

Abstract
G proteins are key mediators of G protein-coupled receptor signalling, which facilitates a plethora of important physiological processes. The cyclic depsipeptides YM-254890 and FR900359 are the only known specific inhibitors of the Gq subfamily of G proteins; however, no synthetic route has been reported previously for these complex natural products and they are not easily isolated from natural sources. Here we report the first total synthesis of YM-254890 and FR900359, as well as of two known analogues, YM-385780 and YM-385781. The versatility of the synthetic approach also enabled the design and synthesis of ten analogues, which provided the first structure–activity relationship study for this class of compounds. Pharmacological characterization of all the compounds at Gq-, Gi- and Gs-mediated signalling provided succinct information on the structural requirements for inhibition, and demonstrated that both YM-254890 and FR900359 are highly potent inhibitors of Gq signalling, with FR900359 being the most potent. These natural products and their analogues represent unique tools for explorative studies of G protein inhibition.
G protein-coupled receptors (GPCRs) are integral membrane proteins that comprise one of the largest classes of proteins and therapeutic targets in the human genome1,2. Agonist binding to a GPCR stabilizes an active conformation of the receptor, which activates intracellular heterotrimeric guanine nucleotide binding proteins (G proteins). G proteins are composed of α, β and γ subunits, which on activation dissociate from GPCRs and modulate a range of intracellular effectors3,4.
In general, the role of G proteins in GPCR-mediated signalling is not as well understood as other aspects of GPCR function, although there is an immense interest in modulating G protein signalling pathways using biased ligands5,6. G proteins are divided into four families, denoted Gs, Gi/o, Gq/11 and G12/13, and only a few compounds are available that can modulate G protein activity, including pertussis toxin and cholera toxin7–9, which enzymatically modulate Gi and Gs proteins, respectively. However, their application is limited by the long incubation time (several hours) required. Thus, fast-acting and subtype-selective modulators of G proteins are in great demand.
The natural product YM-254890 (1 (Fig. 1a)) was isolated from Chromobacterium sp. QS3666 and found to be a unique pharmacological tool as a selective inhibitor of Gq signalling10–14. YM-254890 has been available in very restricted amounts from Yamanochi Pharmaceutical and used, for example, to deconvolute GPCR signalling15. However, the supply of the compound has ended16, and there is currently an urgent need to generate YM-254890 as a valuable tool for studying Gq-mediated signalling.
Figure 1. Structures and retrosynthetic analysis of YM-254890, FR900359 and analogues.
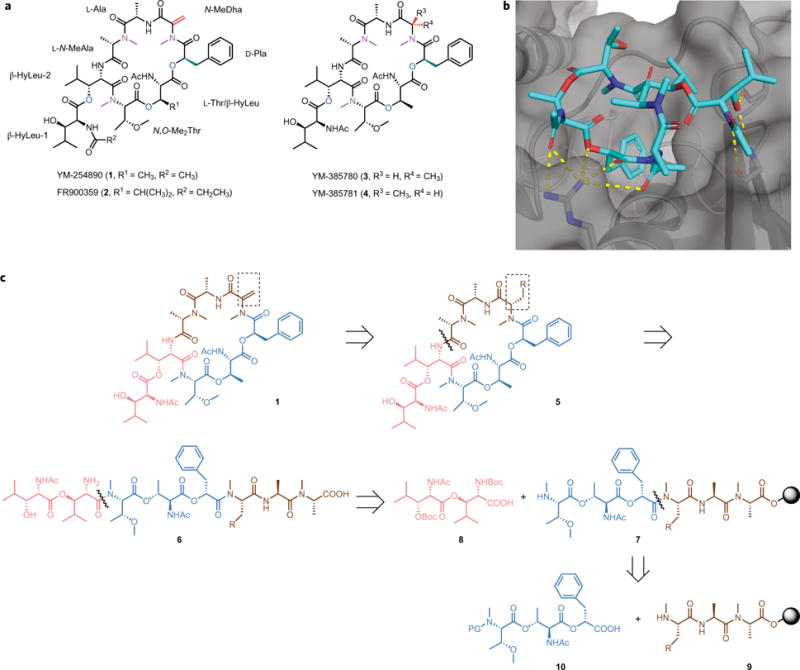
a, Structures of the cyclic depsipeptides YM-254890 (1) and FR900359 (2), isolated from bacteria and plant, respectively, and the only compounds known to inhibit, both potently and selectively, Gq protein activation. In addition, structures of two hydrogenated analogues, YM-385780 (3) and YM-385781 (4), which are also inhibitors of the Gq protein, are shown. b, X-ray crystal structure (Protein Data Bank (PDB) 3AH8) of YM-254890 (1, cyan carbons) bound to a chimeric Gq/i protein (grey cartoon and surface) shows the hydrogen bonds between YM-254890 and the protein (yellow dotted lines). c, Retrosynthetic analysis of YM-254890 (1), which highlights key challenges in the synthesis: preparation of three key building blocks (in brown, blue and red), macrocyclization and generation of the N-MeDha moiety. PG, Protecting group; grey circles, solid support.
YM-254890 is a complex cyclic depsipeptide that has three ester bonds, two proteogenic amino acids (threonine (Thr) and alanine (Ala)), five non-proteogenic amino acids (two residues of β-hydroxyleucine (β-HyLeu-1 and β-HyLeu-2 (Fig. 1a)), N,O-dimethylthreonine (N,O-Me2Thr), N-methylalanine (N-MeAla) and N-methyldehydroalanine (N-MeDha), an α-hydroxy acid (D-3-phenyllactic acid (D-Pla)) and two acetyl groups17. The molecular details of how YM-254890 inhibits Gq-mediated signalling were revealed by an X-ray crystallography structure of YM-254890 bound to a chimeric Gq/i protein (Fig. 1b)18. This demonstrated that YM-254890 binds to a hydrophobic cavity and stabilizes an inactive guanosine diphosphate (GDP)-bound form of the Gq protein.
