

Abstract
Increased osteopontin (OPN) expression in the heart, specifically in myocytes, associates with increased myocyte apoptosis and myocardial dysfunction. Recently, we provided evidence that OPN interacts with CD44 receptor, and induces myocyte apoptosis via the involvement of endoplasmic reticulum stress and mitochondrial death pathways. Here we tested the hypothesis that OPN induces oxidative stress in myocytes and the heart via the involvement of mitochondria and NADPH oxidase−4 (NOX−4). Treatment of adult rat ventricular myocytes (ARVMs) with OPN (20 nM) increased oxidative stress as analyzed by protein carbonylation, and intracellular reactive oxygen species (ROS) levels as analyzed by ROS detection kit and dichlorohydrofluorescein diacetate staining. Pretreatment with NAC (antioxidant), apocynin (NOX inhibitor), MnTBAP (superoxide dismutase mimetic), and mitochondrial KATP channel blockers (glibenclamide and 5-hydroxydecanoate) decreased OPN-stimulated ROS production, cytosolic cytochrome c levels, and apoptosis. OPN increased NOX−4 expression, while decreasing SOD−2 expression. OPN decreased mitochondrial membrane potential as measured by JC−1 staining, and induced mitochondrial abnormalities including swelling and reorganization of cristae as observed using transmission electron microscopy. OPN increased expression of BIK, a pro-apoptotic protein involved in reorganization of mitochondrial cristae. Expression of dominant-negative BIK decreased OPN-stimulated apoptosis. In vivo, OPN expression in cardiac myocyte-specific manner associated with increased protein carbonylation, and expression of NOX−4 and BIK. Thus, OPN induces oxidative stress via the involvement of mitochondria and NOX−4. It may affect mitochondrial morphology and integrity, at least in part, via the involvement of BIK.
Keywords: Osteopontin, Myocyte, Apoptosis, Reactive oxygen species, BIK
Introduction
Matricellular proteins are a class of non-structural extracellular matrix proteins which modulate cellular functions mostly via their interaction with cell surface receptors, other structural proteins and extracellular factors such as growth factors and cytokines [1–3]. The matricellular protein osteopontin (OPN, also called cytokine Eta−1) is a glycosylated phosphoprotein [3]. It has the adhesive arginine-glycine-aspartate (RGD) motif, and is shown to interact with a variety of integrins. Hyaluronan receptor, CD44, is also identified as a receptor for OPN. Tissue injury and inflammation increase OPN expression in a variety of cells, and OPN plays an important role in tissue homeostasis, immune regulation, bone resorption, wound healing, etc. [4–6].
In the heart, OPN is expressed constitutively by myocytes, endothelial cells, and fibroblasts. However, OPN expression increases markedly in the heart under a variety of pathophysiological conditions [2, 3]. Increased OPN expression in myocytes associates with increased myocyte apoptosis and myocardial dysfunction in different models of heart disease [7–10]. Furthermore, expression of OPN in a myocyte-specific manner in the mouse heart led to increased myocyte apoptosis and myocardial dysfunction [11, 12]. Recently, we provided evidence that OPN interacts with CD44 receptors, and OPN acting via CD44 receptors induces apoptosis in cardiac myocyte. This OPN-stimulated apoptosis involves induction of endoplasmic reticulum (ER) stress and mitochondrial death pathways [12].
