

Abstract
Background
Discriminating driver mutations from the ones that play no role in cancer is a severe bottleneck in elucidating molecular mechanisms underlying cancer development. Since protein domains are representatives of functional regions within proteins, mutations on them may disturb the protein functionality. Therefore, studying mutations at domain level may point researchers to more accurate assessment of the functional impact of the mutations.
Results
This article presents a comprehensive study to map mutations from 29 cancer types to both sequence- and structure-based domains. Statistical analysis was performed to identify candidate domains in which mutations occur with high statistical significance. For each cancer type, the corresponding type-specific domains were distinguished among all candidate domains. Subsequently, cancer type-specific domains facilitated the identification of specific proteins for each cancer type. Besides, performing interactome analysis on specific proteins of each cancer type showed high levels of interconnectivity among them, which implies their functional relationship. To evaluate the role of mitochondrial genes, stem cell-specific genes and DNA repair genes in cancer development, their mutation frequency was determined via further analysis.
Conclusions
This study has provided researchers with a publicly available data repository for studying both CATH and Pfam domain regions on protein-coding genes. Moreover, the associations between different groups of genes/domains and various cancer types have been clarified. The work is available at http://www.cancerouspdomains.ir.
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-017-1779-5) contains supplementary material, which is available to authorized users.
Keywords: Cancer, Protein domain, Pfam, Cath, Pan-cancer, Somatic mutation, TCGA exome sequencing data
Background
Cancer refers to a group of diseases characterized by uncontrolled growth and division of cells in the body, and is caused by environmental as well as genetic factors. Genetic factors include, but are not limited to inherited germline mutations, changing DNA methylation rate and microRNA modifications. Cancer is a leading cause of death in most countries. The number of new cases of cancer is 454.8 per 100,000 incidents per year and the number of cancer deaths is 171.2 per 100,000 incidents per year [1–4]. Accordingly, developing methods for detection and treatment of cancer is a main area of interest as well as a challenge.
Several studies have been conducted to find genes that are involved in cancer development [5–8]. Even though there has been some degree of success in identifying genes that are strongly associated with cancer, much is yet to be done for discovering causal genes and variants. In addition, most of those studies disregard the position of those mutations, whereas mutations at different positions of a certain gene may lead to various levels of damage [9, 10].
Proteins are responsible for most cellular functions and their malfunction may undermine cellular performance [11]. Only some of the mutations in coding regions, and not all of them lead to cancer. Therefore, distinguishing mutations with drastic impacts on protein functionality may help discriminate driver mutations from less significant ones. To this end, some researches have focused on mapping genomic positions to protein sequences and tried to distinguish mutations that affect the functionality of proteins [10, 12]. Protein domains are conserved regions of proteins that can fold and act independently [13]. Therefore, it is plausible that mutations within these regions may cause more damage compared to other mutations [13]. To this aim, some efforts have been made to study cancer mutations at the protein domain level. Nehrt et al. [12] mapped non-synonymous somatic mutations of Breast Invasive Carcinoma and Colon Adenocarcinoma Tumor samples to their corresponding protein domains, in order to extract domains with significant mutation frequency. In another study by Yang et al. [10], mutational protein domain hotspots for 21 different cancer types were determined by mapping somatic mutations to protein domains. Regions with high numbers of mutation for each cancer type were called hotspot.
This study represents a method to explore protein domains with significant mutation frequencies, using whole exome sequencing data. Beside analyzing Pfam protein domains as sequence-based domains, CATH protein domains have also been studied as structure-based domains, which were not included in relevant studies to this date. Moreover, in order to more specifically pinpoint the domains of each cancer type, 29 different cancer types as well as pan-cancer were investigated in this study. In addition, the frequency of mutations in mitochondrial genes, stem cell-specific genes and DNA repair genes were examined. These sets of genes are likely to have important roles in cancer development and progression. Furthermore, the interconnectivity of proteins with mutation on causal domains was investigated.
Methods
Data extraction
Whole-exome sequencing data of 7685 cancer patients from 29 different cancer types containing 2,057,977 somatic mutations are downloaded from the TCGA (The Cancer Genome Atlas) data portal [14]. The detailed list of cancer types as well as the number of patients for each type is shown in Table 1. The names of downloaded files (in July 2015) for each cancer type is shown in Additional file 1: Table A1. The data are extracted from non-metastatic patients before giving radiotherapy or chemotherapy and are mapped to the human genome references of the GRCh37 [15].
Table 1.
Prevalence of patients, mutations and domain-specific mutations in different cancer types
| Cancer type | Abbrevation | patients | Somatic Mutations | Mutations on Protein Coding Regions | Mutations on Pfam Domains | Mutations on CATH Domains |
|---|---|---|---|---|---|---|
| Adrenocortical carcinoma | ACC | 451 | 22,679 | 21,355 | 8335 | 1090 |
| Bladder Urothelial Carcinoma | BLCA | 169 | 169,165 | 154,893 | 69,727 | 11,081 |
| Breast invasive carcinoma | BRCA | 983 | 98,882 | 93,405 | 40,390 | 6512 |
| Cholangiocarcinoma | CHOL | 460 | 8593 | 7030 | 3025 | 484 |
| Colon adenocarcinoma | COAD | 412 | 125,386 | 123,917 | 55,913 | 8055 |
| Esophageal carcinoma | ESCA | 202 | 79,536 | 67,566 | 29,249 | 4340 |
| Glioblastoma multiforme | GBM | 92 | 20,932 | 20,225 | 9905 | 1692 |
| Head and Neck squamous cell carcinoma | HNSC | 545 | 151,456 | 131,314 | 59,768 | 9301 |
| Kidney Chromophobe | KICH | 178 | 8739 | 8023 | 3425 | 489 |
| Kidney renal clear cell carcinoma | KIRC | 36 | 51,235 | 47,964 | 21,059 | 3404 |
| Kidney renal papillary cell carcinoma | KIRP | 269 | 33,247 | 31,433 | 13,530 | 2074 |
| Brain Lower Grade Glioma | LGG | 182 | 47,286 | 42,890 | 19,750 | 3489 |
| Liver hepatocellular carcinoma | LIHC | 230 | 89,042 | 82,443 | 36,749 | 5470 |
| Lung adenocarcinoma | LUAD | 275 | 242,542 | 216,730 | 101,616 | 13,522 |
| Lung squamous cell carcinoma | LUSC | 171 | 65,306 | 63,981 | 30,542 | 4206 |
| Ovarian serous cystadenocarcinoma | OV | 524 | 12,751 | 12,214 | 5824 | 998 |
| Pancreatic adenocarcinoma | PAAD | 175 | 82,871 | 72,462 | 29,279 | 4223 |
| Pheochromocytoma and Paraganglioma | PCPG | 425 | 8706 | 7629 | 3115 | 472 |
| Prostate adenocarcinoma | PRAD | 116 | 26,862 | 24,094 | 10,954 | 1657 |
| Rectum adenocarcinoma | READ | 254 | 34,260 | 33,885 | 15,774 | 2313 |
| Sarcoma | SARC | 75 | 83,019 | 71,650 | 30,021 | 4526 |
| Skin Cutaneous Melanoma | SKCM | 387 | 53,086 | 49,645 | 23,347 | 2993 |
| Stomach adenocarcinoma | STAD | 144 | 223,884 | 213,642 | 96,340 | 14,143 |
| Testicular Germ Cell Tumors | TGCT | 356 | 30,293 | 26,170 | 10,902 | 1518 |
| Thyroid carcinoma | THCA | 123 | 16,807 | 15,383 | 7245 | 1243 |
| Thymoma | THYM | 248 | 39,467 | 35,892 | 15,655 | 2379 |
| Uterine Corpus Endometrial Carcinoma | UCEC | 66 | 212,745 | 204,826 | 93,937 | 13,959 |
| Uterine Carcinosarcoma | UCS | 57 | 14,212 | 11,803 | 5266 | 876 |
| Uveal Melanoma | UVM | 80 | 4988 | 4411 | 2087 | 313 |
| pan-cancer | 7685 | 2,057,977 | 2E + 06 | 852,729 | 126,822 | |
Since we are interested in discovering the role of protein domains in cancer, only protein-coding genes were selected among genes reported in TCGA data. Among 2,057,977 somatic mutations reported in this database, 1,896,875 of them occurred in protein-coding regions. Given that synonymous mutations have no effect on protein sequence and no demonstrable impact on phenotype [16, 17], in this study, only non-synonymous mutations within protein coding regions are considered.
Protein domains can be defined in two different ways, either by their sequences or by their structural characteristics. Both these definitions are considered in this study in order to better understand the role of domains in cancer. Pfam [18, 19] and CATH [20, 21] databases are used to extract sequence-based and structure-based domains, respectively, and the UCSC (University of California Santa Cruz) [22] tables and PDB [23] database are exploited to extract the start and end positions of coding regions, exons, and more specifically, domains in genome.
HUGO (HUman Genome Organization) [24] standard gene nomenclature is employed to identify protein-coding genes. The number of protein-coding genes in HUGO is 19,011, all except for 22 of which are linked to PDB and Pfam entries, and 10,913 of them have Pfam domains. All predicted Pfam domains, without any constraints on E-value and bit-rate, were extracted in this study. A CATH domains was selected if it is represented by the same exact sequence in a UniProt record. To map CATH domains position form PDB residue to UniProt sequence, we used SIFTS [25], which is a manually curated database to match the positions of PDB entries to UniProt sequences.
Identification of candidate domains and genes
Once data have been acquired and evaluated, the next step was to extract candidate regions by use of statistical analysis. Candidate regions (domains or genes) are regions in which mutations occur more frequently than expected. If mutations are mutually independent and uniformly distributed over the combined sequences of coding regions within human genome, then for each mutation, the probability of occurring on the i th coding region is p i, which is equal to the length of i th coding region, l i, divided by total length of coding regions, L, in the whole genome, that is, . To extract candidate regions at domain level, l i and L are respectively set to be the length of i th domain and the total length of domains in the whole proteome.