In addition, the natural products YM-254891, YM-254892 and YM-280193 (Supplementary Fig. 1), which are structurally related to YM-254890, have also been isolated from Chromobacterium sp. QS3666 (ref. 19). Moreover, two hydrogenated derivatives, YM-385780 (3 (Fig. 1a)) and YM-385781 (4 (Fig. 1a)), were generated from a reduction of the terminal double bond of the N-MeDha of YM-25489019. Evaluation of these analogues for Gq-inhibitory activity demonstrated that the intact scaffold of YM-254890 is important for potent activity, whereas the terminal double bond is, seemingly, less important (Supplementary Fig. 7)19.
Interestingly, a close structural analogue, FR900359 (2 (Fig. 1a)), isolated from the plant Ardisia crenata by Fujisawa Pharmaceutical20,21, was reported to inhibit the aggregation of human platelets22 and, like YM-254890, to be a specific inhibitor of Gq (refs 23,24). Recently, FR900359 was characterized in a range of biological systems, including a melanoma model system in which it was found to suppress several key malignant features of melanoma cells24. Thus, in addition to their important role as pharmacological tools, FR900359 and YM-254890 provide exciting starting points for new approaches in cancer drug discovery. FR900359 was available under the name UBO-QIC, but is no longer generally available.
These compounds are highly challenging synthetic target molecules and, until now, the total syntheses of neither YM-254890 nor FR900359 has been achieved. Indeed, a worldwide competition provided an award of up to $100,000 for the successful synthesis of 1 mg of YM-254890 (https://www.innocentive.com/ar/challenge/9933017), but no successful synthesis was ever reported. During the course of this work, two groups reported the synthesis of simplified YM-254890 analogues25,26, which, however, did not show a noteworthy inhibitory activity. Taken together, this emphasizes both the pharmacological importance and synthetic challenge of an intact YM-254890 scaffold.
Here we report the first total synthesis of YM-254890 and FR900359 using strategies that employ both solid- and solution-phase syntheses. The versatility of the synthetic procedure was demonstrated by the synthesis of two known analogues, YM-385780 and YM-385781, as well as ten novel analogues, which enabled the first structure–activity relationship (SAR) studies for this class of compounds. All the compounds were examined for the ability to inhibit Gq-mediated signalling, which allowed the first comparative pharmacological characterization of YM-254890 and FR900359, and we then showed that both compounds are highly potent inhibitors of Gq. Moreover, the selectivity of all the compounds was determined by examining their ability to modulate Gs- and Gi-mediated signalling; in addition, the inhibitory effect of YM-254890 on selected oncogenic G11 protein mutants was evaluated.
Results
Retrosynthetic analysis
In the retrosynthetic analysis of YM-254890 (Fig. 1c), one of several anticipated challenges included the formation of the terminal olefin in N-MeDha, which was envisioned to be generated by α,β-elimination via an O-protected serine precursor 5 (Fig. 1c). Another challenge was the macrolactamization, which typically is performed under highly dilute conditions to avoid dimerization and oligomerization. Moreover, N-methylated amino acids are generally challenging to acylate, and thus the disconnection for the cyclization is of utmost importance. We chose to disconnect the cyclic compound 5 (Fig. 1c) between N-MeAla and β-HyLeu to give the linear precursor 6, primarily to avoid an N-methylated terminal amino group in the cyclization. Owing to the challenges of generating ester bonds, combined with their general lack of stability compared with amide bonds, we decided to incorporate the three ester bonds into two building blocks, 8 and 10, which could be synthesized from commercially available amino acids. We could use solid-phase peptide synthesis (SPPS) to prepare resin-bound 9 and assemble it with 10 to deliver 7, which then could be coupled with 8 to afford the linear peptide 6 as a precursor for cyclization to provide the target compound. As FR900359 is structurally similar to YM-254890, we envisioned that simple modifications of the building blocks 8 and 10 could provide FR900359 using the same synthetic strategy and, ideally, that the same strategy could be employed to generate analogues of these compounds.
Towards the total synthesis of YM-254890
The synthesis began with the preparation of building blocks 8 and 10 (Supplementary Fig. 2). Starting from either the commercially available or synthesized fragments D-Pla, Thr/β-HyLeu, N,O-Me2Thr27 or β-HyLeu, and using standard coupling conditions and fine-tuning of protecting groups28,29, we obtained ester building blocks 10a–10c and 8a and 8b (Supplementary Fig. 2). With these building blocks in hand, we initiated the construction of the linear depsipeptide on a solid support, on which a 2-chlorotrityl chloride resin was used because of the relatively mild cleavage conditions required to release the depsipeptide that contained three ester bonds from the resin. Resin-bound peptide 22 was generated using standard SPPS conditions (Supplementary Fig. 3), but the subsequent deprotection of the N-Fmoc (Fmoc, 9-fluorenylmethoxycarbonyl) group provided a mixture of the elimination product 23 as the major product and the desired 7a in trace amounts only. All the subsequent attempts to optimize this step, which included various bases30, failed (Supplementary Fig. 3). We therefore employed the N-Alloc (Alloc, allyloxycarbonyl) group instead, which can be removed smoothly under mild and neutral conditions using palladium, and synthesized building block 10b (Supplementary Fig. 2). Resin-bound peptide 9a was then coupled with 10b (Supplementary Fig. 4) and the N-Alloc group was efficiently removed by treatment with Pd(PPh3)4 and phenylsilane to provide the desired elongated depsipeptide 7a (Supplementary Fig. 4)31. Next, we thought building block 8a should be coupled to the N-methyl resin-bound 7a; however, all the initial attempts under various coupling conditions32 only gave poor yields (<20%). The problem was addressed by increasing the temperature to 35 °C together with increasing the concentration of the reactants, as well as performing the coupling twice, which led to an 80% conversion. Increasing the temperature further to 50 °C led to cleavage of the depsipeptide from the resin.