Reactive oxygen species (ROS) and oxidative stress are important features of cardiovascular disease [13]. Normal cellular metabolism induces accumulation of low levels of ROS, which is attributable to the equilibrium of disposal rates by antioxidants and enzyme systems within the cell [14]. This low level oxidative stress is generally non-pathogenic [15]. However, disruption of this balance by toxic insults can cause overproduction of ROS and create oxidative stress [14]. NADPH oxidase (NOX) and mitochondria are considered as major sources of ROS in cardiovascular system. NOX−4 isoform localizes in the mitochondria of cardiac myocytes, and upregulation of NOX-4 associates with increased ROS production in mitochondria [16]. Increased ROS production induces mitochondrial permeability transition (mPT) [17], leading to matrix swelling, outer membrane rupture, release of apoptotic signaling molecules, such as cytochrome c from the intermembrane space, and irreversible injury to the mitochondria [18,19]. Endoplasmic reticulum (ER; sarcoplasmic reticulum in myocytes) communicates with the mitochondria via focal points interaction called mitochondrial-associated ER membrane (MAM). The MAM is the signaling juncture that facilitates calcium and lipid transfer between the organelles and modulates their function along with cell fate determination [20]. In cells of non-cardiac origin, influx of calcium from ER to the mitochondria is shown to affect mitochondrial morphology and stimulate apoptosis. This calcium influx is promoted by pro-apoptotic proteins such as BAX and BIK [20, 21]. BIK is reported to predominantly localize to the ER [22]. However, it is also shown to localize to the mitochondria [23]. Increased BIK expression induces mitochondrial cristae fragmentation via Drp−1, an important regulator of mitochondrial dynamics [20, 24]. BIK expression is prominent in the heart [23, 25]. However, its role in myocyte apoptosis remains to be investigated.
OPN is suggested to act as a promoter or inhibitor of oxidative stress in renal diseases. [26, 27]. The objective of this study was to examine if OPN induces oxidative stress in myocytes and the heart, and to elucidate the molecular signals involved in OPN-stimulated oxidative stress and apoptosis. The data presented here suggest that OPN induces oxidative stress, mitochondrial depolarization and affects mitochondrial integrity via the involvement of NOX−4, SOD−2, opening of mitochondrial KATP channels (mitoKATP) and BIK.
Materials and methods
Experimental animals
The study used adult male Sprague–Dawley rats and transgenic mice expressing OPN in a cardiac myocyte-specific manner. The investigation conforms to the guide for the Care and Use of Laboratory animals published by the US National Institutes of Health (NIH Publication No. 85–23, Revised 1996). All the experiments were performed in accordance with the protocols approved by the East Tennessee State University Committee on Animal Care.
Cell isolation, culture, and treatments
Calcium-tolerant ARVMs or AMVMs (adult mouse ventricular myocytes) were isolated from the hearts of adult male Sprague–Dawley rats (150–200 g) or C57BL/6 mice (20–25 g) as described [12]. ARVMs or AMVMs, cultured for 24 h, were treated with purified mouse OPN protein (20 nM; R&D system) for various time-points. N-acetyl-l-cysteine (NAC; 1 mM; Enzo Life Sciences), apocynin (APO; 200 μM; Calbiochem), manganese (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP; 10 μM; Calbiochem), glibenclamide (GLIB; 5 μM; SM Biochemicals) or 5-hydroxydecanoate (5-HD; 1 μM; Sigma) were added 30 min prior to OPN treatment. The inhibitors were maintained in the medium during the treatment period with OPN.
OPN transgenic mice
To express OPN in cardiac myocyte-specific manner, two mouse lines (α-MHC-tTA; received from Dr. Anthony Baker, Veterans Affairs Medical Center, San Francisco, CA and TetO-OPN; received from Dr Alain-Pierre Gadeau, University of Bordeaux Victor Segalen, Pessac, France) were used [11, 12]. The transgenic mice were bred and maintained on a doxycycline diet (200 mg/kg, no. S3888; Bio-serve, Frenchtown, NJ). To induce OPN expression in adult heart, doxycycline was withdrawn from the diet when mice were ∼4 months old.
Adenovirus infection
Adenoviruses expressing mouse OPN (courtesy of Dr Toshimitsu Uede, Hokkaido University, Japan) and mutant BIK (courtesy of Dr Gordon C. Shore, McGill University, Montreal, Canada) were propagated using HEK−293 cells. ARVMs were infected with the adenoviruses expressing OPN, mutant BIK or green fluorescent protein (GFP) at a multiplicity of infection of 50–100/cell for 48 h.
RT-PCR
Total RNA (1 μg) was reverse transcribed using SuperScript III RT kit (Thermo Fisher Scientific). Primer sequences for RT-PCR amplification were as follows: BIK; 5′-CCGCTACGCCAGCTTAGCCG-3′and 5′-GACCAGA CGGGGGCTCCGAA-3′, GAPDH; 5′-CTCATGACCA-CAGTCCATGC-3′ and 5′-TTCAGCTCTGGGATGACCT T-3′. The PCR products were analyzed by agarose gel electrophoresis.
