

Abstract
Epilepsy after pediatric traumatic brain injury (TBI) is associated with poor quality of life. This study aimed to characterize post-traumatic epilepsy in a mouse model of pediatric brain injury, and to evaluate the role of interleukin-1 (IL-1) signaling as a target for pharmacological intervention. Male mice received a controlled cortical impact or sham surgery at postnatal day 21, approximating a toddler-aged child. Mice were treated acutely with an IL-1 receptor antagonist (IL-1Ra; 100 mg/kg, s.c.) or vehicle. Spontaneous and evoked seizures were evaluated from video-EEG recordings. Behavioral assays tested for functional outcomes, postmortem analyses assessed neuropathology, and brain atrophy was detected by ex vivo magnetic resonance imaging. At 2 weeks and 3 months post-injury, TBI mice showed an elevated seizure response to the convulsant pentylenetetrazol compared with sham mice, associated with abnormal hippocampal mossy fiber sprouting. A robust increase in IL-1β and IL-1 receptor were detected after TBI. IL-1Ra treatment reduced seizure susceptibility 2 weeks after TBI compared with vehicle, and a reduction in hippocampal astrogliosis. In a chronic study, IL-1Ra-TBI mice showed improved spatial memory at 4 months post-injury. At 5 months, most TBI mice exhibited spontaneous seizures during a 7 d video-EEG recording period. At 6 months, IL-1Ra-TBI mice had fewer evoked seizures compared with vehicle controls, coinciding with greater preservation of cortical tissue. Findings demonstrate this model's utility to delineate mechanisms underlying epileptogenesis after pediatric brain injury, and provide evidence of IL-1 signaling as a mediator of post-traumatic astrogliosis and seizure susceptibility.
SIGNIFICANCE STATEMENT Epilepsy is a common cause of morbidity after traumatic brain injury in early childhood. However, a limited understanding of how epilepsy develops, particularly in the immature brain, likely contributes to the lack of efficacious treatments. In this preclinical study, we first demonstrate that a mouse model of traumatic injury to the pediatric brain reproduces many neuropathological and seizure-like hallmarks characteristic of epilepsy. Second, we demonstrate that targeting the acute inflammatory response reduces cognitive impairments, the degree of neuropathology, and seizure susceptibility, after pediatric brain injury in mice. These findings provide evidence that inflammatory cytokine signaling is a key process underlying epilepsy development after an acquired brain insult, which represents a feasible therapeutic target to improve quality of life for survivors.
Keywords: cytokine, interleukin, neurotrauma, pediatric, seizure, traumatic brain injury
Introduction
Post-traumatic epilepsy (PTE) is a common cause of morbidity after pediatric traumatic brain injury (pTBI). Defined as the occurrence of two or more unprovoked seizures >1 week following TBI (Annegers et al., 1980), PTE has a reported incidence of 10–20%, with a higher risk associated with increased injury severity (up to 35% for severe TBI; Ates et al., 2006), and a younger age at insult (greatest <2 years of age; Ates et al., 2006; Arndt et al., 2013). PTE in the pediatric population is associated with worse functional and cognitive outcomes (Barlow et al., 2000; Arndt et al., 2013). PTE is often resistant to medical management, and no efficacious anti-epileptogenic therapy to prevent its development has been identified (Barlow et al., 2000).
Current understanding of the biological mechanisms underlying epileptogenesis, or the process by which epilepsy is acquired after a brain insult, is based primarily on TBI models in adult animals, despite evidence that seizure susceptibility differs between the immature and adult brain, with developmental changes during early life favoring neuronal excitability (Nardou et al., 2013). Several putative mechanisms of PTE have been proposed, including alterations in neurotransmission, hippocampal cell loss, mossy fiber sprouting, altered calcium-mediated second messenger activity, and neuroinflammation (Hunt et al., 2013). An improved understanding of these mechanisms, particularly in the context of a young brain that is still maturing, may be critical to preventing PTE and improving the outcomes of children with TBI.
Increasing evidence from patient studies and experimental models implicates inflammatory cytokine signaling in seizure induction and propagation via both direct and indirect mechanisms (Webster et al., 2017). In the immature brain, which shows particular vulnerability to neuroinflammation (Potts et al., 2006), even a modest or subthreshold inflammatory response can alter long-term susceptibility to seizures (Galic et al., 2008). Interleukin-1 β (IL-1β), a classically proinflammatory mediator of acute pathogenesis after injury (Brough et al., 2011), has been reported to initiate and exacerbate seizure activity in experimental models of epilepsy (Vezzani et al., 1999, 2000; Dubé et al., 2005). Conversely, preventing IL-1β biosynthesis or blocking signaling via the IL-1R1 receptor can ameliorate seizure severity and incidence (Vezzani et al., 2000; Maroso et al., 2011). To date, however, the potential role of IL-1 signaling in seizure activity after a traumatic insult has not been explored.
One factor that contributes to our limited understanding of PTE mechanisms is a lack of age-appropriate, clinically relevant experimental models (Pitkänen et al., 2009). We have established a reproducible model of unilateral traumatic injury to the mouse brain at postnatal day (P)21, that results in progressive neurodegeneration and behavioral deficits representative of TBI in toddler-aged children (Pullela et al., 2006; Semple et al., 2015). In this study, we first applied the pTBI model to evaluate the effect of early-life brain injury on seizure susceptibility and epilepsy-associated neuropathology over time. Second, to determine whether IL-1 signaling is an underlying mechanism involved in post-traumatic epileptogenesis, we administered a clinically available IL-1R antagonist acutely after pTBI, and examined seizure susceptibility and neurodegeneration across a time course. Together, our findings confirm persistent hyperexcitability after early-life brain injury, and provide evidence of IL-1 signaling as a mediator of post-traumatic epileptogenesis.
Materials and Methods
Experiments were conducted at either the University of California San Francisco (UCSF; see Figs. 1–4) or the University of Melbourne (UoM), Australia (see Figs. 5–8), according to the NIH and Australian guidelines for the care and use of laboratory animals, with approval from Institutional Animal Care and Use Committees (no. AN084572 and 15-016). Analyses were conducted blinded to injury and treatment group. Group allocation was concealed by a third party following randomization at surgery using a random number generator.
Figure 1.

Enhanced susceptibility to evoked seizures and epilepsy-like changes in axonal sprouting after pTBI. Following TBI or sham at P21, mice were administered PTZ at either (A) 2 weeks (50 mg/kg, i.p.) or (B) 3 months (35 mg/kg, i.p.) to evaluate seizure susceptibility (Mann–Whitney tests, *p < 0.05,***p < 0.001; n = 7–10/group). Immunofluorescence staining for ZnT3 mossy fibers, NeuN+ neurons, and DAPI+ nuclei in the injured hippocampus revealed abnormal mossy fiber sprouting at 3 months post-injury (D and E, arrows) but not at 2 weeks (C). n = 7–10/group. Scale bars: C, D, 200 μm; E, 100 μm.
Figure 4.

rIL-1Ra treatment attenuates subacute astrogliosis after pTBI. Quantification of GFAP+ reactivity from immunofluorescent labeling with DAPI (A) at 2 weeks post-injury, revealed a robust increase in astrogliosis in the ipsilateral DG hilus (B; two-way RM ANOVA injury × drug interaction: F(1,17) = 5.04, p = 0.04; post hoc, p < 0.01) and CA1 (C; injury × drug interaction: F(1,17) = 8.76, p < 0.01; post hoc, p < 0.001) of vehicle-TBI mice, which was attenuated in rIL-1Ra-TBI mice. n = 9–10/group. Scale bar, 200 μm. Region-of-interest indicated by dotted white line in A. This was reflected by changes in gene expression of astrocyte markers Gfap (E) and Vim (F), which were robustly elevated at 1 d after injury in the cortex (Gfap: F(2,12) = 28.06, p < 0.0001; Vim: F(2,12) = 17.45, p < 0.01) and hippocampus (Gfap: F(2,12) = 5.04, p = 0.03; post hoc, p < 0.05; Vim: F(2,12) = 6.50, p = 0.01). n = 5/group; *p < 0.01, **p < 0.01, ***p < 0.001.
Figure 5.