With the full sequence assembled on the solid support, we subsequently cleaved the linear depsipeptide from the resin with the concomitant removal of O- and N-Boc groups, which led to 6a (Supplementary Fig. 4). The subsequent macrolactamization33 was achieved by treatment with HATU (1-(bis(dimethylamino) methylene)-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate) and 2,4,6-trimethylpyridine (collidine). The crude cyclized depsipeptide was then treated with hydrogen and Pd/C to remove the benzyl group from the N-MeSer residue and lead to the cyclic YM-254890 precursor 5a (Supplementary Fig. 4), which only needed elimination of the primary alcohol in N-MeSer to generate YM-254890. However, the initial attempts to generate an O-acetyl group did not succeed, as no reaction was observed and 5a was intact. Only when reacting with methanesulfonyl chloride (MsCl) was the formation of YM-254890 observed by liquid chromatography–mass spectrometry, but subsequent attempts to optimize the conversion all failed (Supplementary Fig. 4), as either the conversion was very low or a chloride adduct to the double bond, which is an excellent Michael acceptor, started to appear (Supplementary Fig. 5). Additional attempts with stronger bases led to degradation, and using combinations of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/CuCl34 or disuccinimido carbonate/triethylamine35 (Supplementary Fig. 4) did not provide the target compound either.
Final total synthesis of YM-254890, FR900359 and the analogues
As using N-MeSer as a precursor to generate N-MeDha in the cyclic product 5a failed, it seemed attractive to generate the double bond in a linear depsipeptide before cyclization to avoid issues that result from steric hindrance in the cyclic depsipeptide. It is known that Dha can be accessed from the oxidative elimination of S-aryl- or S-alkyl-protected cysteine derivatives36 or using selenocysteine37. We chose N-MeCys(StBu)-OH (25) as an N-MeDha precursor, as it can be readily introduced into a depsipeptide, and the desired double bond can be generated after deprotection of the thiol under mild conditions using dithiothreitol (DTT) as the reducing reagent and the Cys side chain can undergo elimination, either under oxidative elimination or bisalkylating conditions on the solid support.
We prepared and successfully incorporated N-MeCys(StBu)-OH (25) (ref. 38) into the depsipeptide sequence under standard SPPS procedures to generate resin-bound 9b (Fig. 2a), followed by successive coupling with building blocks 10b and 8a, as described above, to provide the linear depsipeptide 26a. The disulfide bond in 26a was reduced efficiently by treatment with DTT and DIEA (N,N-diisopropylethylamine) to afford the free thiol. Next, the desired double bond should be generated, and O-mesitylenesulfonylhydroxylamine39, 1,4-dibromobutane37 or 2,5-dibromoadipate40 all provided the desired elimination product 27a (Fig. 2a), and treatment with 1,4-dibromobutane led to the highest conversion (about 85%). The resulting depsipeptide was then cleaved from the resin and Boc protecting groups were concomitantly removed to generate a linear precursor of YM-254890, compound 28a. Finally, cyclization of this precursor at dilute concentrations gave the cyclic depsipeptide YM-254890 (Fig. 2a) and characterization by 1H and 13C NMR, high-resolution mass spectroscopy (HRMS) and optical rotation showed this to be identical to the isolated YM-254890 (see the Supplementary Information)17. In addition, synthetic YM-254890 exists as a mixture of two conformers in a ratio of 10:6, which is the same ratio as found for isolated YM-25489017. This could be attributed to the geometry around the amide bond between D-Pla and N-MeDha. Thus, the accomplishment of the total synthesis of YM-254890 confirms the original structural assignment of YM-254890.
Figure 2. Chemical synthesis of YM-254890, FR900359 and analogues.
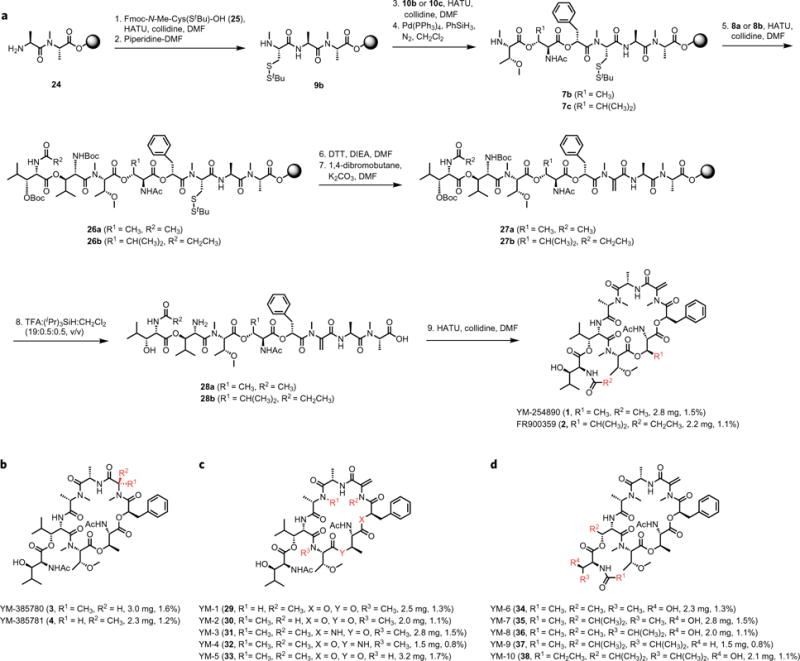
a, The total synthesis of the natural products YM-254890 (1) and FR900359 (2). b, Structures of two reduced analogues, YM-385780 (3) and YM-385781 (4), in which N-Me-Dha is replaced with either N-MeAla or N-Me-D-Ala. c, Structures of backbone-modified YM-254890 analogues (compounds 29–33) in which three N-methylated amide bonds and two ester bonds, respectively, are changed into amide bonds. d, Structures of YM-254890 analogues with modifications in the β-HyLeu moieties (compounds 34–38). DMF, N,N-dimethylformamide; TFA, trifluoroacetic acid.
