

Conspectus
The carbonyl group holds a prominent position in chemistry and biology not only because it allows diverse transformations but also because it supports key intermolecular interactions, including hydrogen bonding. More recently, carbonyl groups have been found to interact with a variety of nucleophiles, including other carbonyl groups, in what we have termed an n→π* interaction. In an n→π* interaction, a nucleophile donates lone-pair (n) electron density into the empty π* orbital of a nearby carbonyl group. Mixing of these orbitals releases energy, resulting in an attractive interaction. Hints of such interactions were evident in small-molecule crystal structures as early as the 1970s, but not until 2001 was the role of such interactions articulated clearly.
These non-covalent interactions were first discovered during investigations into the thermostability of the proline-rich protein collagen, which achieves a robust structure despite a relatively low potential for hydrogen bonding. It was found that by modulating the distance between two carbonyl groups in the peptide backbone, one could alter the conformational preferences of a peptide bond to proline. Specifically, only the trans conformation of a peptide bond to proline allows for an attractive interaction with an adjacent carbonyl group, so when one increases the proximity of the two carbonyl groups, one enhances their interaction and promotes the trans conformation of the peptide bond, which increases the thermostability of collagen.
More recently, attention has been paid to the nature of these interactions. Some have argued that rather than resulting from electron donation, carbonyl interactions are a particular example of dipolar interactions that are well-approximated by classical mechanics. However, experimental evidence has demonstrated otherwise. Numerous examples now exist where an increase in the dipole moment of a carbonyl group decreases the strength of its interactions with other carbonyl groups, demonstrating unequivocally that a dipolar mechanism is insufficient to describe these interactions. Rather, these interactions have important quantum-mechanical character that can be evaluated through careful experimental analysis and judicious use of computation.
Although individual n→π* interactions are relatively weak (∼0.3–0.7 kcal/mol), the ubiquity of carbonyl groups across chemistry and biology gives the n→π* interaction broad impact. In particular, the n→π* interaction is likely to play an important role in dictating protein structure. Indeed, bioinformatics analysis suggests that approximately one-third of residues in folded proteins satisfy the geometric requirements to engage in an n→π* interaction, which is likely to be of particular importance for the α-helix. Other carbonyl-dense polymeric materials like polyesters and peptoids are also influenced by n→π* interactions, as are a variety of small molecules, some with particular medicinal importance. Research will continue to identify molecules whose conformation and activity are affected by the n→π* interaction and will clarify their specific contributions to the structures of biomacromolecules.
1. Introduction
The carbonyl group has received great attention, due in part to its varied reactivity and ubiquity across chemistry and biology. In addition to the enormous catalogue of chemical transformations supported by carbonyl groups, their intermolecular interactions play a paramount role in the organization of biological systems. For example, carbonyl groups participate in the hydrogen bonding that governs the structure of proteins1,2 and nucleic acids.3 More recently, chemists have recognized that carbonyl groups can form attractive interactions with one another. These thoughts had their origin in the seminal analyses of small-molecule crystal structures by Bürgi and Dunitz (Figure 1).4−6 Later, Allen identified intimate contacts between carbonyl groups explicitly.7 Only recently, however, were perturbations of a carbonyl–carbonyl interaction shown to affect the stability of a protein.8 Now substantial effort is being devoted to understanding the nature of these interactions and revealing their contributions to diverse chemical and biological phenomena.9,10
Figure 1.

Recent photographs of Bürgi and Dunitz, whose work in the 1970s laid the foundation for current investigations of n→π* interactions.
2. Nature of Carbonyl Interactions
The charge distribution of the carbonyl group creates the potential for a variety of attractive interactions to exist between two carbonyl groups.11 In particular, one can envisage (1) a Coulombic interaction between point charges on the carbon of one moiety and the oxygen of another, (2) a dipolar interaction between the permanent electric dipoles of the two groups, or (3) a donor–acceptor interaction in which electron density from electron-rich orbitals of one carbonyl group is donated into electron-deficient orbitals of another. Purely Coulombic and dipolar contributions to carbonyl interactions are likely to be well-approximated by the molecular force fields used for interrogating biological phenomena. In contrast, contributions from electron donation are not represented even in sophisticated force fields, which could lead to divergence from the behavior of real biomolecules.
The interactions of carbonyl groups have been explored using a proline model system (Figure 2). Not only does the pyrrolidine ring preorganize the i – 1 and i carbonyl groups for interaction, but these molecules also provide a convenient readout for perturbations. Specifically, peptide bonds to proline populate both the cis and trans conformations, but because an attractive carbonyl interaction exists only in the trans conformation, changes in the conformer populations can report on changes in the strength of a carbonyl interaction. For example, perturbations that enhance the strength of carbonyl interactions cause an increase in the population of the trans conformer.
Figure 2.
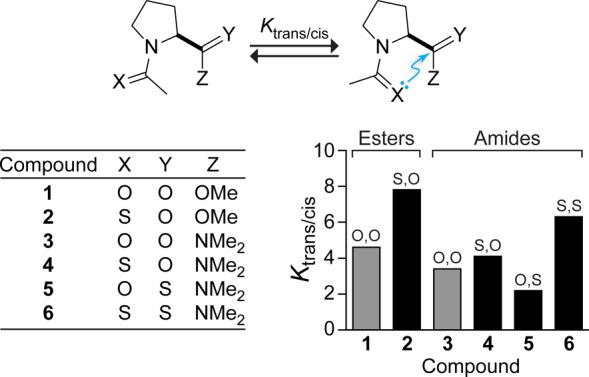
Characterization of carbonyl interactions by torsion balance analysis. Experimental Ktrans/cis values of proline derivatives were measured by NMR spectroscopy in D2O at 25 °C. Adapted from ref (13). Copyright 2013 American Chemical Society.
To probe the nature of carbonyl interactions, thioamides were incorporated strategically into either ester 1 or amide 3.12,13 Thioamides bear less partial negative charge on sulfur than do oxoamides on oxygen, so replacing the N-acetylproline peptide bond with a thioamide should attenuate the Coulombic interactions.14 Nevertheless, an increase in the population of the trans conformation was observed upon thioamide substitution of the i – 1 carbonyl group, indicating a stronger carbonyl interaction in 2 relative to 1 and in 4 relative to 3 (Figure 2). Carbonyl interactions are therefore not well described by simple electrostatics. Amides have higher dipole moments than do esters.14 Accordingly, if a dipolar interaction were dominant, amide 3 would have a stronger carbonyl interaction than ester 1. However, the opposite was observed experimentally. Moreover, the thioamide has a still larger dipole moment than do oxoamides,14 and yet 5 exhibited a weaker carbonyl interaction than did 3. These data demonstrate that carbonyl interactions cannot be described as being purely dipolar.
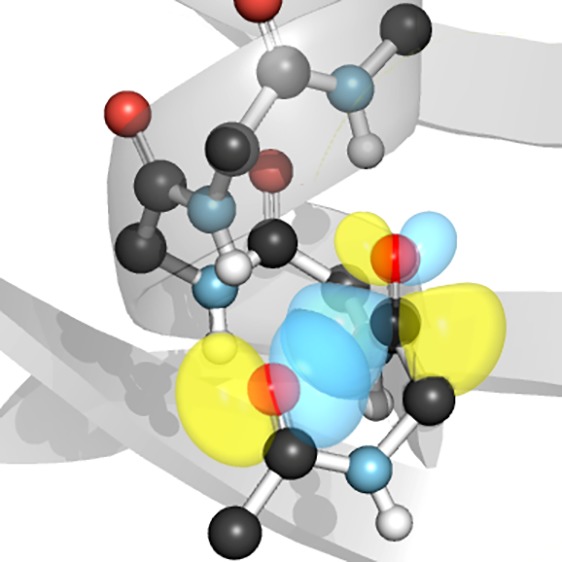
The data on compounds 1–6 are consistent with electron donation. For example, 3 shows a weaker carbonyl interaction than 1 because amides are less electrophilic than esters. Moreover, the divergent consequences of thioamide substitutions (compare 4 and 5 relative to 3) demonstrate that each carbonyl group has a unique role in these interactions, namely, one as a donor and the other as an acceptor. These interactions are reminiscent of the approach of a nucleophile to a carbonyl group during an acyl transfer reaction.4−6 The angle of this approach, which maximizes overlap of the lone pair of the nucleophile with the π* orbital of the acceptor carbonyl group (Figure 3), is known as the Bürgi–Dunitz trajectory.4−6
Figure 3.