rIL-1Ra treatment attenuates chronic cognitive deficits after pTBI. Mice received either rIL-1Ra or vehicle treatment for 7 d following pTBI or sham surgery, and underwent behavioral assessment at 3 months (A). All mice were able to locate a visible platform in the MWM (B; three-way RM ANOVA, within-subjects effect of time: F(1,51) = 199.94, p < 0.0001). In the hidden platform trials (C), all groups again showed an overall improvement with time (F(1,51) = 30.35, p < 0.0001). Direct comparison of vehicle-TBI and rIL-1Ra-TBI mice revealed a trend toward an effect of treatment (two-way RM ANOVA, F(1,32) = 3.37, p = 0.08). However, only vehicle-TBI mice showed a increased latency to locate a hidden platform during subsequent trial days compared with sham controls (F(1,27) = 6.64, p = 0.02). On day 7 of the task, the probe trial revealed an overall effect of injury (F(1,55) = 4.57, p = 0.04), with a priori post hoc analysis revealing that only vehicle-TBI mice spent less time in the target quadrant compared with their sham controls (p < 0.05), indicating a spatial memory deficit which was absent in rIL-1Ra-TBI mice (D). Brain-injured mice showed locomotor hyperactivity compared with sham controls, which was not influenced by rIL-1Ra treatment (E; two-way ANOVA, effect of injury: F(1,53) = 6.93, p = 0.01). All mice showed equivalent ability on an accelerating rotarod (F; two-way ANOVA, effect of time: F(1,53) = 22.92, p < 0.0001) and the elevated plus maze (G). n = 11/group (sham) and 18/group (TBI); *p < 0.05, ***p < 0.001.
Figure 8.
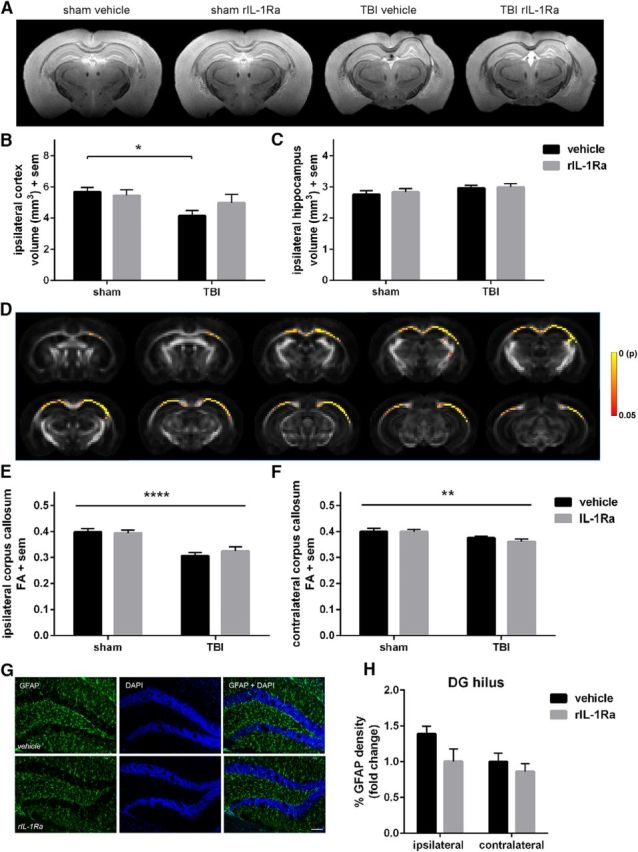
Chronic neuropathology after pTBI. Volumetric analysis of the dorsal cortex and hippocampus at 6 months postsurgery by T2-weighted MRI (A) revealed a significant reduction in the ipsilateral cortex of vehicle-TBI mice compared with their sham controls (B; two-way ANOVA: F(1,28) = 5.98, p = 0.02). Hippocampal volume was unaffected by TBI or drug treatment (C). Track-based spatial statistics analysis of diffusion-weighted imaging demonstrated a significant reduction in fractional anisotropy values in TBI samples compared with sham controls in several brain regions (D; p < 0.05, color bar indicates p value). Region-of-interest quantification of ipsilateral and contralateral corpus callosum fractional anisotropy values similarly detected a reduction in TBI mice (E; two-way ANOVA: F(1,33) =33.99, p < 0.0001, and F; F(1,33) = 12.48, p < 0.01), independent of rIL-1Ra treatment. GFAP immunofluorescence staining in the ipsilateral hippocampus (G, H) suggested a persistent elevation of hippocampal astrogliosis in vehicle-TBI mice at this time (two-way ANOVA: F(1,24) = 4.45, p = 0.05, effect of hemisphere; F(1,24) = 4.31, p < 0.05, effect of treatment). n = 8–13/group; *p < 0.05, **p < 0.01, ****p < 0.0001.
Animal model.
C57BL/6J mice were purchased from the The Jackson Laboratory, or the Animal Resource Centre. Animals were group-housed in individually ventilated cages, and maintained under a 12 h light/dark cycle with ad libitum access to food and water. The controlled cortical impact model of TBI was performed at P21 (±1 d; ∼10 g) in male mice as previously described (Pullela et al., 2006; Semple et al., 2015). Pups were weaned and anesthetized with 1.25% 2,2,2-tribromoethanol in saline (Avertin, Sigma-Aldrich), intraperitoneally at 0.02 ml/g body weight (at UCSF; see Figs. 1–4), or 1.5% isoflurane via a nose cone throughout surgery (at UoM; see Figs. 5–8). Injury parameters were 4.5 m/s velocity, 1.73 mm depth, and 150 ms duration (“severe”) or 4.0 m/s, 1.2 mm depth, and 150 ms duration (“moderate”). Sham-operated mice underwent identical surgical procedures, without receiving the impact.
Figure 2.

IL-1 response after pTBI. IL-1β protein was detected by ELISA across a time course after pTBI, in the ipsilateral cortex (A), hippocampus (C), and serum (B; one-way ANOVA with Dunnett's post hoc). Quantitative PCR revealed a significant increase in Il-1r1 receptor in the cortex (D) at 1 d post-injury, with a trend in the hippocampus (p = 0.09). GFAP, an indicator of astrocyte activation detected by Western blot (E), was also elevated at 1 d post-injury compared with sham controls, in both the injured cortex and hippocampus (F). *p < 0.05, **p < 0.01, ***p < 0.001; n = 4–6/group.
Figure 3.

rIL-1Ra treatment reduces subacute seizure severity after pTBI. Mice underwent severe (A, B) or moderate (C, D) TBI at P21, then received either rIL-1Ra (100 mg/kg, s.c.) or saline vehicle at 2, 8, and 24 h post-injury, followed by a PTZ challenge at 2 weeks. Alternatively, rIL-1Ra was administered 1 h before PTZ (E, F). After both severe and moderate TBI followed by acute drug administration (B, D), but not after delayed administration (F), rIL-1Ra treatment reduced seizure severity compared with vehicle treatment. Red dotted line indicates the median sham score (from Fig. 1A). n = 7–15/group; Mann–Whitney tests, *p < 0.05, **p < 0.01.
Figure 6.
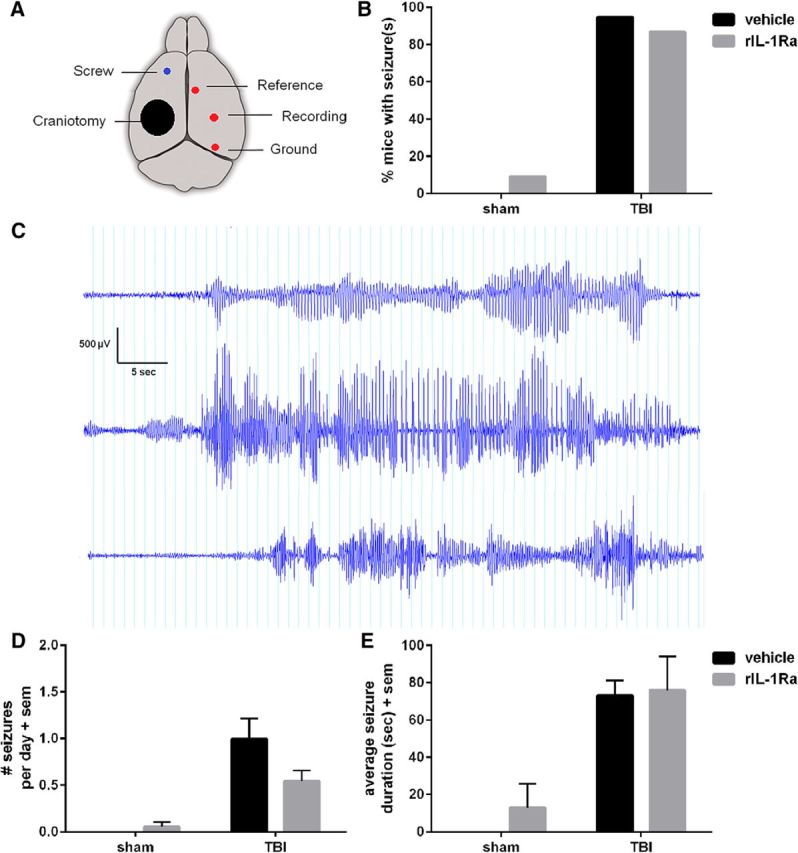
A high incidence of spontaneous seizures at 4–5 months after pTBI was not affected by acute rIL-1Ra treatment. EEG electrodes were implanted over the contralateral hemisphere (A). Following a recovery period, mice underwent continuous video-EEG monitoring. The percentage of mice who displayed at least 1 spontaneous seizure during a 7 d period was quantified (B), revealing late post-traumatic seizures in a majority of TBI mice. Three example events from vehicle-TBI mice are illustrated (C). Number of seizures per day was quantified (D), and found to be higher in TBI mice compared with sham (Kruskal–Wallis test, p < 0.0001). The mean difference in seizures/d between vehicle-TBI and rIL-1Ra-TBI mice did not reach statistical significance (Dunn's multiple comparisons, n.s.). Average seizure duration was also quantified (E). n = 11/group (sham) and n = 15–19/group (TBI).
