

Abstract
Microtubule-stabilizing agents (MSAs) are widely used in chemotherapy. Here, using X-ray crystallography we describe the detailed binding modes of two potent MSAs, (+)-discodermolide (DDM) and the DDM-paclitaxel-hybrid KS-1-199-32, in the taxane pocket of β-tubulin. Both compounds bind in a very similar hairpin conformation as previously observed in solution. However, they differentially stabilize the M-loop of β-tubulin: KS-1-199-32 induces an M-loop helical conformation that is not observed for DDM. In the context of the microtubule structure, both MSAs connect the β-tubulin helices H6 and H7 and loop S9–S10 with the M-loop, which is similar to the structural effects elicited by epothilone A, but distinct from paclitaxel. Together, our data rationalize a differential binding mechanism of DDM and KS-1-199-32 on tubulin.
Keywords: drug discovery, microtubules, molecular mechanism of action, structure elucidation, X-ray crystallography
Table of Contents
Microtubule-stabilizing agents: Discodermolide and KS-1-199-32 are two microtubule-stabilizing agents with potent cytotoxic activity. X-ray crystallography shows that both compounds bind to the taxane site on β-tubulin with distinct features. The data define the mechanism of action of DDM and KS-1-199-32 on tubulin and microtubules.
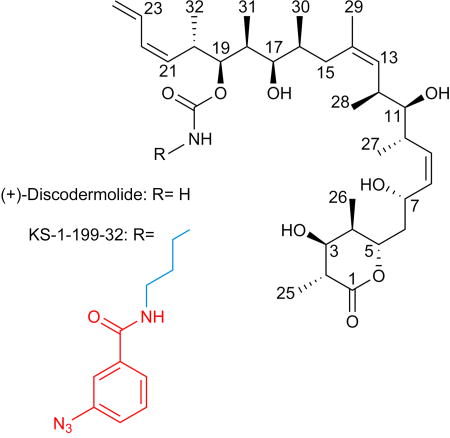
Taxane-site MSAs like paclitaxel and docetaxel are among the most successful classes of chemotherapeutic agents against cancer[1]. However, their clinical application is compromised by their high systemic toxicity and by the development of resistance. (+)-Discodermolide (DDM, Scheme 1), a potent antitumor polyketide from the marine sponge Discoderma dissoluta[2], binds to the taxane site of β-tubulin[3], thereby promoting microtubule stability[4] that is associated with cell death[3, 5], and potent induction of accelerated cell senescence.[6] DDM binds to tubulin more strongly than paclitaxel, is more efficient in promoting tubulin polymerization and has potent cytotoxic activity against a number of human tumor cell lines, including paclitaxel-resistant ovarian and colon carcinoma cells.[5, 7] Furthermore, DDM and paclitaxel have synergistic anti-proliferative efficacy in cancer cell[8] and potentiation effects in an ovarian xenograft tumor model.[7a]
Scheme 1.

The purpose of earlier work was to prepare taxane-based molecules with superior therapeutic activity. To this end a series of DDM-paclitaxel hybrid molecules have been studied[9] and extensive SAR data[10] as well as computational and NMR[11] studies have been performed to define the binding pose and mode of action of DDM. Based on these studies, two conflicting binding modes have been proposed[9a]. The potent DDM-hybrid KS-1-199-32 (Scheme 1)[9a] was designed to fill the aromatic pocket of β–tubulin in the vicinity of the carbamate group of DDM by introducing aromatic groups with optimal tether lengths capable to mimic the AR3 phenyl side chain of paclitaxel (Figure S1). To gain insights into the mechanism of action of this class of compounds and to shed light on the conflicting binding modes we sought to crystallize and solve the high resolution structures of DDM and KS-1-199-32 in complex with tubulin.
Recently, the first high-resolution crystal structures of the taxane-site ligands zampanolide and epothilone A (EpoA) in complex with tubulin have been reported, which shed light on the detailed binding mode and mechanism of action of the two taxane-site MSAs[12]. A common activation mechanism was proposed based on the observed helical organization of the M-loop[12], a critical element involved in the formation of lateral tubulin contacts in microtubules[13]. More recently, a crystal structure and additional molecular modeling study of a tubulin–dictyostatin complex have allowed the rationalization of the structure–activity relationships for a set of synthetic dictyostatin analogues[14].