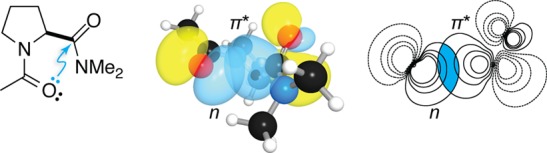
Overlap of the n and π* orbitals (blue) of N-acetylproline dimethyl amide. The overlap integral is 0.1212. Adapted from ref (13). Copyright 2013 American Chemical Society.
Several additional lines of evidence support the quantum-mechanical nature of carbonyl interactions. In analyses of both small-molecule15 and protein16 crystal structures, the angle between the two carbonyl dipoles of interacting pairs varies but the approach of the nucleophile occurs strictly along the Bürgi–Dunitz trajectory, highlighting the importance of the orbitals of these groups in dictating the interaction. Moreover, the n→π* interaction involves population of the π* antibonding orbital and should thereby weaken the carbonyl π bond, which has multiple consequences. First, a red shift in the acceptor carbonyl stretching frequency, corresponding to a weaker carbonyl bond, has been noted repeatedly.8,17−20 Second, weakening of the π bond should reduce the planarity of the carbonyl group, thus engendering pyramidalization (Figure 4) that can be observed in high-resolution crystal structures. Indeed, such signatures of the n→π* interaction have been reported in a wide variety of systems,12,13,15,21−25 including polymers26 and proteins,27 giving strong credence to the notion of carbonyl interactions as fundamentally electronic in nature. This notion has also been supported by changes in electronic spectra observed upon modulation of the n→π* interaction with thioamides.28
Figure 4.
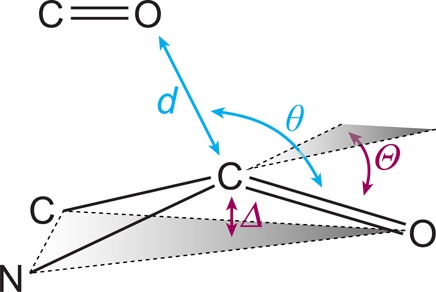
Geometric parameters characterizing an n→π* interaction in a peptide or protein and the ensuing pyramidalization. The O···C distance is d ≤ 3.22 Å, which is the sum of the van der Waals radii of oxygen and carbon; the O···C=O angle is θ = 109° ± 10°, which is near the Bürgi–Dunitz trajectory. The distance Δ and angle Θ report on the extent of pyramidalization.12
The n→π* interaction relies on orbital mixing. Accordingly, two fundamental quantities govern the energy of an n→π* interaction: (1) the degree of orbital overlap and (2) the energy difference between the donor and acceptor orbitals. Greater orbital overlap, generally corresponding to shorter donor–acceptor distances, increases the strength of an n→π* interaction.12 However, as the donor–acceptor distance decreases, the filled donor orbital will also experience Pauli repulsion from filled orbitals of the acceptor. Hence, the acceptor group must be highly polarized, which allows preferential interaction of the electron-pair donor with the unfilled antibonding orbital of the acceptor over the filled bonding orbital (Figure 5). Indeed, carbonyl groups are effective n→π* acceptors, but isosteric alkenes and fluoroalkenes lack sufficient orbital polarization and thus do not engage in substantial n→π* interactions.29−33 As to the effect of the donor–acceptor energy gap, it is known from second-order perturbation theory that the energy released upon the mixing of a filled orbital with an empty one is inversely proportional to the energy gap between the donor and acceptor orbitals. Pairs of thioamides (e.g., in 6) form especially strong n→π* interactions because the donor lone-pair orbital is higher in energy than that of the corresponding amide, whereas the acceptor antibonding orbital is lower in energy, thereby creating a smaller energy gap.13 Alkenes, on the other hand, have π* orbitals of particularly high energy, again making them poor n→π* acceptors.33
Figure 5.
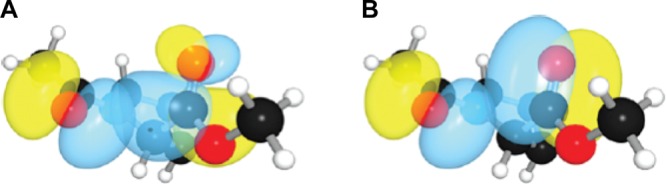
Overlap of the n donor orbital with (A) the π* orbital and (B) the π orbital of N-acetylproline methyl ester. Adapted from ref (33). Copyright 2010 American Chemical Society.
n→π* interactions have a quantum-mechanical nature. Accordingly, computational methods, especially natural bond orbital (NBO) analysis,34 have provided much insight. NBO protocols partition electron density from diffuse molecular orbitals into localized Lewis-type orbitals, from which the energy of mixing can be computed. Extensive calculations have revealed the energies of the n→π* interaction for a wide variety of amide–amide geometries16 and have placed a lower bound on the energy of a typical n→π* interaction between amides at approximately 0.27 kcal/mol,13 though numerous examples of stronger interactions exist (vide infra). These calculations and others35,36 highlight the modest energy of common n→π* interactions. Nevertheless, because of the ubiquity of carbonyl groups, these interactions can make substantial contributions in toto.