Apoptosis
To detect apoptosis, ARVMs were plated on glass cover-slips and stained using in situ cell death detection kit (Roche Biochemicals) [12]. Percentage of TUNEL-positive cells (relative to total ARVMs) was determined by counting ∼200 cells in 10 randomly chosen fields per coverslip for each experiment.
Preparation and analysis of cytosolic cytochrome c
The cytosolic fractions were prepared and analyzed by western blots using anti-cytochrome c antibodies (Santa Cruz) as described [12].
Protein oxidation
Oxidation of proteins was detected using oxyblot protein oxidation detection kit (Thermo Fisher Scientific). Briefly, 15 μg of cell or tissue lysates were treated with 10 μl of DNPH solution. The derivatized proteins were analyzed by western blot using anti-DNP antibodies. Band intensities were quantified using Carestream Molecular Imaging system (Carestream Health Inc.). Protein loading was normalized using SYPRO ruby (Bio-Rad) staining of the Western blots.
Detection of intracellular ROS
ROS levels were detected using the Total ROS detection kit (Enzo Life Sciences) and 2′,7′-dichlorohydrofluorescein diacetate (H2DCFDA; Thermo Fisher Scientific) staining. For total ROS detection kit, ARVMs were treated with OPN for 5 h followed by incubation with ROS-responsive fluorescence probe for 1 h. The cells were then washed with PBS and visualized using a fluorescence microscope (EVOS; Thermo Fisher Scientific). The number of ROS-positive cells exhibiting green fluorescence (relative to total myocytes) was determined by counting ∼50 cells in 10 randomly chosen fields per coverslip for each experiment.
For H2DCFDA staining, cells were pretreated with various inhibitors for 30 min followed by treatment with OPN for 5 h and 30 min. The live cells were then incubated with 50 μM of H2DCFDA in growth medium for 30 min at 37 °C. Cells were washed with PBS, and visualized using fluorescence microscope (EVOS). Identical exposure conditions were used for all fluorescent images to evaluate the mean fluorescent intensity using NIS-Elements image analysis software (Nikon).
Mitochondrial membrane potential
ARVMs were incubated with JC-1 dye (10 μM, Abcam) for 10 min at 37 °C in dark. The cells were then washed in DMEM (without phenol red) and the images of live cells were captured using a fluorescence microscope (EVOS). The fluorescent images, obtained using identical exposure conditions, were analyzed using NIS-Elements image analysis software (Nikon) and the ratio of red (aggregate) to green (monomer) fluorescence (Fλ585/Fλ510) was calculated.
Western blot analysis
Cell or left ventricular (LV) tissue lysates were prepared and analyzed by Western blot as described [12]. The primary antibodies used were as follows-cytochrome c, BIK, SOD−2 (Santa Cruz), and NADPH oxidase−4 (NOX−4; Novus). The membranes were stripped and probed with GAPDH (Santa Cruz) as a protein loading control. Band intensities were quantified using Carestream Molecular Imaging system (Carestream Health Inc.).
Immunoprecipitation assay
Band intensities for NOX−4 in the cell lysates were faint to quantify using Western blot analysis. To enhance the signal intensity, cell lysates (500 μg; prepared in RIPA buffer) were immunoprecipitated using 6 μg of anti-NOX−4 (Santa Cruz) antibodies [12]. The immunoprecipitates were analyzed by Western blot using anti-NOX−4 antibodies (Santa Cruz).
Transmission electron microscopy
To examine mitochondrial morphology, myocytes were postfixed and embedded in Epon plastic as described [28]. Ultrathin sections, prepared using Leica ultracut UCT microtome, were examined using a Philips Tecnai−10 electron microscope (FEI) operating at 60–80 kV.