The related natural product FR900359 differs from YM-254890 only in two positions (Fig. 1a) and required the synthesis of two new building blocks, 8b and 10c, using synthetic transformations similar to those of 8a and 10b (Supplementary Fig. 2). First 27b and then 28b were prepared following the same strategy as for the synthesis of YM-254890 to finalize the total synthesis of FR900359 (Fig. 2a), as confirmed by 1H and 13C NMR and HRMS. We also wanted to synthesize the two reduced analogues YM-385780 (3) and YM-385781 (4), in which N-MeDha is replaced with either N-MeAla or N-Me-D-Ala (Fig. 1a), which could be generated in slightly fewer synthetic steps compared with YM-254890 and, according to the literature, provide compound 4 equipotent to YM-25489019. Synthetically, this was achieved from 29a and 29b by introducing either N-MeAla or N-Me-D-Ala, instead of N-MeCys(StBu), and coupling with building blocks 10b and 8a to provide 31a and 31b, which were cyclized to give the two target compounds (Fig. 1a). The structural identity was confirmed by 1H and 13C NMR, optical rotation and HRMS, and the ratio of conformers corresponded to previous reports19.
Having established a versatile synthetic strategy for this class of compounds, we wanted to exploit it to perform SAR studies. We therefore designed and synthesized two sets of analogues. First, we wanted to explore the possibility of replacing either N-Me amides or ester bonds in the YM-254890 with amides, compounds YM-1 to YM-5 (29–33 (Fig. 2c)), to examine how this would affect the pharmacological activity, and subsequently as a potential means to generate simplified YM-254890 analogues. Next, we also wanted to explore SARs related to the β-HyLeu moiety in YM-254890, which is known to be crucial for pharmacological activity. Thus we designed and synthesized five analogues with a systematic variation in this moiety, compounds YM-6 to YM-10 (34–38 (Fig. 2d)).
Characterization of inhibitory activity
The inhibitory effects of YM-254890 (1), FR900359 (2), isolated FR900359, YM-385780 (3) and YM-385781 (4) on Gq-mediated signalling were evaluated in Chinese hamster ovary (CHO) cells that stably express the M1 muscarinic receptor. The M1 receptor is activated by carbachol, which leads to the Gq-mediated generation of inositol monophosphate (IP1), and Gq inhibition is measured as a decrease in the IP1 production. We first examined the synthesized YM-254890 and observed the full inhibition of Gq activity with an IC50 (half-maximum inhibitory concentration) value of 0.095 μM (Fig. 3a and Table 1), which correlates well with previously determined inhibitory activity of YM-254890 in related assays19. Next, we examined our synthesized FR900359, as well as the isolated natural product FR900359 (purchased from the Kostenis group, University of Bonn), and observed that both compounds inhibited IP1 production with IC50 values of 0.033 and 0.032 μM, respectively (Fig. 3a and Table 1). The analogues YM-385780 and YM-385781 inhibited IP1 production with IC50 values of 15.3 and 1.54 μM, respectively (Fig. 3a and Table 1).
Figure 3. Pharmacological properties of the Gq inhibitors.
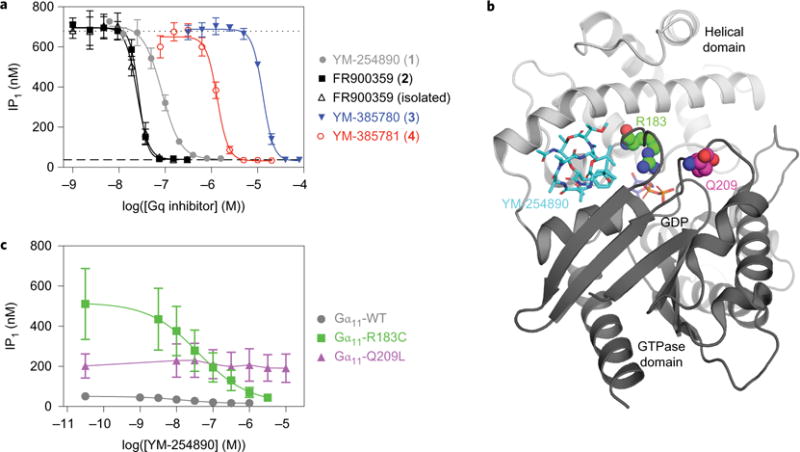
a, Inhibition of carbachol-induced IP1 production in CHO cells that stably express the M1 muscarinic receptor. The IP1 production induced by buffer and by 3 μM carbachol are represented by dashed and dotted lines, respectively. Data represent one of four independent experiments performed in triplicate. Error bars represent the s.d. of each data point. b, X-ray crystal structure of chimeric Gq/i in complex with YM-254890 (PDB 3AH8) that highlights the two Gα domains (light and dark grey cartoon), the two sites of oncogenic mutations (R183 (green carbons) and Q209 (magenta carbons)), GDP (purple carbons) and YM-254890 (cyan carbons) in a space-filled representation. c, Effects of YM-254890 on basal IP1 levels in HEK293 cells transfected with Gα11-WT, Gα11-R183C or Gα11-Q209L; pIC50 values ± s.e.m. derived for Gα11-WT and Gα11-R183C were 8.2 ± 0.5 and 7.4 ± 0.1, respectively. Data are means from three experiments, each performed in duplicate. Error bars represent the s.e.m. of each data point.
Table 1.