Figure 7.

Acute rIL-1Ra treatment reduces the chronic evoked seizure response. PTZ results in a stereotypical pattern of brief, abnormal epileptiform events paired with isolated myoclonic contractions (A, black arrows), which was observed in some mice to culminate in a generalized convulsive seizure (black bracket). Fifty-eight percent of vehicle-TBI mice exhibited an evoked seizure in response to PTZ, compared with only 21% of IL-1Ra-TBI mice (B; Fisher's exact test, p = 0.07). Quantification of EEG seizures (C) revealed an increase in vehicle-TBI mice but not in rIL-1Ra-TBI mice compared with their respective sham controls (Kruskal–Wallis test, p < 0.01; post hoc, p < 0.01), but no increase in rIL-1Ra-TBI mice compared with their sham controls. Average seizure duration (D) was similarly increased specifically in vehicle-TBI mice compared with sham controls (p < 0.01). The overall Seizure Severity Score (E) revealed a significant effect of injury (F(1,50) = 4.96, p = 0.03) and a treatment × injury interaction (F(1,50) = 4.65, p = 0.04), with post hoc analysis finding an elevation only in vehicle-TBI mice compared with IL-1Ra-TBI mice (p < 0.01). n = 11/group (sham) and 15–19/group (TBI); *p < 0.05, **p < 0.01.
rIL-1Ra administration.
The human recombinant IL-1R antagonist (rIL-1Ra), commercially available as Kineret (Anakinra, Amgen), was received as a stock solution of 100 mg/0.67 ml and diluted to 10 mg/ml in sterile saline for subcutaneous injection. For the subacute study, conducted at UCSF, 100 mg/kg rIL-1ra (or sterile saline as the vehicle) was administered at 2, 8, and 24 h post-injury (see Fig. 3A). This dosage is known to penetrate the rodent BBB (Greenhalgh et al., 2010), afford neuroprotection in models of CNS damage (McCann et al., 2016), and show safety in adult TBI patients (Helmy et al., 2014). For the chronic study, conducted at UoM, a bolus of 100 mg/kg rIL-1ra or vehicle was administered at 2 h post-injury (see Fig. 5A). In addition, a primed ALZET osmotic mini-pump loaded with rIL-1Ra or vehicle was implanted subcutaneously at the shoulder during surgery, to provide continuous release of 100 mg/kg/d for 7 d. Mice were then briefly anesthetized for explantation of the pump, and drug delivery confirmed by quantification of residual fluid.
PTZ seizure induction.
The proconvulsant pentylenetetrazol (PTZ; Sigma-Aldrich) was administered to induce evoked seizures, at doses of 30–50 mg/kg depending on age/time post-injury as determined from pilot and published studies (Nehlig and Pereira de Vasconcelos, 1996; Bao et al., 2011). At these doses, sham-operated mice typically showed minor, subconvulsive behaviors. Individual mice were placed into a standard cage and responses were video-recorded using a side-mounted camera. Based upon the Racine score (Racine, 1972) and previous adaptations (Cole et al., 2000; Lüttjohann et al., 2009), a modified “Seizure Severity Score” was used to most accurately capture the overall PTZ response, where 0 = no response; 1 = behavioral arrest; loss of posture; 2 = isolated, focal twitches; 3 = forelimb clonus and head nodding; 4 = rearing; 5 = generalized convulsions (loss of posture, rearing and falling); 6 = repeated and/or extended generalized seizures; and 7 = status epilepticus resulting in death. The latency to demonstrate key behaviors were also calculated from the time of PTZ injection. Group sizes were n = 7–10 mice/group for subacute experiments (2 weeks post-injury), and n = 8–13/group for the chronic experiment (6 months post-injury).
Gene expression.
Fresh dissected brain samples (n = 4–6/group) were homogenized for RNA extraction using the Purelink RNA Mini Kit (Invitrogen), and prepared for real-time quantitative PCR using a StepOnePlus Real-Time PCR System (Applied Biosystems) as previously described (Frugier et al., 2010). TaqMan gene expression assays were used to detect 18S (Hs99999901_s1), Il1r1 (IL-1R1; Mm00434237_m1), Gfap (GFAP; Mm01253033_m1), and Vim (vimentin; Mm01333430_m1). Relative gene expression was determined by the 2ΔΔCT method and normalized to 18S.
Protein analysis.
Serum was obtained by centrifugation of blood samples collected by cardiac puncture from a subset of terminally anesthetized mice, and fresh dissected brain tissue was snap frozen (n = 4–6/group). IL-1β levels in serum, ipsilateral cortex, and hippocampal tissue were determined using a mouse IL-1β Quantikine ELISA kit (R&D Systems) per the manufacturer's instructions. Western blots were performed using antibodies against GFAP (polyclonal rabbit, DAKO, Z0334; 1:5000) or the internal control GAPDH (mouse monoclonal, Abcam, ab8245; 1:10,000; Semple et al., 2015).
Immunofluorescence.
Following transcardial perfusion with 4% paraformaldehyde, brains were cryosectioned for collection of serial coronal sections (40 μm). Mossy fiber sprouting was detected by the zinc transporter protein ZnT3 (polyclonal anti-rabbit, Synaptic Systems, 197003; 1:1000), double-labeled with NeuN (mouse monoclonal; Millipore, MAB377; 1:1000) and the nuclear counterstain DAPI, followed by the application of secondary antibodies (Cy3 goat ant-rabbit IgG, The Jackson Laboratory, 111-165-144; 1:500; and AF488 goat anti-mouse IgG, Invitrogen, A11029; 1:500). The astroglial response was evaluated by immunofluorescence for GFAP (rabbit polyclonal, DAKO, Z0334; 1:1000), followed by secondary antibody detection (FITC goat anti-rabbit IgG, Jackson ImmunoResearch, 111-095-003; 1:1000) and a DAPI counterstain (n = 9–10/group). The microglial response was evaluated by immunofluorescence for Iba-1 (goat polyclonal, Abcam, ab5076; 1:750), followed by secondary antibody detection (AF488 donkey anti-goat IgG, Life Technologies, A-11055; 1:200), and a DAPI counterstain. Images were captured at 20× magnification (5 sections/brain), then calibrated and thresholded using MetaMorph v7.7.11.0 for quantification of fluorescent intensity in key regions of interest. Percentage GFAP or Iba-1 density was defined as (threshold pixel intensity/total pixel intensity) × 100, and expressed as fold-change from contralateral vehicle-treated samples.
Behavior.
