

Abstract
HIV-associated neurocognitive disorder (HAND) affects approximately half of HIV-infected patients. Loss of synaptic connections is a hallmark of many neurocognitive disorders, including HAND. The HIV-1 protein transactivator of transcription (Tat) disrupts synaptic connections both in vitro and in vivo and has been linked to impaired neurocognitive function in humans. In vitro studies have shown that ifenprodil, an antagonist selective for GluN2B-containing NMDARs, reverses synapse loss when applied after Tat. Here, we tested the hypothesis that Tat-induced loss and ifenprodil-mediated rescue of synaptic spines in vivo would predict impairment and rescue of cognitive function. Using intracranial multiphoton imaging, we found that infusion of 100 ng of HIV-1 Tat into the lateral ventricle of yellow fluorescent protein-expressing transgenic mice produced a 17 ± 1% loss of dendritic spines in layer 1 of retrosplenial cortex. Repeated imaging of the same dendrites over 3 weeks enabled longitudinal experiments that demonstrated sustained spine loss after Tat infusion and transient rescue after ifenprodil administration (10 mg/kg, i.p.). Parallel trace fear conditioning experiments showed that spine loss predicted learning deficits and that the time course of ifenprodil-induced rescue of spine density correlated with restoration of cognitive function. These results show for the first time that, during exposure to an HIV-1 neurotoxin in vivo, alteration of GluN2B-containing NMDAR signaling suppresses spine density and impairs learning. Pharmacological inhibition of these NMDARs rescued spines and restored cognitive function. Drugs that rescue synapses may improve neurocognitive function in HAND.
SIGNIFICANCE STATEMENT Synaptodendritic damage correlates with cognitive decline in HIV-associated neurocognitive disorder (HAND) patients. We developed an in vivo imaging approach for longitudinal tracking of spine density that enabled correlation of synaptic changes with behavioral outcomes in a model of HAND. We show for the first time that spine loss after exposure to an HIV-1 protein can be reversed pharmacologically and that loss and recovery of dendritic spines predict impairment and restoration of cognitive function, respectively. Therefore, synapse loss, the hallmark of cognitive decline in HAND, is reversible. Drugs that restore spine density may have broad application for improving cognitive function during the early phases of neurodegenerative diseases.
Keywords: dendritic spines, GluN2B, HIV neurotoxicity, ifenprodil, multiphoton imaging, NMDAR
Introduction
HIV associated neurocognitive disorder (HAND) is a significant public health burden. Despite systemic suppression of viral replication with antiretroviral therapy, almost half of HIV-infected individuals develop HAND (Saylor et al., 2016). The severity of HAND correlates with the loss of synaptic markers (Ellis et al., 2007) and patients with mild cognitive deficits have an increased risk of progressing to more severe neurocognitive impairment (Saylor et al., 2016). Therefore, models of the early stages of HAND are critical to develop treatments to prevent or delay disease progression.
Because HIV does not infect neurons, HIV neurotoxicity is mediated by factors released from infected non-neuronal cells. The HIV protein transactivator of transcription (Tat) is a potent neurotoxin (Li et al., 2009) that is highly neuroinflammatory (Chivero et al., 2017). Tat is present in the CSF of HIV-infected individuals and, once HIV DNA integrates into the host genome, Tat expression continues even in the presence of antiretroviral drugs (Johnson et al., 2013). The titer of antibodies to Tat negatively correlates with HAND symptoms (Bachani et al., 2013), suggesting that an antibody response to Tat may be neuroprotective. Neuronal exposure to HIV-1 Tat replicates key aspects of HAND in both in vitro and in vivo models. Tat potentiates NMDA receptors (Green and Thayer, 2016), followed by an adaptive response that results in the loss of excitatory synapses (Kim et al., 2008; Bertrand et al., 2013), an increase in inhibitory synapses (Hargus and Thayer, 2013), and eventually neuronal death (Eugenin et al., 2007). Expression of Tat in the CNS produces loss of synaptic spines and impaired cognitive function (Fitting et al., 2006; Carey et al., 2012; Fitting et al., 2013). Therefore, treatment with Tat appears to mimic key aspects of HAND neurotoxicity.
Experiments with primary neuronal cultures describe a changing role for NMDAR subtypes during exposure to Tat. Tat initially potentiates both GluN2A- and GluN2B-containing NMDARs via a Src-dependent pathway (Haughey et al., 2001; Krogh et al., 2014). Then, excitatory synapses are lost via a process dependent on GluN2A-containing NMDARs (Shin et al., 2012). Ifenprodil, an antagonist for GluN2B-containing NMDARs, has no effect on Tat-induced loss of synapses; however, when applied after synapse loss and in the maintained presence of Tat, ifenprodil induces a recovery of synapses (Shin et al., 2012). This observation suggests that sustained activity of GluN2B-containing NMDARs during exposure to Tat suppresses synaptic number. Because synapse loss correlates with cognitive decline in HAND, we hypothesized that Tat-induced loss and ifenprodil-induced rescue of synaptic spines in vivo would predict impairment and rescue of cognitive function, respectively.
To test this hypothesis, we used intracranial multiphoton imaging to show that infusion of HIV-1 Tat into the lateral ventricle of yellow fluorescent protein (YFP)-expressing transgenic mice produced a significant loss of dendritic spines in layer 1 of the retrosplenial cortex (RSC). Repeated imaging of the same dendrites over 3 weeks enabled longitudinal experiments to track Tat-induced loss and ifenprodil-induced rescue of spines. Parallel trace fear conditioning experiments found that spine loss predicted learning deficits and that the time course of ifenprodil-induced rescue of spine density correlated with restoration of cognitive function. These results demonstrate that synapse loss early in neurodegenerative disease predicts loss of function and that this impairment is reversible. Drugs that restore spine density may improve cognitive function in individuals with neuroinflammatory diseases such as HAND.
Materials and Methods
Materials.
Ifenprodil was from Tocris Bioscience. HIV-1 Tat1–86 (Clade B, recombinant) was obtained through the National Institutes of Health AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH from Dr. John Brady.
Subjects.
Experiments were conducted with 104 male mice from three strains, C57BL/6j, B6;CBA-Tg(Thy1-EGFP)SJrs/NdivJ (stock #011070), and B6.Cg-Tg(Thy1-YFP)HJrs/J (stock #003782). C57BL/6j mice were ordered directly from The Jackson Laboratory and assigned randomly to treatment groups for behavioral experiments. Transgenic strains were obtained from The Jackson Laboratory and bred in-house; imaging experiments were performed in parallel on littermates randomly assigned to treatment groups. Mice were 8–12 weeks of age at the beginning of experimental procedures. All animals were housed on a 14/10 light/dark cycle in standard-sized shoebox caging with ad libitum access to food (standard rodent diet) and water. To facilitate recovery and to decrease surgery-related attrition, mice were singly housed. Transgenic expression was confirmed via PCR. All procedures were approved by the University of Minnesota Institutional Care and Use Committee and were conducted in accordance with the ethical guidelines and policy of the Society for Neuroscience.
Surgical methods.
Cranial windows were placed over the RSC. The RSC was chosen for study for several reasons: (1) it is accessible to multiphoton imaging, which is limited to ∼1 mm below the cranial window; (2) this region participates in fear conditioning, a behavior shown previously to be affected by HIV Tat (Fitting et al., 2013); and (3) the thickness of the RSC is decreased in HAND (Shin et al., 2017). Surgical procedures were based largely on previous methodological reports (Paukert and Bergles, 2012; Goldey et al., 2014). Briefly, 1 h before surgery, mice were dosed with the preoperative analgesic and anti-inflammatory drugs ketoprofen (2.5 mg/kg; Fort Dodge Animal Health) and dexamethasone (0.1 mg/kg; Bimeda). Mice were then anesthetized with 5% isoflurane (1–2% maintenance), their eyes protected with ophthalmic ointment, and their scalp shaved and scrubbed with povidone iodine antiseptic. Subsequently, the scalp was excised over the target location and a 26 gauge intracranial chronic indwelling guide cannula was placed over the lateral ventricle targeted to the infusion site at A/P 1.0, L/M 0.5, D/V 2.75 mm with respect to bregma (Paxinos and Franklin, 2012). The cannula and a custom titanium head plate (see Fig. 1A,B) were then secured to the skull with luting cement (Alpha-Dent; Dental Technologies). Once the cement set, a 4 mm craniotomy was performed with a dental drill and then a 5 mm #1 cover glass (Warner Instruments) was gently placed over the craniotomy and secured with cyanoacrylate topical tissue adhesive (Zoetis), followed by dental acrylic (Jet-Liquid/Tooth Shade; Lang Dental). For behavioral experiments, cannulas were implanted at the same coordinates, but the head plate, craniotomy, and coverslip were omitted. After surgery, mice were returned to home cages, placed on ibuprofen-treated drinking water (0.4 mg/ml) for at least 3 d, and allowed 2–3 weeks recovery before beginning experiments.