3. Contributions to Protein Structure
Carbonyl groups constitute half of the non-hydrogen atoms in the backbone of peptides and proteins. This prevalence suggests that the impact of the n→π* interaction could be substantial. Early on, molecular modeling suggested that many conformations of the peptide backbone allow for the close contact of adjacent carbonyl groups,37 which was confirmed subsequently through detailed computational and bioinformatics analysis.9 In a later study,16 the energy of the n→π* interaction was calculated for the entire conformational space of the peptide backbone, which showed clear areas of Ramachandran space with significant (>0.5 kcal/mol) n→π* interactions. In addition, a survey of high-resolution crystal structures from the Protein Data Bank found that a large fraction (∼34%) of residues were oriented properly for an n→π* interaction (Oi–1···Ci′ distance within the sum of the van der Waals radii and Oi–1···Ci′=Oi angle approximately along the Bürgi–Dunitz trajectory). Importantly, those residues found to be oriented for an n→π* interaction had backbone dihedral angles consistent with those predicted computationally to engage in n→π* interactions of significant energy.16 A later analysis of protein crystal structures determined at subangstrom resolution found that carbonyl groups that accept n→π* interactions exhibit greater pyramidalization than do other carbonyl groups, demonstrating that these interactions have measurable consequences for protein structure.27 Even considering only the approximate lower bound to the energy of a typical n→π* interaction (0.27 kcal/mol),13 the fact that a third of residues in folded proteins engage in an n→π* interaction means that their contributions could be nearly 10 kcal/mol for a 100-residue protein. To put this contribution in context, the conformational stability of typical globular proteins is ΔG ≈ 5–15 kcal/mol relative to their unfolded states.38
3.1. Collagen
The pyrrolidine ring of proline preorganizes adjacent carbonyl groups in a peptide for the formation of n→π* interactions. Accordingly, these interactions play a large role in the conformation of proline-rich peptides and proteins. Indeed, the discovery that n→π* interactions contribute to protein structure was made with collagen-mimetic peptides.8 Collagen is the predominant protein in animals and consists of three intertwined polyproline II-type (PPII) helices. Its unique structure is enabled by a distinctive amino acid sequence: an Xaa-Yaa-Gly repeat, in which Xaa is often (2S)-proline (Pro) and Yaa is often (2S,4R)-4-hydroxyproline (Hyp).39 The presence of a hydroxy group at a 4R-configured stereogenic center of the Yaa residue is important for the thermostability of collagen because that electron-withdrawing substituent elicits a gauche effect that enforces the exo pucker of the pyrrolidine ring (Figure 6A).8,40,41 An electron-withdrawing substituent at a 4S-configured center of the Yaa residue, which enforces the endo pucker, decreases the thermostability. The different pyrrolidine conformations modulate the attraction between adjacent carbonyl groups and thus the trans/cis ratio of prolyl peptide bonds (Figure 6B). Specifically, prolines with 4R-configured electron-withdrawing substituents, either hydroxy or fluoro, have a higher preference for the trans conformation of the prolyl peptide bond, indicating a stronger carbonyl interaction. Conversely, proline residues with 4S-configured electron-withdrawing substituents have a weaker preference. Analogous results were obtained for prolines with 4-azido substituents.17 Crystallographic and computational analyses,12 microwave spectroscopy,42 and experiments with methanoprolines43,44 have since established that in the absence of complicating hydrogen bonds, the endo pucker of the pyrrolidine ring generally increases the distance between the donor oxygen and the acceptor carbon, leading to weaker n→π* interactions. These results explain the destabilization of collagen-mimetic peptides by proline residues with electron-withdrawing substituents at 4S-configured centers in the Yaa position. In a collagen triple helix, all of the peptide bonds are in the trans conformation. Hence, the strength of the n→π* interaction that enforces the trans conformation correlates with the thermostability of collagen. This insight has been applied in the design of collagens with a wide variety of physical and chemical properties,45−49 especially by Wennemers and co-workers.50−53
Figure 6.
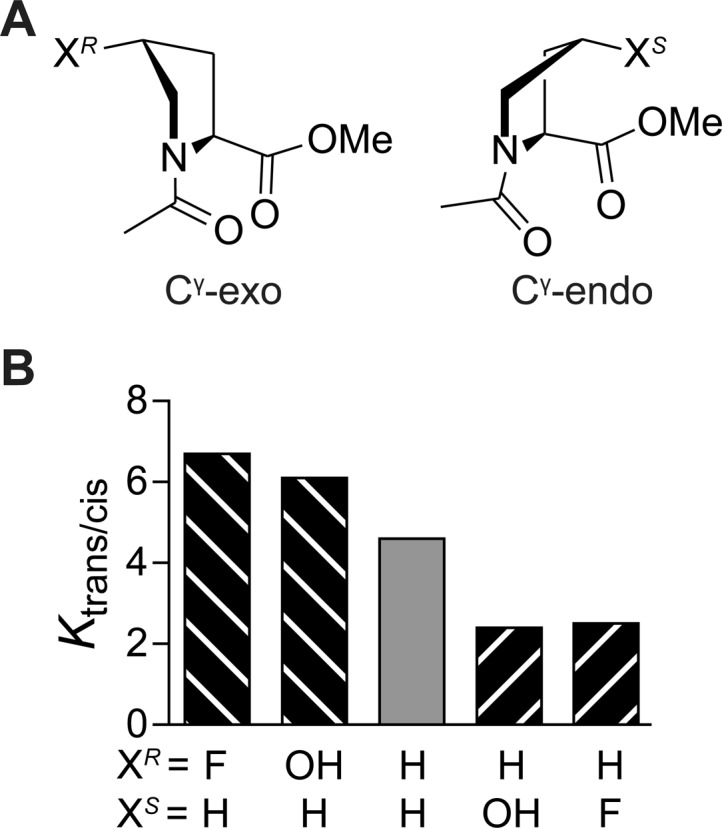
(A) Cγ-exo and Cγ-endo puckers of the pyrrolidine ring. (B) Experimental Ktrans/cis values of proline derivatives measured with NMR spectroscopy in D2O at 25 °C.8
3.2. Polyproline II-Type Helices
The importance of the n→π* interaction for controlling the conformational preferences of peptide bonds to proline is magnified in polyproline peptides, which, because of the lack of backbone hydrogen bonding, rely heavily on the n→π* interaction. Polyproline adopts two distinct helical conformations: the polyproline I-type (PPI) helix with exclusively cis peptide bonds and the aforementioned PPII helix with exclusively trans peptide bonds. Polymers of proline with 4R-configured hydroxy, fluoro, or azido substituents have a stronger preference for the PPII conformation than do polymers of unsubstituted proline.54,55 Conversely, polymers of the diastereomeric (i.e., 4S-configured) prolines show a weaker preference for the PPII conformation than does polyproline. Moreover, the presence of a strong n→π* interaction, enforced by 4R-configured electron-withdrawing substituents, also increases the barrier to interconversion of PPI and PPII helices.56 The n→π* interaction has been implicated further in the PPII structures of other sequences,57−59 demonstrating its ability to control peptide conformation. Recently, Wennemers and co-workers determined the first high-resolution crystal structure of an oligoproline.60 In the crystalline state, the oligoproline adopted a PPII helix with unequivocal hallmarks of n→π* interactions: short donor–acceptor distances and significant pyramidalization of carbonyl groups. Moreover, the lack of water in the crystal precludes the importance of hydration for stabilizing a PPII helix, underscoring the stability conferred by n→π* interactions. Notably, the PPII conformation has been observed in the unfolded states of some peptides and proteins.61 Thus, the n→π* interaction, which operates between adjacent residues (i → i + 1), could direct the peptide chain toward folding into the PPII conformation prior to the formation of a native hydrogen-bonding pattern that enlists residues more distant in sequence.
3.3. α-Helices
Most of the residues engaged in n→π* interactions in proteins are located within α-helices. Initial analysis of protein crystal structures demonstrated that over 70% of residues in α-helices are aligned to participate in an n→π* interaction.16 Strong evidence for the influence of n→π* interactions has come from analyses of high-resolution crystal structures, which showed pyramidalization of residues in α/β peptides that adopt helical conformations similar to that of an α-helix.23 Although α-amino acids can position adjacent amide carbonyl groups within close proximity, β-amino acids (which contain an extra methylene group in their backbone) cannot. In these α/β helices, only the carbonyl groups from α-amino acids exhibited pyramidalization toward their putative n→π* donors. These results provide compelling evidence that n→π* interactions are not only extant in α-helices but also alter their structure. Likewise, n→π* interactions appear to contribute to the stability of 310 helices,16 as α-aminoisobutyric acid residues, which strongly enforce the 310 conformation, also induce strong n→π* interactions.62
n→π* interactions enable carbonyl groups in the backbone of helices to utilize both of their lone pairs simultaneously.63 This concurrence creates interplay between these two interactions. The geometry of a hydrogen bond to an n→π* donor affects the ensuing n→π* interaction by controlling the demixing of the carbonyl lone pairs. When hydrogen-bond donors approach along the axis of the carbonyl bond, they encourage demixing of the carbonyl lone pairs into s- and p-type orbitals (Figure 7A,B).64 The s-like orbital engages in an i → i + 4 hydrogen bond (Figure 7C), while the p-like orbital engages in an n→π* interaction (Figure 7D). When hydrogen-bond donors approach at ∼120° with respect to the carbonyl-bond axis, they encourage mixing of the s- and p-like orbitals, which then adopt the “rabbit ears” geometry that is all too familiar65 to chemists.