Behavioral testing was conducted in the Florey Institute animal facility in allocated mouse rooms, staggered over four cohorts (final n = 11/sham group and n = 18/TBI group). Mice were separated into individual cages 3–5 d before testing. An accelerating rotarod (Ugo Basile) assessed general motor function, coordination and motor learning, in four trials/d across 2 consecutive days (Semple et al., 2015). General locomotor activity was evaluated in a 30 min period in individual Activity Monitor cells (Med Associates; Semple et al., 2017). Anxiety-like behavior was evaluated using an elevated plus maze (Semple et al., 2017). Spatial learning and memory was assessed in the Morris water maze (MWM; Semple et al., 2015), comprised of six trials/d for 6 consecutive days. During days 1–2, the platform was raised above the water surface, clearly labeled with a flag (“visible platform”), and rotated to different quadrants. During days 3–6 (“hidden platform”), the platform was hidden just below the water surface and maintained in a constant location, requiring the use of external spatial cues to locate it. On day 7, a 60 s “probe trial” assessed spatial memory retention as the time spent in the target quadrant (where the platform was previously located).
24/7 video-EEG recording.
Implantation of extradural EEG electrodes were performed under isoflurane anesthesia (Bolkvadze and Pitkänen, 2012). A three-channel stainless steel electrode system connected to a plastic pedestal (Plastics One) was fixed to the skull by dental acrylic cement and stabilized by an extradural screw (see Fig. 7A). Two mice died during implantation and one mouse was excluded due to dislodgement of electrodes before EEG recording (final n = 11/sham group and n = 15–19/TBI group). Seizure-like behaviors were recorded by a side-mounted video camera accompanied by an infrared light.
For analysis, entire digital files were reviewed and manually annotated in Profusion EEG software (Compumedics v.5.0). An electrographic event (“EEG seizure”) was defined as a high-amplitude (at least 3× baseline) rhythmic discharge, frequency >5 Hz and lasting longer than 5 s, that clearly represented an evolving, oscillatory, and atypical EEG pattern (Ziyatdinova et al., 2011; Bolkvadze and Pitkänen, 2012; Van Nieuwenhuyse et al., 2015; Liu et al., 2016). All events quantified from EEG were associated with a simultaneous behavioral event. For spontaneous events, the average number of seizures per day as well as average seizure duration were quantified. The responses to PTZ (at 6 months) were scored according to the modified Seizure Severity Score as described earlier. A random selection of observed epileptiform events were independently reviewed by an experienced epileptologist (T.J.O.) for verification. Inter- and intra-scorer consistency of PTZ-evoked events was verified by two independent investigators, and variation found to be <6%.
Neuroimaging.
At 6 months post-injury, a subset of mice underwent ex vivo magnetic resonance imaging (MRI; n = 8–13/group). Mice were transcardially perfused within 1 h of PTZ and the collected brains postfixed in 4% paraformaldehyde overnight, rinsed, and embedded in 3% agar. MRI was performed on a 4.7 Tesla Avance III scanner with a cryogenically cooled surface coil (Bruker Biospec; Shultz et al., 2013). T2-weighted images were acquired using a 3D RARE sequence with the parameters: TR = 2000 ms; RARE factor = 8, TE = 60 ms; FOV = 15.4 × 15.4 × 7.68 mm3; matrix size = 192 × 192 × 96; and isotropic spatial resolution = 60 × 60 × 60 μm3. Diffusion weighted imaging was acquired using an echoplanar imaging sequence with the parameters: TR = 11,000 ms; TE = 69 ms; shots = 3; FOV = 17.28 × 17.28 mm2; matrix size = 96 × 96; spatial resolution = 180 × 180 μm2; and slice thickness = 180 μm. Diffusion weighting was performed in 81 non-collinear directions with diffusion duration (δ) = 8 ms; diffusion gradient separation (Δ) = 15 ms; and b values = 1000, 3000, 5000, and 7000 s/mm2.
Analysis was performed on T2-weighted images using ITK-SNAP software (http://www.itksnap.org; Shultz et al., 2013). Regions of interest were outlined on 18 coronal slices, and volumetric measurements were confined to the dorsal hemispheres containing the impacted region (Semple et al., 2015). For diffusion-weighted imaging, a region-of-interest analysis evaluated fractional anisotropy in the corpus callosum, and template images for each group were generated then combined using mrtrix (http://www.mrtrix.org/) for track-based spatial statistics. Voxelwise analyses were performed with 5000 permutations, familywise error correction, and threshold-free cluster enhancement.
Experimental design and statistical analysis.
Sample sizes were calculated from preliminary and published data (Semple et al., 2015; Liu et al., 2016) to achieve 60% power with a significance level of 0.05. Statistical analyses were performed using GraphPad Prism 6.07 or SPSS v.21. ANOVAs were used to compare two or more normally-distributed groups or factors (e.g., injury, treatment) followed by a priori post hoc analyses (specifically, to compare vehicle-TBI versus IL-1Ra-TBI groups), with time as a repeated measure where appropriate (e.g., for MWM data). Results are expressed as mean ± SEM. Data that were not normally distributed (e.g., Seizure Severity Scores and EEG data) were analyzed by nonparametric tests (Mann–Whitney or Kruskal–Wallis) and presented as dot plots with median values compared. Two-tailed Fisher's exact tests evaluated the percentage of animals exhibiting a particular phenotype (e.g., seizure).
Results
Pediatric TBI leads to increased seizure susceptibility and mossy fiber sprouting
TBI and sham mice were challenged with PTZ to evaluate seizure susceptibility. At 2 weeks post-injury, TBI mice exhibited a more severe response compared with sham controls (Mann–Whitney test, U = 3.5, p < 0.0001; Fig. 1A). TBI mice also showed more rapid progression to a myoclonic jerk (106 vs 193 s; Mann–Whitney U = 20, p = 0.02), and precipitation to a generalized convulsive seizure (289 vs 600 s; U = 7, p < 0.01). 100% of TBI mice displayed a generalized seizure within the 10 min observation period versus 20% of shams (Fisher's exact test, p < 0.01). At 3 months, TBI mice again showed a more severe response compared with age-matched sham controls (Mann–Whitney test, U = 10.5, p = 0.03; Fig. 1B). In addition, TBI mice developed generalized convulsions more rapidly compared with sham (268 vs 600 s; U = 7, p = 0.04), and showed an increased likelihood of exhibiting this behavior (86% vs 38%; p = 0.04).
Mossy fiber sprouting is a pathological hallmark of abnormal hippocampal connectivity associated with epileptogenesis (Santhakumar et al., 2005). Intense ZnT3 staining of mossy fiber terminals was restricted to the stratum lucidum of the CA3 and hilus of all brains at 2 weeks post-injury/sham (Fig. 1C). At 3 months, however, all TBI brains presented with aberrant ZnT3 immunoreactivity in the ipsilateral hippocampus, extending through the granule cell layer into the inner molecular layer (Fig. 1D,E). The highest density of abnormal sprouting was observed at the lesion epicenter.
Acute inflammatory response after pediatric TBI
Based on evidence implicating inflammatory cytokines in the process of epileptogenesis (Webster et al., 2017), we next characterized the IL-1 response after pTBI. IL-1β protein levels were robustly elevated in the injured cortex, peaking from 4 h until 4 d (F(9,29) = 1.01, p < 0.0001; post hoc, p < 0.01; Fig. 2A). Similarly, IL-1β was elevated in the injured hippocampus, exhibiting a biphasic response to peak at 8 h and 7 d (F(9,29) = 3.41, p < 0.0001; post hoc, p < 0.01; Fig. 2C). IL-1β was detected in serum at days 1–2 after pTBI compared with sham controls (F(7,23) = 3.52, p < 0.0001, post hoc, p < 0.05; Fig. 2B). Expression of the Il-1r1 receptor, quantified by qPCR, was robustly elevated in the cortex at 1 d post-injury (t(8) = 6.05, p < 0.001; Fig. 2D). Last, we confirmed concurrent astroglial activation at 1 d post-injury, as an increase in GFAP protein by Western blot in both the cortex (t(6) = 3.25, p < 0.05) and hippocampus (t(6) = 3.31, p < 0.05; Fig. 2E,F).
rIL-1Ra reduces subacute seizure susceptibility after pTBI
We next administered rIL-1Ra across an acute time course post-injury corresponding to elevated IL-1β levels (Fig. 3A). After 2 weeks, mice were challenged with PTZ. rIL-1Ra-TBI mice showed lower Seizure Severity Scores compared with vehicle-TBI mice (Mann–Whitney test, p = 0.01; Fig. 3B). Eighty-six percent of vehicle-TBI mice developed a generalized convulsive seizure within 10 min, compared with 53% of rIL-1Ra-TBI mice (Fisher's exact test, p = 0.10). rIL-1Ra-TBI mice also showed a trend toward increased latency to first myoclonic jerk (143 vs 132 s) and a generalized convulsive seizure (579 vs 282 s; p > 0.05). We replicated this finding in a model of moderate TBI severity (Fig. 3C), after which rIL-1Ra-TBI mice again showed reduced Seizure Severity Scores compared with vehicle-TBI mice (p < 0.01; Fig. 3D), as well as an increased latency to first myoclonic jerk (94 vs 73 s; p < 0.05) and generalized convulsive seizure (234 vs 112 s; p < 0.05). The effect of rIL-1Ra on seizure susceptibility was dependent upon acute post-injury administration, as treatment at 2 weeks post-injury, 1 h before the PTZ challenge, failed to influence seizure severity (p > 0.05, Fig. 3E,F).