Figure 1.
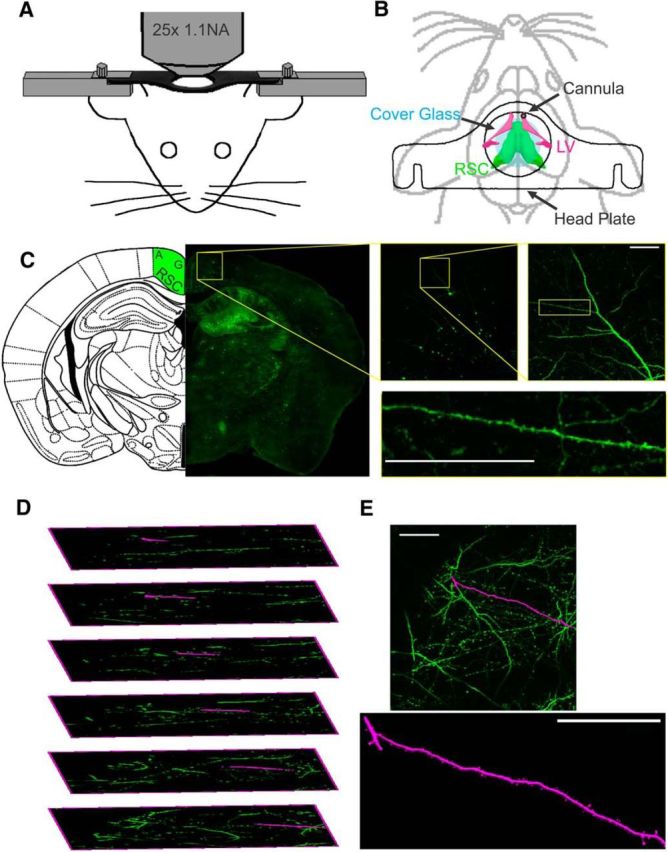
In vivo multiphoton imaging of dendritic spines. A, Schematic representation of custom stage mount to stabilize mouse for multiphoton imaging. B, Diagram of location of head plate (outline), ICV-directed guide cannula (black), and glass coverslip (blue) in relation to RSC (green) and lateral ventricle (LV, magenta). C, Location and example dendrite from fixed Thy1-GFP brain section showing source of spine images in RSC agranular cortex. D, Expanded image stack demonstrating dendrite extraction (magenta) from a complex 3D structure (green). E, Extracted dendrite segment (magenta) shown as an overlay on max-Z projection of image stack (green) and as individual image. Scale bars, 50 μm.
Multiphoton imaging.
In vivo multiphoton imaging was performed with a Nikon A1R MP equipped with a plan apo LWD 25×, water-immersion, 1.1 numerical aperture objective (see Fig. 1A). Multiphoton excitation was generated with a Spectra Physics 15 W Mai Tai eHP tunable Ti:sapphire femtosecond pulsed infrared laser. EGFP was excited at 920 nm and YFP was excited at 960 nm. Emission light was directed with a dichroic mirror through a 650 nm short-pass filter to enhanced hybrid photomultiplier tubes via fluorophore specific filters. For EGFP, light was reflected off of a 560 nm dichroic mirror through a 510 nm filter (80 nm band-pass). For YFP, light was reflected off of a 580 nm dichroic through a 540 nm filter (80 nm band-pass). Imaging sites in time-lapse experiments were aligned based on vasculature landmarks (visible against the fluorescent background under wide-field epifluorescence) and by the multiphoton imaged dendritic structure. Images were collected with a 2.4 μs pixel dwell time, with the laser attenuated to 5% of total power, a pixel size of 0.24 μm (XY), and a step size of 1 μm (Z); 100 μm z-stacks, starting at the dorsal extremity of dendrite-bearing tissue and reaching through layer 1 of RSC, were collected with a piezo-driven objective (see Fig. 1C). Initial studies were performed with Thy1-EGFP mice and then switched to Thy1-YFP mice because of the improved image quality obtained with the longer-wavelength YFP fluorophore. Other than making it easier to identify high-quality images, the data did not differ qualitatively or statistically so the datasets were combined.
Imaging experiments were begun after allowing mice 2–3 weeks to recover from surgery. Four baseline imaging sessions were performed over the course of the week preceding infusion (days −7, −5, −3, 0). Mice were infused with HIV-1 Tat1–86 (100 ng in 2 μl PBS) or vehicle on day 0. The precise amount of Tat that leaves the ventricle and reaches layer 1 of retrosplenial cortex is not known. Factors that decrease the steady-state concentration at the site of imaging include dilution in the CSF, the need to cross the ventricular lining, metabolism, and uptake of Tat by glia and neurons. Tat concentrations in the CSF of HIV-infected patients on combination antiretroviral therapy are difficult to relate to the amount of Tat in the brain parenchyma because of the following considerations: Tat concentrations may be higher at the sites where it is produced relative to the CSF, Tat is taken up by neurons and glia, Tat forms multimers, and there are problems with antibody specificity for Tat. Previous studies in which Tat was administered by intracerebroventricular (ICV) infusion range from 40 ng (Lawson et al., 2011) to >10 μg (Philippon et al., 1994). The 100 ng of Tat infused in these studies was chosen to be near the bottom of this range. Mice were imaged on days 2, 4, and 7 after infusion. On days 7 and 8, saline or ifenprodil (10 mg/kg, i.p.) were administered. Mice were imaged again on days 9, 11, and 14 after infusion. Treatment groups were cohorts of four to six littermates run in parallel.
Spine quantification.
To quantify spine density, dendritic segments were first extracted from source images with the Fiji plugin Simple Neurite Tracer (see Fig. 1D). Segments were selected for analysis based on minimal overlying processes, the presence of discrete landmarks to allow extraction of the same section of dendrite over time, and image stability. The length of the extracted segment was recorded and later used to calculate spine density. Spines were counted using a semiautomated analysis of extracted dendrites (see Fig. 1E). A custom ImageJ macro was used to measure total image intensity (integrated density) of maximum Z-projections for the time series of a single dendritic segment; the average integrated density was then used to generate a correction factor that was applied across the time series to compensate for changes in intensity. Subsequently, the time course of corrected max-Z projections of a segment were randomized and presented to two blinded observers who counted spines based on the criteria of Peters and Kaiserman-Abramof (1970), which includes a minimum length of 0.5 μm. Spine counts were recorded by the macro and the average from the two observers used to compute spine densities. Resolution was not sufficiently high across all images to allow reliable characterization of spines into different subtypes. For display purposes, images were processed equally; they were cropped, rotated, scaled (5×), aligned, and passed through a three-pixel Gauss filter.
Behavior.