Figure 7.
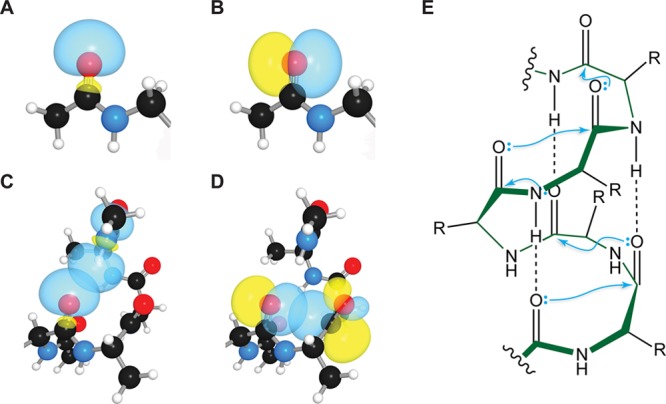
(A) s-Type lone pair of a carbonyl oxygen. (B) p-Type lone pair of a carbonyl oxygen. (C) Hydrogen bond to the s-type carbonyl lone pair in an α-helix. (D) n→π* interaction with the p-type carbonyl lone pair in an α-helix. Panels A–D were adapted with permission from ref (16). Copyright 2010 Nature Publishing Group. (E) Structure of an α-helix showing i → i + 4 hydrogen bonds1 and i → i + 1 n→π* interactions, which have d ≈ 3.0 Å and θ ≈ 103°.16
In an α-helix, a hydrogen bond and an n→π* interaction avail electron density from a single oxygen atom. Consistent with expectations, an n→π* interaction has been shown to antagonize hydrogen bonding in a peptidic system.66 Thus, the canonical hydrogen bonds in an α-helix1 are undermined by concurrent n→π* interactions. Moreover, like the hydrogen bonds,67 the n→π* interactions in an α-helix could be cooperative.37 An n→π* interaction increases both the length of the C=O bond in the carbonyl acceptor and the charge on its oxygen,40 effects that make this C=O bond a better donor for another n→π* interaction.
3.4. Side Chains
Like those in the backbone, carbonyl groups in amino acid side chains are capable of forming n→π* interactions. For example, aspartate residues interact with their own backbone carbonyl groups.68 These interactions were first identified by their relatively short oxygen–oxygen distances, which seemed counterintuitive. Upon examining the geometries of these interactions, a side-chain oxygen was often found to approach the backbone carbonyl group along the Bürgi–Dunitz trajectory. Moreover, the arrangement of the carbonyl dipoles in these cases is likely to be destabilizing, suggesting that the n→π* interaction stabilizes what would otherwise be an unfavorable self-contact.
Similar contacts are made between the carbonyl groups of asparagine residues.64 Moreover, hydrogen bonds to a carbonyl oxygen affect its ability to serve as an n→π* donor. Self-contacting n→π* interactions are much more common when a side-chain carbonyl group accepts a hydrogen bond along the carbonyl-bond axis. When the hydrogen-bond donor approaches at an angle of ∼120° with respect to the carbonyl-bond axis, the prevalence and calculated energy of self-contacting n→π* interactions diminish, consistent with experimental evidence that these two interactions compete with one another.66 These data provide independent support for the demixing of the carbonyl lone pairs into s- and p-type orbitals (Figure 7A,B) upon hydrogen-bond formation along the carbonyl-bond axis.
4. Contributions to Other Polymers
4.1. Poly(lactic acid)
Although their roles in protein structure have received the most attention, n→π* interactions contribute to the conformations of other polymers as well. Consider poly(lactic acid) (PLA), a biodegradable polyester (Figure 8A).26 Fiber diffraction has shown that the backbone dihedral angles in PLA resemble those of the PPII helix of peptides, which takes advantage of numerous n→π* interactions (vide supra). Computation placed the average energy of an n→π* interaction in PLA at 0.44 kcal/mol, and analysis of small-molecule crystal structures demonstrated characteristic pyramidalization of the acceptor carbonyl group that results from accepting an n→π* interaction. Like polyproline, PLA has no potential for hydrogen bonding, so the observation of n→π* interactions in this polymer demonstrates not only that the n→π* interaction can operate independently of hydrogen bonding but also that it is sufficient to dictate molecular conformation even in the absence of a preorganizing ring, implicating n→π* interactions further in organizing early protein-folding intermediates.
Figure 8.
n→π* interactions in polymers: (A) poly(lactic acid);26 (B) a peptoid.70
4.2. Peptoids
n→π* interactions are especially important for controlling the conformation of polymers of N-substituted glycine residues, which are also known as “peptoids” (Figure 8B).69 Analogously to polyproline and PLA, a peptoid lacks hydrogen bonds within its backbone, so its conformation must be controlled by other forces such as the n→π* interaction. In addition to forming typical n→π* interactions, peptoids can also form n→π* interactions with aryl rings on side chains. Whereas backbone amide–amide n→π* interactions favor the trans conformation of the tertiary amide, the amide–aryl n→π* interaction between the backbone and side chain favors the cis conformation.70,71 These two interactions can therefore be exploited to tune peptoid structure. For example, decoration of side-chain phenyl rings with electron-withdrawing fluoro or nitro groups increases the electrophilicity of the aromatic π* orbitals and thereby encourages the cis conformation of the tertiary amide; adding electron-donating hydroxy groups reverses this preference.72 Similarly, Huisgen azide–alkyne 1,3-dipolar cycloaddition can be used to construct an electron-deficient triazolium ring, which is a potent n→π* acceptor that enforces a cis conformation upon the tertiary amide of a peptoid.73 This tack is complementary to tuning the ability of a carbonyl group to be an n→π* donor, which can be either enhanced with a thioamide (vide supra)12−14,28,74−76 or selenoamide77 or attenuated with electron-withdrawing groups.74,78
5. Contributions to Small Molecules
5.1. Amino Acids
In principle, many compounds (Figure 9) are likely to engage in n→π* interactions, especially since the lone-pair donor need not be a carbonyl group. For example, n→π* interactions between the hydroxy group of (2S,4S)-4-hydroxyproline and its carbonyl group in the gas phase have been observed using microwave spectroscopy.42 A comparable interaction has been observed in crystal structures of a wide variety of substituted proline residues, including an especially strong one in an S-oxide for N-acetyl-4-thiaproline methyl ester (7).22,79 Indeed, many functional groups can interact with carbonyl groups in an n→π* manner, including halide ions,15 thiols/thioethers/disulfides,80 and a variety of nitrogen heterocycles.81,82 Similar types of electronic interactions involving carbonyl groups have been studied computationally and include complexes of SO2 with carbon dioxide or formaldehyde.83
Figure 9.