rIL-1Ra attenuates subacute hippocampal astrogliosis after pTBI
As IL-1β has known effects on astrocyte activation (John et al., 2004), and astrocytes have been implicated in neuronal excitability and seizures (Ortinski et al., 2010; Robel et al., 2015), we examined the effect of rIL-1Ra on astrocyte activation after pTBI. Robust GFAP+ immunofluorescence was evident at 2 weeks, particularly in the ipsilateral hippocampus (Fig. 4A). Quantification of fluorescence confirmed a significant increase in the ipsilateral DG hilus, CA1 and CA2/3 of vehicle-TBI mice, which was considerably reduced in rIL-1Ra-TBI mice in the hilus (two-way RM ANOVA injury × drug interaction: F(1,17) = 5.04, p = 0.04; post hoc, p < 0.01) and CA1 (injury × drug interaction: F(1,17) = 8.76, p < 0.01; post hoc, p < 0.001; Fig. 4B–D). In contrast, whereas the degree of Iba-1+ microglial staining in the DG hilus ipsilateral to the injury was significantly elevated compared with the contralateral side (two-way RM ANOVA effect of hemisphere: F(1,13) = 82.64, p < 0.0001), rIL-1Ra treatment did not influence this (effect of drug: F(1,13) = 0.05, p = 0.83; hemisphere × drug interaction: F(1,13) = 0.03, p = 0.86).
Astrogliosis observed at the protein level was reflected by gene expression, Gfap. mRNA was robustly upregulated at 1 d post-injury (F(2,12) = 28.06, p < 0.0001), more so in vehicle-TBI compared with rIL-1Ra-TBI mice (Fig. 4E). In the hippocampus, only vehicle-TBI mice showed a significant elevation in Gfap expression at 1 d compared with sham controls (F(2,12) = 5.04, p = 0.03; post hoc, p < 0.05). By 14 d, hippocampal Gfap mRNA levels had returned to baseline, but remained elevated above sham in vehicle-TBI mice (F(2,12) = 4.46, p = 0.04; post hoc, p < 0.05). Vimentin (Vim), another marker of glial activation, was similarly upregulated at 1 d in vehicle-TBI mice, with an attenuated response in rIL-1Ra-TBI mice (cortex: F(2,12) = 17.45, p < 0.01; hippocampus: F(2,12) = 6.50, p = 0.01; Fig. 4F).
rIL-1Ra attenuates chronic cognitive deficits
We next conducted a chronic time course experiment to evaluate acute rIL-1Ra treatment on long-term post-injury outcomes (Fig. 5A). Functional outcomes were assessed at adulthood (3 months post-injury). On days 1–2 of the MWM all groups showed equivalent ability to locate a visible platform (three-way RM ANOVA, within-subjects effect of time: F(1,51) = 199.94, p < 0.0001; Fig. 5B). On days 3–6 (Fig. 5C; hidden platform), all groups again showed an overall improvement with time (three-way RM ANOVA: F(1,51) = 30.35, p < 0.0001). There was also no difference between groups in terms of the learning rate, calculated as the difference between escape latency on day 3 versus day 6, normalized to performance on day 3 (two-way ANOVA effect of injury: F(1,51) = 0.15, p = 0.71; effect of treatment: F(1,51) = 0.001, p = 0.98; injury × treatment interaction: F(1,51) = 1.30, p = 0.26). However, direct comparison of vehicle-TBI and rIL-1Ra-TBI mice across the hidden sessions revealed a trend toward an effect of treatment (three-way RM ANOVA: F(1,32) = 3.37, p = 0.08). A spatial memory deficit was detected specifically in vehicle-TBI mice compared with their sham controls (two-way RM ANOVA: F(1,27) = 6.64, p = 0.02). In contrast, rIL-1Ra-TBI mice did not exhibit an injury deficit (F(1,24) = 0.20, p = 0.66). This was confirmed in the probe trial on day 7 (Fig. 5D), when vehicle-TBI mice spent less time in the target quadrant compared with sham controls (effect of injury: F(1,55) = 4.57, p = 0.04; post hoc, p < 0.05). In contrast, rIL-1Ra-TBI mice did not exhibit a deficit compared with sham controls (post hoc, p > 0.05).
Consistent with previous studies (Pullela et al., 2006; Semple et al., 2015), pTBI resulted in general hyperactivity at adulthood (two-way ANOVA effect of injury: F(1,53) = 6.93, p = 0.01). However, rIL-1Ra did not influence this behavior (Fig. 5E). All mice showed improvement in rotarod performance over time (two-way ANOVA effect of time: F(1,53) = 22.92, p < 0.0001), independent of both injury or treatment (Fig. 5F). Finally, in the elevated plus maze, all groups spent similar time in the open arms (Fig. 5G).
Spontaneous seizures at 4–5 months post-injury
Following behavioral testing, mice underwent 7 d of video-EEG monitoring to evaluate spontaneous seizure activity. The majority of TBI mice (>87%) displayed at least one generalized motor seizure during this period, compared with >10% of shams (Fisher's exact test, p = 0.57; Fig. 6A–C). The average number of seizures per day was greater in TBI mice compared with shams (Kruskal–Wallis test, p < 0.0001), and tended to be higher for vehicle-TBI mice (1 seizure/d) compared with rIL-1Ra-TBI mice (0.5 seizures/d); however, this did not reach statistical significance (Fig. 6D). Average seizure duration did not differ between vehicle-TBI and rIL-1Ra-TBI mice (Fig. 6E). Similarly, seizure severity was not affected by rIL-1Ra treatment (Mann–Whitney U = 137, p = 0.85; median seizure score of 3.9 and 4.0 for vehicle-TBI and rIL-1Ra-TBI mice, respectively).
rIL-1Ra reduces the chronic evoked seizure response
At 6 months post-injury, PTZ was administered to evaluate chronic seizure susceptibility (Fig. 7A). Fifty-eight percent of vehicle-TBI mice exhibited a PTZ-evoked convulsive seizure, versus 21% of IL-1Ra-TBI mice (Fisher's exact test, p = 0.07; Fig. 7B). Quantification of EEG seizures revealed an increase in vehicle-TBI mice compared with vehicle-sham mice specifically (Kruskal–Wallis test, p < 0.01; post hoc, p < 0.01; Fig. 7C). The average duration of EEG seizures was also longer in vehicle-TBI mice compared with shams (post hoc, p < 0.01), which was not the case for rIL-1Ra-TBI mice (p > 0.05; Fig. 7D). The overall Seizure Severity Score (Fig. 7E) revealed a significant effect of injury (F(1,50) = 4.96, p = 0.03) and a treatment × injury interaction (F(1,50) = 4.65, p = 0.04),with post hoc analysis finding an elevation only in vehicle-TBI mice compared with IL-1Ra-TBI mice (p < 0.01), in line with the EEG seizure data indicating a protective effect of rIL-1Ra treatment on chronic seizure susceptibility.
rIL-1Ra partially rescues chronic neuropathology after pediatric TBI
By 6 months, pronounced tissue deformation and volumetric loss was evident in all TBI mice, particularly in the ipsilateral cortex (two-way ANOVA: F(1,28) = 5.98, p = 0.02; Fig. 8A). A priori post hoc analyses indicated that vehicle-TBI mice had a smaller cortex volume compared with vehicle-sham mice (p < 0.05), whereas rIL-1Ra-TBI were not significantly different to their sham controls (p > 0.05; Fig. 8B). Neither injury nor drug treatment affected the volume of the ipsilateral hippocampus (Fig. 8C), contralateral hippocampus or contralateral cortex (data not shown).