Trace fear conditioning was conducted as described previously (Tipps et al., 2014). Briefly, mice were allowed 2–3 weeks to recover from ICV cannulation and to acclimate to colony conditions. Timing of behavioral experiments was designed to match changes in spine density observed in RSC using multiphoton imaging. After recovery, mice were infused with HIV-1 Tat1–86 (100 ng in 2 μl PBS) or vehicle on day 0. On days 7 and 8, mice were prehandled and administered saline or ifenprodil (10 mg/kg, i.p.). Twenty-four hours after the last drug administration, mice were trained in trace fear conditioning. Mice were allowed 120 s to explore the training apparatus (MED-VFC-SCT-M; Med Associates) and then presented with two conditioned stimulus (CS)–unconditioned stimulus (US) pairings separated by a 90 s intertrial interval; after the last US, mice remained in the training apparatus for 60 s. The stimulus pair comprised a CS (30 s 80 dB white noise), a 30 s stimulus-free trace interval, followed by a US (2 s 0.5 mA foot shock). To examine baseline exploratory activity and US-induced locomotion, locomotor activity during training was quantified with Med PC software. On day 10, mice were placed in the training apparatus and contextual freezing was assessed for 5 min. Freezing was quantified with time sampling in which an observer blinded to treatment assessed freezing during a 1 s period at 10 s intervals from a video recording of the testing sessions (Wehner and Radcliffe, 2004; Tipps et al., 2014). On day 11, mice were placed in an altered context for a 15 min CS test. During the CS test, freezing to the altered context was assessed during the first 3 min, the CS was presented (3 min, 80 dB white noise), freezing was measured for another 3 min, a second CS (3 min) was presented, and then freezing was measured for the final 3 min. Twenty-eight days after testing (39 d after infusion), mice were again tested for remote retention of contextual, altered, and cued freezing. In the “shifted” experiment, manipulations were identical except that behavioral procedures were shifted 2 d later to begin on day 11.
Analysis.
Spine density data were collected from three to five segments per subject and final analysis was performed on a mean generated for each subject per time point. Raw spine densities (spines/micrometer) for each time point were converted to percentage change from baseline, where baseline was the average of all baseline sessions. This approach preserved variability during the three to four baseline measures and scaled changes from dendritic segments, which varied in baseline spine density, to common levels for analysis and presentation. To capture the complexity of these longitudinal data, analysis was performed at multiple levels. A priori comparisons were used to examine within-group (paired t test) and across-group (t test) changes in spine levels at key experimental time points (see Fig. 2B). ANOVA was used to examine between-group effects on binned data. The combined dataset (see Fig. 2G) was analyzed using a repeated-measures multiway ANOVA of binned data from all four groups to compare effects across all conditions, followed by analysis of individual binned time points. Behavioral data were analyzed with two-way ANOVA. When appropriate, omnibus tests were followed by Tukey's post hoc comparison. Statistical analyses were performed with Excel (t tests) or R under Rcmdr (Baier and Neuwirth, 2007). Data are reported as mean ± the SEM or with individual subjects means (see Fig. 2E).
Figure 2.

Infusion of HIV-1 Tat induces loss of dendritic spines and ifenprodil rescues Tat-induced spine loss. A, Schematic representation of time course of experiment illustrates timing of surgery, imaging sessions, and ifenprodil administration relative to vehicle or Tat infusion. B, Time course showing changes in spine density (%) relative to mean density of 4 baseline images (day −7, −5, −3, 0). Control subjects (n = 5, open circles) received vehicle infusion (PBS) on day 0 and received saline injections on days 7 and 8. Tat subjects (n = 5, solid circles) received 100 ng of Tat in 2 μl of PBS on day 0 and received saline injections on days 7 and 8. Ifenprodil control subjects (n = 5; ifen; open triangles) received vehicle infusion on day 0 and received ifenprodil injections (10 m/kg, i.p.) on days 7 and 8. Subjects that received Tat followed by ifenprodil (n = 5; Tat + ifen; solid triangles) received 100 ng of Tat in 2 μl of PBS on day 0 and received ifenprodil injections (10 m/kg, i.p.) on days 7 and 8. †Significant within-group change in spine density relative to previous imaging session. *Significant difference relative to control group. Data are shown as mean ± SEM. C, Representative image sequence demonstrating the stability of spines during baseline imaging sessions and continued stability after PBS infusion (vehicle). D, Representative micrographs showing Tat-induced loss of dendritic spines. White arrows denote locations of spines present on day 0 that were lost after Tat infusion (days 2, 4, and 7). Scale bars, 50 μm. n = 5 for all groups. E, Scatter plot showing dose-dependent effects of ifenprodil administered at the indicated dose twice by intraperitoneal injection on spine density. Data are shown as mean (solid black diamonds) and individual data points (hollow circles). Abscissa is log scale, with 0 mg/kg inserted for reference. *Significant difference from saline-treated control group (n = 2–4 for each concentration). Inset, Representative micrographs of dendritic segment showing ifenprodil-induced gain of spines from before ifenprodil (0), after the first injection of 100 mg/kg ifenprodil (1), and after the last injection of ifenprodil (2). White arrows indicate new spines. Scale bar, 5 μm. F, Representative micrographs of Tat-induced loss and ifenprodil induced recovery of dendritic spines. White arrows denote locations of spines present at baseline and lost after Tat infusion and new spines that appear after ifenprodil rescue that were not present after infusion. Note loss of spines after rescue on day 11. Scale bar, 5 μm. n = 5 for all groups. G, Binned data from all groups analyzed with repeated-measures ANOVA to evaluate loss and rescue effects. Gray shading in B shows time periods analyzed in the following groups: baseline (days −7 through 0), infusion (days 2–7), rescue (day 9), and postinfusion (days 11–14). Data are shown as means ± SEM, *Significant difference between control and Tat-infused treatment conditions. #Significant difference from Tat-infused treatment group.
Results
Expression of HIV-1 Tat protein produces synapse loss and impairs cognitive function (Kim et al., 2008; Fitting et al., 2013). In in vitro studies, we found that the antagonist for GluN2B-containing NMDARs, ifenprodil, reversed Tat-induced synapse loss (Shin et al., 2012). Here, we examine the effects of HIV-1 Tat1–86 on synaptic spines in vivo with the objective of correlating Tat- and drug-induced changes in spines with changes in behavior. Our overall strategy was to image individual dendrites in Thy1-YFP transgenic mice using multiphoton imaging of dendritic spines emanating from layer 2/3 pyramidal neurons in the RSC. To take advantage of the longitudinal imaging of spines afforded by this technique, we administered Tat via ICV infusion to enable precise timing of the onset of Tat-induced effects on spine density. In behavioral studies, Tat was given by the same route of administration before assessing performance in contextual and trace fear conditioning, two behaviors that have been shown to rely on the RSC (Kwapis et al., 2015).
HIV-1 Tat decreases spine density
Our first objective was to determine the stability of spine measurements using multiphoton imaging. Imaging started 2 weeks after surgical placement of the cranial window and ICV cannula. The treatment groups were littermates imaged in parallel. Animals were anesthetized with isoflurane and fixed to the stage of the multiphoton microscope using a custom-machined head plate that minimized movement induced artifacts (Fig. 1A). Precise imaging locations were determined using a map of cortical surface vasculature. Three to five dendrites were imaged in each mouse and the mean change relative to baseline defined as a single subject (n = 1). As shown in Figure 2, A–C, spine density was stable across 4 baseline imaging sessions (days −7, −5, −3, 0) and did not change significantly over time. The mean baseline spine densities for control, ifenprodil, Tat, and Tat then ifenprodil-treated animals were 0.53 ± 0.05, 0.49 ± 0.03, 0.48 ± 0.02, and 0.52 ± 0.03 spines/μm dendrite, respectively. Immediately after imaging on day 0, each subject received ICV infusion of 100 ng of HIV-1 Tat1–86 (in 2 μl of PBS) or vehicle (Fig. 2A). Tat infusion decreased spine density significantly (Fig. 2B,D), as demonstrated with a paired t test comparing preinfusion and postinfusion spine counts, day 0 versus day 2 in Tat-treated mice (t(9) = 2.65, p < 0.05). Comparisons at individual time points showed that Tat-infused mice had significantly lower spine density relative to mice that received vehicle (control) on days 4 (p < 0.05), 7 (p < 0.005), and 9 (p < 0.01) after infusion. A maximal depression of spine density of 19 ± 3% was observed on day 9 after Tat infusion. Overall, Tat infusion decreased spine density significantly during the 9 d after infusion relative to the vehicle-infused control group or the paired baseline group (Fig. 2B). For a small cohort of animals, data collection was extended to 28 d after infusion. On day 28, the Tat-infused cohort remained lower than vehicle controls, but this trend did not reach significance (p = 0.084). Therefore, a single infusion of Tat decreased spine density significantly within 2 d and this loss persisted for at least 9 d.