Suppose that n is the number of mutations occurred in all protein-coding regions and k i is the number of mutations happened in the i th coding region, then the probability of having k i mutations on the i th coding region can be modeled by the binomial distribution, as follows [26].
| 1 |
To determine whether a protein-coding region is a potential candidate for a specific cancer, the number of observed mutations on that region is compared with what would be expected by the binomial distribution model, and a p-value threshold of 0.05 is adopted to test the null hypothesis [27]. To this aim, for k observed mutations on each region, the hypothesis is rejected if p(X < k) > 0.95, where
| 2 |
Since multiple independent hypothesis tests are conducted in all cases, to maintain the family-wise error rate (FWER) [27], a post hoc Bonferroni [28] test is applied. Accordingly, when m independent hypothesis tests are performed, the criterion for rejecting the null hypothesis is divided by m. In other words, when the significance level for the whole family of tests is set to be 0.05, then with Bonferroni correction each individual hypothesis is evaluated at a significance level of.
To eliminate the possibility of overflow or underflow in computing values such as, and , log Pr (X = k) is calculated instead of Pr(X = k). Accordingly, computations are performed using eq. 3 instead of eq. 1:
| 3 |
In addition, to avoid numerical problems in computing in eq. 3, Stirling’s approximation [29] is applied.
Aims and objectives of the study
There are more than 100 types of cancer [30] and despite their differences, they present underlying biological (genetic) similarities. Pan-cancer study aims to uncover similarities and differences between various cancer types [31]. With this background, all the data downloaded from different cancer types are assembled together in this study to form a pan-cancer dataset for further investigations.
The main focus of this study is to assess the frequency of mutations on domain regions. However, we are also interested in evaluating the frequency of mutations in protein coding regions of three particular sets of genes: mitochondrial genes, stem cell-specific genes and DNA repair genes. Mitochondria are responsible for producing energy in almost all cell types and have their own DNA. Since mitochondrial DNA mutations are known to be highly associated with human cancer [32], mutations within the mitochondrial genome are investigated in this study. Most of cancerous cells possess the classical characteristics of normal stem cells, including extensive capacity of self-renewal and acquired resistance to apoptosis [33, 34]. Therefore, genes responsible for the maintenance of stem cells are appropriate candidates for our goal. Mutation in genes that are associated with DNA repair function in a cell may induce partial loss of gene functionality [35, 36]. In this light, studying the presence of mutations in these genes may also be informative for cancer research.
Results and discussion
This study covers four areas of assessment, namely, mutations in protein coding regions of mitochondrial, stem cell-specific and DNA repair genes, and mutations in protein domain regions. The results of each assessment are described in the following subsections.
Mitochondrial genes
Several studies have reported the presence of somatic mitochondrial mutations in cancer cells. Even though many of these studies have demonstrated the role of mitochondrial mutations in human cancers such as Kidney Renal Cell Carcinoma [37], Breast Invasive Carcinoma [38], Gastric Carcinoma [39], Prostate Adenocarcinoma [40], Ovarian Carcinoma [41] and Thyroid Carcinoma [42], such an association was not identified in all relevant studies. For instance, studies on Glioblastoma Multiforme [43] and Liver Hepatocellular Carcinoma [44] have not been able to pinpoint the role of mitochondrial mutations in cancer. In this light, it is worthwhile to further investigate the role of mitochondria in cancer development.
To examine the role of mitochondria in cancer development, the observed somatic mutations in all mitochondrial genes are studied. There are 37 different genes in mitochondrial DNA, which are assigned to six groups of complexes, based on their roles (shown in Additional file 1: Table A2). For instance, the genes MT-RNR1 and MT-RNR2, which are responsible for making rRNAs, are assigned to rRNA complex [45]. Mutations within each group are identified to better understand its role in developing cancer.
To identify mitochondrial candidate genes associated with each of the 29 cancer types as well as pan-cancer (30 cancer types in total), the statistical analysis is performed on two levels. In the first level of analysis, each mitochondrial gene is considered individually, while in the second level, genes are analyzed in their group (complex) context. Accordingly, in the Bonferroni correction, the parameter m is set to 37 × 30 and 6 × 30 for the first and second level, respectively. With a corrected p-value threshold of , there are 13 cancer types and pan-cancer (shown in Table 2) for which at least one mitochondrial candidate gene or complex is identified. In Table 2, the number in parentheses next to each gene shows the percentage of patients for which this gene is mutated. All in all, nine mitochondrial genes have been identified as candidate ones: MT-CO2, MT-CYB, MT-ND1, MT-ND5, MT-RNR1, MT-RNR2, MT-TL1, MT-TT and MT-TV. Additional file 1: Table A3 shows the number of patients with mitochondrial mutations and the number of mutations for each.
Table 2.
Candidate mitochondrial genes and complexes for each cancer type
| Cancer Type | Genes (Percentage) | Complexes (Percentage) |
|---|---|---|
| ACC | MT-TL1 (32.6) | - |
| BRCA | MT-RNR1 (3.6), MT-RNR2 (5.6), MT-TT (0.8) | rRNA (8.4) |
| CHOL | MT-RNR2 (33.3) | rRNA (33.3) |
| ESCA | MT-CYB (13.7) | COMPLEX III (13.7) |
| HNSC | MT-ND5 (1) | - |
| KICH | MT-ND1 (13.6) | - |
| LIHC | MT-ND5 (12.9) | COMPLEX I (18.3) |
| LUAD | - | COMPLEX III (1.1) |
| PRAD | MT-RNR2 (1.9) | - |
| SARC | MT-RNR2 (12.6), MT-CO2 (7.1) | rRNA (14.2), COMPLEX IV (15.7) |
| TGCT | - | COMPLEX III (6.3) |
| THYM | MT-RNR2 (27.6) | rRNA (27.6) |
| UCEC | MT-RNR1 (11.7), MT-RNR2 (11.7), MT-TV (12) | rRNA (0.41.7) |
| Pan Cancer | MT-RNR2, MT-CYB | rRNA, COMPLEX III |
Among six mitochondrial groups of complexes, ATP synthesis and tRNA complexes have not been chosen as candidate for any cancer type. In particular, no significant mutation was observed in genes MT-ATP6 and MT-ATP8 in any of the cancer types. This result is consistent with the assumption that more energy is required for rapid reproduction in cancerous cells. The results also show that two mitochondrial genes, namely MT-RNR2 and MT-CYB are significantly mutated in pan-cancer.
Stem cell-specific genes
Researches have pointed out a number of similarities between stem cells and cancer cells, including their self-renewal potential and their ability to migrate to other regions of the body [46–48]. Moreover, the ability of stem cells to differentiate into various types of cells increases the risk of malignant transformations. Accordingly, stem cell-specific gene analysis is expected to provide a foundation for better understanding of their role in cancer.
The stem cell-specific gene set studied in this research (shown in Additional file 1: Table A4), which is first identified by Palmer et al. [49], contains 182 protein-coding genes. To extract candidate stem cell-specific genes associated with each of the 29 cancer types as well as with pan-cancer, statistical analysis was performed and subsequently, in the Bonferroni correction, the parameter m was set to 182 × 30. With a corrected p-value threshold of , 57 stem cell-specific genes were selected as candidates for at least one cancer type. The most significant genes among them are CHEK2 and KMT2C, which are associated with 20 and 18 different cancer types respectively. The other genes are related to at most seven types. Given that some researches have already demonstrated the role of CHEK2 [50, 51] and KMT2C [52] in different cancer types, their identified association with a large number of cancer types is not surprising. Rectum Adenocarcinoma and Lung Squamous Cell Carcinoma cancers are the only cancer types for which no candidate stem cell-specific gene has been identified. In Table 3, the list of candidate stem cell-specific genes for each cancer type is shown. Similar to Table 2, the numbers in this table also show the percentage of patients in which the genes are mutated.
Table 3.
Candidate stem cell-specific genes for each cancer type
| Cancer Type(Percentage) | Genes(Percentage) |
|---|---|
| ACC (56.5) | HDAC2 (5.4), ERCC2 (20.7), GARS (38.0), PRR34 (8.7) |
| BLCA (30.3) | CHEK2 (6.1), ERCC2 (9.7),KMT2C (20.9) |
| BRCA (8.2) | KMT2C (6.9), PILRB (0.8), HLA_DRB5 (0.7) |
| CHOL (33.3) | CHEK2 (8.3),KMT2C (25),GIMAP8 (2.8) |
| COAD (1.5) | HLA_DPA1 (1.5), |
| ESCA (9.3) | NREP (2.2),BRINP1 (7.7) |
| GBM (4.4) | CHEK2 (1.8),TSHZ2 (2.5) |
| HNSC (20.4) | CHEK2 (3.8), LIN28B (1.5), BRINP1 (3.1), KMT2C (12.0), NPR3 (2.7) |
| KICH.21.2) | DIMT1 (1.5),KMT2C (13.6),HLA_DRB5 (7.6), HLA_DQA1 (3) |
| LIHC (5) | HTR7 (5) |
| KIRC (11.1) | DNMT3B (3.1), CHEK2 (2.2), RRAS2 (1.8), NREP (0.7), TNFSF10 (1.3), FYB (2.9), SMARCC2 (3.1), RCSD1 (2), HLA_DRB5 (1.8) |
| KIRP (7.1) | CHEK2 (5.9), DPH3(1.2) |
| LGG (9.3) | CHEK2(3.9),HDAC2(1.7),ZBTB20(4.6) |
| LUAD (42.0) | SPDL1 (1.8), CHEK2 (7.2), TRPC4 (7.2), CDH6 (7.2), GIMAP1 (2.2), KMT2C (17.8), PILRB (2.2), TSHZ2 (6.8), NPR3 (4.6), FYB (5.5) |
| OV (2.6) | BOD1 (0.9), HAS2 (1.7) |
| PAAD (57.3) | CHEK2 (17.0), BBS9 (9.4), GARS (5.8), SLC24A1 (9.4), KMT2C (17), SMARCC2 (13.5), NPR3 (8.8), AFTPH (13.5) |
| PCPG (14.3) | CHEK2 (5.1), NUSAP1 (4.0), KMT2C (5.1), HLA_DRB5(1.1) |
| PRAD (8.9) | CHEK2(3.5),KMT2C(5.4) |
| SARC (4.3) | ZNF788 (2.8), BRINP1 (2.0) |
| SKCM (37.3) | GDF3 (8.0), CCDC90B (4.0), CDH6 (10.7), KMT2C (16.0), GIMAP5 (6.7) ,GIMAP7 (6.7),GIMAP1 (6.7),GIMAP8 (12) |
| STAD (32.3) | CHEK2 (5.4), SOHLH2 (4.4), BRINP1 (5.9), KMT2C (16.5), TSHZ2(7.2),ZBTB20(9) |
| TGCT (2.8) | C10orf128 (2.1), HLA_DRB5 (2.1), HLA_DQA1 (1.4) |
| THCA (4.8) | CHEK2 (1.4), GDF3 (0.8), RIOK2 (0.8), HLA_DRB5 (1.7) |
| THYM (5.7) | CHEK2 (5.7) |
| UCEC (9.7) | ATP11C (9.7) |
| UCS(15.8) | CHEK2(7),KMT2C(10.) |
| UVM(13.8) | CHEK2(7.5),NUSAP1(5),HLA_DRB5(5) |
| Pan Cancer(24.3) | CHEK2 (4.1), SOHLH2 (1), BRINP1 (2.2), TRPC4 (2.4), CDH6 (2.1), KMT2C (10.6), PILRB (0.8) ,HLA_DRB5 (1.1), TSHZ2 (2.6), NPR3 (1.7),GIMAP1 (0.9), GIMAP8(1.7),ZBTB20(2.1) |
DNA repair genes
DNA repair genes are responsible for recognizing and correcting damages in the replication of DNA. Hence, mutations in DNA repair genes can be expected to alter the efficiency of repairing mechanism, which in turn can be associated with severe health issues such as cancer. Moreover, it has been reported that DNA repair genes are frequently mutated in cancer [53]. Accordingly, studying mutations within DNA repair genes may be helpful for revealing their role in cancer.