Immunofluorescence
ARVMs were fixed with 2 % paraformaldehyde for 10 min followed by quenching with 100 mM glycine (pH 7.4) for 5 min at room temperature. After permeabilization using 0.4 % Triton X−100 in PBS for 10 min, the cells were incubated with Image-iT® FX Signal Enhancer ReadyProbes® Reagent (Thermo Fisher Scientific) for 30 min. The cells were then blocked with 10 % donkey serum for 1 h and incubated overnight at room temperature with anti-BIK (1:50, Santa Cruz) and anti-cyclophilin D (1:100, GeneX) antibodies. After washing, the cells were incubated sequentially with Alexa 594-conjugated (to detect BIK; 1:200 dilution) and Alexa 488-conjugated (to detect cyclophilin D; 1:200 dilution, Thermo Fisher Scientific) secondary antibodies for 1 h. The cells were then washed, mounted and visualized using con-focal microscopy (Leica TCS SP8).
Statistical analyses
All data are expressed as mean ± SE. Statistical analysis was performed using Student's t test or one-way ANOVA and a post hoc Tukey's test. Probability (p) values <0.05 were considered to be significant.
Results
OPN induces oxidative stress and increases intracellular ROS in ARVMs
The increased plasma OPN concentration in heart failure patients amounts up to ∼16.6 nM (∼1145 ng/ml) assuming molecular weight of OPN as 69 kDa. Previously, we have shown that 2.0 and 20 nM concentrations of OPN increase the magnitude of apoptosis by ∼2- and 3-fold, respectively (12). Since the magnitude of apoptosis was greater with 20 nM, this concentration of OPN was used in this study. Protein carbonylation, an irreversible and non-enzymatic form of post-translational protein modification, is commonly used as a marker of oxidative stress [29]. To investigate if OPN induces oxidative stress, we first examined protein carbonylation using OxyBlot. For this, ARVMs were treated with purified OPN protein (20 nM) for 24 h. Quantitative analysis of OxyBlot indicated a significant increase in protein carbonylation in OPN-treated ARVMs versus control (p < 0.05; n = 4; Fig. 1a).
Fig. 1.
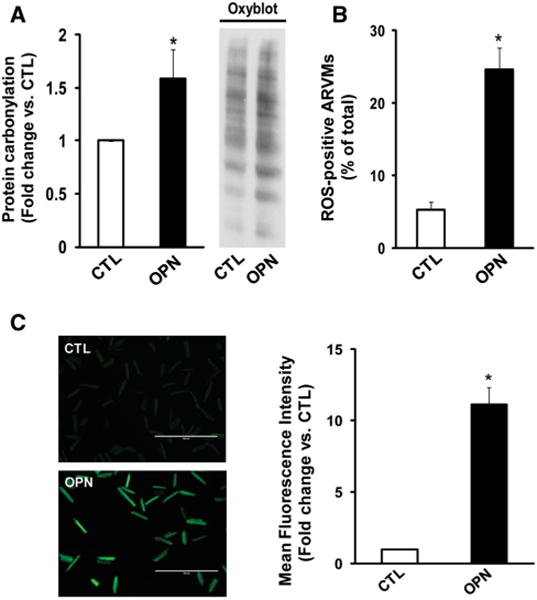
OPN induces oxidative stress in ARVMs. a ARVMs were treated with OPN (20 nM) for 24 h. Total cell lysates were analyzed using oxyblot assay. Representative oxyblot is depicted in the right panel. The left panel depicts the mean data normalized to SYPRO ruby staining, *p < 0.05 versus CTL; n = 4. b ARVMs were treated with OPN for 6 h and stained using ROS detection kit. The number of ROS-positive cells was counted using fluorescence microscope. *p < 0.05 versus control (CTL); n = 3. c ARVMs were treated with OPN for 6 h and stained with H2DCFDA. The left panel exhibits representative images of H2DCFDA-stained cells, while right panel exhibits mean fluorescence intensity. *p < 0.05 versus CTL; n = 6
To investigate if OPN increases intracellular ROS, ARVMs were treated with OPN for 5 h followed by incubation with ROS-responsive fluorescence probe for 1 h. Analysis of ROS-positive ARVMs using fluorescent microscopy indicated that OPN increases the number of ROS-positive cells (CTL, 5.30 ± 1.02; OPN, 24.59 ± 2.99*;*p < 0.05 vs. CTL; n = 3; Fig. 1b). Increased ROS levels were confirmed using H2DCFDA staining. The mean fluorescence intensity was significantly greater in OPN-treated ARVMs versus control (*p < 0.05; n = 6; Fig. 1c). OPN treatment also increased ROS production in AMVMs as analyzed by H2DCFDA staining (data not shown). Due to higher myocyte yield, further experiments were carried out using ARVMs.