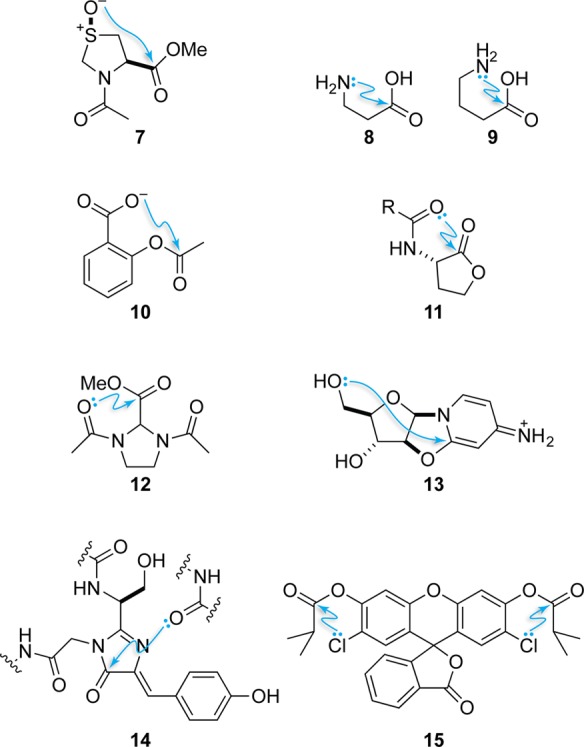
A selection of molecules that manifest n→π* interactions.
n→π* interactions have also been detected in amino acids other than proline. In particular, gas-phase microwave spectroscopy identified conformations consistent with the presence of an n→π* interaction between the amino nitrogen and carboxylic acid of β-alanine (8).500 The population of this conformer was similar to those for hydrogen-bonded conformations, suggesting that the energy of the n→π* interaction might be similar to that of a typical hydrogen bond. Similar results were obtained in an analysis of γ-aminobutyric acid (GABA, 9), an important neurotransmitter.84 Here the n→π* interaction was observed in the conformation predicted as the global minimum.
5.2. Drugs
Other medicinal implications of n→π* interactions have become apparent. For example, an n→π* interaction in aspirin (10) was revealed through both crystallographic24 and spectroscopic85 investigations. Donation of electron density from the anionic carboxylate into the ester carbonyl group is predicted to shield and disperse negative charge on the molecule and thereby to improve its entry into cells. An n→π* interaction was also observed in N-acyl homoserine lactones (AHLs, 11), which mediate quorum sensing in Gram-negative bacteria.25,86 Crystallography and computation established the presence of an n→π* interaction in the free AHL, whereas examination of protein crystal structures with bound AHLs demonstrated that AHL receptors break that n→π* interaction upon binding. Attenuation of the n→π* interaction, which can be accomplished by appending electron-withdrawing substituents to the acyl group,74,78 could preorganize the ligand for receptor binding and thereby increase potency.
5.3. Synthetic Intermediates
n→π* interactions also affect carbonyl reactivity. In an imidazolidine-based model system (12), a carbonyl group was able to be an acceptor of one or two n→π* interactions with identical donor carbonyl groups.87 As only one n→π* interaction was observed, n→π* donation apparently reduces the electrophilicity of the acceptor carbonyl group. The consequences for the reactivity of carbonyl groups that accept n→π* interactions are profound. For example, Houk and co-workers have identified carbonyl interactions as determinants of stereoselectivity, such as in the dihydroxylation of cis-bicyclo[3.0.0]octenes88 and the kinetic resolution of azlactones by benzotetramisole catalysis.89 An analogous stereoelectronic interaction was also proposed to explain the observed preference of an anhydroarabinonucleoside (13) for phosphorylation at the 3′ oxygen rather than the less sterically encumbered 5′ oxygen.21 This reaction is a key step in a proposed prebiotic route toward nucleotide synthesis, and regioselectivity in phosphorylation is essential for generating cyclic phosphates of cytidine for polymerization. A similar interaction was observed in cycloadditions of 3-hydroxyflavones.90 The n→π* interaction induced by proline was also shown to be the cause of the sluggish native chemical ligation with proline thioesters.91 The pyrrolidine ring preorganizes the prolyl peptide bond to form an n→π* interaction with the thioester, decreasing its electrophilicity.87
5.4. Fluorophores
Finally, n→π* interactions can modulate fluorescence. In natural fluorescent proteins, such as green fluorescent protein (GFP), an n→π* interaction forms between a backbone oxygen and the imidazolidine chromophore (14).92 The presence of this n→π* interaction is consistent with the red shift in the vibrational frequency of the imidazolidine carbonyl group in the protein-bound chromophore relative to small-molecule mimics in solution. Moreover, analyses of protein crystal structures with premature chromophores suggest that this n→π* interaction preorganizes the chromophore for cyclization and precludes bond rotations that would lower the quantum yield. In small-molecule fluorogenic probes, such as 2′,7′-dichlorofluorescein diisobutyrate (15), the reactivity can be tuned by n→π* interactions.93 The two Cl···C=O n→π* interactions deter solvent water from gaining access to the π* orbitals of the proximal carbonyl groups (cf. 12), though an esterase can still do so.
6. Conclusions and Outlook
The n→π* interaction is an emergent interaction that contributes to biomolecular structure and function. Its discovery has not only refined our understanding but also inspired new thoughts. For example, characterization of the n→π* interaction expedited the identification of an unappreciated type of hydrogen bond within the backbones of peptides and proteins.94 These so-called “C5” hydrogen bonds confer stability to β-strands just as n→π* interactions stabilize α-helices—by allowing the protein backbone to exploit both carbonyl lone pairs. Like n→π* interactions, C5 hydrogen bonds are distributed broadly and contribute to protein structure.
We anticipate that n→π* interactions will be found in an ever-expanding array of molecules, particularly those with a high density of carbonyl groups, such as proteins. Revealing their impact will provoke clever experiments, and interpretations will be guided by computational methodology. In view of the current limitations in protein structure prediction and design, a thorough understanding of these ubiquitous interactions is likely to enhance countless efforts with peptides and proteins.
Acknowledgments
We are grateful to Dr. M. R. Aronoff for his photograph of Professor Dunitz and to Dr. C. L. Jenkins for comments on the manuscript. Work in our laboratory on the n→π* interaction has been supported by Grants R01 AR044276 (NIH) and CHE-1124944 (NSF). R.W.N. was supported by Biotechnology Training Grant T32 GM008349 (NIH) and an ACS Division of Organic Chemistry Graduate Fellowship.
Biographies
Robert W. Newberry received his B.S. degree in biochemistry from The University of Texas at Austin in 2011 and his Ph.D. degree in chemistry from the University of Wisconsin–Madison in 2016. He is now a postdoctoral researcher at the University of California, San Francisco.
Ronald T. Raines is the Henry Lardy Professor of Biochemistry, Linus Pauling Professor of Chemical Biology, and a Professor of Chemistry at the University of Wisconsin–Madison. His research is focused on the chemistry and biology of proteins. In summer 2017 he will become the Firmenich Professor of Chemistry at his alma mater, the Massachusetts Institute of Technology.
The authors declare no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Chemical Biology of Peptides”.