High-dose but not low-dose ifenprodil increases dendritic spine density
Before testing ifenprodil on Tat-induced changes in spine density, we first examined the effects of different doses of the drug on spines in naive animals. We imaged Thy1-EGFP mice for 3 baseline sessions and then began daily administration of ifenprodil (0, 1, 10, or 100 mg/kg, i.p.) for 4 d. To capture the time point at which drug effects emerge, mice were imaged daily during ifenprodil administration. After 2 d of treatment, the high dose (100 mg/kg) increased spine density significantly, but the lower doses did not. Figure 2E shows dose-dependent effects of ifenprodil on spine density after 2 d of treatment. One-way ANOVA (F(3,9) = 7.1, p < 0.01) with post hoc examination showed that the high dose of ifenprodil enhanced spine density significantly compared with the control (saline) (p < 0.05) or 1 mg/kg (p < 0.05) treatment groups. Based on these data, we selected a subthreshold regimen of 2 d of 10 mg/kg ifenprodil as treatment for Tat-induced spine loss in the following studies.
Ifenprodil reverses Tat-induced spine loss
To determine whether spine loss induced by Tat could be reversed by ifenprodil, we administered drug intraperitoneally to mice during the period immediately preceding the peak of Tat-induced spine loss (Fig. 2B). Therefore, after imaging on day 7, mice received 10 mg/kg ifenprodil or saline and a second injection 24 h later (day 8). On day 9 after Tat infusion (24 h after the last ifenprodil injection), mice were imaged and spine densities from that day examined for potential rescue from Tat-induced loss or stand-alone effects of ifenprodil. Mice that were infused with Tat before receiving ifenprodil showed within-group Tat-induced spine loss (t(9) = 3.8, p < 0.01) and were significantly different from the vehicle-infused group on the 3 sessions after infusion on day 2 (p < 0.005), day 4 (p < 0.01), and day 7 (p < 0.05) (Fig. 2B). Ifenprodil treatment had no effect on spine density in mice that received vehicle infusions. However, in Tat-infused mice, ifenprodil increased spine density significantly, as demonstrated by a significant within-group increase in spine density from day 7 to day 9 (t(9) = 4.2, p < 0.005). Interestingly, this increase was temporary because spine density in these mice decreased significantly (t(9) = 3.5, p < 0.01) from day 9 to day 11. The transient effects of ifenprodil are consistent with its rapid clearance in mice (t1/2 = 4.4 h; Tallaksen-Greene et al., 2010).
To compare the effects of Tat infusion and ifenprodil treatment across all four treatment conditions, we used repeated-measures ANOVA of the binned data (individual subject averages) for experimental time periods, baseline (days −7, −5, −3, and 0), infusion (days 2, 4, and 7), rescue (day 9), and postinfusion (days 11 and 14) (Fig. 2G). Repeated-measures analysis of binned data, from baseline (days −7 through 0), infusion (days 2 through 7), rescue (day 9), and post-infusion (days 11 and 14) with infusion (vehicle or Tat), drug (saline or ifenprodil), and experimental time period (baseline, infusion, rescue, and postinfusion) as factors, revealed main effects of infusion (F(1,14) = 17.9, p < 0.005) and time period (F(3,45) = 12.9, p < 0.001) and interactions between infusion and time (F(3,45) = 8.0, p < 0.0005), drug and time (F(3,45) = 5.9, p < 0.005), and infusion by drug by time (F(3,445) = 9.0, p < 0.0001).
To more closely examine individual time bins, each time point was analyzed with a two-way ANOVA with infusion and drug treatment as factors (Fig. 2G). At the infusion time points, 2-way ANOVA showed a main effect of infusion (F(1,16) = 28.0, p < 0.0001), demonstrating a significant decrease in spine density induced by Tat infusion. Post hoc analysis showed that, at infusion time points (days 2–7), Tat significantly decreased spine density (p < 0.05) in both of the Tat-infused groups. At rescue (day 9), 2-way ANOVA showed a main effect of drug treatment (saline vs ifenprodil) (F(1,16) = 13.0, p < 0.005) and a drug by infusion interaction (F(1,16) = 12.6, p < 0.005). Post hoc analysis attributed these effects to ifenprodil, which significantly increased spine density to levels comparable to vehicle-infused control mice in Tat-treated mice (p < 0.05). Two-way ANOVA of postinfusion (days 11–14) showed a main effect of infusion (F(1,16) = 13.8, p < 0.005), but no other effects. Post hoc analysis showed that the Tat-infused/ifenprodil-treated group was significantly lower than control groups (p < 0.05), suggesting that the Tat-induced decrease in spine density reemerged in the days after rescue. Therefore, ifenprodil, at a dose without effect in naive animals, restored spine density to control levels in Tat-treated mice. Expression of Tat in the CNS of mice produces spine loss and impairs fear conditioning (Fitting et al., 2006, 2013; Carey et al., 2012), suggesting that ifenprodil might reverse the effects of Tat on behavior.
Ifenprodil rescues Tat-induced impairment of fear conditioning
To determine whether loss and recovery of dendritic spines in the RSC are accompanied by cognitive effects, we used the time course of Tat- and ifenprodil-induced changes in spine density to design fear conditioning experiments (Fig. 3A). C57B6 mice received surgical implantation of chronic indwelling intracranial cannula directed toward the right rostral portion of the lateral ventricle, identical to the placement of cannula for imaging studies (Fig. 3B). Before behavioral testing, mice were allowed 2–3 weeks to recover from surgery, matching the recovery period used in imaging studies and allowing acclimation to colony housing. Mice were infused with 100 ng of HIV-1 Tat1–86 or vehicle (2 μl PBS) on day 0. Prehandling and drug administration (saline or ifenprodil, 10 mg/kg, i.p.) began 7 d later to match the timing of the imaging experiments. Briefly, mice were weighed, acclimated to procedural cues (procedure room, cage location, experimenter handling), and administered ifenprodil or saline on each of 2 d (days 7 and 8) before fear conditioning. Twenty-four hours after the second prehandling day, mice were subject to standard trace fear conditioning procedures (two CS–US pairings, 30 s trace interval; Fig. 3A).
Figure 3.
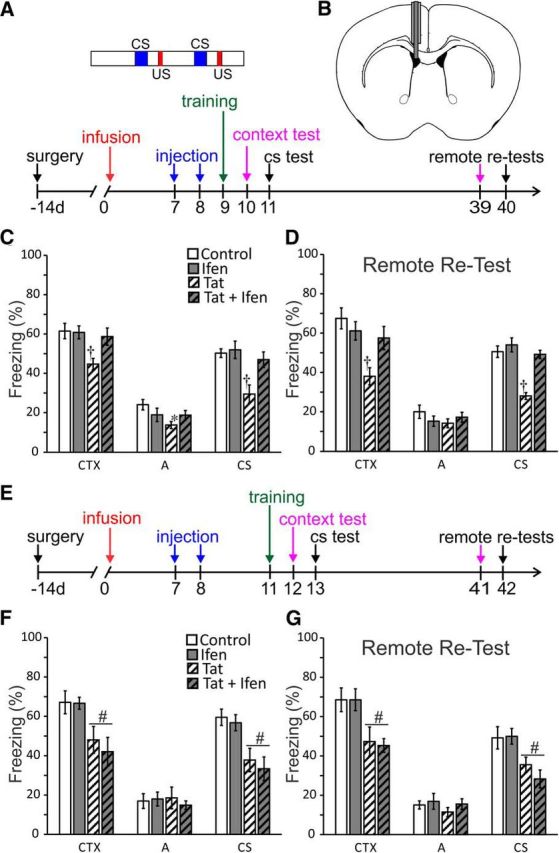
Ifenprodil rescues Tat-induced learning deficits. A, Schematic illustrating the timing of surgical cannula implantation, ifenprodil or saline administration (injection), and training and testing of context and trace (CS) fear conditioning relative to ICV vehicle or Tat infusion. Inset, Training scheme showing 120 s baseline, then 2 CS–US pairings (30 s CS, 30 s trace interval, and 2 s US), followed by a 60 s posttraining period. B, Diagram of coronal mouse brain illustrating placement of chronic indwelling guide cannula directed at lateral ventricle and infusion cannula used to administer Tat or vehicle. C, D and F, G, Bar graphs showing percentage freezing in context test (CTX) altered context test (A) and conditioned stimulus test (CS) for subject groups that received PBS (2 μl ICV) followed by intraperitoneal saline on days 7 and 8 (control), PBS then 10 mg/kg ifenprodil intraperitoneally (ifen), Tat (100 ng ICV) then saline (Tat) and Tat then ifenprodil (Tat + ifen). C, Infusion of Tat induced deficits in contextual and trace-cued fear conditioning. Ifenprodil rescued learning in mice treated with Tat, but had no effect on its own. D, Mice were retested 28 d later at a remote time point (days 39 and 40). n = 10 in all groups (C,D). All data are shown as mean ± SEM. *Significant difference from control. †Significant difference from all other treatment conditions. E, Diagram of timeline of shifted experiment. Training was shifted to after transient rescue of spine density by ifenprodil (day 11; Fig. 2B). F, Mice trained on day 11 (3 d after administration of ifenprodil or saline) showed deficits in contextual and trace-cued learning; ifenprodil failed to rescue cognitive function. G, Retesting 28 d later showed the same pattern of effects, a Tat-infusion induced deficit in contextual and trace-cued fear, but no effects of ifenprodil administration. n = 7 in groups treated with saline and n = 5 in groups treated with ifenprodil. All data are mean ± SEM. #Significant effect of Tat infusion.