Inhibition of Gq-mediated signalling.
| Compound | IC50 (μM) | pIC50 ± s.e.m. |
|---|---|---|
| YM-254890 (1) | 0.095 | 7.03 ± 0.02 |
| FR900359 (2, synthetic) | 0.033 | 7.49 ± 0.04 |
| FR900359 (2, isolated) | 0.032 | 7.50 ± 0.04 |
| YM-385780 (3) | 15.3 | 4.82 ± 0.03 |
| YM-385781 (4) | 1.54 | 5.82 ± 0.04 |
The inhibition of carbachol-induced IP1 production in CHO cells that stably express the M1 muscarinic receptor. IC50 values represent the mean of four independent experiments performed in triplicate.
We subsequently characterized the ten new analogues pharmacologically (Table 2). In the series of five compounds in which backbone modifications of N-Me amides and ester bonds were replaced with amides, compounds 29–33 (Fig. 2c), we observed very distinct effects, with the most pronounced being replacement of the N-Me amide next to the dehydroalanine moiety, as in 30, which resulted in a 670-fold loss of potency (Table 2). In contrast, replacing the N-Me amide between the two Ala moieties (as in 29) only led to a ninefold decrease in potency. Replacing the two ester bonds with amides resulted in a 107- and 323-fold loss of potency for 31 and 32, respectively (Table 2). In the other series of five compounds, the β-HyLeu moieties were modulated, compounds 34–38 (Fig. 2d), and the largest effect was observed when both isopropyl groups in the β-HyLeu moieties were replaced with methyl groups (as in 34), which led to a 340-fold loss in potency (Table 2). Replacing each of these groups individually has much less effect, with a ten- and 24-fold loss of potency for 35 and 36, respectively. Interestingly, removal of the hydroxyl group, which displays two hydrogen bonds to the G protein in the YM-254890/Gq/i co-crystal structure (Fig. 1b)18, as in 37, led to a 103-fold decrease in potency (Table 2).
Table 2.
Gq inhibition by YM-254890 analogues.
| Compound | IC50 (μM) | pIC50 ± s.e.m. |
|---|---|---|
| YM-1 (29) | 0.87 | 6.08 ± 0.05 |
| YM-2 (30) | 63.7 | 4.20 ± 0.06 |
| YM-3 (31) | 10.2 | 4.99 ± 0.02 |
| YM-4 (32) | 30.7 | 4.51 ± 0.01 |
| YM-5 (33) | 47.0 | 4.33 ± 0.03 |
| YM-6 (34) | 32.3 | 4.49 ± 0.01 |
| YM-7 (35) | 0.99 | 6.03 ± 0.03 |
| YM-8 (36) | 2.31 | 5.65 ± 0.03 |
| YM-9 (37) | 9.80 | 5.01 ± 0.02 |
| YM-10 (38) | 0.18 | 6.77 ± 0.15 |
The inhibition of carbachol-induced IP1 production in CHO cells that stably express the M1 muscarinic receptor. IC50 values represent the mean of at least three independent experiments performed in triplicate.
As YM-254890 and FR900359 are characterized by being selective inhibitors of Gq proteins, we examined these compounds, as well as the 12 analogues, for their ability to inhibit Gs- and Gi-mediated signalling (Supplementary Table 2). This was accomplished by examining the inhibition of isoproterenol-induced cyclic adenosine monophosphate (cAMP) production in human embryonic kidney (HEK) 293 cells that endogenously express the β2-adrenergic receptor (Gs signalling) and the inhibition of glutamic acid-induced cAMP reduction in CHO cells that stably express the rat metabotropic glutamate receptor 2 (mGlu2 receptor, Gi signalling). We observed that, in all cases, the compounds were selective Gq inhibitors, as most compounds did not display inhibitory activity of Gi and Gs signalling in the concentrations tested (Supplementary Table 2).
Finally, we wanted to assess the capacity of YM-254890 to inhibit activating mutations of the Gq-related G11 protein that have been associated with cancers. Specifically, we examined the effect of YM-254890 on the two most-prevalent oncogenic Gα11 mutants, Q209L and R183C (Fig. 3b), which are found in more than 2% of all human cancers41. The capacity of YM-254890 to inhibit the constitutive signalling activity of these two mutants was assessed in HEK293 cells. The basal IP1 levels were considerably higher in cells that express these mutants, as compared with cells that express Gα11-WT (WT, wild type), particularly for the Gα11-Q209L mutant, for which a tenfold lower amount of DNA was thus used for transfection (Fig. 3c). YM-254890 inhibited the accumulation of IP1 levels in cells that express Gα11-R183C, but not in cells that express Gα11-Q209L. The compound thus exhibited selectivity between these two mutant G proteins.
Discussion
We have described a versatile synthesis of the only known potent and selective Gq protein inhibitors, YM-254890 and FR900359, which are complex cyclic depsipeptide natural products, and thereby provide the first total synthesis of the complete bioactive structures of YM-254890 and FR900359. A combination of solution-phase synthesis for depsipeptide building blocks and solid-phase approaches was used to generate both YM-254890 and FR900359, which provided a confirmation of the original structural assignment of the corresponding natural products. Here we generated sufficient material for pharmacological characterization and, gratifyingly, a comparison of the inhibitory activities of synthetic FR900359 with the isolated natural product showed identical IC50 values (Fig. 3a and Table 1), which confirms that synthetic FR900359 is pharmacologically equivalent to the natural product. In previous studies using the natural products, it was shown that YM-254890 and FR900359 inhibit Gq-mediated signalling with IC50 values in the range 0.03–0.20 μM (refs 12,18,21,42), which correlates very well with our results (Table 1). Here we also performed the first head-to-head comparison of the two compounds and revealed that FR900359 is threefold more potent than YM-254890.