Track-based spatial statistics analysis of diffusion-weighted imaging revealed an overall effect of injury but not drug treatment on fractional anisotropy, a measure of white matter integrity. Decreased fractional anisotropy in TBI brains was most evident in the ipsilateral corpus callosum between ∼bregma 0.7 mm to −3.8 mm, extending into the contralateral hemisphere, and ipsilateral internal capsule (p < 0.05; Fig. 8D). Region-of-interest analysis confirmed an overall reduction in fractional anisotropy in TBI brains compared with sham, in both the ipsilateral (two-way ANOVA: F(1,33) = 33.99, p < 0.0001) and contralateral corpus callosum (F(1,33) = 12.48, p < 0.01; Fig. 8E,F). Treatment with rIL-1Ra did not influence these measures (p > 0.05).
IL-1R antagonism attenuates chronic hippocampal astrogliosis after pTBI
Following ex vivo neuroimaging, brains were processed to evaluate the effect of rIL-1Ra on chronic glial reactivity in the hippocampus. Immunofluorescence for GFAP was elevated in the ipsilateral DG hilus compared with the contralateral hilus (two-way ANOVA: F(1,24) = 4.45, p = 0.05), and an overall effect of treatment (F(1,24) = 4.31, p < 0.05), suggesting that rIL-1Ra treatment attenuated chronic astrogliosis (Fig. 8G,H). In contrast, Iba-1 reactivity was no longer elevated in the ipsilateral compared with contralateral hemisphere at this time, nor affected by rIL-1Ra treatment (two-way ANOVA, effect of hemisphere: F(1,24) = 0.37, p = 0.55; effect of drug: F(1,24) = 0.65, p = 0.43; hemisphere × drug interaction: F(1,24) = 0.01, p = 0.97).
Discussion
The development of novel anti-epileptic therapeutics for PTE is, in part, predicated upon the use of age appropriate, well characterized, and clinically relevant animal models. In this study, we first characterized the neuropathological and seizure-like hallmarks of PTE in a mouse model of TBI to the pediatric brain, which reproduces many of the risk factors associated with PTE clinically (e.g., early age, severity, hemorrhage; Hahn et al., 1988; Annegers and Coan, 2000). Compared with sham-operated controls, pTBI mice exhibited multiple features that have been associated with epileptogenesis from studies with postmortem patient tissue and experimental models of adult PTE, including robust astrogliosis and increased susceptibility to spontaneous and evoked seizures. Of note, at 3 months post-injury, we found evidence of aberrant mossy fiber sprouting in the ipsilateral hippocampus of pTBI mice. Sprouting represents long-term structural reorganization in the dentate gyrus, via the appearance of new recurrent excitatory circuitry which may contribute to hippocampal hyperexcitability (Santhakumar et al., 2005), and has been reported in the DG of epilepsy patients with a history of head trauma (Jeub et al., 1999), in human temporal lobe epilepsy (Sutula et al., 1989; Babb, 1997), and in epileptic experimental animals (Tauck and Nadler, 1985; Babb, 1997). Consistent with our findings, mossy fiber sprouting in adult models of experimental TBI has been reported to develop over weeks after injury, and is associated with an increase in epileptiform activity (Hunt et al., 2009, 2010).
Spontaneous and evoked seizures after pTBI
In adult TBI rodent models, the reported incidence of spontaneous seizures up to 9 months post-injury has varied from 9 to 50% (Hunt et al., 2009, 2010; Bolkvadze and Pitkänen, 2012; Shultz et al., 2013), with seizures being fairly infrequent (∼0.3 seizures/d), consistent with what is seen in patients with PTE (Pitkänen et al., 2009). The only other preclinical study to examine seizures in a pediatric population evaluated a small group of rats 4–11 months following controlled cortical impact at P17 (n = 8), observing spontaneous seizures in 1 rat and abnormal EEG discharges in 7/8 rats (Statler et al., 2009). In the current study, at 4–5 months post-injury, we observed on average 0.5–1 seizure/d and at least 1 seizure within a 7 d period in 95% of vehicle-TBI mice. It is unclear whether this high incidence and frequency is due to the age at injury, model severity, or other factors such as species or strain, and we seek to replicate this finding in ongoing and future studies.
Administration of PTZ, a noncompetitive antagonist of the GABAA receptor, triggers the well characterized, progressive development of limbic seizures, culminating in generalized tonic-clonic seizures depending upon the dosage used (Cole et al., 2000). Here, we considered the response to PTZ as an indicator of network excitability, whereby an increased response implicates a primed neuronal environment with a reduced threshold for seizure activity (Löscher and Nolting, 1991). Although the clinical relevance of increased susceptibility to a proconvulsant remains controversial, several models of adult TBI and temporal lobe epilepsy have demonstrated a link between hyperexcitability in a PTZ test and the development of spontaneous seizures (Blanco et al., 2009; Rattka et al., 2011; Bolkvadze and Pitkänen, 2012). An increased PTZ response was evident in young brain-injured mice compared with shams as early as 2 weeks post-injury, suggesting that epileptogenesis is already underway at this time, and persists to at least 6 months post-injury. This finding builds upon previous data from adult TBI models (Bolkvadze and Pitkänen, 2012; Huusko et al., 2015), and is in alignment with the hypothesis that an early increase in neuronal excitability signifies an environment primed for the development of PTE.
IL-1R signaling mediates post-traumatic seizure susceptibility
Whether immune signaling plays a role in post-traumatic alterations in network excitability has been largely theoretical to date. A robust IL-1 response was evident after pTBI, including upregulation of IL-1β in the injured brain and serum. The latter increase in serum was temporally delayed, suggesting a predominantly cerebral source of the cytokine. Based upon evidence that IL-1β can initiate and exacerbate seizure activity in experimental models of epilepsy (Vezzani et al., 1999, ., 2000; Dubé et al., 2005), and blockage of IL-1β biosynthesis or signaling via the receptor IL-1R ameliorates seizure severity/incidence (Vezzani et al., 2000; Maroso et al., 2011), we evaluated the effect of a commercially available rIL-1Ra on chronic seizures, neuropathology and functional outcomes after pTBI.
Our findings demonstrate that acute rIL-1Ra reduces the PTZ-evoked seizure response compared with vehicle treatment, both subacutely (2 weeks) and more chronically post-injury (3 and 6 months). Importantly, this anti-seizure effect was replicated across two independent cohorts under moderate and severe injury parameters. Second, the effect of rIL-1Ra was dependent upon acute post-injury administration to presumably prevent acute cytokine signaling, as treatment at 2 weeks, just before the PTZ challenge, failed to modify seizure severity. This finding is consistent with the hypothesis that acute inflammation response can alter long-term susceptibility of the brain to seizure activity (Galic et al., 2008).
The influence of rIL-1Ra on PTZ-evoked seizures, however, failed to translate into a hypothesized reduction in the percentage of mice to develop spontaneous seizures, although there was a nonsignificant trend toward a reduced seizure frequency in rIL-1Ra-TBI compared with vehicle-TBI mice. Together, these findings support a role for IL-1β/IL-1R in neuronal excitability after pTBI, suggesting that inflammatory signaling is a key process underlying post-traumatic epileptogenesis; however, additional mechanisms are evidently involved in the initiation of spontaneous epileptic seizures long-term.
It remains unclear from our findings whether IL-1 has a direct or indirect effect on neurotransmission to modulate neuronal excitability. Almost all CNS cells express IL-1 receptors and components of the signaling pathways, and therefore can respond to IL-1β and IL-1α. For example, a direct interaction between IL-1R and NMDA receptors to modulate excitatory receptor structure and function has been proposed (Viviani et al., 2003). In addition, IL-1β acts via astrocytic IL-1R to promote glial activation, proliferation and further cytokine release (Pinteaux et al., 2009). Reactive astrocytes have recently been implicated in the modulation of neuronal excitability and promotion of epileptogenesis (Ortinski et al., 2010; Robel et al., 2015; Webster et al., 2017). Indeed, hippocampal sclerosis involving the loss of pyramidal neurons, interneurons, and astrogliosis is one of the most common pathologies associated with increased network excitability in both temporal lobe epilepsy and PTE. Our findings, that rIL-1Ra treatment reduces markers of hippocampal astrocyte activation (Gfap and Vim), but not microglial activation, concurrent with a reduction in seizure susceptibility during the subacute post-injury period, implicates post-traumatic astrocytosis as an indirect mechanism by which IL-1R mediates neuronal hyperexcitability underlying the observed effects on PTZ-evoked seizures.
IL-1R as a broad neuroprotectant?