To determine whether general exploration or US sensitivity was affected by Tat infusion or ifenprodil treatment, we analyzed locomotor activity during training (Table 1). All treatment groups showed similar locomotor activity during the training session, reported as averages of the first 120 s (baseline) and the last 60 s (post-US) of activity during the training context, with relatively high levels of activity at the beginning of the session and a general decrease in activity by the end of the session. In addition, examination of peak locomotion from the 30 s time bin during the first US presentation showed that all groups had similar responses to the foot shock (US). Lack of differences in activity during training suggests that Tat-induced effects on fear conditioning were not due to altered exploration of the training context or sensitivity to the US.
Table 1.
Locomotion data from training
| Baseline | US | Post-US | |
|---|---|---|---|
| Control | 1065 ± 91 | 2215 ± 258 | 520 ± 66 |
| Ifen | 1087 ± 102 | 2108 ± 181 | 471 ± 76 |
| Tat | 1090 ± 95 | 2204 ± 296 | 527 ± 68 |
| Tat + Ifen | 1011 ± 84 | 1987 ± 291 | 552 ± 56 |
Locomotor activity during baseline (first 120 s of training), two US presentations (30 s time period after each US), and the final 60 s of training (post-US). Data are shown as mean (max motion index units) ± SEM.
Two-way ANOVA of context freezing with infusion treatment (Tat or PBS) and drug treatment (ifenprodil or saline) as factors showed a main effect of Tat infusion (F(1,36) = 7.4, p < 0.01), no effect of drug treatment, and an infusion by drug treatment interaction (F(1,36) = 4.4, p < 0.05). Post hoc analysis revealed these effects to be driven by a significant deficit in the Tat-infused saline-treated group, which showed significantly less context freezing than all other groups (Fig. 3C). Therefore, Tat infusion resulted in a 27% deficit in context-induced freezing and ifenprodil rescued this behavior. The same analysis of freezing to the altered context showed a main effect of Tat infusion (F(1,36) = 4.5, p < 0.05) and an infusion by drug treatment interaction (F(1,36) = 4.2, p = 0.05). Post hoc analysis showed that these effects were driven by the Tat-infused saline-treated group, which was significantly lower than the PBS-infused saline-treated group (Fig. 3C). Lack of group differences during training (Table 1) suggests that this effect may reflect context generalization, where the weaker context learning in the Tat-infusion group generalizes to lower freezing in the altered context as well. Analysis of cued freezing data showed main effects of both infusion (F(1,36) = 12.2, p = 0.001) and drug treatment (F(1,36) = 6.7, p = 0.0.01), as well as an infusion by drug treatment interaction (F(1,36) = 4.6, p = 0.039) (Fig. 3C). Post hoc analysis isolated these effects to a significant deficit in the Tat-infused saline-treated group, which froze significantly less than all other groups. Therefore, Tat infusion resulted in a 41% deficit in trace-cued freezing and ifenprodil treatment rescued this behavior.
To determine whether the effects of Tat and ifenprodil on trace fear conditioning were long lasting, we retested mice at a remote time point (28 d after training). The use of remote testing is a standard approach to distinguish effects on learning from those on retrieval of fear memories and to assess changes in consolidation over time (Frankland and Bontempi, 2005). As shown in Figure 3D, the results were similar. Two-way ANOVA of context freezing, with infusion treatment (Tat or PBS) and drug treatment (ifenprodil or saline) as factors showed a main effect of Tat infusion (F(1,36) = 12.6, p < 0.005), no effect of drug treatment, and an infusion by drug treatment interaction (F(1,36) = 7.1, p < 0.05). Post hoc analysis revealed these effects to be driven by a significant deficit in the Tat-infused saline-treated group, which showed significantly less context freezing than all other groups (Fig. 3D). The same analysis of freezing in the altered context failed to detect an effect of treatment on freezing in the altered context (Fig. 3D). Analysis of cued freezing data from the remote tests showed main effects of both infusion (F(1,36) = 33.1, p < 0.0001) and drug treatment (F(1,36) = 26.9, p < 0.0001), as well as an interaction (F(1,36) = 12.9, p < 0.01) (Fig. 3D). Post hoc analysis isolated these effects to a significant deficit in the Tat-infused saline-treated group, which froze significantly less than all other groups. The persistence of behavioral deficits for at least 28 d suggests that the Tat-induced impairment results from impaired learning rather than expression of fear memories.
Ifenprodil rescued learning coincides with increased spine density
Because decreased spine density in medial prefrontal cortex and hippocampus correlates with impaired learning (Fitting et al., 2013; Leuner and Shors, 2013), we hypothesized that rescue of Tat-induced learning deficits by ifenprodil would coincide with ifenprodil-induced recovery of dendritic spines. To test this hypothesis, we shifted training to day 11 after Tat infusion (Fig. 3E). At this time, the effects of ifenprodil on spine density had subsided, but Tat-induced spine loss was still pronounced (Fig. 2B). We found that Tat-induced impairment of contextual and trace-cued conditioning remained at the shifted training time and, consistent with our hypothesis, ifenprodil failed to rescue cognitive function at this shifted training time.
Two-way ANOVA of context freezing with infusion treatment (Tat or PBS) and drug treatment (ifenprodil or saline) as factors showed a main effect of Tat infusion (F(1,20) = 17.8, p < 0.0005), but no other effects, demonstrating a clear effect of Tat infusion on contextual learning (Fig. 3F). There was no effect of treatment on freezing in the altered context (Fig. 3F). Analysis of cued freezing data showed a main effect of Tat infusion (F(1,20) = 25.1, p < 0.0001), but no other effects, indicating a clear effect of Tat infusion on trace-cued learning (Fig. 3F). Therefore, Tat-induced learning deficits persist on day 11 after infusion, but the ifenprodil treatment effects are no longer present. Therefore, the effects of ifenprodil on Tat-induced behavioral deficits track with changes in spine density.
To determine whether this pattern of effects was long lasting, we retested mice at a remote time point (28 d) after training. Retesting showed that deficits induced by Tat infusion remained (Fig. 3G). Two-way ANOVA of context freezing with infusion treatment (Tat or PBS) and drug treatment (ifenprodil or saline) as factors showed a main effect of Tat infusion (F(1,20) = 16.5, p < 0.001), but no effect of drug treatment and no interaction. Similarly, analysis of freezing during the CS showed a main effect of Tat treatment (F(1,20) = 15.9, p < 0.001), but no other effects. In addition, there were no effects of Tat infusion or drug treatment on freezing to the altered context. These findings confirm the above result that Tat-induced deficits in learning are persistent and that shifting behavioral training until after washout of the transient rescue of dendritic spines by ifenprodil prevents behavioral recovery.