Involvement of NOX and mitochondria in OPN-stimulated ROS production, cytosolic cytochrome c release, and apoptosis
To examine if NAC (antioxidant) can counteract OPN-stimulated increases in ROS levels, cells were pretreated with NAC (1 mM) for 30 min followed by treatment with OPN. Quantitative analysis of ROS using H2DCFDA-stained cells revealed that NAC significantly counteracts OPN-stimulated ROS levels (Fig. 2a). Pretreatment with apocynin (APO; 200 μM; NOX inhibitor), MnTBAP (10 μM; SOD mimetic), glibenclamide (GLIB; 5 μM; mitochondrial KATP channel blocker) or 5-hydroxydecanoate(5-HD; 1 μM; mitochondrial KATP channel blocker) also significantly negated OPN-stimulated ROS levels (*p < 0.05 vs. CTL;$p < 0.05 vs. OPN, n = 3–7; Fig. 2a, b), suggesting a role for NOX and mitochondria. NAC, APO, MnTBAP, GLIB or 5-HD alone had no effect on ROS levels.
Fig. 2.
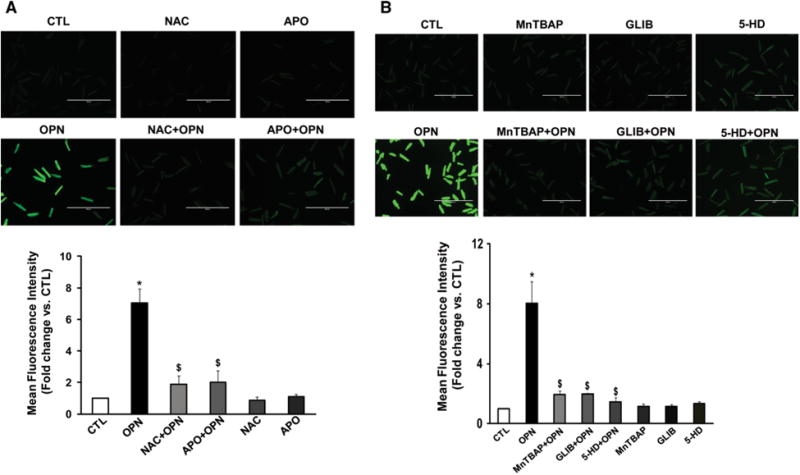
Involvement of NADPH oxidase and mitochondria in ROS production. ARVMs were pretreated with NAC or APO (a), MnTBAP, GLIB or 5-HD (b) for 30 min followed by treatment with OPN for 6 h. The cells were then stained with H2DCFDA. The upper panels exhibit representative images of H2DCFDA-stained cells, while lower panels exhibit mean fluorescence intensity, *p < 0.05 versus CTL; $p < 0.05 versus OPN; n = 3–7
To investigate whether these inhibitors affect mitochondrial death pathway, ARVMs were pretreated with NAC, APO, MnTBAP, or GLIB for 30 min followed by treatment with OPN for 6 h. Analysis of cytosolic fractions by Western blot using anti-cytochrome c antibodies indicated that OPN treatment significantly increases the levels of cytosolic cytochrome c. Pretreatment with NAC, APO, MnTBAP, or GLIB significantly inhibited OPN-stimulated increases in cytosolic cytochrome c levels (Fig. 3a). These inhibitors alone had no effect on the levels of cytosolic cytochrome c (data not shown).
Fig. 3.
