1996;184:1349–1355. doi: 10.1084/jem.184.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell DG, Dant J, Sturgill-Koszycki S. Mycobacterium avium- and Mycobacterium tuberculosis-containing vacuoles are dynamic, fusion-competent vesicles that are accessible to glycosphingolipids from the host cell plasmalemma. J Immunol. 1996;156:4764–4773. [PubMed] [Google Scholar]
- 9.Sturgill-Koszycki S, Schaible UE, Russell DG. Mycobacterium-containing phagosomes are accessible to early endosomes and reflect a transitional state in normal phagosome biogenesis. EMBO J. 1996;15:6960–6968. [PMC free article] [PubMed] [Google Scholar]
- 10.Via LE, Fratti RA, McFalone M, Pagan-Ramos E, Deretic D, Deretic V. Effects of cytokines on mycobacterial phagosome maturation. J Cell Sci. 1998;111(Pt 7):897–905. doi: 10.1242/jcs.111.7.897. [DOI] [PubMed] [Google Scholar]
- 11.Schaible UE, Sturgill-Koszycki S, Schlesinger PH, Russell DG. Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J Immunol. 1998;160:1290–1296. [PubMed] [Google Scholar]
- 12.Abramovitch RB, Rohde KH, Hsu FF, Russell DG. aprABC: a Mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Mol Microbiol. 2011;80:678–694. doi: 10.1111/j.1365-2958.2011.07601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rohde KH, Abramovitch RB, Russell DG. Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe. 2007;2:352–364. doi: 10.1016/j.chom.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Rohde KH, Veiga DF, Caldwell S, Balazsi G, Russell DG. Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog. 2012;8:e1002769. doi: 10.1371/journal.ppat.1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan S, Sukumar N, Abramovitch RB, Parish T, Russell DG. Mycobacterium tuberculosis responds to chloride and pH as synergistic cues to the immune status of its host cell. PLoS Pathog. 2013;9:e1003282. doi: 10.1371/journal.ppat.1003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sukumar N, Tan S, Aldridge BB, Russell DG. Exploitation of Mycobacterium tuberculosis reporter strains to probe the impact of vaccination at sites of infection. PLoS Pathog. 2014;10:e1004394. doi: 10.1371/journal.ppat.1004394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reyes-Lamothe R, Possoz C, Danilova O, Sherratt DJ. Independent positioning and action of Escherichia coli replisomes in live cells. Cell. 2008;133:90–102. doi: 10.1016/j.cell.2008.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carroll P, Schreuder LJ, Muwanguzi-Karugaba J, Wiles S, Robertson BD, Ripoll J, Ward TH, Bancroft GJ, Schaible UE, Parish T. Sensitive detection of gene expression in mycobacteria under replicating and non-replicating conditions using optimized far-red reporters. PLoS One. 2010;5:e9823. doi: 10.1371/journal.pone.0009823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yates RM, Russell DG. Real-time spectrofluorometric assays for the lumenal environment of the maturing phagosome. Methods in molecular biology. 2008;445:311–325. doi: 10.1007/978-1-59745-157-4_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yates RM, Russell DG. Phagosome maturation proceeds independently of stimulation of toll-like receptors 2 and 4. Immunity. 2005;23:409–417. doi: 10.1016/j.immuni.2005.09.007. [DOI] [PubMed] [Google Scholar]