The preparation of derivatives YM-385780 and YM-385781 was motivated by previous studies showing that YM-385781 was almost equipotent to YM-25489019 and, consequently, may provide an attractive alternative starting point for Gq inhibitor synthesis. However, although YM-385781 was synthesized in slightly fewer steps, we demonstrated that in our hands it is ~16-fold less potent than YM-254890. The N-MeDha functionality prompts a different binding conformation relative to that of the hydrogenated depsipeptides (Supplementary Fig. 7). This conformational change can influence the potency in two ways, either by affecting the cell permeability or the G protein binding directly.
We took advantage of the versatility of the synthetic methodology and designed and synthesized ten new analogues of YM-254890, which constitute two small, systematic SAR studies, and examined either the backbone modifications (29–33 (Fig. 2c)) or changes in the key β-HyLeu moieties (34–38 (Fig. 2d)). This outlined key moieties in retaining potent Gq activity and demonstrated that, in general, even very small changes in the structure can have substantial pharmacological effects, but also that certain modifications do not compromise activity too much. All the compounds were also evaluated for their effect on Gs- and Gi-mediated signalling, and they showed only weak or no activity, in line with the previously established Gq selectivity of YM-254890 and FR900359.
Finally, YM-254890 was examined for its effect on the two most-prevalent oncogenic Gα11 mutations, R183C and Q209L. YM-254890 fully inhibited one of these mutants, Gα11-R183C, but was ineffective on Gα11-Q209L. This selectivity profile, which has been shown previously for these mutations in Gαq (refs 12,24), could be related to the distinct locations of the two mutations relative to YM-254890 (Fig. 3b) and/or to differences in the mechanisms by which they modulate G protein function12.
Collectively, these efforts not only provide access to selective Gq inhibitors, YM-254890 and FR900359, but also allow ample opportunities to perform further SAR studies, which could optimize the pharmacological and physicochemical properties of these compounds. Moreover, YM-254890 and FR900359 may be used as synthetic templates for the structure-based design of highly warranted inhibitors for other G protein classes, as well as being explored as templates for drug design directed towards the treatment of various forms of cancer.
Methods
Chemical synthesis
Detailed procedures for the synthesis of all the compounds and their characterization are provided in the Supplementary Information. The cell lines CHO-k1, native HEK293 and GS-22A tested negative for mycoplasma infection in our lab, and the mGluR2-CHO cell lines were verified pharmacologically in our lab.
Cell culturing
CHO-k1 cells that stably express the muscarinic M1 receptor were purchased from the cDNA Resource Center (www.cdna.org, catalogue No. CEM100TN00). The CHO cell line, which stably expresses the rat mGlu2 receptor, was a generous gift from S. Nakanishi (Kyoto University). CHO-M1 cells were maintained in Ham’s F12 media (Life Technologies) supplemented with 10% (v/v) fetal bovine serum (FBS) (Life Technologies), 1% (v/v) penicillin-streptomycin (10.000 units ml–1 (Life Technologies)) and 0.25 mg ml–1 G418 (Life Technologies). HEK293 cells that endogenously express the β2 adrenergic receptor and CHO-mGlu2 receptor cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies) supplemented with 10% (v/v) FBS or dialysed FBS (Life Technologies), respectively, and 1% (v/v) penicillin-streptomycin (10.000 units ml–1 (Life Technologies)). CHO-mGlu2 media was also supplemented with 1% (v/v) L-proline. GS-22A cells, derived from HEK293 purchased from ACTT, were cultured in DMEM supplemented with 10% (v/v) FBS. All the cells were kept at 37 °C in a humidified atmosphere (95% air and 5% CO2).
Functional assays
The IP1 HTRF assay (Cisbio) was used to quantify Gq/11 signalling activity, and the cAMP dynamic 2 assay (Cisbio) was used to quantify the Gs and Gi signalling. G protein inhibitors were dissolved in 20 mM DMSO stock solutions, diluted in assay buffer (Hanks’ balanced salt solution buffer, 20 mM HEPES pH 7.4, 1 mM CaCl2, 1 mM MgCl2) and added to a 384-well Optiplate (PerkinElmer) in triplicate unless stated otherwise. Subconfluent cells were detached from the culture dish with non-enzymatic cell-dissociation solution (Sigma).
IP1 assay of the M1 receptor
CHO-M1 cells were resuspended in assay buffer supplemented with 0.2% bovine serum albumin (BSA) to achieve a cell density of 2 million cells ml–1. G protein inhibitors and cells (10.000 cells per well) were incubated at 37 °C for one hour. After the incubation, carbamoylcholine chloride (carbachol (Sigma)) was dissolved in assay buffer supplemented with 200 mM LiCl (final concentration of LiCl in the assay was 20 mM). Carbachol was added to each well at a final concentration of 3 μM (~EC80 (drug concentration that induces 80% of the maximal response)) and the plate was incubated at 37 °C for one hour followed by 15 minutes of incubation at room temperature. The detection solution was prepared as follows: IP1 conjugate and lysis buffer (Cisbio) + 2.5% anti-IP1 cryptate Tb conjugate (Cisbio) + 2.5% D-myo-IP1-d2 conjugate (Cisbio).
cAMP assay
HEK293 cells or CHO-mGlu2 cells were resuspended in assay buffer supplemented with 0.2% BSA and 100 μM 3-isobutyl-1-methylxanthine (Sigma) to achieve a cell density of 1 million cells ml–1. G protein inhibitors and cells (5.000 cells per well) were incubated at 37 °C for one hour. To stimulate Gs signalling, isoproterenol bitartrate salt (Sigma) was dissolved in the assay buffer and added to each well containing HEK293 cells at a final concentration of 17 nM (~EC80). For Gi activity, L-glutamate (Sigma) was dissolved in assay buffer supplemented with forskolin (Sigma) (the final concentration of forskolin in the assay was 15 μM) and added to each well containing CHO-mGlu2 cells at a final concentration of 16 μM (~EC80). The plate was incubated at room temperature for 30 minutes on a plate shaker. The detection solution was prepared as follows: cAMP conjugate and lysis buffer (Cisbio) + 2.5% anti-cAMP cryptate conjugate (Cisbio) + 2.5% cAMP-d2 conjugate (Cisbio). For both assays, the detection solution was added to the plate (10 μl per well) and the plate was incubated in the dark for one hour at room temperature. The plate was read on an EnVision Multilabel Reader (PerkinElmer Life and Analytical Sciences), with excitation at 340 nm and measurements of emission at 615 and 665 nm. The fluorescence resonance energy transfer ratios (665/615 nm) were converted into IP1 or cAMP concentrations, respectively, by interpolating values from an IP1 or cAMP standard curve.