To identify DNA repair genes which are associated with a certain type of cancer, a statistical analysis similar to that performed in previous subsections was applied. 174 known DNA repair genes, reported in [54–56], are shown in Additional file 1: Table A5. By setting the parameter m to 174 × 30 in the Bonferroni correction, 27 DNA repair genes were identified as candidate for at least one cancer type. The results show that the most significant DNA repair gene is TP53, which was identified as candidate for 25 cancer types as well as for pan-cancer. This conforms with the previous findings about the crucial role of TP53 mutations in cancer development [57, 58]. This further endorses the reliability of the other results in this study. For each cancer type other than Testicular Germ Cell Tumors, at least one candidate DNA repair gene was identified. In particular, Pancreatic Adenocarcinoma has eight candidate DNA repair genes, and ATM, TCG, TP53 and CHEK2 are the candidate DNA repair genes for pan-cancer. Table 4 shows the candidate DNA repair genes for each cancer type and the number next to each gene shows the percentage of patients in which this gene is mutated.
Table 4.
Candidate DNA repair genes for each cancer type
| Cancer Type(Percentage) | Genes(Percentage) |
|---|---|
| ACC (37) | MSH3(6.5),TP53(19.6),ERCC2(20.7) |
| BLCA (63.6) | ATM (13.6), TP53 (49.8) ,ERCC2 (9.7), CHEK2 (6.1) |
| BRCA (33.4) | TP53 (33.4) |
| CHOL (22.2) | TP53 (13.9), CHEK2 (8.3) |
| COAD (52) | TP53 (52.0) |
| ESCA (87.9) | TP53 (87.9) |
| GBM (28.7) | TP53 (28.7) |
| HNSC (71.6) | TP53 (71.2), CHEK2 (3.8) |
| KICH (33.3) | TP53 (33.3) |
| KIRC(10.9) | FANCE (4.4), DDB1 (4.9), RPA1 (2.2), TP53 (4.2), CHEK2 (2.2) |
| KIRP (17.2) | OGG1 (2.4), MSH3 (4.1), TDG (3.6), TP53 (4.1), CHEK2 (5.9) |
| LGG (50.4) | TP53 (48), CHEK2 (3.9) |
| LIHC (32.2) | TP53(32.2) |
| LUAD (57.8) | ERCC5 (3.3), TP53 (54.7), CHEK2 (7.2) |
| LUSC (79.2) | TP53 (79.2) |
| OV (84.8) | TP53 (84.8) |
| UCEC (34.7) | MSH4 (7.3), TP53 (29) |
| PAAD (84.2) | ERCC3 (8.8), XPC (9.9), WRN (14.6), TDG (9.9), FAN1 (9.9), EME2(11.1), TP53 (67.3), CHEK2 (17) |
| PCPG (17.7) | FANCD2 (5.1), ERCC8 (1.1), TDG (7.4), CHEK2 (5.1) |
| PRAD (19.3) | ATM (4.5), TP53 (10.8), POLI (1.4), CHEK2 (3.5) |
| READ (67.2) | TP53 (67.2) |
| SARC(37.0) | TP53 (37) |
| SKCM(22.7) | BLM (6.7), MPG (4.0), TP53 (10.7), CHEK2 (09.3) |
| STAD (57.9) | UVSSA (4.4), SLX4 (6.7), TP53 (49.9), CHEK2 (5.4) |
| THCA (4.5) | SMUG1 (0.8), TDG (2.2), TP53 (0.8), CHEK2 (1.4) |
| THYM (5.7) | CHEK2 (5.7) |
| UCS (91.2) | TP53 (91.2) |
| UVM(20) | FANCD2 (6.3), CCNH (2.5), TDG (3.7), CHEK2(7.5) |
| Pan Cancer(45.2) | ATM(5.5),TDG(1.7),TP53 (39.1),CHEK2 (4.0) |
To identify cancer-associated genes within mitochondrial, stem cell-specific and DNA repair genes, not only the mutations on domain regions but all those on full protein coding regions are included in the assessment. To be more confident in extracting cancer-associated genes within each biological process, its related candidate genes were restricted to those which also contain at least one candidate domain. Upon studying the mitochondrial genes, we found no candidate domains (defined in the following sections) associated with those genes. Among candidate stem cell-specific genes, 51% and 46% of them contain at least one Pfam and one CATH candidate domain, respectively, as shown in Fig. 1a and b. For each cancer type, the entire list of stem cell-specific genes with Pfam and CATH candidate domains are presented in Additional file 1: Tables A6 and A7. Similarly, 25% and 26% of candidate repair genes consist of at least one Pfam and one CATH candidate domain, respectively, as shown in Fig. 1c and d. More details on repair genes with Pfam and CATH domains are given in Additional file 1: Tables A8 and A9.
Fig. 1.
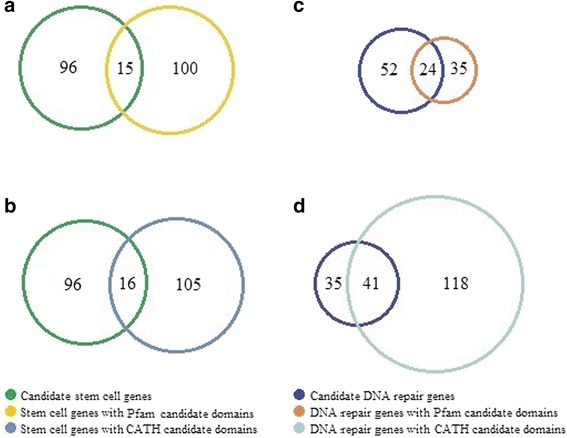
Comparison of candidate genes and genes with candidate domains. (a) Comparison of stem cell genes and genes with Pfam candidate domains. (b) Comparison of stem cell genes and genes with CATH candidate domains. (c) Comparison of DNA repair genes and genes with Pfam candidate domains. (d) Comparison of DNA repair genes and genes with CATH candidate domains
CATH candidate domains
A key objective of this study is to identify CATH candidate domains, which have gone unnoticed in the previous researches conducted in this field. There are 759 CATH-reported domains which are located in 2993 human proteins. Detailed information for each CATH-reported domain can be found in Additional file 1: Table A10. In addition, the position of each CATH domain on each protein-coding gene is available in Additional file 2: Table B1.
To assess CATH domains, the significance level of was used. The results indicate that each cancer type has a number of associated CATH candidate domains ranging from 1 to 19, while pan-cancer analysis reveals 93 related CATH candidate domains. Some domains seemed to not be associated with any individual cancer type, yet they were identified as significant candidates in the pan-cancer study. We say a candidate domain “covers” a particular patient, if the patient has at least one mutation in that specific candidate domain. Surveying the results, we realize that each CATH candidate domain of each cancer type covers various percentages of patients in that cancer type, ranging from 0.02% to 95%. Moreover, all CATH candidate domains of each cancer type cover 28% to 98% of patients of that cancer type. The CATH candidate domains identified for Breast Invasive Carcinoma, Ovarian Serous Cyst Adenocarcinoma and pan-cancer are presented in Table 5. The number next to each domain shows the percentage of patients which are covered by this domain. Additional file 1: Table A11 shows CATH candidate domains in each cancer type. To assess the statistical significance of an identified candidate domain, the percentage of patients covered by that domain can theoretically be used as a selection attribute.
Table 5.