Collectively, the effects of Tat infusion and ifenprodil treatment on both the contextual and cued components of trace fear conditioning suggest that the spine loss demonstrated with in vivo imaging is accompanied by deficits in learning and memory and that both spine density and behavioral function can be rescued by the GluN2B antagonist ifenprodil. In addition, the persistence of both deficit and pharmacological rescue suggest that Tat infusion and ifenprodil administration act on acquisition or consolidation of contextual and trace-cued learning, rather than by affecting retrieval or expression. Further, ifenprodil rescues cognitive function with a time course that indicates rescued learning coincides with the time course of increased spine density, suggesting that recovery of lost spines may mediate restoration of cognitive function.
Discussion
Using in vivo longitudinal multiphoton imaging, we found that ICV administration of HIV-1 Tat decreased dendritic spine density in layer 1 of RSC and that ifenprodil, an antagonist of GluN2B-containing NMDARs, rescued spine loss. Therefore, synapse loss, the hallmark of cognitive decline in HAND, is reversible. Tat infusion impaired trace fear conditioning, an associative conditioning task that relies on RSC (Kwapis et al., 2015). Ifenprodil restored cognitive function in Tat-treated animals coincident with restoration of spine density. This is the first report showing recovery of spine density and cognitive function in a model of HAND. These findings suggest that rescue of dendritic spines restores cognitive function and may be a viable target for treatment of HAND (Fig. 4).
Figure 4.

Effects of Tat and ifenprodil on learning and dendritic spines. Diagram shows hypothetical relationship of Tat and ifenprodil-induced changes in spine density to the ability to learn. Shortly after Tat infusion, a deficit in learning and decreased spine density are evident. Loss of spines may be driven by homeostatic compensation to the excitotoxic effects of Tat, resulting in decreased ability to encode new memories. Ifenprodil administration temporarily rescues spine density and the ability to learn.
This is the first description of HIV-1 Tat-associated spine loss in RSC, which may be of particular relevance to HAND because the thickness of the RSC is decreased in these patients (Shin et al., 2017) and because of its role in spatial cognition and associative learning and memory (Vann et al., 2009; Bucci and Robinson, 2014), functions that are impaired in HAND patients (Heaton et al., 2011). In addition, RSC dysfunction occurs early in the development of Alzheimer's-related learning deficits (Aggleton, 2014). Therefore, like a “canary in a coal mine,” the RSC's susceptibility to early damage may make it a valuable brain region for assessing disease progression in neuroinflammatory models.
HIV Tat affects multiple brain regions and behaviors. Tat is detectable in hippocampus relatively soon after ICV infusion (Li et al., 2004), suggesting that the toxic protein moves through brain tissue. However, actual disposition after ICV infusion is difficult to determine. The 100 ng Tat infusion used in the present studies would be expected to induce central cytokine activation (Lawson et al., 2011). Indeed, Tat activates microglia (Chivero et al., 2017) and produces astrogliosis (Jones et al., 1998; Hahn et al., 2015; Harricharan et al., 2015), possibly accounting for the long-lasting spine loss described here. The degree to which a Tat-induced neuroinflammatory response contributes synaptodendritic damage in HAND patients is not known. Doses of Tat >20 times that used in the present study produce lesions near the ventricle (Philippon et al., 1994). Transgenic expression of HIV-1 Tat increases anxiety-like behavior in mice (Paris et al., 2014). However, it is unlikely that the present findings are due to increases in anxiety because we show no effects of Tat infusion on locomotion during training (Table 1) and increases in anxiety would be expected to enhance fear conditioning instead of producing the deficits described here. Expression of Tat in the CNS or its direct intracranial infusion decreases dendritic spine density in hippocampus and medial prefrontal cortex, impairs spatial and contextual learning, and inhibits long-term potentiation (Li et al., 2004; Carey et al., 2012; Fitting et al., 2013). Tat disrupts the dopaminergic system (Zauli et al., 2000), which is critically implicated in trace conditioning involving the RSC (Runyan and Dash, 2004; Katche et al., 2013). Therefore, whereas the present findings show clear temporal association of the effects of Tat on spine density in RSC with changes in cognitive function, this may reflect the distributed effects of Tat on multiple brain systems and a broad-based disruption may be ultimately responsible for cognitive dysfunction. Interestingly, if Tat-induced spine loss is symptomatic of actions throughout the CNS, then rescue by ifenprodil may also represent a broad-based restoration of function.
This is the first report showing that an NMDAR antagonist administered after an HIV protein reverses functional loss. The rescue of spine density and learning produced by ifenprodil is surprising given the acute effects of antagonists for GluN2B-containing NMDARs, which generally impair cognitive function (Weed et al., 2016). For example, the GluN2B antagonist Ro25-6981 blocks extinction of fear conditioning (Corcoran et al., 2013) and inhibits trace fear conditioning (Gilmartin et al., 2013). This discrepancy is explained by noting that, in the present experiments, ifenprodil was cleared before imaging or training of trace fear conditioning because it was administered 24 h before the experiment and has a half-life of 4.4 h in mice (Tallaksen-Greene et al., 2010). The increased ability to learn 24 h after administration of an antagonist of GluN2B-containing NMDARs is consistent with enhanced induction of LTP in rat hippocampal slices after treatment with the GluN2B antagonist CP-101,606 (Graef et al., 2015). Indeed, treatments that increase hippocampal dendritic spines facilitate induction of LTP and enhance hippocampus dependent learning (Leuner and Shors, 2013). For example, in the APP transgenic mouse, insulin-like growth factor 2 restored spine density and reversed memory deficits (Pascual-Lucas et al., 2014). The timing of ifenprodil-induced recovery of cognitive function suggests that cognitive rescue is optimal when spine density is maximal and that, once learned, the conditioned association is stable, persisting through remote testing. However, whether Tat-induced spine loss affects retrieval is not clear. Tat and ifenprodil might affect a subset of spines that support acquisition without affecting more stable spines involved in long-term consolidation or perhaps the RSC is not the final locus of these memories. It is intriguing that ifenprodil, at the dose used in this study, did not increase spines or affect cognitive function in animals that had not first been treated with HIV Tat. This dependence on disease state shares some features with the ability of antagonists of GluN2B-containing NMDARs to rescue spines and reverse depression-like behavior (Miller et al., 2014). Indeed, stress and inflammation induce spine loss in animals (Duman and Duman, 2015) and trigger depression in humans, leading some researchers to describe depression as a neurodegenerative disease (Abdallah et al., 2015). Depression is common in HIV-1-infected patients (Heikinheimo et al., 2015). Clearly, treatments that restore spine density to “normal” levels are preferable to treatments that lead to the formation of excess excitatory synapses that could lower the seizure threshold (Takahashi et al., 2016).
HIV Tat-induced loss of excitatory synapses and their rescue by ifenprodil was initially described in primary neuronal cultures (Kim et al., 2008; Shin et al., 2012). The same mechanisms may underlie the effects described here. Tat binds to lipoprotein receptors on neurons, subsequently activating Src, followed by phosphorylation-mediated potentiation of NMDARs (Haughey et al., 2001; Krogh et al., 2014; Green and Thayer, 2016). Other HIV proteins and inflammatory cytokines similarly potentiate NMDARs via different upstream pathways (Kim et al., 2011; Mishra et al., 2012). Synapse loss requires activation of GluN2A-containing NMDARs. Blocking GluN2B-containing NMDARs with ifenprodil fails to prevent synapse loss, but restores synaptic density after 24 h of exposure to Tat (Shin et al., 2012). There is precedent for activity-dependent suppression of synapse formation by GluN2B-containing NMDARs during development (Gray et al., 2011). We interpret these findings to indicate that, after exposure to Tat, synapse loss is a homeostatic mechanism that compensates for excessive activation of NMDARs and that chronic activation of GluN2B-containing NMDARs suppresses synapse recovery. The idea that loss of excitatory synapses is part of a homeostatic mechanism is consistent with the increase in inhibitory synapses induced by Tat exposure (Hargus and Thayer, 2013). In addition, experiments using microfluidic chambers to separate direct from spreading excitotoxicity show that activation of GluN2A-containing NMDARs protects neural networks from spreading cell death and that the pharmacology changes after the insult, providing a new therapeutic window (Samson et al., 2016). Going forward, it will be important to determine whether restoring spine density in the presence of HIV neurotoxins, which, as shown here, coincides with improved cognitive function, puts rescued networks at risk for excitotoxic damage.