Measuring the effects of Gα11 mutants
HEK293 cells were transfected transiently in 96-well plates with pcDNA3.1-derived plasmids that encoded human Gα11-WT (100 ng per well), Gα11-R183C (100 ng per well) or Gα11-Q209L (10 ng per well) using Fugene HD. Two days later, the cells were treated with different concentrations of YM-254890 for 30 minutes in the presence of LiCl and then analysed for IP1 content (IP-One HTRF assay kit (Cisbio)), as described above.
Supplementary Material
Acknowledgments
We thank C. A. Olsen for constructive comments on the manuscript, and D. Stærk for performing the HRMS. H.B.-O. and K.S. acknowledge financial support from the Lundbeck Foundation. X.-F.X. thanks the Novo Nordisk Foundation, H.Z. thanks the Chinese Scholarship Council, C.R.U. acknowledges financial support from the Lundbeck Foundation, D.E.G. thanks the European Research Commission (DE-ORPHAN 639125) and the Lundbeck Foundation (R163-2013-16327), M.M. was supported by the National Institutes of Health (NIH) grant R01-DK100584, T.J.G. by NIH P01-DK11794, Project I, and M.M. and T.J.G. by the Center for Skeletal Research Core (NIH P30 AR066261).
Footnotes
Author contributions
H.B.-O. and K.S. conceived the project. X.-F.X., H.Z. and K.S. designed the synthesis, and X.-F.X. and H.Z. performed the synthesis. C.R.U. and M.F.W. performed pharmacological characterization. T.J.R. and M.M. performed the characterization of mutant G proteins. K.H. performed molecular modelling. K.S., H.B.-O., D.E.G. and M.M. supervised the research. X.-F.X. and K.S. wrote the manuscript, with input from all the authors.
Additional information
Supplementary information and chemical compound linking are available in the online version of the paper.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304–1350. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 2.Kumari P, Ghosh E, Shukla AK. Emerging approaches to GPCR ligand screening for drug discovery. Trends Mol Med. 2015;21:687–701. doi: 10.1016/j.molmed.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- 4.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nature Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 5.Wisler JW, Xiao KH, Thomsen ARB, Lefkowitz RJ. Recent developments in biased agonism. Curr Opin Cell Biol. 2014;27:18–24. doi: 10.1016/j.ceb.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shukla AK. Biasing GPCR signaling from inside. Sci Signal. 2014;7:e3. doi: 10.1126/scisignal.2005021. [DOI] [PubMed] [Google Scholar]
- 7.Katada T, Ui M. ADP ribosylation of the specific membrane-protein of C6 cells by islet-activating protein associated with modification of adenylate-cyclase activity. J Biol Chem. 1982;257:7210–7216. [PubMed] [Google Scholar]
- 8.West RE, Moss J, Vaughan M, Liu T, Liu TY. Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor site. J Biol Chem. 1985;260:4428–4430. [PubMed] [Google Scholar]
- 9.Gill DM, Meren R. ADP-ribosylation of membrane proteins catalyzed by cholera toxin: basis of activation of adenylate cyclase. Proc Natl Acad Sci USA. 1978;75:3050–3054. doi: 10.1073/pnas.75.7.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taniguchi M, et al. YM-254890, a novel platelet aggregation inhibitor produced by Chromobacterium sp QS3666. J Antibiot. 2003;56:358–363. doi: 10.7164/antibiotics.56.358. [DOI] [PubMed] [Google Scholar]
- 11.Kawasaki T, et al. Pharmacological properties of YM-254890, a specific Gαq/11 inhibitor, on thrombosis and neointima formation in mice. Thromb Haemost. 2005;94:184–192. doi: 10.1160/TH04-09-0635. [DOI] [PubMed] [Google Scholar]
- 12.Takasaki J, et al. A novel Gαq/11-selective inhibitor. J Biol Chem. 2004;279:47438–47445. doi: 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- 13.Uemura T, et al. Biological properties of a specific Gαq/11 inhibitor, YM-254890, on platelet functions and thrombus formation under high-shear stress. Br J Pharmacol. 2006;148:61–69. doi: 10.1038/sj.bjp.0706711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uemura T, et al. Effect of YM-254890, a specific Gαq/11 inhibitor, on experimental peripheral arterial disease in rats. Eur J Pharmacol. 2006;536:154–161. doi: 10.1016/j.ejphar.2006.02.048. [DOI] [PubMed] [Google Scholar]
- 15.Schröder R, et al. Deconvolution of complex G protein-coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nature Biotechnol. 2010;28:943–950. doi: 10.1038/nbt.1671. [DOI] [PubMed] [Google Scholar]
- 16.Kamato D, et al. Structure, function, pharmacology, and therapeutic potential of the G protein, Gα/q,11. Front Cardiovasc Med. 2015;2:14. doi: 10.3389/fcvm.2015.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taniguchi M, et al. Structure of YM-254890, a novel Gq/11 inhibitor from Chromobacterium sp QS3666. Tetrahedron. 2003;59:4533–4538. [Google Scholar]