Candidate domains for Breast Invasive Carcinoma and Ovarian Serous Cystadenocarcinoma
| Cancer Type (Percentage) | CATH Domains (Percentage) |
|---|---|
| BRCA (77.3) | 1.10.1070.11 (33.88), 1.10.220.60 (0.31), 1.10.437.10 (1.73), 1.10.510.10 (25.84), 2.170.260.10 (0.71), 2.40.250.10 (2.03), 2.60.200.10 (1.83), 2.60.40.10 (20.24), 2.60.40.1110 (4.27), 2.60.40.60 (4.48), 2.60.40.720 (33.27)4.10.365.10 (0.71) |
| OV (80) | 1.10.287.650 (0.87), 2.60.40.720 (80.00), 3.30.450.40 (0.87) |
| Pan Cancer (91.5) | 1.10.10.10 (7.90), 1.10.10.440 (0.92), 1.10.10.60 (4.23), 1.10.101.10 (1.59), 1.10.1070.11 (15.52), 1.10.1300.10 (5.87), 1.10.1380.10 (2.34), 1.10.150.210 (0.78), 1.10.150.50 (3.03), 1.10.150.60 (1.30), 1.10.1520.10 (0.55), 1.10.1540.10 (1.47), 1.10.167.10 (3.70), 1.10.246.10 (2.17), 1.10.287.450 (0.94), 1.10.437.10 (2.25), 1.10.472.10 (4.68), 1.10.490.10 (2.46), 1.10.506.10 (0.78), 1.10.510.10 (44.89), 1.10.555.10 (3.85), 1.10.565.10 (10.70), 1.10.630.10 (10.98), 1.10.640.10 (0.98), 1.10.720.50 (0.64), 1.10.750.10 (3.32), 1.10.800.10 (1.60), 1.20.1050.10 (4.98), 1.20.1250.10 (5.37), 1.20.1260.10 (1.29), 1.20.1280.50 (1.17), 1.20.1340.10 (1.61), 1.20.245.10 (0.95), 1.20.5.100 (1.17), 1.20.5.110 (0.48), 1.20.5.50 (1.86), 1.20.58.60 (2.32), 1.20.82.10 (1.01), 1.20.920.10 (6.57), 1.20.930.40 (4.19), 1.25.10.10 (8.64), 1.25.40.20 (8.26), 2.10.220.10 (7.52), 2.10.25.10 (6.69), 2.10.310.10 (0.46), 2.10.60.10 (1.76), 2.10.70.10 (6.31), 2.130.10.10 (7.43), 2.120.10.80 (1.91), 2.140.10.30 (3.99), 2.130.10.130 (3.15), 2.170.270.10 (4.18), 2.170.8.10 (1.20), 2.30.30.190 (1.21), 2.30.39.10 (8.76), 2.30.42.10 (8.87), 2.40.128.20 (4.23), 2.40.20.10 (1.94), 2.40.250.10 (0.38), 2.40.50.40 (4.42), 2.60.120.200 (4.42), 2.60.120.260 (4.65), 2.60.20.10 (2.49), 2.60.200.10 (3.99), 2.60.210.10 (2.78), 2.60.40.10 (33.88), 2.60.40.1110 (6.69), 2.60.40.1120 (1.57), 2.60.40.60 (2.32), 2.60.40.720 (36.55), 2.60.60.20 (1.59), 2.70.98.20 (2.38), 2.80.10.50 (5.22), 3.10.100.10 (5.87), 3.10.20.230 (0.94), 3.10.200.10 (3.60), 3.10.50.10 (2.64), 3.10.620.10 (0.44), 3.20.20.100 (4.55), 3.20.20.140 (4.49), 3.30.1370.10 (2.34), 3.30.1490.20 (2.13), 3.30.300.30 (1.70), 3.30.450.40 (0.48), 3.30.450.50 (0.87), 3.30.70.1230 (0.88), 3.30.70.1470 (0.91), 3.30.70.330 (12.00), 3.30.800.10 (1.98), 3.30.9.10 (0.77), 3.40.190.10 (3.68), 3.40.50.10140 (1.54), 3.40.470.10 (1.13), 3.40.50.10190 (4.22), 3.40.50.1370 (0.75), 3.40.50.2300 (1.51), 3.40.50.300 (25.67), 3.40.718.10 (5.78), 3.90.1170.10 (0.43), 3.90.1230.10 (1.63), 3.90.190.10 (13.32), 4.10.280.10 (1.08), 4.10.365.10 (0.34), 4.10.75.10 (0.72) |
Pfam candidate domains
There are 6009 predicted Pfam domains located in 17,722 human proteins. Detailed information for Pfam domains can be found in Additional file 1: Table A12. In addition, the position of each Pfam domain on each protein-coding gene is available in Additional file 2: Table B2. The significance level of was used to perform statistical assessment, the results of which show that each cancer type has a different number of Pfam candidate domains, ranging from 3 to 93. For pan-cancer, the number of identified Pfam candidate domains is 202, which indicates a large number of domains are significant to pan-cancer but not to individual cancer types. The results are consistent with those of CATH domains.
Each Pfam Candidate domain of each cancer type covers different percentages of patients with a minimum of 0.2% and a maximum of 98%. Overall, all Pfam candidate domains of each cancer type cover 74% to 100% of patients of that cancer type. Table 6 shows Pfam candidate domains of Breast Invasive Carcinoma and Ovarian Serous Cyst Adenocarcinoma and the number next to each domain shows the percentage of patients, which are covered by this domain. Additional file 1: Table A13 shows Pfam candidate domains in each cancer type. Similar to CATH candidate domains, the percentage of patients covered by a candidate domain can be used as a proper measure. For instance, P53 and tm_4 cover the first and the second highest percentages (42% and 28%) of Breast invasive carcinoma patients, respectively, which shows their significant role in this particular cancer.
Table 6.
Pfam candidate domains for Breast Invasive Carcinoma and Ovarian Serous Cystadenocarcinoma
| Cancer Type (Percentage) | Pfam Domains (Percentage) |
|---|---|
| BRCA (78.5) | 7tm_4 (41.51), ATP-synt_A (0.81), Atrophin-1 (2.85), CBF_beta (2.24), COX1 (2.03), COX3 (1.12), Cadherin (23.91), Cytochrom_B_N_2 (1.12), DUF4647 (1.32), FAM219A (0.51), FRG1 (1.32), GATA (3.97), G_path_suppress (1.12), H-K_ATPase_N (0.31), Histone (7.32), NADH5_C (0.92), NADHdh (1.63), Oxidored_q4 (0.71), Oxidored_q5_N (0.81), P53 (28.48), P53_tetramer (2.03), PI3K_C2 (2.54), PI3K_P85_iSH2 (2.34), PI3K_p85B (1.02), PI3Ka (13.22), PTEN_C2 (2.03), Proton_antipo_M (3.56), Runt (2.44), T-box (4.17), TMEM247 (1.12), Tis11B_N (1.02) |
| OV (88.3) | 7tm_4 (49.57), DUF2462 (0.43), DUF4552 (1.30), MRP-S32 (0.87), NtCtMGAM_N (2.17), ODAM (1.30), P53 (72.61), P53_tetramer (4.78), PTCRA (0.87), Sam68-YY (1.30), UPF0054 (0.87) |
The statistical analysis conducted in this study is different to that used by Nehrt et al. [12]. Moreover, different data sources were exploited in these two studies. Therefore, it is no surprise that the results of the two studies are dissimilar. To further emphasize the difference between these approaches, we remark that the number of Pfam domains examined in our study is much larger than that of Nehrt et al. [12] due to the cut-off used in that study for minimum protein or domain length (150 amino acids) and due to Pfam E-value threshold used for inclusion (0.001). The comprehensive comparison performed over Pfam and CATH regions (discussed in the next section) clearly indicates the high reliability of Pfam-reported domains, regardless of their associated E-values. Furthermore, 5918 out of 6009 investigated Pfam domains have E-value less than threshold of 0.001. Also, among 769 identified Pfam candidate domains, 754 (98%) satisfy the threshold condition. Accordingly, we decided not to exclude any Pfam-reported domain. In addition, the statistical method used by Nehrt et al. [12], is extremely sensitive to the number of patients having mutations within the domain region of each protein. This is due to the fact that the number of mutations in each domain is normalized by the cumulative length of all its associated proteins, wherein at least one patient had mutation. Hence, if a new patient with a mutation on an associated protein is added, for which no previous mutation is reported, this would significantly impact the normalizing factor, and subsequently, the statistic used. Moreover, the threshold level of 0.1 is applied in Nehrt et al. [12] for determining significantly mutated domains, by using local false discovery rate (LFDR). As shown in Fig. 2, Nehrt et al. [12] reported 41 and 45 Pfam domains as significantly mutated in Breast Invasive Carcinoma and Colon Adenocarcinoma Tumor, respectively, while our results identified 31 Pfam candidate domains for Breast Invasive Carcinoma and 35 ones for Colon Adenocarcinoma Tumor. Comparing the results of the two studies shows that they share nine domains for Breast Invasive Carcinoma including CBF_beta, FRG1, GATA, P53, PI3K_p85B, PI3Ka, PTEN_C2, T-box and Tis11B_N. Moreover, the four domains of APC_crr, MH2, P53 and PI3K_p85B are reported by both studies for Colon Adenocarcinoma Tumor.
Fig. 2.
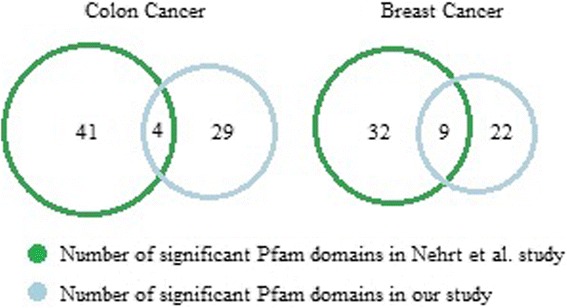
The comparison between our study and Nehrt et al. [13]
In another study by Yang et al. [10] mutations were obtained from COSMIC database [59] and the analysis was restricted to potentially damaging missense mutations, predicted by IntOGen-mutation platform. To determine significantly mutated domains in a given cancer type, Fisher’s exact test was exploited in that study. Accordingly, the results obtained by Yang et al. [10] are different from those of this study, as expected. The list of cancer types investigated in Yang et al. [11] and those considered in this study share 13 in common. For each of these 13 cancer types, significantly mutated domains obtained by both studies are shown in Table 7. Based on these two studies, seven cancer types share P53 as one of their significant domains.
Table 7.
Shared significant domains in our study and Yang et al. [11]
| Cancer type | Shared domains | |||
|---|---|---|---|---|
| BRCA | Cytochrom_B_C | P53 | ||
| COAD | MH2 | |||
| ESCA | P53 | |||
| HNSC | P53 | |||
| KIRC | Bromodomain | Oxidored_q3 | VHL | |
| LIHC | P53 | |||
| LUAD | P53 | Pkinase_Tyr | Ras | Sushi |
| LUSC | P53 | Sushi | ||
| OV | P53 | |||
| PAAD | Ras | |||
| PRAD | MATH | |||
| SKCM | Pkinase_Tyr | |||
| UCEC | DSPc | NADHdh | PI3K_p85B | PI3Ka |
CATH vs. Pfam protein domains
There is a gap between the number of sequenced proteins and that of proteins with known structure, which can also be observed at the level of protein domains. On the other hand, structure-based protein domains are biologically more informative and reliable. Therefore, to benefit from the high number of sequence-based protein domains as well as from the accuracy of structure-based protein domains, both sequence-based and structure-based domains are studied in this research. CATH and Pfam databases are used to extract structure-based and sequence-based domains, respectively.