Here, using a crystal system of a protein complex composed of bovine brain αβ-tubulin, the rat stathmin-like protein RB3 and chicken tubulin tyrosine ligase (T2R-TTL)[12, 15], we report the crystal structures of both the tubulin-bound DDM and the DDM-paclitaxel hybrid, KS-1-199-32, to 1.9 and 2.2 Å resolution, respectively (Figure S2A, Table S1). The overall structure of tubulin in the two T2R-TTL-MSA complexes superimposed well with the one obtained in the absence of ligands[12] (PDB ID 4I55; rmsdDDM T2R-TTL: 0.28 Å, 2059 Cα-atoms; rmsdDDM β-tubulin: 0.14 Å, 363 Cα-atoms; rmsdKS-1-199-32 T2R-TTL: 0.43 Å, 2044 Cα-atoms; rmsdKS-1-199-32 β-tubulin: 0.18 Å, 354 Cα-atoms), suggesting that binding of DDM or KS-1-199-32 does not affect the global conformation of tubulin. Both MSAs share a common binding mode in the taxane pocket with distinct M-loop stabilization effects, consistent with previously reported photolabeling-[16], HDX-MS-[17] and NMR studies[11a].
In the tubulin-DDM complex, the ligand is deeply buried into the taxane pocket (Figure 1A); its conformation is very similar to the hairpin structure observed in both solid state and solution NMR studies[11d, 18] (rmsd 0.31 Å, 23 core atoms (CSD-ID VINTAN01) and rmsd 1.15 Å, 42 atoms, respectively), which only differ in the orientation of the lactone ring that is in the half-chair conformation[11d, 19] (Figure S2B). The binding mode is in close agreement with the one previously suggested from docking studies[19] (rmsd 1.01 Å, 29 Cα atoms of the taxane binding site; rmsd 0.43 Å, 41 core atoms; Figure S2C), but differs substantially from the binding modes of other proposals[9a, 11b]. Five main hydrogen bonds are observed between the C1 ester carbonyl and Ser232 through a water molecule, the C3 hydroxyl and the backbone carbonyl of Arg369, the C11 hydroxyl and Asp226, the C17 hydroxyl and both the backbone carbonyl and amide of Pro274 and Thr276, respectively (Figure 1B). The carbamate group is exposed to the solvent facing the S9–S10 loop, and together with the C2, C4 and the C18 methyl groups it embraces the side chain of Leu371. The terminal diene group of C21–C24 forms a weak hydrogen bond to the main chain carbonyl of Thr276 via C23, and a π-π interaction to the guanidyl moiety of Arg278 thereby stabilizing the amino-terminal section of the M-loop. The DDM-tubulin interaction is completed by van der Waals and hydrophobic contacts to Cys213, Leu217, Leu219, His229, Leu230, Ala233, Phe272, Pro274, Leu275, Leu371 and Pro360 (Figure 1B).
Figure 1. Structures of tubulin-DDM and tubulin-KS-1-199-32.

Overall views of (A) tubulin–DDM and (C) tubulin-KS-1-199-32 interactions. The β-tubulin –––subunits (chain B) are shown in surface representation. The M-loop residues are highlighted in yellow. All ligands are in stick representation and are colored in purple (DDM) and green (KS-1-199-32). The SigmaA-weighted 2mFo-DFc (grey mesh) and mFo-DFc (green mesh) omit maps are contoured at + 1.0 σ and + 3.0 σ, respectively. Close-up view of the interactions observed between (B) DDM, (D) KS-1-199-32 and β-tubulin in stick and ribbon representation, respectively.
The characteristic binding mode of DDM to tubulin supports previously published SAR studies[20]. The C1 carbonyl and both the C3 and C7 hydroxyls of DDM are exposed to the solvent, explaining why substitutions at these positions are favorable to retention of potent cytotoxicity. The structure further reveals why variations at the C2, C14 and the carbamate carbons are tolerable, while changes in other portions of the molecule, such as C11 or C17 significantly affect biological activity[10, 21]. Although C2 and C14 are pointing inside the binding pocket, there is sufficient amount of space to accommodate larger substituents at these locations. The C2 methyl is anchored by three hydrophobic contacts to Ala233, Pro360 and Leu371 4.2 Å above the CZ atom of the Phe272 side chain, explaining why DDM conserves the full activity on paclitaxel resistant cell lines presenting a Phe270Val mutation (Phe272 according to the numbering used here). Moreover, of the β1-tubulin mutations that have been reported to confer resistance to paclitaxel only Leu217, Leu219 and Leu230 are in direct contact with DDM. All other residues are not, which is in agreement with the observation that DDM is active in such mutated cell lines[22].