References
- Pauling L.; Corey R. B.; Branson H. R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. U. S. A. 1951, 37, 205–211. 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L.; Corey R. B. Configurations of polypeptide chains with favored orientations around single bonds: Two new pleated sheets. Proc. Natl. Acad. Sci. U. S. A. 1951, 37, 729–740. 10.1073/pnas.37.11.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J. D.; Crick F. H. C. Molecular structure of nucleic acids: A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- Bürgi H. B.; Dunitz J. D.; Shefter E. Geometric reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 1973, 95, 5065–5067. 10.1021/ja00796a058. [DOI] [Google Scholar]
- Bürgi H. B.; Dunitz J. D.; Shefter E. Chemical reaction paths. IV. Aspects of O···C=O interactions in crystals. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1974, 30, 1517–1527. 10.1107/S0567740874005188. [DOI] [Google Scholar]
- Bürgi H. B.; Dunitz J. D.; Lehn J. M.; Wipff G. Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 1974, 30, 1563–1572. 10.1016/S0040-4020(01)90678-7. [DOI] [Google Scholar]
- Allen F. H.; Baalham C. A.; Lommerse J. P. M.; Raithby P. R. Carbonyl–carbonyl interactions can be competitive with hydrogen bonds. Acta Crystallogr., Sect. B: Struct. Sci. 1998, 54, 320–329. 10.1107/S0108768198001463. [DOI] [Google Scholar]
- Bretscher L. E.; Jenkins C. L.; Taylor K. M.; DeRider M. L.; Raines R. T. Conformational stability of collagen relies on a stereoelectronic effect. J. Am. Chem. Soc. 2001, 123, 777–778. 10.1021/ja005542v. [DOI] [PubMed] [Google Scholar]
- Fufezan C. The role of Buergi–Dunitz interactions in the structural stability of proteins. Proteins: Struct., Funct., Genet. 2010, 78, 2831–2838. 10.1002/prot.22800. [DOI] [PubMed] [Google Scholar]
- Singh S. K.; Das A. The n→π* interaction: A rapidly emerging non-covalent interaction. Phys. Chem. Chem. Phys. 2015, 17, 9596–9612. 10.1039/C4CP05536E. [DOI] [PubMed] [Google Scholar]
- Persch E.; Dumele O.; Diederich F. Molecular recognition in chemical and biological systems. Angew. Chem., Int. Ed. 2015, 54, 3290–3327. 10.1002/anie.201408487. [DOI] [PubMed] [Google Scholar]
- Choudhary A.; Gandla D.; Krow G. R.; Raines R. T. Nature of amide carbonyl–carbonyl interactions in proteins. J. Am. Chem. Soc. 2009, 131, 7244–7246. 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry R. W.; VanVeller B.; Guzei I. A.; Raines R. T. n→π* interactions of amides and thioamides: Implications for protein stability. J. Am. Chem. Soc. 2013, 135, 7843–7846. 10.1021/ja4033583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Raines R. T. An evaluation of peptide-bond isosteres. ChemBioChem 2011, 12, 1801–1807. 10.1002/cbic.201100272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer K. J.; Choudhary A.; Raines R. T. Intimate interactions with carbonyl groups: Dipole–dipole or n→π*?. J. Org. Chem. 2013, 78, 2099–2103. 10.1021/jo302265k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett G. J.; Choudhary A.; Raines R. T.; Woolfson D. N. n→π* Interactions in proteins. Nat. Chem. Biol. 2010, 6, 615–620. 10.1038/nchembio.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag L.-S.; Schweizer S.; Ochsenfeld C.; Wennemers H. The “azido gauche effect”—implications for the conformation of azidoprolines. J. Am. Chem. Soc. 2006, 128, 14697–14703. 10.1021/ja0654938. [DOI] [PubMed] [Google Scholar]
- Kuemin M.; Nagel Y. A.; Schweizer S.; Monnard F. W.; Ochsenfeld C.; Wennemers H. Tuning the cis/trans conformer ratio of Xaa–Pro amide bonds by intramolecular hydrogen bonds: The effect on PPII helix stability. Angew. Chem., Int. Ed. 2010, 49, 6324–6327. 10.1002/anie.201001851. [DOI] [PubMed] [Google Scholar]
- Shoulders M. D.; Kotch F. W.; Choudhary A.; Guzei I. A.; Raines R. T. The aberrance of the 4S diastereomer of 4-hydroxyproline. J. Am. Chem. Soc. 2010, 132, 10857–10865. 10.1021/ja103082y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. K.; Mishra K. K.; Sharma N.; Das A. Direct spectroscopic evidence for an n→π* interaction. Angew. Chem., Int. Ed. 2016, 55, 7801–7805. 10.1002/anie.201511925. [DOI] [PubMed] [Google Scholar]
- Choudhary A.; Kamer K. J.; Powner M. W.; Sutherland J. D.; Raines R. T. A stereoelectronic effect in prebiotic nucleotide synthesis. ACS Chem. Biol. 2010, 5, 655–657. 10.1021/cb100093g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Pua K. H.; Raines R. T. Quantum mechanical origin of the conformational preferences of 4-thiaproline and its S-oxides. Amino Acids 2011, 41, 181–186. 10.1007/s00726-010-0504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Raines R. T. Signature of n→π* interactions in α-helices. Protein Sci. 2011, 20, 1077–1081. 10.1002/pro.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Kamer K. J.; Raines R. T. An n→π* interaction in aspirin: Implications for structure and reactivity. J. Org. Chem. 2011, 76, 7933–7937. 10.1021/jo201389d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry R. W.; Raines R. T. A key n→π* interaction in N-acyl homoserine lactones. ACS Chem. Biol. 2014, 9, 880–883. 10.1021/cb500022u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry R. W.; Raines R. T. n→π* Interactions in poly(lactic acid) suggest a role in protein folding. Chem. Commun. 2013, 49, 7699–7701. 10.1039/c3cc44317e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry R. W.; Bartlett G. J.; VanVeller B.; Woolfson D. N.; Raines R. T. Signatures of n→π* interactions in proteins. Protein Sci. 2014, 23, 284–288. 10.1002/pro.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Ferrie J. J.; Chen X.; Zhang Y.; Szantai-Kis D. M.; Chenoweth D. M.; Petersson E. J. Electronic interactions of i, i + 1 dithioamides: Increased fluorescence quenching and evidence for n→π* interactions. Chem. Commun. 2016, 52, 7798–7801. 10.1039/C6CC00105J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins C. L.; Vasbinder M. M.; Miller S. J.; Raines R. T. Peptide bond isosteres: Ester or (E)-alkene in the backbone of the collagen triple helix. Org. Lett. 2005, 7, 2619–2622. 10.1021/ol050780m. [DOI] [PubMed] [Google Scholar]
- Gao J.; Kelly J. W. Toward quantification of protein backbone–backbone hydrogen bonding energies: An energetic analysis of an amide-to-ester mutation in an α-helix within a protein. Protein Sci. 2008, 17, 1096–1011. 10.1110/ps.083439708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai N.; Wang X. J.; Etzkorn F. A. The effect of a trans-locked Gly–Pro alkene isostere on collagen triple helix stability. J. Am. Chem. Soc. 2008, 130, 5396–5397. 10.1021/ja711021m. [DOI] [PubMed] [Google Scholar]
- Dai N.; Etzkorn F. A. Cis–trans proline isomerization effects on collagen triple-helix stability are limited. J. Am. Chem. Soc. 2009, 131, 13728–13732. 10.1021/ja904177k. [DOI] [PubMed] [Google Scholar]
- Jakobsche C. E.; Choudhary A.; Miller S. J.; Raines R. T. n→π* Interaction and n)(π Pauli repulsion are antagonistic for protein stability. J. Am. Chem. Soc. 2010, 132, 6651–6653. 10.1021/ja100931y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed A. E.; Curtiss L. A.; Weinhold F. Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. 10.1021/cr00088a005. [DOI] [Google Scholar]