Through further investigation, for each protein which has both Pfam and CATH annotations (2974 proteins), the overlap between its Pfam domain region and CATH domain region is computed. For instance, as it is shown in Fig. 3a, for gene VPS25 which contains two homologous domain superfamilies with CATH IDs 1.10.10.10 (amino acids 102–176) and 1.10.10.570 (amino acids 1–176) as well as one Pfam domain of ESCRT-II (amino acids 10–145), the computed overlap is from amino acid 10 to 145. This overlap covers 77% of CATH domain region and 100% of Pfam domain region. Overall, for all 2974 proteins with both Pfam and CATH annotations, computed overlaps cover 79% of CATH domain regions and 75% of Pfam domain regions, on average, as shown in Fig. 3b. This suggests that for a protein with no annotation in CATH database, it is reasonable to study its Pfam domain region as a representative of its functional unit.
Fig. 3.
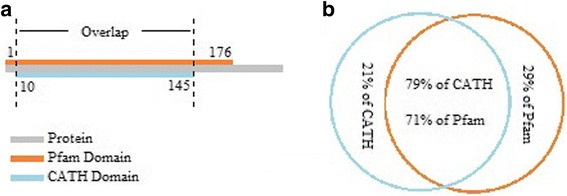
The overlap between Pfam domain region and CATH domain region. (a) The overlap between Pfam domain region and CATH domain region for gene VPS25. (b) The average overlap between Pfam domain regions and CATH domain regions
In addition, the percentage of patients in each cancer type, which are covered by Pfam candidate domains are compared with the ones covered by CATH candidate domains (shown in Fig. 4). As it is shown in Fig. 4, for several cancer types including Bladder Urothelial Carcinoma, Breast Invasive Carcinoma, Uterine Corpus Endometrial Carcinoma and Uterine Carcinosarcoma, Pfam and CATH candidate domains cover the same percentage of patients, while in some other types such as Adrenocortical Carcinoma, there is a huge gap between the two. The considerably high level of overlap between Pfam and CATH domain regions suggest that wherever CATH candidate domains are incapable of covering patients, Pfam candidate domains are suitable substitutions.
Fig. 4.
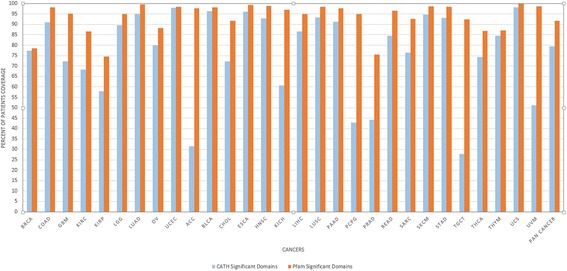
CATH vs. Pfam candidate domain coverage for patients
Among 6009 investigated Pfam domains, 769 are identified as candidate domains in at least one cancer type. Candidate domains are observed to be significantly mutated in varying numbers of cancer types (more details are given in Additional file 1: Table A14). To assess the contribution of each candidate domain in different types of cancer, the list of 769 candidate domains were sorted in decreasing order based on the number of associated cancer types. The 17 top-listed domains, presented in Additional file 1: Table A15, are found to be the least number of candidates that each studied cancer type is associated with at least three candidate domains within them. Given that P53 is one of the most commonly mutated domains in all cancers, it is no surprise that it is placed at the top of the list, above other domains. The second domain in the sorted list, tm_4, is identified as a candidate domain for 22 cancer types and for pan-cancer. The tm_4 domain, which is present in a large number of proteins (376), has not previously been implicated in cancer susceptibility, hence can be seen as a newly found candidate.
Proteins of keratin family contain six domains, all except Keratin_assoc are found to be candidate in different numbers of cancer types, ranging from 6 to 17. Interestingly, three of keratin-related domains (Keratin_B2, Keratin_B2_2 and Keratin_2_tail) are placed in our list of top 17 domains. The great contribution of keratin-related domains to cancer may be due to their role in protecting epithelial cells from damage or stress [60].
Similar investigations performed on CATH domains show that among 759 CATH domains, 181 are identified as candidate ones. Detailed information on their associated cancer types are given in Additional file 1: Table A16. Going through the sorted list of CATH candidate domains shows that the 15 top-listed domains, presented in Additional file 1: Table A17, are found to be the least number of candidates that each studied cancer type is associated with at least one candidate domain within them.
Besides, this study sheds some light on the role of domains in cancer. For instance, there are in total 181 CATH and 769 Pfam candidate domains associated to at least one cancer type or to pan-cancer. 94% of Pfam domains and 95% of CATH domains have mutations in more than 95% of their corresponding proteins. However, a high percentage of proteins with mutations on a particular domain does not necessarily imply that domain as a significant candidate. As an example, Pkinase is a domain involved in 348 proteins, for which the number of occurrences on those proteins is 369. Based on the data available, the total number of mutations on this domain in different cancer types is 346, yet it is not identified as a candidate domain for any cancer type. In contrast to Pkinase, Phostensin_N is a domain which is located on two different proteins with mutations on only one of them, yet it is identified as a candidate domain for Pancreatic Adenocarcinoma.
Further investigation was performed to determine specific domains of each cancer type. As an example, there are 30 Pfam candidate domains for Breast Invasive Carcinoma among which four are only specific to this cancer type, including Atrophin-1, CBF_beta, GATA and Runt. The entire list of specific domains for all cancer types is available in Additional file 1: Table A18. Subsequently, a protein is termed “Pfam specific protein” to a cancer type, if it contains only those Pfam candidate domains which are specific to the cancer of interest. Additional file 1: Table A19 shows all proteins with mutations on Pfam candidate domains and Additional file 1: Table A20 presents Pfam specific proteins of all cancer types. Similar tables (Additional file 1: Tables A21, A22 and A23) are provided for CATH candidate domains. For each cancer type, the number of Pfam candidate domains, proteins with mutations on Pfam candidate domain, Pfam specific domains and specific proteins are summarized in Table 8. Table 9 shows similar results for CATH candidate domains. On average, the number of CATH specific domains for a cancer type is 2.4, whereas this number is 15.8 for Pfam specific domains.
Table 8.
Number of Pfam Candidate domains, proteins with candidate domains and specific domains and proteins for each cancer type
| Cancer type | Number of Pfam candidate domains | Number of Proteins with Pfam candidate domains | Number of Pfam specific candidate domains | Number of Pfam specific proteins |
|---|---|---|---|---|
| ACC | 57 | 323 | 23 | 47 |
| BLCA | 51 | 1076 | 15 | 64 |
| BRCA | 31 | 495 | 4 | 11 |
| CHOL | 39 | 45 | 19 | 21 |
| COAD | 35 | 955 | 6 | 7 |
| ESCA | 25 | 355 | 4 | 5 |
| GBM | 24 | 474 | 6 | 8 |
| HNSC | 42 | 743 | 5 | 42 |
| KICH | 55 | 200 | 21 | 23 |
| KIRC | 90 | 494 | 54 | 157 |
| KIRP | 36 | 84 | 22 | 44 |
| LGG | 25 | 366 | 3 | 15 |
| LIHC | 25 | 432 | 5 | 9 |
| LUAD | 115 | 2328 | 27 | 248 |
| LUSC | 31 | 1128 | 5 | 8 |
| OV | 11 | 188 | 6 | 6 |
| PAAD | 143 | 793 | 70 | 151 |
| PCPG | 70 | 151 | 20 | 44 |
| PRAD | 34 | 353 | 13 | 20 |
| READ | 29 | 266 | 10 | 17 |
| SARC | 25 | 383 | 7 | 11 |
| SKCM | 40 | 1116 | 11 | 25 |
| STAD | 52 | 1476 | 10 | 35 |
| TGCT | 66 | 366 | 29 | 43 |
| THCA | 38 | 304 | 15 | 19 |
| THYM | 18 | 41 | 5 | 5 |
| UCEC | 23 | 1094 | 3 | 16 |
| UCS | 44 | 57 | 19 | 27 |
| UVM | 63 | 99 | 22 | 34 |
| Pan-cancer | 222 | 3685 | 27 | 315 |
Table 9.
Number of CATH Candidate domains, proteins with candidate domains and specific domains and proteins for each cancer type
| Cancer type | Number of CATH candidate domains | Number of proteins with CATH candidate domains | Number of CATH specific candidate domains | Number of CATH specific proteins |
|---|---|---|---|---|
| ACC | 7 | 15 | 4 | 5 |
| BLCA | 22 | 649 | 1 | 1 |
| BRCA | 12 | 315 | 1 | 2 |
| CHOL | 9 | 39 | 2 | 4 |
| COAD | 16 | 377 | 0 | 0 |
| ESCA | 13 | 191 | 2 | 2 |
| GBM | 9 | 28 | 0 | 0 |
| HNSC | 14 | 372 | 0 | 0 |
| KICH | 10 | 18 | 6 | 7 |
| KIRC | 19 | 49 | 3 | 3 |
| KIRP | 11 | 110 | 8 | 10 |
| LGG | 7 | 21 | 1 | 2 |
| LIHC | 14 | 307 | 2 | 4 |
| LUAD | 32 | 590 | 3 | 24 |
| LUSC | 11 | 154 | 0 | 0 |
| OV | 3 | 6 | 1 | 2 |
| PAAD | 20 | 249 | 6 | 27 |
| PCPG | 9 | 56 | 3 | 3 |
| PRAD | 13 | 113 | 4 | 5 |
| READ | 7 | 116 | 0 | 0 |
| SARC | 8 | 285 | 1 | 2 |
| SKCM | 17 | 281 | 5 | 16 |
| STAD | 26 | 575 | 2 | 6 |
| TGCT | 9 | 78 | 3 | 5 |
| THCA | 5 | 91 | 1 | 3 |
| THYM | 5 | 146 | 2 | 11 |
| UCEC | 26 | 449 | 4 | 5 |
| UCS | 11 | 22 | 3 | 4 |
| UVM | 6 | 13 | 2 | 2 |
| Pan-cancer | 104 | 456 | 18 | 48 |
To evaluate the reliability of the proposed methodology, the following process was performed. For each cancer type, the genes that contain identified candidate domains and have mutations in that cancer were compared with experimentally verified cancer genes from COSMIC [59]. A list of 616 unique genes for which mutations have been causally implicated in cancer, were downloaded from COSMIC database (in May 2017). Since some of the cancer causal genes are associated to more than one cancer type, the cumulative number (counting with repetition) of genes, for all 29 cancer types investigated in this study tallied 967. Considering all 29 cancer types (shown in Fig. 5), 814 COSMIC genes have mutations on Pfam domains, out of which, 413 genes contain at least one Pfam candidate domain. Therefore, among COSMIC genes with at least one Pfam domain, 51% have Pfam candidate domains. For each cancer type, the list of its specific COSMIC genes which also have Pfam candidate domain is given in Additional file 1: Table A24.