- 18.Nishimura A, et al. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc Natl Acad Sci USA. 2010;107:13666–13671. doi: 10.1073/pnas.1003553107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taniguchi M, et al. YM-254890 analogues, novel cyclic depsipeptides with Gαq/11 inhibitory activity from Chromobacterium sp QS3666. Bioorg Med Chem. 2004;12:3125–3133. doi: 10.1016/j.bmc.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 20.Fujioka M, Koda S, Morimoto Y, Biemann K. Structure of FR900359, a cyclic depsipeptide from Ardisia crenata sims. J Org Chem. 1988;53:2820–2825. [Google Scholar]
- 21.Miyamae A, Fujioka M, Koda S, Morimoto Y. Structural studies of FR900359, a novel cyclic depsipeptide from Ardisia crenata sims (Myrsinaceae) J Chem Soc Perkin Trans. 1989;1:873–878. [Google Scholar]
- 22.Zaima K, et al. Vasorelaxant effect of FR900359 from Ardisia crenata on rat aortic artery. J Nat Med. 2013;67:196–201. doi: 10.1007/s11418-012-0644-0. [DOI] [PubMed] [Google Scholar]
- 23.Inamdar V, Patel A, Manne BK, Dangelmaier C, Kunapuli SP. Characterization of UBO-QIC as a Gαq inhibitor in platelets. Platelets. 2015:771–778. doi: 10.3109/09537104.2014.998993. [DOI] [PubMed] [Google Scholar]
- 24.Schrage R, et al. The experimental power of FR900359 to study Gq-regulated biological processes. Nature Commun. 2015;6:10156. doi: 10.1038/ncomms10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rensing DT, Uppal S, Blumer KJ, Moeller KD. Toward the selective inhibition of G proteins: total synthesis of a simplified YM-254890 analog. Org Lett. 2015;17:2270–2273. doi: 10.1021/acs.orglett.5b00944. [DOI] [PubMed] [Google Scholar]
- 26.Kaur H, Harris PW, Little PJ, Brimble MA. Total synthesis of the cyclic depsipeptide YM-280193, a platelet aggregation inhibitor. Org Lett. 2015;17:492–495. doi: 10.1021/ol503507g. [DOI] [PubMed] [Google Scholar]
- 27.Bühlmayer P, et al. Synthesis and biological activity of some transition-state inhibitors of human renin. J Med Chem. 1988;31:1839–1846. doi: 10.1021/jm00117a027. [DOI] [PubMed] [Google Scholar]
- 28.Isidro-Llobet A, Alvarez M, Albericio F. Amino acid-protecting groups. Chem Rev. 2009;109:2455–2504. doi: 10.1021/cr800323s. [DOI] [PubMed] [Google Scholar]
- 29.Hanessian S, Vakiti RR, Dorich S, Banerjee S, Deschenes-Simard B. Total synthesis of pactamycin and pactamycate: a detailed account. J Org Chem. 2012;77:9458–9472. doi: 10.1021/jo301638z. [DOI] [PubMed] [Google Scholar]
- 30.Coin I, Beerbaum M, Schmieder P, Bienert M, Beyermann M. Solid-phase synthesis of a cyclodepsipeptide: cotransin. Org Lett. 2008;10:3857–3860. doi: 10.1021/ol800855p. [DOI] [PubMed] [Google Scholar]
- 31.Pelay-Gimeno M, et al. The first total synthesis of the cyclodepsipeptide pipecolidepsin A. Nature Commun. 2013;4:2352. doi: 10.1038/ncomms3352. [DOI] [PubMed] [Google Scholar]
- 32.El-Faham A, Albericio F. Peptide coupling reagents, more than a letter soup. Chem Rev. 2011;111:6557–6602. doi: 10.1021/cr100048w. [DOI] [PubMed] [Google Scholar]
- 33.White CJ, Yudin AK. Contemporary strategies for peptide macrocyclization. Nature Chem. 2011;3:509–524. doi: 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]
- 34.Jimenez JC, et al. Synthesis of peptides containing α,β-didehydroamino acids. Scope and limitations. Lett Pept Sci. 2002;9:135–141. [Google Scholar]
- 35.Ogura H, Sato O, Takeda K. β-Elimination of β-hydroxyamino acids with disuccinimido carbonate. Tetrahedron Lett. 1981;22:4817–4818. [Google Scholar]
- 36.Chalker JM, et al. Methods for converting cysteine to dehydroalanine on peptides and proteins. Chem Sci. 2011;2:1666–1676. [Google Scholar]
- 37.Levengood MR, van der Donk WA. Dehydroalanine-containing peptides: preparation from phenylselenocysteine and utility in convergent ligation strategies. Nature Protocols. 2006;1:3001–3010. doi: 10.1038/nprot.2006.470. [DOI] [PubMed] [Google Scholar]
- 38.Ruggles EL, Flemer S, Jr, Hondal RJ. A viable synthesis of N-methyl cysteine. Biopolymers. 2008;90:61–68. doi: 10.1002/bip.20889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bernardes GJL, Chalker JM, Errey JC, Davis BG. Facile conversion of cysteine and alkyl cysteines to dehydroalanine on protein surfaces: versatile and switchable access to functionalized proteins. J Am Chem Soc. 2008;130:5052–5053. doi: 10.1021/ja800800p. [DOI] [PubMed] [Google Scholar]
- 40.Chalker JM, Lercher L, Rose NR, Schofield CJ, Davis BG. Conversion of cysteine into dehydroalanine enables access to synthetic histones bearing diverse post-translational modifications. Angew Chem Int Ed. 2012;51:1835–1839. doi: 10.1002/anie.201106432. [DOI] [PubMed] [Google Scholar]
- 41.O’Hayre M, et al. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nature Rev Cancer. 2013;13:412–424. doi: 10.1038/nrc3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kenakin T. A holistic view of GPCR signaling. Nature Biotechnol. 2010;28:928–929. doi: 10.1038/nbt0910-928. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.