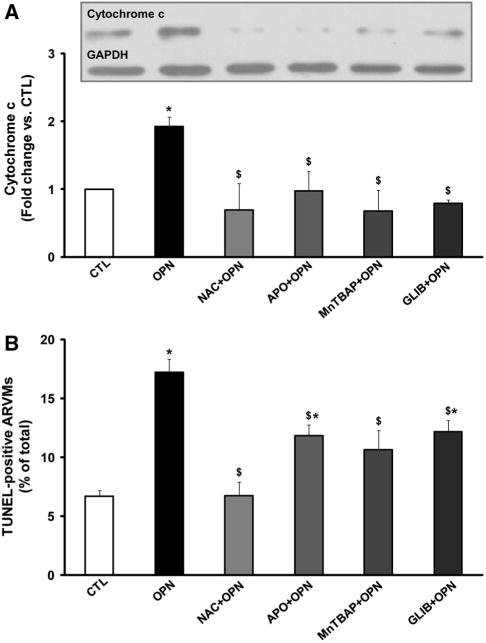
Involvement of NADPH oxidase and mitochondria in cytosolic cytochrome c release and apoptosis. ARVMs were pretreated with NAC, APO, MnTBAP, or GLIB for 30 min followed by treatment with OPN for 6 h (a) or 24 h (b). a Cytosolic fractions were analyzed by Western blots using anti-cytochrome c antibodies. The lower panel exhibits the mean data normalized to GAPDH, *p < 0.05 versus CTL; $p < 0.05 versus OPN; n = 3–7. b Apoptosis was measured using TUNEL-staining assay, *p < 0.05 versus CTL; $p < 0.05 versus OPN; n = 6–8
Analysis of apoptosis using TUNEL-assay indicated that pretreatment with NAC, APO, MnTBAP, or GLIB significantly inhibits OPN-stimulated apoptosis. (CTL, 7.34 ± 0.61; OPN, 17.22 ± 1.09*; NAC + OPN, 8.38 ± 1.94; APO + OPN, 11.87 ± 0.89$*; MnTBAP + OPN, 10.64 ± 1.67; GLIB + OPN 12.85 ± 1.08 *; *p < 0.05 vs. CTL; $p < 0.05 vs. OPN, n = 6-8; Fig. 3b). NAC, APO, MnTBAP or GLIB alone had no effect on apoptosis (data not shown).
OPN affects expression of oxidative stress-related proteins
In cardiac myocytes, NOX-4 isoform localizes to the mitochondria and aids in mitochondrial production of ROS [16]. SOD-2 is a mitochondrial matrix enzyme that scavenges ROS produced by mitochondria [30]. Immunoprecipitation analysis of cell lysates using anti-NOX-4 antibodies indicated that OPN treatment significantly increases NOX-4 protein levels (*p < 0.05 vs. CTL; n = 3; Fig. 4a). In contrast, OPN treatment significantly decreased SOD-2 protein levels as analyzed by Western blot (*p < 0.05 vs. CTL; n = 5; Fig. 4b).
Fig. 4.
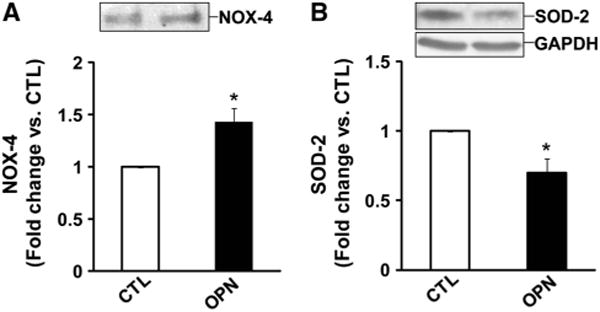
OPN affects expression of NOX−4 and SOD−2. ARVMs were treated with OPN for 24 h. a Cell lysates were immunoprecipitated using anti-NOX−4 antibodies and analyzed by Western blot using anti-NOX−4 antibodies, *p < 0.05 versus CTL; n = 3. b Cell lysates were analyzed by western blot using anti-SOD−2 antibodies. The lower panel exhibits the mean data normalized to GAPDH, *p < 0.05 versus CTL; n = 5
OPN induces mitochondrial depolarization and remodeling
Depolarization of mitochondrial transmembrane potential is implicated in ROS production and apoptosis [31]. Therefore, we next measured mitochondrial transmembrane potential using JC−1 staining. A reduction in the ratio of red to green fluorescence indicates a decrease in mitochondrial transmembrane potential. OPN treatment significantly decreased the red-to-green fluorescence ratio (*p < 0.05; n = 3; Fig. 5a).