Fig. 5.
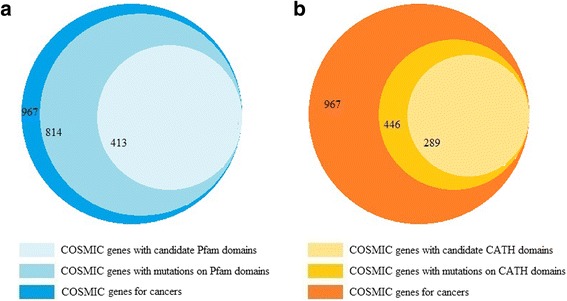
Comparison of causal genes in COSMIC with genes having candidate domains. (a) Comparison of COSMIC genes with genes having Pfam candidate domains. (b) Comparison of COSMIC genes with genes having CATH candidate domains
Similar analysis for CATH domains indicates that among 967 cancer causal genes, there are 446 genes with mutations on their CATH domains, out of which, 289 genes have mutations on CATH candidate domains. Detailed information for each cancer type is presented in Additional file 1: Table A25. Even though 52% of the cancer causal genes reported by COSMIC have no CATH domains, among those with at least one CATH domain, 65% have the candidate ones. This suggests that CATH domains are superior indicators compared to Pfam domains, in identification of cancer causal genes.
Curiously, studying mutations at domain level provides an interesting insight into the role of gene families in cancer development. For instance, in Uterine Corpus Endometrial Carcinoma, eight out of nine genes in TCEAL gene family have mutations on identified Pfam candidate domains. Members of this family had already been identified as nuclear phosphoproteins that modulate transcription in a promoter context-dependent manner [61]. Besides, for 19 out of 22 genes of FGF gene family, mutations occur on Pfam candidate domains in Stomach adenocarcinoma. FGF’s are known to play a key role in the processes of proliferation and differentiation of a wide variety of cells and tissues [62].
We also evaluated the impact of domain mutations in cancer using SnpEff [63], which annotates the effects of variants on genes and classifies them as low, moderate and high impact. The results indicate that 13.4% of mutations on Pfam and CATH domains are classified as high impact mutations and the rest are reported as moderate ones. Restricting the analysis to candidate domains shows that 14.3% and 17.7% of mutations on Pfam and CATH candidate domains are considered as high impact ones, respectively. Moreover, we calculated the frequency of different types of variants, including nucleotide variants, insertions and deletions, on domain regions. Our analysis reveals that all variants has similar frequencies on domains and candidate domains.
Protein-protein interactions
Protein-Protein Interactions (PPIs) commonly refer to physical contact between two or more proteins [64] and offer a wealth of molecular information, which exists in various molecular pathways. Besides, domains are usually responsible for mediating protein-protein interactions [65]. Understanding specific interaction map of a disease is becoming increasingly crucial in elucidating its underlying molecular mechanisms. Therefore, it is reasonable to study PPI for proteins with candidate domains. To this end, the specific proteins of each cancer type are exploited to determine specific PPIs of that cancer type. The interactome database STRING [66] was used to extract all interactions with the highest confidence score (0.9 and above) among specific proteins of each cancer type. The results show that in 20 out of 29 cancer types and also in pan-cancer, specific proteins are significantly connected. The connectivity significance of proteins was determined by the PPI enrichment p-value, reported by STRING. This measure quantifies whether the set of input proteins have more interactions among themselves than a random set of proteins of similar size.
Subsequently, for each cancer type in which a significant interaction network is not formed on its specific proteins, the same analysis was performed on the entire list of proteins with mutation on Pfam candidate domains of that cancer type. The entire lists of proteins are significantly interconnected for all cancer types other than Ovarian serous cystadenocarcinoma. Table 10 gives some information about the number of nodes and edges in the interaction networks of each cancer type, as well as their enrichment p-values. Furthermore, for each cancer type the interactome analysis was performed on the entire list of proteins with mutation on CATH candidate domains of that cancer type. The results show that in all cancer types except Uveal Melanoma, proteins with mutations on CATH candidate domains are significantly connected. Some information including the number of nodes and edges in each interaction network, and its enrichment p-value is presented in Table 11. Due to the small number of proteins in the CATH specific lists, interactome analysis was not performed on them.
Table 10.
The result of interactome analysis for Pfam candidate domains in different cancer types
| Cancer type | Pfam specific proteins | Proteins with Pfam candidate domain | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| #Nodes | #Edges | Expected number of edges | PPI enrichment p-value | Significant or Not | #Nodes | #Edges | Expected number of edges | PPI enrichment p-value | Significant or Not | |
| BRCA | 16 | 14 | 5 | 0.002 | YES | |||||
| COAD | 7 | 1 | 0 | 0.04 | YES | |||||
| GBM | 8 | 1 | 0 | 0.126 | NO | 471 | 2687 | 558 | 0 | YES |
| KIRC | 156 | 40 | 16 | 4E-07 | YES | |||||
| KIRP | 43 | 30 | 3 | 0 | YES | |||||
| LGG | 14 | 2 | 0 | 7E-04 | YES | |||||
| LUAD | 247 | 282 | 9 | 0 | YES | |||||
| OV | 6 | 0 | 0 | 1 | NO | 186 | 3 | 1 | 0.0738 | NO |
| UCEC | 16 | 1 | 0 | 7E-04 | YES | |||||
| ACC | 46 | 3 | 1 | 0.069 | NO | 319 | 113 | 14 | 0 | YES |
| BLCA | 64 | 14 | 2 | 1E-07 | YES | |||||
| CHOL | 21 | 4 | 0 | 2E-04 | YES | |||||
| ESCA | 5 | 1 | 0 | 0.002 | YES | |||||
| HNSC | 42 | 25 | 1 | 0 | YES | |||||
| KICH | 23 | 1 | 2 | 0.79 | NO | 198 | 105 | 33 | 0 | YES |
| LIHC | 9 | 1 | 0 | 0.016 | YES | |||||
| LUSC | 8 | 0 | 0 | 1 | NO | 1132 | 9242 | 2583 | 0 | YES |
| PAAD | 151 | 59 | 21 | 6E-12 | YES | |||||
| PCPG | 43 | 5 | 1 | 0.01 | YES | |||||
| PRAD | 20 | 7 | 1 | 2E-06 | YES | |||||
| READ | 17 | 4 | 0 | 1E-04 | YES | |||||
| SARC | 11 | 3 | 0 | 2E-05 | YES | |||||
| SKCM | 25 | 22 | 4 | 7E-11 | YES | |||||
| STAD | 35 | 69 | 1 | 0 | YES | |||||
| TGCT | 42 | 8 | 1 | 3E-06 | YES | |||||
| THCA | 19 | 1 | 0 | 0.149 | NO | 301 | 132 | 29 | 0 | YES |
| THYM | 5 | 0 | 0 | 1 | NO | 40 | 32 | 0 | 0 | YES |
| UCS | 26 | 1 | 1 | 0.462 | NO | 55 | 45 | 8 | 0 | YES |
| UVM | 34 | 3 | 3 | 0.609 | NO | 98 | 22 | 14 | 0.039 | YES |
| Pan-cancer | 312 | 231 | 34 | 0 | YES | |||||
Table 11.
The result of interactome analysis for CATH candidate domains in different cancer types
| Proteins with at least one CATH candidate domain | |||||
|---|---|---|---|---|---|
| Cancer type | #Nods | #Edges | Expected number of edges | PPI enrichment p-value | Significant or not |
| BRCA | 315 | 1136 | 261 | 0 | YES |
| COAD | 377 | 1325 | 363 | 0 | YES |
| GBM | 28 | 61 | 9 | 0 | YES |
| KIRC | 49 | 37 | 13 | 1.62E-08 | YES |
| KIRP | 110 | 199 | 57 | 0 | YES |
| LGG | 21 | 26 | 4 | 2.55E-13 | YES |
| LUAD | 588 | 2173 | 652 | 0 | YES |
| OV | 6 | 3 | 0 | 0.000256 | YES |
| UCEC | 449 | 2159 | 671 | 0 | YES |
| ACC | 15 | 1 | 0 | 0.152 | YES |
| BLCA | 648 | 3067 | 1487 | 0 | YES |
| CHOL | 39 | 21 | 6 | 4.84E-06 | YES |
| ESCA | 191 | 703 | 180 | 0 | YES |
| HNSC | 370 | 1322 | 366 | 0 | YES |
| KICH | 18 | 15 | 2 | 7.81E-10 | YES |
| LIHC | 307 | 1065 | 292 | 0 | YES |
| LUSC | 154 | 177 | 40 | 0 | YES |
| PAAD | 249 | 610 | 231 | 0 | YES |
| PCPG | 56 | 35 | 10 | 5.99E-10 | YES |
| PRAD | 113 | 249 | 72 | 0 | YES |
| READ | 116 | 285 | 68 | 0 | YES |
| SARC | 285 | 902 | 223 | 0 | YES |
| SKCM | 281 | 498 | 145 | 0 | YES |
| STAD | 575 | 2181 | 762 | 0 | YES |
| TGCT | 78 | 91 | 9 | 0 | YES |
| THCA | 91 | 248 | 136 | 0 | YES |
| THYM | 146 | 411 | 83 | 0 | YES |
| UCS | 22 | 15 | 6 | 0.00156 | YES |
| UVM | 13 | 3 | 1 | 0.186 | NO |
| Pan-cancer | 457 | 1821 | 610 | 0 | YES |
Overall, there are 4968 proteins with mutations on Pfam candidate domains, each of which is associated with 4 cancer types on average. About 31% of them are specific to only one cancer type and more than 7% of them (353 proteins) are linked to at least half of cancer types. Given these figures, one can expect the specific proteins to be not significantly interconnected in some cancer types, even though their entire list of proteins with mutations on candidate domains form highly connected networks. Additional file 1: Table A26 presents the number of associated cancer types for each protein. To have a more comprehensive picture of CATH candidate domains, similar results for 1379 proteins with mutations on CATH candidate domains are given in Additional file 1: Table A27.