In the tubulin-KS-1-199-32 and tubulin DDM structures, the interactions of DDM are identical (Figure 1C,D). In addition to these interactions, the m-azidophenyl moiety occupies a totally different space compared to the aromatic rings of paclitaxel (Figure 2A,B): It extends towards the M-loop and stabilizes the M-loop laterally into a helical conformation. This is in contrast to the original design idea[9a] where the hybrid side chain was modeled to occupy the aromatic pocket on β-tubulin in the vicinity of the carbamate group of DDM, which in the case of paclitaxel is occupied by the AR3 phenyl side chain (Figure S1). The interaction comprises one hydrogen bond to Asn281 and hydrophobic interactions of the m-azidophenyl substituent with both the apolar moieties of the Gln281 and Gln282 side chains (Figure 1C,D). Although the overall stabilization effect on the M-loop is comparable to the one observed for EpoA[12], the stabilization is distinct as it is accomplished through lateral contacts to the M-loop helix, and not to the base, as observed for EpoA. Superposition of the binding site of tubulin-EpoA with the corresponding one of tubulin-KS-1-199-32 reveals minor variations in the relative orientation between the individual M-loops and the remaining portion of the taxane site (Figure 2B), indicating that the two molecules share a common mechanism of tubulin activation.
Figure 2. Tubulin binding of DDM and KS-1-199-32 compared to paclitaxel and EpoA.

(A) Close-up view of the superimposed paclitaxel-stabilized microtubule (dark grey ribbon, white surface, PDB ID 3J6G) and tubulin–DDM (light gray ribbon) complexes. The black ellipsoids denote the regions of the taxane site where differences are observed. (B) Same close-up view as in (A), but with superimposed KS-1-199-32 (green) and EpoA (slate, PDB ID 4I50). The M-loop of the superimposed KS-1-199-32-structure is highlighted in yellow.
DDM is one of the most potent natural compounds that target tubulin (apparent Kb 872 ± 82 × 107M−1)[23]. To find a structural relationship for the observed differences in binding affinities to other classical taxane-site binders, we compared the binding modes of DDM and KS-1-199-32 with the ones of paclitaxel and EpoA (Figure 2). For this purpose, we superimposed the crystal structures of the DDM, KS-1-199-32 and EpoA complexes onto the taxane pocket of the intermediate domain of β-tubulin in the high-resolution cryo-EM structure of a paclitaxel-stabilized microtubule[13] (PDB ID 3J6G; DDM: rmsd 1.15 Å, 150 Cα atoms; KS-1-199-32: 1.18 Å, 150 Cα atoms; EpoA, PDB ID 4I50: rmsd 0.92 Å, 147 Cα atoms). Compared to paclitaxel (Kb 2.64 ± 0.17 × 107M−1)[23], DDM completely fills the cavity formed by helices H6 and H7, the H6–H7 loop, and the N-terminal section of the M-loop with the C10, C12, C14, C16, C18 and C20 methyl groups, and with the diene C21–C24 side chain. Moreover, the C11–hydroxyl forms a hydrogen bond to Asp226 and the C21–C24-diene stabilizes the terminal section of the M-loop by interacting with Arg278 (Figure 2A). These interactions tightly connect the M-loop to helices H6 and H7 of β-tubulin and fix their relative orientation. In contrast, paclitaxel packs much loser in that pocket. It has neither a substituent at the C19 carbon nor on the C2-benzoyl-ring that could interact to the same extent with the cavity described above (Figure 2A). However, distinct interactions are formed by paclitaxel with helices H1 and H7 via the C13 benzamide and the AR3 phenyl side chains, which connect the base of the M-loop to H1, H7 and the S9–S10 loop. The importance of these interactions is underpinned by the fact that Baccatin III, which represents the core of paclitaxel lacking both the C13 benzamide and the AR3 phenyl side chains, displays the poorest binding affinity (Kb 0.015 ± 0.005 × 107M−1)[24].