- Hodges J. A.; Raines R. T. Energetics of an n→π* interaction that impacts protein structure. Org. Lett. 2006, 8, 4695–4697. 10.1021/ol061569t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari U.; Scheiner S. Preferred configurations of peptide–peptide interactions. J. Phys. Chem. A 2013, 117, 489–496. 10.1021/jp310942u. [DOI] [PubMed] [Google Scholar]
- Hinderaker M. P.; Raines R. T. An electronic effect on protein structure. Protein Sci. 2003, 12, 1188–1194. 10.1110/ps.0241903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace C. N. Conformational stability of globular proteins. Trends Biochem. Sci. 1990, 15, 14–17. 10.1016/0968-0004(90)90124-T. [DOI] [PubMed] [Google Scholar]
- Shoulders M. D.; Raines R. T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. 10.1146/annurev.biochem.77.032207.120833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRider M. L.; Wilkens S. J.; Waddell M. J.; Bretscher L. E.; Weinhold F.; Raines R. T.; Markley J. L. Collagen stability: Insights from NMR spectroscopic and hybrid density functional computational investigations of the effect of electronegative substituents on prolyl ring conformations. J. Am. Chem. Soc. 2002, 124, 2497–2505. 10.1021/ja0166904. [DOI] [PubMed] [Google Scholar]
- Newberry R. W.; Raines R. T. 4-Fluoroprolines: Conformational analysis and effects on the stability and folding of peptides and proteins. Top. Heterocycl. Chem. 2017, 48, 1–25. 10.1007/7081_2015_196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesarri A.; Cocinero E. J.; López J. C.; Alonso J. L. Shape of 4(S)- and 4(R)-hydroxyproline in the gas phase. J. Am. Chem. Soc. 2005, 127, 2572–2579. 10.1021/ja045955m. [DOI] [PubMed] [Google Scholar]
- Jenkins C. L.; Lin G.; Duo J.; Rapolu D.; Guzei I. A.; Raines R. T.; Krow G. R. Substituted 2-azabicyclo[2.1.1]hexanes as constrained proline analogues: Implications for collagen stability. J. Org. Chem. 2004, 69, 8565–8573. 10.1021/jo049242y. [DOI] [PubMed] [Google Scholar]
- Krow G. R.; Edupuganti R.; Gandla D.; Yu F.; Sender M.; Sonnet P. E.; Zdilla M. J.; DeBrosse C.; Cannon K. C.; Ross C. W.; Choudhary A.; Shoulders M. D.; Raines R. T. Synthesis of conformationally constrained 5-fluoro- and 5-hydroxymethanopyrrolidines. Ring-puckered mimics of gauche- and anti-3-fluoro- and 3-hydroxypyrrolidines. J. Org. Chem. 2011, 76, 3626–3634. 10.1021/jo200117p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges J. A.; Raines R. T. Stereoelectronic effects on collagen stability: The dichotomy of 4-fluoroproline diastereomers. J. Am. Chem. Soc. 2003, 125, 9262–9263. 10.1021/ja035881z. [DOI] [PubMed] [Google Scholar]
- Shoulders M. D.; Hodges J. A.; Raines R. T. Reciprocity of steric and stereoelectronic effects in the collagen triple helix. J. Am. Chem. Soc. 2006, 128, 8112–8113. 10.1021/ja061793d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotch F. W.; Guzei I. A.; Raines R. T. Stabilization of the collagen triple helix by O-methylation of hydroxyproline residues. J. Am. Chem. Soc. 2008, 130, 2952–2953. 10.1021/ja800225k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders M. D.; Satyshur K. A.; Forest K. T.; Raines R. T. Stereoelectronic and steric effects in side chains preorganize a protein main chain. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 559–564. 10.1073/pnas.0909592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Kamer K. J.; Shoulders M. D.; Raines R. T. 4-Ketoproline: An electrophilic proline analog for bioconjugation. Biopolymers 2015, 104, 110–115. 10.1002/bip.22620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann R. S.; Wennemers H. Functionalizable collagen model peptides. J. Am. Chem. Soc. 2010, 132, 13957–13959. 10.1021/ja103392t. [DOI] [PubMed] [Google Scholar]
- Erdmann R. S.; Wennemers H. Importance of ring puckering versus interstrand hydrogen bonds for the conformational stability of collagen. Angew. Chem., Int. Ed. 2011, 50, 6835–6838. 10.1002/anie.201008118. [DOI] [PubMed] [Google Scholar]
- Erdmann R. S.; Wennemers H. Effect of sterically demanding substituents on the conformational stability of the collagen triple helix. J. Am. Chem. Soc. 2012, 134, 17117–17124. 10.1021/ja3066418. [DOI] [PubMed] [Google Scholar]
- Siebler R. S.; Erdmann R. S.; Wennemers H. Switchable proline derivatives: Tuning the conformational stability of the collagen triple helix by pH changes. Angew. Chem., Int. Ed. 2014, 53, 10340–10344. 10.1002/anie.201404935. [DOI] [PubMed] [Google Scholar]
- Horng J.-C.; Raines R. T. Stereoelectronic effects on polyproline conformation. Protein Sci. 2006, 15, 74–83. 10.1110/ps.051779806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kümin M.; Sonntag L.-S.; Wennemers H. Azidoproline containing helices: Stabilization of the polyproline II structure by a functionalizable group. J. Am. Chem. Soc. 2007, 129, 466–467. 10.1021/ja067148o. [DOI] [PubMed] [Google Scholar]
- Chiang Y.-C.; Lin Y.-J.; Horng J.-C. Stereoelectronic effects on the transition barrier of polyproline conformational interconversion. Protein Sci. 2009, 18, 1967–1977. 10.1002/pro.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielska A. A.; Zondlo N. J. Hyperphosphorylation of Tau induces local polyproline II helix. Biochemistry 2006, 45, 5527–5537. 10.1021/bi052662c. [DOI] [PubMed] [Google Scholar]
- Brown A. M.; Zondlo N. J. A propensity scale for type II polyproline helices (PPII): Aromatic amino acids in proline-rich sequences strongly disfavor PPII due to proline–arromatic interactions. Biochemistry 2012, 51, 5041–5051. 10.1021/bi3002924. [DOI] [PubMed] [Google Scholar]
- Elbaum M. B.; Zondlo N. J. OGlcNAcylation and phosphorylation have similar structural effects in α-helices: Post-translational modifications as inducible start and stop signals in α-helices, with greater structural effects on threonine modification. Biochemistry 2014, 53, 2242–2260. 10.1021/bi500117c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm P.; Lewandowski B.; Trapp N.; Wennemers H. A crystal structure of an oligoproline PPII-helix, at last. J. Am. Chem. Soc. 2014, 136, 15829–15832. 10.1021/ja507405j. [DOI] [PubMed] [Google Scholar]
- Shi Z.; Chen K.; Liu Z.; Kallenbach N. R. Conformation of the backbone in unfolded proteins. Chem. Rev. 2006, 106, 1877–1897. 10.1021/cr040433a. [DOI] [PubMed] [Google Scholar]
- Reddy D. N.; George G.; Prabhakaran E. N. Crystal-structure analysis of cis-X–Pro-containing peptidomimetics: Understanding the steric interactions at cis X–Pro amide bonds. Angew. Chem., Int. Ed. 2013, 52, 3935–3939. 10.1002/anie.201209517. [DOI] [PubMed] [Google Scholar]
- Bartlett G. J.; Woolfson D. N. On the satisfaction of backbone–carbonyl lone pairs of electrons in protein structures. Protein Sci. 2016, 25, 887–897. 10.1002/pro.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett G. J.; Newberry R. W.; VanVeller B.; Raines R. T.; Woolfson D. N. Interplay of hydrogen bonds and n→π* interactions in proteins. J. Am. Chem. Soc. 2013, 135, 18682–18688. 10.1021/ja4106122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauss A. D.; Nelsen S. F.; Ayoub M.; Moore J. W.; Landis C. R.; Weinhold F. Rabbit-ears hybrids, VSEPR sterics, and other orbital anachronisms. Chem. Educ. Res. Pract. 2014, 15, 417–434. 10.1039/C4RP00057A. [DOI] [Google Scholar]