Website
As previously noted, data integration is an essential requirement for this study and related fields of research. Several data sources were used to incorporate different types of information for human protein-coding genes, including gene symbols and protein identifiers from multiple resources, the start and end positions of each CATH and Pfam domain within a given protein, and the positions of exons and introns in the human genome. This comprehensive data integration provides researchers with a unified data source, which can be accessed via http://www.cancerouspdomains.ir, as well as from Additional file 2: Table B3. All previously mentioned tables in Additional file 1 are also downloadable from the website. An example of using the embedded search engine to extract the integrated information for gene TP53 is depicted in Fig. 6.
Fig. 6.
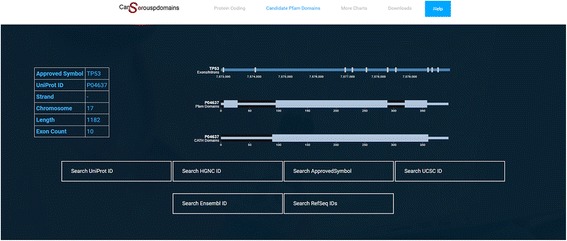
The result of searching TP53 in website
Some of the highly beneficial data provided on the website are the five graph charts, which show the associations between different groups of genes or domains to various cancer types. These graph charts are represented by bipartite graphs in which one set of nodes corresponds to the genes/domains and the other set of nodes corresponds to cancer type. In each bipartite graph, an edge connects a gene/domain to a cancer type, if this gene/domain is identified as a candidate in the cancer type. For instance, one set of nodes represents Pfam candidate domains and the other set represents different cancer types. An edge connects two nodes in these sets, if the corresponding domain is a candidate for the corresponding cancer type. The graph chart of Pfam candidate domains is illustrated in Fig. 7.
Fig. 7.
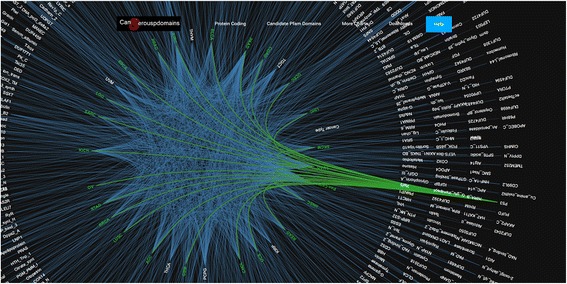
The graph chart for Pfam candidate domains
A help file is also provided for detailed description of the information embedded in the website. Some tailored interface options such as “moving”, “zoom in” and “zoom out” are available to control the size of the display. These options are specifically helpful due to the large number of CATH and Pfam candidate domains. Furthermore, by clicking on each cancer type, all related candidate domain or genes can be distinguished via a change of edge colors. The same option is also provided for each candidate domain or gene.
Conclusion
Distinguishing mutations on protein-coding regions which impact the functionality of coded proteins, is one of the main obstacles in the study of cancer genomics. The exome-wide study of somatic mutations for several cancer types and the available structural information of proteins provide invaluable resources for specifically studying cancer genomics at functional level. Both sequence-based and structure-based domains, which are available in Pfam and CATH databases respectively, are promising representatives of functional regions within proteins. Accordingly, extracting domain regions which are significantly mutated in a cancer type reveals critical information required for validating the impact of mutations.
In this paper, a comprehensive investigation is conducted on all 29 TCGA cancer types and pan-cancer to identify sequence-based and structure-based domains in which mutations occur with high statistical significance. The domains identified for each cancer type offer an explanation for the functional impact of mutations in that cancer type. It is shown that each cancer type has its specific set of candidate domains, which in turn suggests a specific set of proteins associated to that cancer type. The interactome analysis showed that the specific proteins of each cancer type are significantly connected. This interconnectivity supports the idea that leveraging domain regions can improve accurate identification of functionally related causal proteins. Through further investigation, the frequency of mutations in mitochondrial genes, stem cell-specific genes and DNA repair genes was determined to examine their role in cancer development and progression.
Additionally, this study provides other researchers with a comprehensive and unified data repository for studying both CATH and Pfam domain regions on protein-coding genes. Moreover, it has clarified the associations between different groups of genes or domains and various cancer types. All this information is accessible via our website.
Additional files
It contains 27 supplementary tables, Table A1–27 with the data described in the text. (XLSX 1032 kb)
It contains three tables, Tables B1-B3 with the data gathered from different datasets. All these data are used in study. (XLSX 55782 kb)
Acknowledgements
The authors would like to thank Farshad Jafari for helping to design the website. In addition, the authors wish to express their gratitude to the anonymous reviewers, whose constructive comments improved the manuscript.
Funding
Not applicable.
Availability of data and materials
All information is accessible via our well-designed website http://www.cancerouspdomains.ir.
Abbreviations
- HUGO
HUman Genome Organization
- TCGA
The Cancer Genome Atlas
- UCSC
University of California Santa Cruz
Authors’ contributions
Initial idea of the research was proposed by SH, AND, AMB, AJ and ZR. The framework was designed, implemented, and tested by SH, AND, AJ, and ZR. All authors participated in designing the structure and organization of the manuscript equally. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12859-017-1779-5) contains supplementary material, which is available to authorized users.
Contributor Information
Seirana Hashemi, Email: seirana.hashemi@ut.ac.ir.
Abbas Nowzari Dalini, Email: nowzari@ut.ac.ir.
Adrin Jalali, Email: ajalali@mpi-inf.mpg.de.
Ali Mohammad Banaei-Moghaddam, Email: am_banaei@ut.ac.ir.
Zahra Razaghi-Moghadam, Phone: +989151161725, Email: razzaghi@ut.ac.ir.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society, "Cancer facts & figures," American Atlanta, 2016.
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 4.Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin D, Forman D, Bray F. GLOBOCAN 2012 v1. 0, Cancer incidence and mortality worldwide: IARC CancerBase No. 11. [Online]. Lyon: International Agency for Research on Cancer; 2013. Available from: globocan.iarc.fr. Accessed 24 Nov 2014.
- 5.T. Sjöblom, S. Jones, LD. Wood, DW. Parsons, J. Lin, TD. Barber, D. Mandelker, RJ. Leary, J. Ptak, N. Silliman and others, "The consensus coding sequences of human breast and colorectal cancers," Science, vol. 314, no. 5797, pp. 268–274, 2006. [DOI] [PubMed]
- 6.DW. Parsons, M. Li, X. Zhang, S. Jones, RJ. Leary, JCH. Lin, SM. Boca, H. Carter, J. Samayoa, Ch. Bettegowda and others, "The genetic landscape of the childhood cancer medulloblastoma," Science, vol. 331, no. 6061, pp. 435–439, 2011. [DOI] [PMC free article] [PubMed]
- 7.LD. Wood, DW. Parsons, S. Jones, J. Lin, T. Sjöblom, RJ. Leary, D. Shen, SM. Boca, T. Barber, J. Ptak and others, "The genomic landscapes of human breast and colorectal cancers," Science, vol. 318, no. 5853, pp. 1108–1113, 2007. [DOI] [PubMed]
- 8.MF. Berger, MS. Lawrence, F. Demichelis, Y. Drier, K. Cibulskis, AY. Sivachenko, A. Sboner, R. Esgueva, D. Pflueger, C. Sougnez and others, "The genomic complexity of primary human prostate cancer," Nature, vol. 470, no. 7333, pp. 214–220, 2011. [DOI] [PMC free article] [PubMed]
- 9.Yue P, Forrest WF, Kaminker JS, Lohr S, Zhang Z, Cavet G. Inferring the functional effects of mutation through clusters of mutations in homologous proteins. Hum Mutat. 2010;31(3):264–271. doi: 10.1002/humu.21194. [DOI] [PubMed] [Google Scholar]
- 10.Yang F, Petsalaki E, Rolland T, Hill DE, Vidal M, Roth FP. Protein domain-level landscape of cancer-type-specific somatic mutations. PLoS Computational Biology. 2015;11(3):e1004147. doi: 10.1371/journal.pcbi.1004147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.AJF. Griffiths, Modern genetic analysis: integrating genes and genomes, Macmillan, 2002.
- 12.Nehrt NL, Peterson TA, Park D, Kann MG. Domain landscapes of somatic mutations in cancer. BMC Genomics. 2012;13(4):1. doi: 10.1186/1471-2164-13-S4-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular biology of the cell (5th edit.) garland science. New York: Garland Science; 2007. [Google Scholar]
- 14.Tomczak K, Czerwinska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol. 2015;19(1A):A68. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schneider V, Church D. Genome reference consortium. US: National Center for Biotechnology Information; 2013. [Google Scholar]
- 16.Goymer P. Synonymous mutations break their silence. Nature Reviews Genetics. 2007;8(2):92. [Google Scholar]
- 17.T. Zhou, A.E. Ko, W. Gu, I. Lim, H. Bang and J. Ko, "Non-silent story on synonymous sites in voltage-gated ion channel genes," PloS One, vol. 7, no. 10, p. e48541, 2012. [DOI] [PMC free article] [PubMed]
- 18.RD. Finn, A. Bateman, J. Clements, P. Coggill, RY. Eberhardt, SR. Eddy, A. Heger, K. Hetherington, L. Holm and J. Mistry, "Pfam: the protein families database," Nucleic Acids Research, p. gkt1223, 2013. [DOI] [PMC free article] [PubMed]
- 19.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer ELL. The Pfam protein families database. Nucleic Acids Res. 2004;32(suppl 1):D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH--a hierarchic classification of protein domain structures. Structure. 1997;5(8):1093–1109. doi: 10.1016/S0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 21.I. Sillitoe, TE. Lewis, A. Cuff, S. Das, P. Ashford, NL. Dawson, N. Furnham, RA. Laskowski, D. Lee and JG. Lees, "CATH: comprehensive structural and functional annotations for genome sequences," Nucleic acids research, p. gku947, 2014. [DOI] [PMC free article] [PubMed]
- 22.Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC table browser data retrieval tool. Nucleic Acids Res. 2004;32(suppl 1):D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bernstein FC, Koetzle TF, Williams GB, Meyer EF, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The protein data bank. Eur J Biochem. 1977;80(2):319–324. doi: 10.1111/j.1432-1033.1977.tb11885.x. [DOI] [PubMed] [Google Scholar]
- 24.KA. Gray, B. Yates, RL. Seal, MW. Wright and EA. Bruford, "Genenames. org: the HGNC resources in 2015," Nucleic Acids Research, p. gku1071, 2014. [DOI] [PMC free article] [PubMed]
- 25.S. Velankar, JM. Dana, J. Jacobsen, G.V. Ginkel, PJ. Gane, J. Luo, TJ. Oldfield, C. O’Donovan, M. Martin and GJ. Kleywegt, "SIFTS: structure integration with function, taxonomy and sequences resource," Nucleic Acids Research, p. gks1258, 2012. [DOI] [PMC free article] [PubMed]
- 26.L. Devore, Probability and Statistics for Engineering and the Sciences, Cengage Learning, 2015.