KS-1-199-32 (Kb 9.3 ± 2.0 × 107M−1) binds to tubulin with a similar binding constant as described for EpoA (Kb 7.48 ± 1.00 × 107M−1)[23]. It forms the same interactions as DDM and additionally induces the structuring of the M-loop with the m-azidophenyl moiety (Figures 1D and 2B). A comparison of KS-1-199-32 and EpoA binding to tubulin highlights how both molecules exploit common tubulin stabilizing features. The two orders of magnitude difference in binding affinity compared to DDM suggest that both molecules pay a penalty for M-loop structuring. Moreover, the observed structural features discussed above suggest that either the hydrophobic interactions with the cavity that harbors the four leucine residues Leu217, Leu219, Leu230 and Leu275 or the interactions with helices H1 and H7 and the S9–S10 loop are required for high-affinity binding of taxane-site MSAs (Figures 2A,B and S2).
To relate the observed structural features to the potencies in enhancing tubulin polymerization in vitro, the critical concentrations for tubulin assembly in GAB buffer (Cr) were determined for all the five MSAs described above (Figure 3). The differential degree of M-loop stabilization observed in the individual structures of DDM, KS-1-199-32 and EpoA is in agreement with their corresponding Cr values. DDM (Cr: 0.65 ± 0.07 µM) only interacts with the N-terminal segment of the M-loop without contacting the M-loop helix. KS-1-199-32 (Cr: 0.39 ± 0.06 µM) forms the same core interactions as DDM, but in addition establishes lateral contacts with the M-loop helix via its m-azidophenyl moiety. EpoA (Cr: 0.26 ± 0.04 µM) features the most extensive interactions with the M-loop by contacting the linkers of the M-loop helix as well as the helix itself[12]. Interestingly, despite lacking the close contacts to helix H6 and despite interacting only with the base of the M-loop, paclitaxel (Cr: 0.47 ± 0.06 µM) reduces the critical concentration of tubulin to a similar extent as DDM. The increased Cr measured for Baccatin III (Cr: 2.90 ± 0.3 µM) together with the observed structural features discussed in the previous paragraph suggest that the tubulin assembly promoted by paclitaxel follows a distinct mechanism.
Figure 3. Critical concentrations for tubulin assembly.

Critical concentration of the GTP-tubulin assembly in GAB buffer[23] in the presence of the individual ligands compared to DMSO (control; Cr 3.3 ± 0.1 µM[23]). The error bars indicate the standard error.
Taken together, our results provide detailed insights into the molecular mechanism of binding of DDM and KS-1-199-32 to the taxane site of β-tubulin. We find that the major consequences of DDM binding to tubulin are (1) the bridging of the four adjacent tubulin structural elements H6, H7, S9–S10 with the M-loop, and (2) the stabilization of the β-tubulin M-loop conformation. Importantly, our experimental data resolve the conflicting binding modes that have been proposed based on modelling studies[9a, 11b, 19]. Our analysis further suggests that the superior antiproliferative activity of the hybrid arises through M-loop stabilization rather than through interaction with the aromatic pocket with its paclitaxel moiety, which is in contrast to the original design idea based on a DDM docking model[9a]. Because helical structuring of the M-loop is a critical molecular process for the establishment of lateral tubulin contacts between adjacent protofilaments in microtubules[12–13], our observations explain the promoting effect of DDM and KS-1-199-32 on microtubule assembly and stability[10]. Our results are in agreement with the recently published SAR study of dictyostatin[14]. The comparison of the poses of DDM, KS-1-199-32, paclitaxel and EpoA in the context of the microtubule lattice[13] highlights that these features can hold true for microtubules as well (Figure 4A–D).
Figure 4. Binding of taxane-site ligands in the context of the microtubule.

Superposition of the intermediate domains of (A) DDM, (C) KS-1-199-32 and (D) EpoA (PDB ID 4I50) onto the corresponding domain of (B) the paclitaxel-bound microtubule (PDB ID 3J6G). The helices shaping the taxane site of the microtubule are displayed as cylinders, the β-tubulin subunits of the flanking protofilament are in surface representation. The M-loop is highlighted in yellow. The ligands are in spheres representation. Key interactions are marked with a black arrow. (E) Schematic representation of the structural features shown in panels (A) – (D) highlighting the secondary structural elements contacted by the individual ligands. The ligand contacted elements are framed with the same color code as the individual ligands. The thick black lines denote the connection between the secondary structural elements.