The ifenprodil-mediated restoration of cognitive function described here was transient, raising the question of whether chronic drug administration could sustain improved function. Our longitudinal imaging experiments and carefully timed behavioral testing were designed to determine whether Tat-induced loss and ifenprodil-induced recovery of spines predicted behavioral outcomes. Chronic neuroinflammation more accurately reflects the conditions of HAND and prolonged drug administration is a more realistic therapeutic approach. We look forward to pursuing such studies. However, the longitudinal studies described here demonstrate that GluN2B-containing NMDARs, signaling pathways downstream from NMDARs, as well as the numerous other mechanisms that regulate synapse loss and formation are all targets of potential interest for improving cognitive function in neuroinflammatory diseases. Although drugs that selectively inhibit NMDAR subtypes may prove unsuitable for chronic use, their success in experimental models such as Tat-induced neurotoxicity bodes well for strategies that rescue excitatory synapses and may prove broadly applicable to neuroinflammatory and neurodegenerative diseases.
HIV-1 Tat induces NMDAR potentiation, which drives adaptive changes, including the loss of dendritic spines described here. Homeostatic mechanisms exist to normalize neuronal excitability, but in some disease states (such as chronic stress, drug addiction, and neuroinflammation), these mechanisms may generate allostatic state shifts that contribute to long-term neural and cognitive dysfunction (Koob and Le Moal, 1997). Better understanding of neural disease states induced by exposure to neurotoxins such as HIV-1 Tat may reveal therapeutic targets through which we may be able to restore function. Indeed, the pharmacological rescue of synapses lost during exposure to HIV-1 neurotoxins sets an encouraging precedent: synapse loss that correlates with neurocognitive impairment in diseases such as HAND can be reversed.
Footnotes
This work was supported by the National Institutes of Health (National Institute on Drug Abuse Grants DA07304 and DA035663 to SAT). Multiphoton imaging was conducted with support of the University Imaging Centers at the University of Minnesota. We thank Dr. Guillermo Marques for help in designing the imaging assay; Drs. Mark Thomas and Michael Benneyworth for helpful discussions regarding behavior experiments; and Mustafa Mithaiwala and Ansel Shuster for assistance with colony maintenance and data quantification.
The authors declare no competing financial interests.
References
- Abdallah CG, Sanacora G, Duman RS, Krystal JH (2015) Ketamine and rapid-acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annu Rev Med 66:509–523. 10.1146/annurev-med-053013-062946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggleton JP. (2014) Looking beyond the hippocampus: old and new neurological targets for understanding memory disorders. Proc Biol Sci 281: pii: 20140565. 10.1098/rspb.2014.0565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachani M, Sacktor N, McArthur JC, Nath A, Rumbaugh J (2013) Detection of anti-tat antibodies in CSF of individuals with HIV-associated neurocognitive disorders. J Neurovirol 19:82–88. 10.1007/s13365-012-0144-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier T, Neuwirth E (2007) Excel::COM::R. Computational Statistics 22:91–108. 10.1007/s00180-007-0023-6 [DOI] [Google Scholar]
- Bertrand SJ, Aksenova MV, Mactutus CF, Booze RM (2013) HIV-1 Tat protein variants: critical role for the cysteine region in synaptodendritic injury. Exp Neurol 248:228–235. 10.1016/j.expneurol.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci DJ, Robinson S (2014) Toward a conceptualization of retrohippocampal contributions to learning and memory. Neurobiol Learn Mem 116:197–207. 10.1016/j.nlm.2014.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey AN, Sypek EI, Singh HD, Kaufman MJ, McLaughlin JP (2012) Expression of HIV-Tat protein is associated with learning and memory deficits in the mouse. Behav Brain Res 229:48–56. 10.1016/j.bbr.2011.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivero ET, Guo ML, Periyasamy P, Liao K, Callen SE, Buch S (2017) HIV-1 Tat primes and activates microglial NLRP3 inflammasome-mediated neuroinflammation. J Neurosci 37:3599–3609. 10.1523/JNEUROSCI.3045-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran KA, Leaderbrand K, Radulovic J (2013) Extinction of remotely acquired fear depends on an inhibitory NR2B/PKA pathway in the retrosplenial cortex. J Neurosci 33:19492–19498. 10.1523/JNEUROSCI.3338-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman CH, Duman RS (2015) Spine synapse remodeling in the pathophysiology and treatment of depression. Neurosci Lett 601:20–29. 10.1016/j.neulet.2015.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E (2007) HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci 8:33–44. 10.1038/nrn2040 [DOI] [PubMed] [Google Scholar]
- Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW (2007) HIV-tat induces formation of an LRP-PSD-95-NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A 104:3438–3443. 10.1073/pnas.0611699104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Booze RM, Hasselrot U, Mactutus CF (2006) Intrahippocampal injections of Tat: effects on prepulse inhibition of the auditory startle response in adult male rats. Pharmacol Biochem Behav 84:189–196. 10.1016/j.pbb.2006.04.014 [DOI] [PubMed] [Google Scholar]
- Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, Knapp PE, Guido W, Hauser KF (2013) Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 tat transgenic mice. Biol Psychiatry 73:443–453. 10.1016/j.biopsych.2012.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B (2005) The organization of recent and remote memories. Nat Rev Neurosci 6:119–130. [DOI] [PubMed] [Google Scholar]
- Gilmartin MR, Kwapis JL, Helmstetter FJ (2013) NR2A- and NR2B-containing NMDA receptors in the prelimbic medial prefrontal cortex differentially mediate trace, delay, and contextual fear conditioning. Learn Mem 20:290–294. 10.1101/lm.030510.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldey GJ, Roumis DK, Glickfeld LL, Kerlin AM, Reid RC, Bonin V, Schafer DP, Andermann ML (2014) Removable cranial windows for long-term imaging in awake mice. Nat Protoc 9:2515–2538. 10.1038/nprot.2014.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef JD, Newberry K, Newton A, Pieschl R, Shields E, Luan FN, Simmermacher J, Luchetti D, Schaeffer E, Li YW, Kiss L, Bristow LJ (2015) Effect of acute NR2B antagonist treatment on long-term potentiation in the rat hippocampus. Brain Res 1609:31–39. 10.1016/j.brainres.2015.03.019 [DOI] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, Nicoll RA (2011) Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron 71:1085–1101. 10.1016/j.neuron.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MV, Thayer SA (2016) NMDARs adapt to neurotoxic HIV protein Tat downstream of a GluN2A-ubiquitin ligase signaling pathway. J Neurosci 36:12640–12649. 10.1523/JNEUROSCI.2980-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn YK, Podhaizer EM, Farris SP, Miles MF, Hauser KF, Knapp PE (2015) Effects of chronic HIV-1 Tat exposure in the CNS: heightened vulnerability of males versus females to changes in cell numbers, synaptic integrity, and behavior. Brain Struct Funct 220:605–623. 10.1007/s00429-013-0676-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus NJ, Thayer SA (2013) Human immunodeficiency virus-1 Tat protein increases the number of inhibitory synapses between hippocampal neurons in culture. J Neurosci 33:17908–17920. 10.1523/JNEUROSCI.1312-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harricharan R, Thaver V, Russell VA, Daniels WM (2015) Tat-induced histopathological alterations mediate hippocampus-associated behavioural impairments in rats. Behav Brain Funct 11:3. 10.1186/s12993-014-0047-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD (2001) HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem 78:457–467. 10.1046/j.1471-4159.2001.00396.x [DOI] [PubMed] [Google Scholar]