- 27.Field A. Discovering statistics using SPSS: Sage publications; 2009.
- 28.Goeman JJ, Solari A. Multiple hypothesis testing in genomics. Stat Med. 2014;33(11):1946–1978. doi: 10.1002/sim.6082. [DOI] [PubMed] [Google Scholar]
- 29.Robert B. Ash. Information Theory, New York: Dover Publications; 1990. [Google Scholar]
- 30.Pories S. Cancer. Biographies of Disease Series: Greenwood Press, Portsmouth; 2009. [Google Scholar]
- 31.Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM, Cancer Genome Atlas Research Network and others The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45(10):1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.GS. Stein, M. Borowski, MX. Luong, M. Shi, KP. Smith and P. Vazquez, Human Stem Cell Technology and Biology: A Research Guide and Laboratory Manual, Weinheim: John Wiley & Sons, 2011.
- 34.Y. Liang, I. Russell, C. Walworth and C. Chen, "Gene expression in stem cells," Critical Reviews™ in Eukaryotic Gene Expression, vol. 19, no. 4, 2009. [DOI] [PubMed]
- 35.Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer. 2016;16(1):35–42. doi: 10.1038/nrc.2015.4. [DOI] [PubMed] [Google Scholar]
- 36.Lahtz C, Pfeifer GP. Epigenetic changes of DNA repair genes in cancer. J Mol Cell Biol. 2011;3(1):51–58. doi: 10.1093/jmcb/mjq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Horton TM, Petros JA, Heddi A, Shoffner J, Kaufman AE, Graham SD, Gramlich T, Wallace DC. Novel mitochondrial DNA deletion found in a renal cell carcinoma. Genes Chromosom Cancer. 1996;15(2):95–101. doi: 10.1002/(SICI)1098-2264(199602)15:2<95::AID-GCC3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 38.Parrella P, Xiao Y, Fliss M, Sanchez-Cespedes M, Mazzarelli P, Rinaldi M, Nicol T, Gabrielson E, Cuomo C, Cohen D. Detection of mitochondrial DNA mutations in primary breast cancer and fine-needle aspirates. Cancer Res. 2001;61(20):7623–7626. [PubMed] [Google Scholar]
- 39.Maximo V, Soares P, Seruca R, Rocha AS, Castro P, Sobrinho-Simoes M. Microsatellite instability, mitochondrial DNA large deletions, and mitochondrial DNA mutations in gastric carcinoma. Genes Chromosom Cancer. 2001;32(2):136–143. doi: 10.1002/gcc.1175. [DOI] [PubMed] [Google Scholar]
- 40.Jessie BC, Sun CQ, Irons HR, Marshall FF, Wallace DC, Petros JA. Accumulation of mitochondrial DNA deletions in the malignant prostate of patients of different ages. Exp Gerontol. 2001;37(11):169–174. doi: 10.1016/S0531-5565(01)00153-X. [DOI] [PubMed] [Google Scholar]
- 41.Liu VWS, Shi H, Cheung ANY, Chiu PM, Leung TW, Nagley P, Wong LC, Ngan HYS. High incidence of somatic mitochondrial DNA mutations in human ovarian carcinomas. Cancer Res. 2001;61(16):5998–6001. [PubMed] [Google Scholar]
- 42.Maximo V, Soares P, Lima J, Cameselle-Teijeiro J, Sobrinho-Simoes M. Mitochondrial DNA somatic mutations (point mutations and large deletions) and mitochondrial DNA variants in human thyroid pathology: a study with emphasis on Hurthle cell tumors. Am J Pathol. 2002;160(5):1857–1865. doi: 10.1016/S0002-9440(10)61132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang BC, Hays L. Mitochondrial DNA copy number changes in human gliomas. Cancer Lett. 1996;105(2):167–173. doi: 10.1016/0304-3835(96)04276-0. [DOI] [PubMed] [Google Scholar]
- 44.Nomoto S, Yamashita K, Koshikawa K, Nakao A, Sidransky D. Mitochondrial D-loop mutations as clonal markers in multicentric hepatocellular carcinoma and plasma. Clin Cancer Res. 2002;8(2):481–487. [PubMed] [Google Scholar]
- 45.John JC, Mitochondrial DNA. Mitochondria. Disease and Stem Cells, London: Springer Science & Business Media; 2012. [Google Scholar]
- 46.López-Lázaro M. Stem cell division theory of cancer. Cell Cycle. 2015;14(16):2547–2548. doi: 10.1080/15384101.2015.1062330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 48.López-Lázaro M. The migration ability of stem cells can explain the existence of cancer of unknown primary site. Rethinking metastasis. Oncoscience. 2015;2(5):467–475. doi: 10.18632/oncoscience.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palmer N, Schmid PR, Berger B, Kohane I. A gene expression profile of stem cell pluripotentiality and differentiation is conserved across diverse solid and hematopoietic cancers. Genome Biology. 2012;13(8):R71. doi: 10.1186/gb-2012-13-8-r71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.C. Cybulski, B. Gorski, T. Huzarski, B. Masoj'c, M. Mierzejewski, J. Dkebniak, U. Teodorczyk, T. Byrski, J. Gronwald, J. Matyjasik and others, "CHEK2 is a multiorgan cancer susceptibility gene," Am J Hum Genet, vol. 75, no. 6, pp. 1131–1135, 2004. [DOI] [PMC free article] [PubMed]
- 51.M. Siojek, C. Cybulski, D. Gkasior-Perczak, A. Kowalik,B. Kozak-Klonowska, A. Kowalska, M. Chlopek, W. Klu'zniak, D. Wokolorczyk and Palyg, "CHEK2 mutations and the risk of papillary thyroid cancer," Int J Cancer, vol. 137, no. 3, pp. 548–552, 2015. [DOI] [PubMed]
- 52.Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15(6):334–346. doi: 10.1038/nrc3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.hae, Young Kwang and Anker, Jonathan F and Carneiro, Benedito A and Chandra, Sunandana and Kaplan, Jason and Kalyan, Aparna and Santa-Maria, Cesar A and Platanias, Leonidas C and Giles, Francis J, "Genomic landscape of DNA repair genes in cancer," Oncotarget, vol. 7, no. 17, p. 23312, 2016. [DOI] [PMC free article] [PubMed]
- 54.Wood RD, Mitchell M, Lindahl T. Human DNA repair genes. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2005;577(1):275–283. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 55.Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer. 2011;11(2):96–110. doi: 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.EC. Friedberg, GC. Walker, W. Siede and RD. Wood, DNA repair and mutagenesis, New York: American Society for Microbiology Press, 2005.
- 57.Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor perspectives in biology. 2010;2(1):a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petitjean A, Achatz M, Borresen-Dale A, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26(15):2157–2165. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 59.Forbes, Simon A and Beare, David and Gunasekaran, Prasad and Leung, Kenric and Bindal, Nidhi and Boutselakis, Harry and Ding, Minjie and Bamford, Sally and Cole, Charlotte and Ward, Sari and others, "COSMIC: exploring the world's knowledge of somatic mutations in human cancer," Nucleic Acids Res, vol. 43, no. D1, pp. D805-D811, 2015. [DOI] [PMC free article] [PubMed]
- 60.Wang B, Yang W, McKittrick J, Meyers MA. Keratin: Structure, mechanical properties, occurrence in biological organisms, and efforts at bioinspiration. Progress in Materials Science. 2016;76:229–318. doi: 10.1016/j.pmatsci.2015.06.001. [DOI] [Google Scholar]
- 61.TCEAL1 transcription elongation factor A like 1 [Homo sapiens (human)] - Gene - NCBI. National Center for Biotechnology Information, U.S. National Library of Medicine, 25 May 017. [Online]. Available: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=ShowDetailView&TermToSearch=9338. Accessed 31 May 2017.
- 62.Ornitz David M, Itoh Nobuyuki. Fibroblast growth factors. Genome biol. 2001;2(3):reviews3005.1-reviews3005.12. [DOI] [PMC free article] [PubMed]
- 63.Cingolani P, Platts A, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6(2):80-92. [DOI] [PMC free article] [PubMed]
- 64.De Las Rivas J, Fontanillo C. Protein--protein interactions essentials: key concepts to building and analyzing interactome networks. PLoS Computational Biology. 2010;6(6):e1000807. [DOI] [PMC free article] [PubMed]
- 65.Deng M, Mehta S, Sun F, Chen T. Inferring domain--domain interactions from protein--protein interactions. Genome Research. 2002;12(10):1540-1548. [DOI] [PMC free article] [PubMed]
- 66.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou K, et al. STRING v10: protein--protein interaction networks, integrated over the tree of life. Nucleic acids research. 2014. p. gku1003. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
It contains 27 supplementary tables, Table A1–27 with the data described in the text. (XLSX 1032 kb)
It contains three tables, Tables B1-B3 with the data gathered from different datasets. All these data are used in study. (XLSX 55782 kb)
Data Availability Statement
All information is accessible via our well-designed website http://www.cancerouspdomains.ir.