However, the analysis also suggests that the activation promoted by DDM likely occurs through a mechanism, which is distinct from the one promoted by paclitaxel. While DDM connects the β-tubulin helices H6 and H7 with the M-loop base, the C13 side chain of paclitaxel via helices H1 and H7 and loop S9–S10 achieves a similar effect on M-loop stabilization (Figure 4E). The proposed model further shows that KS-1-199-32 can form a direct lateral contact with the β-tubulin subunit from a neighboring protofilament (Figure 4C), which cannot be established by the other taxane site MSAs compared in this study (Figure 4A,B,D). The features discussed above together with the direct lateral contact open up new prospectives for the design of DDM analogs with superior therapeutic activity that fully exploit the landscape of the taxane pocket and potentially glue the lateral contacts of adjacent protofilaments in microtubules.
Supplementary Material
Acknowledgments
We thank V. Olieric and M. Wang for excellent technical assistance with the collection of X-ray data at beamline X06DA of the Swiss Light Source (Paul Scherrer Institut, Villigen PSI, Switzerland). We also thank Dr. Onur Atasoylu for valuable discussion. This work was supported by grants from the Ministerio de Economia y Competitividad (BFU2016-75319-R (AEI/FEDER, UE), to J.F.D), the National Cancer Institute Grant CA077263 (A.B.S, H.M.D. and S.B.H), the Breast Cancer Research Foundation (H.M.D. and S.B.H.) and the Swiss National Science Foundation (31003A_166608; to M.O.S.). The authors acknowledge networking contribution by the COST Action CM1407 “Challenging organic syntheses inspired by nature - from natural products chemistry to drug discovery” and the COST action CM1470.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Coordinates have been deposited at the Protein Data Bank (PDB) under accession numbers 5LXT (T2R-TTL-DDM) and 5LXS (T2R-TTL-KS-1-199-32). Note that tubulin residue numbering is as defined in Löwe et al.[25]
References
- 1.Dumontet C, Jordan MA. Nat Rev Drug Discov. 2010;9:790–803. doi: 10.1038/nrd3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gunasekera M SPG, Longley RE, Schulte GK. J Org Chem. 1990;55:4912–4915. [Google Scholar]
- 3.Hung DT, Chen J, Schreiber SL. Chem Biol. 1996;3:287–293. doi: 10.1016/s1074-5521(96)90108-8. [DOI] [PubMed] [Google Scholar]
- 4.ter Haar E, Kowalski RJ, Hamel E, Lin CM, Longley RE, Gunasekera SP, Rosenkranz HS, Day BW. Biochemistry. 1996;35:243–250. doi: 10.1021/bi9515127. [DOI] [PubMed] [Google Scholar]
- 5.Kowalski RJ, Giannakakou P, Gunasekera SP, Longley RE, Day BW, Hamel E. Mol Pharmacol. 1997;52:613–622. [PubMed] [Google Scholar]
- 6.Klein LE, Freeze BS, Smith AB, III, Horwitz SB. Cell Cycle. 2005;4:501–507. doi: 10.4161/cc.4.3.1550. [DOI] [PubMed] [Google Scholar]
- 7.a Huang GS, Lopez-Barcons L, Freeze BS, Smith AB, III, Goldberg GL, Horwitz SB, McDaid HM. Clin Cancer Res. 2006;12:298–304. doi: 10.1158/1078-0432.CCR-05-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kar S, Florence GJ, Paterson I, Amos LA. FEBS Lett. 2003;539:34–36. doi: 10.1016/s0014-5793(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 8.a Honore S, Kamath K, Braguer D, Horwitz SB, Wilson L, Briand C, Jordan MA. Cancer Res. 2004;64:4957–4964. doi: 10.1158/0008-5472.CAN-04-0693. [DOI] [PubMed] [Google Scholar]; b Martello LA, McDaid HM, Regl DL, Yang CP, Meng D, Pettus TR, Kaufman MD, Arimoto H, Danishefsky SJ, Smith AB, III, Horwitz SB. Clin Cancer Res. 2000;6:1978–1987. [PubMed] [Google Scholar]