- Heaton RK, et al. (2011) HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol 17:3–16. 10.1007/s13365-010-0006-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikinheimo T, Poutiainen E, Salonen O, Elovaara I, Ristola M (2015) Three-decade neurological and neurocognitive follow-up of HIV-1-infected patients on best-available antiretroviral therapy in Finland. BMJ Open 5:e007986. 10.1136/bmjopen-2015-007986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TP, Patel K, Johnson KR, Maric D, Calabresi PA, Hasbun R, Nath A (2013) Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc Natl Acad Sci U S A 110:13588–13593. 10.1073/pnas.1308673110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M, Olafson K, Del Bigio MR, Peeling J, Nath A (1998) Intraventricular injection of human immunodeficiency virus type 1 (Hiv-1) Tat protein causes inflammation, gliosis, apoptosis, and ventricular enlargement. J Neuropath Exp Neurol 57:563–570. 10.1097/00005072-199806000-00004 [DOI] [PubMed] [Google Scholar]
- Katche C, Dorman G, Gonzalez C, Kramar CP, Slipczuk L, Rossato JI, Cammarota M, Medina JH (2013) On the role of retrosplenial cortex in long-lasting memory storage. Hippocampus 23:295–302. 10.1002/hipo.22092 [DOI] [PubMed] [Google Scholar]
- Kim HJ, Martemyanov KA, Thayer SA (2008) Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci 28:12604–12613. 10.1523/JNEUROSCI.2958-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Shin AH, Thayer SA (2011) Activation of cannabinoid type 2 receptors inhibits HIV-1 envelope glycoprotein gp120-induced synapse loss. Mol Pharmacol 80:357–366. 10.1124/mol.111.071647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M (1997) Drug abuse: hedonic homeostatic dysregulation. Science 278:52–58. 10.1126/science.278.5335.52 [DOI] [PubMed] [Google Scholar]
- Krogh KA, Wydeven N, Wickman K, Thayer SA (2014) HIV-1 protein Tat produces biphasic changes in NMDA-evoked increases in intracellular Ca concentration via activation of Src kinase and nitric oxide signaling pathways. J Neurochem 130:642–656. 10.1111/jnc.12724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwapis JL, Jarome TJ, Lee JL, Helmstetter FJ (2015) The retrosplenial cortex is involved in the formation of memory for context and trace fear conditioning. Neurobiol Learn Mem 123:110–116. 10.1016/j.nlm.2015.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson MA, Kelley KW, Dantzer R (2011) Intracerebroventricular administration of HIV-1 Tat induces brain cytokine and indoleamine 2,3-dioxygenase expression: a possible mechanism for AIDS comorbid depression. Brain Behav Immun 25:1569–1575. 10.1016/j.bbi.2011.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuner B, Shors TJ (2013) Stress, anxiety, and dendritic spines: what are the connections? Neuroscience 251:108–119. 10.1016/j.neuroscience.2012.04.021 [DOI] [PubMed] [Google Scholar]
- Li ST, Matsushita M, Moriwaki A, Saheki Y, Lu YF, Tomizawa K, Wu HY, Terada H, Matsui H (2004) HIV-1 Tat inhibits long-term potentiation and attenuates spatial learning. Ann Neurol 55:362–371. 10.1002/ana.10844 [DOI] [PubMed] [Google Scholar]
- Li W, Li G, Steiner J, Nath A (2009) Role of Tat Protein in HIV Neuropathogenesis. Neurotox Res 16:205–220. 10.1007/s12640-009-9047-8 [DOI] [PubMed] [Google Scholar]
- Miller OH, Yang L, Wang CC, Hargroder EA, Zhang Y, Delpire E, Hall BJ (2014) GluN2B-containing NMDA receptors regulate depression-like behavior and are critical for the rapid antidepressant actions of ketamine. eLife 3:e03581. 10.7554/eLife.03581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Kim HJ, Shin AH, Thayer SA (2012) Synapse loss induced by interleukin-1beta requires pre- and post-synaptic mechanisms. J Neuroimmune Pharmacol 7:571–578. 10.1007/s11481-012-9342-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris JJ, Singh HD, Ganno ML, Jackson P, McLaughlin JP (2014) Anxiety-like behavior of mice produced by conditional central expression of the HIV-1 regulatory protein, Tat. Psychopharmacology (Berl) 231:2349–2360. 10.1007/s00213-013-3385-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Lucas M, Viana da Silva S, Di Scala M, Garcia-Barroso C, González-Aseguinolaza G, Mulle C, Alberini CM, Cuadrado-Tejedor M, Garcia-Osta A (2014) Insulin-like growth factor 2 reverses memory and synaptic deficits in APP transgenic mice. EMBO Mol Med 6:1246–1262. 10.15252/emmm.201404228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paukert M, Bergles DE (2012) Reduction of motion artifacts during in vivo two-photon imaging of brain through heartbeat triggered scanning. J Physiol 590:2955–2963. 10.1113/jphysiol.2012.228114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin K (2012) The mouse brain in stereotaxic coordinates. San Diego: Academic. [Google Scholar]
- Peters A, Kaiserman-Abramof IR (1970) The small pyramidal neuron of the rat cerebral cortex: the perikaryon, dendrites and spines. Am J Anat 127:321–355. 10.1002/aja.1001270402 [DOI] [PubMed] [Google Scholar]
- Philippon V, Vellutini C, Gambarelli D, Harkiss G, Arbuthnott G, Metzger D, Roubin R, Filippi P (1994) The basic domain of the lentiviral Tat protein is responsible for damages in mouse brain: involvement of cytokines. Virology 205:519–529. 10.1006/viro.1994.1673 [DOI] [PubMed] [Google Scholar]
- Runyan JD, Dash PK (2004) Intra-medial prefrontal administration of SCH-23390 attenuates ERK phosphorylation and long-term memory for trace fear conditioning in rats. Neurobiol Learn Mem 82:65–70. 10.1016/j.nlm.2004.04.006 [DOI] [PubMed] [Google Scholar]
- Samson AJ, Robertson G, Zagnoni M, Connolly CN (2016) Neuronal networks provide rapid neuroprotection against spreading toxicity. Sci Rep 6:33746. 10.1038/srep33746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, Mankowski JL, Brown A, Volsky DJ, McArthur JC (2016) HIV-associated neurocognitive disorder: pathogenesis and prospects for treatment. Nat Rev Neurol 12:234–248. 10.1038/nrneurol.2016.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin AH, Kim HJ, Thayer SA (2012) Subtype selective NMDA receptor antagonists induce recovery of synapses lost following exposure to HIV-1 Tat. Br J Pharmacol 166:1002–1017. 10.1111/j.1476-5381.2011.01805.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin NY, Hong J, Choi JY, Lee SK, Lim SM, Yoon U (2017) Retrosplenial cortical thinning as a possible major contributor for cognitive impairment in HIV patients. Eur Radiol. In press. 10.1007/s00330-017-4836-6 [DOI] [PubMed] [Google Scholar]
- Takahashi DK, Gu F, Parada I, Vyas S, Prince DA (2016) Aberrant excitatory rewiring of layer V pyramidal neurons early after neocortical trauma. Neurobiol Dis 91:166–181. 10.1016/j.nbd.2016.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallaksen-Greene SJ, Janiszewska A, Benton K, Ruprecht L, Albin RL (2010) Lack of efficacy of NMDA receptor-NR2B selective antagonists in the R6/2 model of Huntington disease. Exp Neurol 225:402–407. 10.1016/j.expneurol.2010.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipps ME, Raybuck JD, Buck KJ, Lattal KM (2014) Delay and trace fear conditioning in C57BL/6 and DBA/2 mice: issues of measurement and performance. Learn Mem 21:380–393. 10.1101/lm.035261.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vann SD, Aggleton JP, Maguire EA (2009) What does the retrosplenial cortex do? Nat Rev Neurosci 10:792–802. 10.1038/nrn2733 [DOI] [PubMed] [Google Scholar]
- Weed MR, Bookbinder M, Polino J, Keavy D, Cardinal RN, Simmermacher-Mayer J, Cometa FN, King D, Thangathirupathy S, Macor JE, Bristow LJ (2016) Negative allosteric modulators selective for the NR2B subtype of the NMDA receptor impair cognition in multiple domains. Neuropsychopharmacology 41:568–577. 10.1038/npp.2015.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner JM, Radcliffe RA (2004) Cued and contextual fear conditioning in mice. Curr Protoc Neurosci Chapter 8:Unit 8.5C. 10.1002/0471142301.ns0805cs27 [DOI] [PubMed] [Google Scholar]
- Zauli G, Secchiero P, Rodella L, Gibellini D, Mirandola P, Mazzoni M, Milani D, Dowd DR, Capitani S, Vitale M (2000) HIV-1 Tat-mediated inhibition of the tyrosine hydroxylase gene expression in dopaminergic neuronal cells. J Biol Chem 275:4159–4165. 10.1074/jbc.275.6.4159 [DOI] [PubMed] [Google Scholar]