- Newberry R. W.; Orke S. J.; Raines R. T. n→π* interactions are competitive with hydrogen bonds. Org. Lett. 2016, 18, 3614–3617. 10.1021/acs.orglett.6b01655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp D. S. Peptidomimetics and the template approach to nucleation of β-sheets and α-helices in peptides. Trends Biotechnol. 1990, 8, 249–255. 10.1016/0167-7799(90)90187-3. [DOI] [PubMed] [Google Scholar]
- Pal T. K.; Sankararamakrishnan R. Quantum chemical investigations on intraresidue carbonyl–carbonyl contacts in aspartates of high-resolution protein structures. J. Phys. Chem. B 2010, 114, 1038–1049. 10.1021/jp909339r. [DOI] [PubMed] [Google Scholar]
- Butterfoss G. L.; Renfrew P. D.; Kuhlman B.; Kirshenbaum K.; Bonneau R. A preliminary survey of the peptoid folding landscape. J. Am. Chem. Soc. 2009, 131, 16798–16807. 10.1021/ja905267k. [DOI] [PubMed] [Google Scholar]
- Gorske B. C.; Bastian B. L.; Geske G. D.; Blackwell H. E. Local and tunable n→π* interactions regulate amide isomerism in the peptoid backbone. J. Am. Chem. Soc. 2007, 129, 8928–8929. 10.1021/ja071310l. [DOI] [PubMed] [Google Scholar]
- Stringer J. R.; Crapster J. A.; Guzei I. A.; Blackwell H. E. Construction of peptoids with all trans-amide backbones and peptoid reverse turns via the tactical incorporation of N-aryl side chains capable of hydrogen bonding. J. Org. Chem. 2010, 75, 6068–6078. 10.1021/jo101075a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorske B. C.; Stringer J. R.; Bastian B. L.; Fowler S. A.; Blackwell H. E. New strategies for the design of folded peptoids revealed by a survey of noncovalent interactions in model systems. J. Am. Chem. Soc. 2009, 131, 16555–16567. 10.1021/ja907184g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caumes C.; Roy O.; Faure S.; Taillefumier C. The click triazolium peptoid side chain: A strong cis-amide inducer enabling chemical diversity. J. Am. Chem. Soc. 2012, 134, 9553–6. 10.1021/ja302342h. [DOI] [PubMed] [Google Scholar]
- Laursen J. S.; Engel-Andreasen J.; Fristrup P.; Harris P.; Olsen C. A. Cis–trans amide bond rotamers in β-peptoids and peptoids: Evaluation of stereoelectronic effects in backbone and side chains. J. Am. Chem. Soc. 2013, 135, 2835–2844. 10.1021/ja312532x. [DOI] [PubMed] [Google Scholar]
- Gorske B. C.; Nelson R. C.; Bowden Z. S.; Kufe T. A.; Childs A. M. “Bridged” n→π* interactions can stabilize peptoid helices. J. Org. Chem. 2013, 78, 11172–11183. 10.1021/jo4014113. [DOI] [PubMed] [Google Scholar]
- Newberry R. W.; VanVeller B.; Raines R. T. Thioamides in the collagen triple helix. Chem. Commun. 2015, 51, 9624–9627. 10.1039/C5CC02685G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzei I. A.; Choudhary A.; Raines R. T. Pyramidalization of a carbonyl C atom in (2S)-N-(selenoacetyl)proline methyl ester. Acta Crystallogr., Sect. E: Struct. Rep. Online 2013, 69, o805–o806. 10.1107/S1600536813011112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Fry C. G.; Raines R. T. Modulation of an n→π* interaction with α-fluoro groups. ARKIVOC 2010, 2010 (viii), 251–262. 10.3998/ark.5550190.0011.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Newberry R. W.; Raines R. T. n→π* interactions engender chirality in carbonyl groups. Org. Lett. 2014, 16, 3421–3423. 10.1021/ol5012967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari U.; Scheiner S. Contributions of various noncovalent bonds to the interaction between an amide and S-containing molecules. ChemPhysChem 2012, 13, 3535–3541. 10.1002/cphc.201200412. [DOI] [PubMed] [Google Scholar]
- Reddy D. N.; Thirupathi R.; Tumminakatti S.; Prabhakaran E. N. A method for stabilizing the cis prolyl peptide bond: Influence of an unusual n→π* interaction in 1,3-oxazine and 1,3-thiazine containing peptidomimetics. Tetrahedron Lett. 2012, 53, 4413–4417. 10.1016/j.tetlet.2012.06.031. [DOI] [Google Scholar]
- Singh S. K.; Kumar S.; Das A. Competition between n→π* and conventional hydrogen bonding (N-HN) interactions: An ab initio study of the complexes of 7-azaindole and fluorosubstituted pyridines. Phys. Chem. Chem. Phys. 2014, 16, 8819–8827. 10.1039/C3CP54169J. [DOI] [PubMed] [Google Scholar]
- Azofra L. M.; Scheiner S. Complexes containing CO2 and SO2. Mixed dimers, trimers and tetramers. Phys. Chem. Chem. Phys. 2014, 16, 5142–5149. 10.1039/c3cp55489a. [DOI] [PubMed] [Google Scholar]
- Sanz M. E.; Lesarri A.; Peña M. I.; Vaquero V.; Cortijo V.; López J. C.; Alonso J. L. The shape of β-alanine. J. Am. Chem. Soc. 2006, 128, 3812–3817. 10.1021/ja058194b. [DOI] [PubMed] [Google Scholar]
- Blanco S.; López J. C.; Mata S.; Alonso J. L. Conformations of γ-aminobutyric acid (GABA): The role of the n→π* interaction. Angew. Chem., Int. Ed. 2010, 49, 9187–9192. 10.1002/anie.201002535. [DOI] [PubMed] [Google Scholar]
- Cabezas C.; Alonso J. L.; López J. C.; Mata S. Unveiling the shape of aspirin in the gas phase. Angew. Chem., Int. Ed. 2012, 51, 1375–1378. 10.1002/anie.201106621. [DOI] [PubMed] [Google Scholar]
- Newberry R. W.; Raines R. T. Crystal structure of N-(3-oxobutanoyl)-l-homoserine lactone. Acta Crystallogr. 2016, 72, 136–139. 10.1107/S2056989015024913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary A.; Fry C. G.; Kamer K. J.; Raines R. T. An n→π* interaction reduces the electrophilicity of the acceptor carbonyl group. Chem. Commun. 2013, 49, 8166–8168. 10.1039/c3cc44573a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Kohler P.; Overman L. E.; Houk K. N. Origins of stereoselectivities of dihydroxylations of cis-bicyclo[3.3.0]octenes. J. Am. Chem. Soc. 2012, 134, 16054–16058. 10.1021/ja3075538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.; Yang X.; Birman V. B.; Houk K. N. Origin of enantioselectivity in benzotetramisole-catalyzed dynamic kinetic resolution of azlactones. Org. Lett. 2012, 14, 3288–3291. 10.1021/ol301243f. [DOI] [PubMed] [Google Scholar]
- Lajkiewicz N. J.; Roche S. P.; Gerard B.; Porco J. A. Jr. Enantioselective photocycloaddition of 3-hydroxyflavones: Total syntheses and absolute configuration assignments of (+)-ponapensin and (+)-elliptifoline. J. Am. Chem. Soc. 2012, 134, 13108–13113. 10.1021/ja305342f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock S. B.; Kent S. B. H. An investigation into the origin of the dramatically reduced reactivity of peptide-prolyl-thioesters in native chemical ligation. Chem. Commun. 2011, 47, 2342–2344. 10.1039/C0CC04120C. [DOI] [PubMed] [Google Scholar]
- Choudhary A.; Kamer K. J.; Raines R. T. A conserved interaction with the chromophore of fluorescent proteins. Protein Sci. 2012, 21, 171–177. 10.1002/pro.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chyan W.; Kilgore H. R.; Gold B.; Raines R. T. Electronic and steric optimization of fluorogenic probes for biomolecular imaging. J. Org. Chem. 2017, 82, 4297–4304. 10.1021/acs.joc.7b00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry R. W.; Raines R. T. A prevalent intraresidue hydrogen bond stabilizes proteins. Nat. Chem. Biol. 2016, 12, 1084–1088. 10.1038/nchembio.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]