- 9.a Smith AB, III, Sugasawa K, Atasoylu O, Yang CP, Horwitz SB. J Med Chem. 2011;54:6319–6327. doi: 10.1021/jm200692n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Smith AB, III, Freeze BS, Lamarche MJ, Hirose T, Brouard I, Rucker PV, Xian M, Sundermann KF, Shaw SJ, Burlingame MA, Horwitz SB, Myles DC. Org Lett. 2005;7:311–314. doi: 10.1021/ol047686a. [DOI] [PubMed] [Google Scholar]
- 10.Smith AB, III, Freeze BS. Tetrahedron. 2008;64:261–298. doi: 10.1016/j.tet.2007.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a Canales A, Rodriguez-Salarichs J, Trigili C, Nieto L, Coderch C, Andreu JM, Paterson I, Jimenez-Barbero J, Diaz JF. ACS Chem Biol. 2011;6:789–799. doi: 10.1021/cb200099u. [DOI] [PubMed] [Google Scholar]; b Jogalekar AS, Kriel FH, Shi Q, Cornett B, Cicero D, Snyder JP. J Med Chem. 2010;53:155–165. doi: 10.1021/jm9015284. [DOI] [PubMed] [Google Scholar]; c Sanchez-Pedregal VM, Kubicek K, Meiler J, Lyothier I, Paterson I, Carlomagno T. Angew Chem Int Ed Engl. 2006;45:7388–7394. doi: 10.1002/anie.200602793. [DOI] [PubMed] [Google Scholar]; d Smith AB, III, LaMarche MJ, Falcone-Hindley M. Org Lett. 2001;3:695–698. doi: 10.1021/ol006967p. [DOI] [PubMed] [Google Scholar]
- 12.Prota AE, Bargsten K, Zurwerra D, Field JJ, Diaz JF, Altmann KH, Steinmetz MO. Science. 2013;339:587–590. doi: 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- 13.a Alushin GM, Lander GC, Kellogg EH, Zhang R, Baker D, Nogales E. Cell. 2014;157:1117–1129. doi: 10.1016/j.cell.2014.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang R, Alushin GM, Brown A, Nogales E. Cell. 2015;162:849–859. doi: 10.1016/j.cell.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trigili C, Barasoain I, Sánchez-Murcia PA, Bargsten K, Redondo-Horcajo M, Nogales A, Gardner NM, Meyer A, Naylor GJ, Gómez-Rubio E, Gago F, Steinmetz MO, Paterson I, Prota AE, Díaz JF. ACS Omega. 2016;1:1192–1204. doi: 10.1021/acsomega.6b00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prota AE, Magiera MM, Kuijpers M, Bargsten K, Frey D, Wieser M, Jaussi R, Hoogenraad CC, Kammerer RA, Janke C, Steinmetz MO. J Cell Biol. 2013;200:259–270. doi: 10.1083/jcb.201211017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia S, Kenesky CS, Rucker PV, Smith AB, III, Orr GA, Horwitz SB. Biochemistry. 2006;45:11762–11775. doi: 10.1021/bi060497a. [DOI] [PubMed] [Google Scholar]
- 17.Khrapunovich-Baine M, Menon V, Verdier-Pinard P, Smith AB, III, Angeletti RH, Fiser A, Horwitz SB, Xiao H. Biochemistry. 2009;48:11664–11677. doi: 10.1021/bi901351q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith AB, III, Kaufman MD, Beauchamp TJ, LaMarche MJ, Arimoto H. Org Lett. 1999;1:1823–1826. doi: 10.1021/ol9910870. [DOI] [PubMed] [Google Scholar]
- 19.Canales A, Matesanz R, Gardner NM, Andreu JM, Paterson I, Diaz JF, Jimenez-Barbero J. Chemistry. 2008;14:7557–7569. doi: 10.1002/chem.200800039. [DOI] [PubMed] [Google Scholar]
- 20.Smith AB, III, Freeze BS. Tetrahedron. 2007;64:261–298. doi: 10.1016/j.tet.2007.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunasekera SP, Longley RE, Isbrucker RA. J Nat Prod. 2002;65:1830–1837. doi: 10.1021/np0203234. [DOI] [PubMed] [Google Scholar]
- 22.Kanakkanthara A, Teesdale-Spittle PH, Miller JH. Anticancer Agents Med Chem. 2013;13:147–158. [PubMed] [Google Scholar]
- 23.Buey RM, Barasoain I, Jackson E, Meyer A, Giannakakou P, Paterson I, Mooberry S, Andreu JM, Diaz JF. Chem Biol. 2005;12:1269–1279. doi: 10.1016/j.chembiol.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 24.Andreu JM, Barasoain I. Biochemistry. 2001;40:11975–11984. doi: 10.1021/bi010869+. [DOI] [PubMed] [Google Scholar]
- 25.Lowe J, Li H, Downing KH, Nogales E. J Mol Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.