Targeting IL-1R signaling in the acutely injured brain was postulated to have a broad neuroprotective effect, based on an abundance of experimental studies implicating IL-1β in leukocyte infiltration, BBB damage and cell death in models of ischemic stroke and TBI (Pinteaux et al., 2009; Brough et al., 2011). For example, antibody-mediated neutralization of IL-1β has been reported to yield neuroprotection against cognitive deficits after focal or diffuse experimental TBI in adult rodents (Clausen et al., 2009, 2011; Ekmark-Lewén et al., 2016). A recent meta-analysis of preclinical studies in stroke confirmed the high biological plausibility of peripherally administered rIL-1Ra as a novel therapeutic agent in the injured brain (McCann et al., 2016). However, few studies to date have considered the long-term consequences of IL-1 signaling, nor had the IL-1 response after injury to the pediatric brain been characterized. Here, we found that acute treatment with rIL-1Ra was effective at reducing cognitive impairments, the degree of cortical loss, and astrogliosis, alongside a reduction in evoked seizure susceptibility. In contrast, rIL-1Ra did not appear to influence the degree of white matter damage, nor rescue injury-induced hyperactivity. Together, these findings support IL-1 signaling as a mediator of long-term pathogenesis and outcomes after early-life injury.
Conclusion
There is an urgent need for novel therapeutic agents to prevent and slow the development of epileptogenesis after pTBI, with scientific progress hindered in part due to our poor understanding of the pathological mechanisms that underlie PTE. In this study, we first demonstrated that pTBI in the young mouse results in a heightened response to evoked seizures as well as many of the neuropathological hallmarks previously associated with epilepsy in both patients and adult animals. This model therefore provides a suitable paradigm to elucidate potential mechanisms of post-traumatic epileptogenesis in the pediatric injured brain. Pharmacological targeting of IL-1Ra represents a feasible therapeutic approach to reduce chronic morbidity for survivors of pTBI, although optimization or complementary application of another therapeutic agent may be needed to maximize neuroprotection, and prevent the onset of spontaneous seizures chronically.
Footnotes
This work was supported by an NHMRC Early Career Fellowship, a UoM Early Career Grant, NIH/NINDS R01 NS077767, and funding from the Rebecca L. Cooper Medical Research Foundation. We thank Ann Turnley (UoM) for providing access to the controlled cortical impactor device, Leigh Johnson (UoM) for guidance and oversight of neuroimaging, and the scientific and technical assistance of the animal MRI facility at the Florey Institute of Neuroscience and Mental Health, a node of the National Imaging Facility.
The authors declare no competing financial interests.
References
- Annegers JF, Coan SP (2000) The risks of epilepsy after traumatic brain injury. Seizure 9:453–457. 10.1053/seiz.2000.0458 [DOI] [PubMed] [Google Scholar]
- Annegers JF, Grabow JD, Groover RV, Laws ER Jr, Elveback LR, Kurland LT (1980) Seizures after head trauma: a population study. Neurology 30:683–689. 10.1212/WNL.30.7.683 [DOI] [PubMed] [Google Scholar]
- Arndt DH, Lerner JT, Matsumoto JH, Madikians A, Yudovin S, Valino H, McArthur DL, Wu JY, Leung M, Buxey F, Szeliga C, Van Hirtum-Das M, Sankar R, Brooks-Kayal A, Giza CC (2013) Subclinical early posttraumatic seizures detected by continuous EEG monitoring in a consecutive pediatric cohort. Epilepsia 54:1780–1788. 10.1111/epi.12369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ates O, Ondül S, Onal C, Büyükkiraz M, Somay H, Cayli SR, Göğüsgeren MA, Orakdöğen M, Koçak A, Yoloğlu S, Berkman Z, Tevrüz M (2006) Post-traumatic early epilepsy in pediatric age group with emphasis on influential factors. Childs Nerv Syst 22:279–284. [DOI] [PubMed] [Google Scholar]
- Babb TL. (1997) Axonal growth and neosynaptogenesis in human and experimental hippocampal epilepsy. Adv Neurol 72:45–51. [PubMed] [Google Scholar]
- Bao YH, Bramlett HM, Atkins CM, Truettner JS, Lotocki G, Alonso OF, Dietrich WD (2011) Post-traumatic seizures exacerbate histopathological damage after fluid-percussion brain injury. J Neurotrauma 28:35–42. 10.1089/neu.2010.1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow KM, Spowart JJ, Minns RA (2000) Early posttraumatic seizures in non-accidental head injury: relation to outcome. Dev Med Child Neurol 42:591–594. 10.1017/S0012162200001110 [DOI] [PubMed] [Google Scholar]
- Blanco MM, dos Santos JG Jr, Perez-Mendes P, Kohek SR, Cavarsan CF, Hummel M, Albuquerque C, Mello LE (2009) Assessment of seizure susceptibility in pilocarpine epileptic and nonepileptic Wistar rats and of seizure reinduction with pentylenetetrazole and electroshock models. Epilepsia 50:824–831. 10.1111/j.1528-1167.2008.01797.x [DOI] [PubMed] [Google Scholar]
- Bolkvadze T, Pitkänen A (2012) Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. J Neurotrauma 29:789–812. 10.1089/neu.2011.1954 [DOI] [PubMed] [Google Scholar]
- Brough D, Tyrrell PJ, Allan SM (2011) Regulation of interleukin-1 in acute brain injury. Trends Pharmacol Sci 32:617–622. 10.1016/j.tips.2011.06.002 [DOI] [PubMed] [Google Scholar]
- Clausen F, Hånell A, Björk M, Hillered L, Mir AK, Gram H, Marklund N (2009) Neutralization of interleukin-1beta modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci 30:385–396. 10.1111/j.1460-9568.2009.06820.x [DOI] [PubMed] [Google Scholar]
- Clausen F, Hånell A, Israelsson C, Hedin J, Ebendal T, Mir AK, Gram H, Marklund N (2011) Neutralization of interleukin-1beta reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur J Neurosci 34:110–123. 10.1111/j.1460-9568.2011.07723.x [DOI] [PubMed] [Google Scholar]
- Cole TB, Robbins CA, Wenzel HJ, Schwartzkroin PA, Palmiter RD (2000) Seizures and neuronal damage in mice lacking vesicular zinc. Epilepsy Res 39:153–169. 10.1016/S0920-1211(99)00121-7 [DOI] [PubMed] [Google Scholar]
- Dubé C, Vezzani A, Behrens M, Bartfai T, Baram TZ (2005) Interleukin-1beta contributes to the generation of experimental febrile seizures. Ann Neurol 57:152–155. 10.1002/ana.20358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekmark-Lewén S, Flygt J, Fridgeirsdottir GA, Kiwanuka O, Hånell A, Meyerson BJ, Mir AK, Gram H, Lewén A, Clausen F, Hillered L, Marklund N (2016) Diffuse traumatic axonal injury in mice induces complex behavioural alterations that are normalized by neutralization of interleukin-1beta. Eur J Neurosci 43:1016–1033. 10.1111/ejn.13190 [DOI] [PubMed] [Google Scholar]
- Frugier T, Morganti-Kossmann MC, O'Reilly D, McLean CA (2010) In situ detection of inflammatory mediators in post mortem human brain tissue after traumatic injury. J Neurotrauma 27:497–507. 10.1089/neu.2009.1120 [DOI] [PubMed] [Google Scholar]
- Galic MA, Riazi K, Heida JG, Mouihate A, Fournier NM, Spencer SJ, Kalynchuk LE, Teskey GC, Pittman QJ (2008) Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci 28:6904–6913. 10.1523/JNEUROSCI.1901-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhalgh AD, Galea J, Dénes A, Tyrrell PJ, Rothwell NJ (2010) Rapid brain penetration of interleukin-1 receptor antagonist in rat cerebral ischaemia: pharmacokinetics, distribution, protection. Br J Pharmacol 160:153–159. 10.1111/j.1476-5381.2010.00684.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn YS, Fuchs S, Flannery AM, Barthel MJ, McLone DG (1988) Factors influencing posttraumatic seizures in children. Neurosurgery 22:864–867. 10.1227/00006123-198805000-00008 [DOI] [PubMed] [Google Scholar]
- Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ (2014) Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: a phase II randomized control trial. J Cereb Blood Flow Metab 34:845–851. 10.1038/jcbfm.2014.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN (2009) Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol 215:243–252. 10.1016/j.expneurol.2008.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Scheff SW, Smith BN (2010) Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy. J Neurophysiol 103:1490–1500. 10.1152/jn.00957.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Boychuk JA, Smith BN (2013) Neural circuit mechanisms of post-traumatic epilepsy. Front Cell Neurosci 7:89. 10.3389/fncel.2013.00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huusko N, Römer C, Ndode-Ekane XE, Lukasiuk K, Pitkänen A (2015) Loss of hippocampal interneurons and epileptogenesis: a comparison of two animal models of acquired epilepsy. Brain Struct Funct 220:153–191. 10.1007/s00429-013-0644-1 [DOI] [PubMed] [Google Scholar]
- Jeub M, Lie A, Blümcke I, Elger CE, Beck H (1999) Loss of dynorphin-mediated inhibition of voltage-dependent Ca2+ currents in hippocampal granule cells isolated from epilepsy patients is associated with mossy fiber sprouting. Neuroscience 94:465–471. 10.1016/S0306-4522(99)00249-3 [DOI] [PubMed] [Google Scholar]
- John GR, Chen L, Rivieccio MA, Melendez-Vasquez CV, Hartley A, Brosnan CF (2004) Interleukin-1β induces a reactive astroglial phenotype via deactivation of the Rho GTPase–Rock axis. J Neurosci 24:2837–2845. 10.1523/JNEUROSCI.4789-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SJ, Zheng P, Wright DK, Dezsi G, Braine E, Nguyen T, Corcoran NM, Johnston LA, Hovens CM, Mayo JN, Hudson M, Shultz SR, Jones NC, O'Brien TJ (2016) Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain 139:1919–1938. 10.1093/brain/aww116 [DOI] [PubMed] [Google Scholar]
- Löscher W, Nolting B (1991) The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs: IV. Protective indices. Epilepsy Res 9:1–10. 10.1016/0920-1211(91)90041-D [DOI] [PubMed] [Google Scholar]
- Lüttjohann A, Fabene PF, van Luijtelaar G (2009) A revised Racine's scale for PTZ-induced seizures in rats. Physiol Behav 98:579–586. 10.1016/j.physbeh.2009.09.005 [DOI] [PubMed] [Google Scholar]
- Maroso M, Balosso S, Ravizza T, Iori V, Wright CI, French J, Vezzani A (2011) Interleukin-1β biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics 8:304–315. 10.1007/s13311-011-0039-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann SK, Cramond F, Macleod MR, Sena ES (2016) Systematic review and meta-analysis of the efficacy of interleukin-1 receptor antagonist in animal models of stroke: an update. Transl Stroke Res 7:395–406. 10.1007/s12975-016-0489-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardou R, Ferrari DC, Ben-Ari Y (2013) Mechanisms and effects of seizures in the immature brain. Semin Fetal Neonatal Med 18:175–184. 10.1016/j.siny.2013.02.003 [DOI] [PubMed] [Google Scholar]
- Nehlig A, Pereira de Vasconcelos A (1996) The model of pentylenetetrazol-induced status epilepticus in the immature rat: short- and long-term effects. Epilepsy Res 26:93–103. 10.1016/S0920-1211(96)00045-9 [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA (2010) Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci 13:584–591. 10.1038/nn.2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinteaux E, Trotter P, Simi A (2009) Cell-specific and concentration-dependent actions of interleukin-1 in acute brain inflammation. Cytokine 45:1–7. 10.1016/j.cyto.2008.10.008 [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Immonen RJ, Gröhn OHJ, Kharatishvili I (2009) From traumatic brain injury to posttraumatic epilepsy: what animal models tell us about the process and treatment options. Epilepsia 50:21–29. 10.1111/j.1528-1167.2008.02007.x [DOI] [PubMed] [Google Scholar]
- Potts MB, Koh SE, Whetstone WD, Walker BA, Yoneyama T, Claus CP, Manvelyan HM, Noble-Haeusslein LJ (2006) Traumatic injury to the immature brain: inflammation, oxidative injury, and iron-mediated damage as potential therapeutic targets. NeuroRx 3:143–153. 10.1016/j.nurx.2006.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullela R, Raber J, Pfankuch T, Ferriero DM, Claus CP, Koh SE, Yamauchi T, Rola R, Fike JR, Noble-Haeusslein LJ (2006) Traumatic injury to the immature brain results in progressive neuronal loss, hyperactivity and delayed cognitive impairments. Dev Neurosci 28:396–409. 10.1159/000094166 [DOI] [PubMed] [Google Scholar]
- Racine RJ. (1972) Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroenceph Clin Neurophysiol 32:281–294. 10.1016/0013-4694(72)90177-0 [DOI] [PubMed] [Google Scholar]
- Rattka M, Brandt C, Bankstahl M, Bröer S, Löscher W (2011) Enhanced susceptibility to the GABA antagonist pentylenetetrazole during the latent period following a pilocarpine-induced status epilepticus in rats. Neuropharmacology 60:505–512. 10.1016/j.neuropharm.2010.11.005 [DOI] [PubMed] [Google Scholar]
- Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, Riedemann T, Sutor B, Sontheimer H (2015) Reactive astrogliosis causes the development of spontaneous seizures. J Neurosci 35:3330–3345. 10.1523/JNEUROSCI.1574-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Aradi I, Soltesz I (2005) Role of mossy fiber sprouting and mossy cell loss in hyperexcitability: a network model of the dentate gyrus incorporating cell types and axonal topography. J Neurophysiol 93:437–453. 10.1152/jn.00777.2004 [DOI] [PubMed] [Google Scholar]
- Semple BD, Trivedi A, Gimlin K, Noble-Haeusslein LJ (2015) Neutrophil elastase mediates acute pathogenesis and is a determinant of long-term behavioral recovery after traumatic injury to the immature brain. Neurobiol Dis 74:263–280. 10.1016/j.nbd.2014.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple BD, Dixit S, Shultz SR, Boon WC, O'Brien TJ (2017) Sex-dependent changes in neuronal morphology and psychosocial behaviors after pediatric brain injury. Behav Brain Res 319:48–62. 10.1016/j.bbr.2016.10.045 [DOI] [PubMed] [Google Scholar]
- Shultz SR, Cardamone L, Liu YR, Hogan RE, Maccotta L, Wright DK, Zheng P, Koe A, Gregoire MC, Williams JP, Hicks RJ, Jones NC, Myers DE, O'Brien TJ, Bouilleret V (2013) Can structural or functional changes following traumatic brain injury in the rat predict epileptic outcome? Epilepsia 54:1240–1250. 10.1111/epi.12223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statler KD, Scheerlinck P, Pouliot W, Hamilton M, White HS, Dudek FE (2009) A potential model of pediatric posttraumatic epilepsy. Epilepsy Res 86:221–223. 10.1016/j.eplepsyres.2009.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L (1989) Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol 26:321–330. 10.1002/ana.410260303 [DOI] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV (1985) Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci 5:1016–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nieuwenhuyse B, Raedt R, Sprengers M, Dauwe I, Gadeyne S, Carrette E, Delbeke J, Wadman WJ, Boon P, Vonck K (2015) The systemic kainic acid rat model of temporal lobe epilepsy: long-term EEG monitoring. Brain Res 1627:1–11. 10.1016/j.brainres.2015.08.016 [DOI] [PubMed] [Google Scholar]
- Vezzani A, Conti M, De Luigi A, Ravizza T, Moneta D, Marchesi F, De Simoni MG (1999) Interleukin-1β immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J Neurosci 19:5054–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Moneta D, Conti M, Richichi C, Ravizza T, De Luigi A, De Simoni MG, Sperk G, Andell-Jonsson S, Lundkvist J, Iverfeldt K, Bartfai T (2000) Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc Natl Acad Sci U S A 97:11534–11539. 10.1073/pnas.190206797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M (2003) Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 23:8692–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster KM, Sun M, Crack P, O'Brien TJ, Shultz SR, Semple BD (2017) Inflammation in epileptogenesis after traumatic brain injury. J Neuroinflammation 14:10. 10.1186/s12974-016-0786-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziyatdinova S, Gurevicius K, Kutchiashvili N, Bolkvadze T, Nissinen J, Tanila H, Pitkänen A (2011) Spontaneous epileptiform discharges in a mouse model of Alzheimer's disease are suppressed by antiepileptic drugs that block sodium channels. Epilepsy Res 94:75–85. 10.1016/j.eplepsyres.2011.01.003 [DOI] [PubMed] [Google Scholar]