Fig. 5.
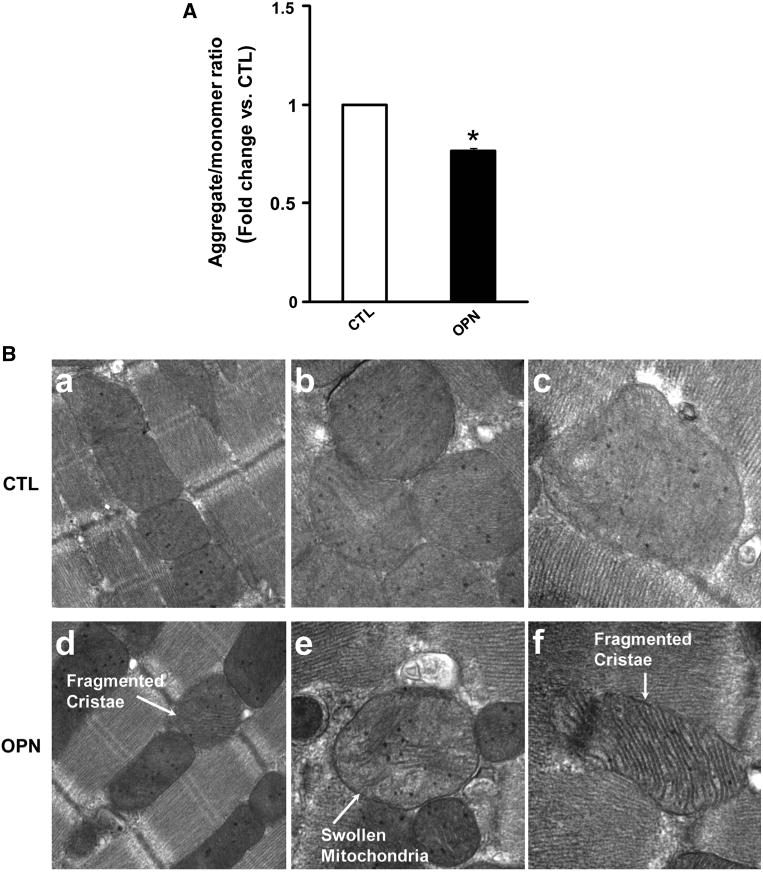
OPN induces mitochondrial depolarization and remodeling. a ARVMs were treated with OPN for 6 h and incubated with JC−1 dye for 10 min at 37 °C. The cells were visualized using epifluorescence microscope and the ratio of red-to-green fluorescence was calculated. *p < 0.05 versus CTL; n = 3. b ARVMs were treated with OPN for 48 h. After fixing and processing, the cells were visualized using transmission electron microscope. Photomicrographs illustrate ultrastructural features of mitochondria in control (CTL; a ×944,000; b, c ×973,000) and OPN-treated (d ×44,000; e, f × 73000) ARVMs
Structural analysis of mitochondria using transmission electron microscopy revealed that OPN induces mitochondrial remodeling. Mitochondria in OPN-treated ARVMs were giant polygon-shaped megamitochondria with decreased matrix density. The mitochondrial cristae were fragmented and disorganized, and formed concave loops (Fig. 5b).
OPN increases expression of BIK
In cells of non-cardiac origin, BIK is shown to induce remodeling of mitochondrial cristae and apoptosis by mobilizing calcium from the ER to the mitochondria [22, 24]. Western blot and RT-PCR analyses demonstrated that OPN significantly increases BIK expression (Fig. 6a, b). Adenoviral-mediated expression of OPN also led to increased BIK expression in ARVMs (Fig. 6c).
Fig. 6.
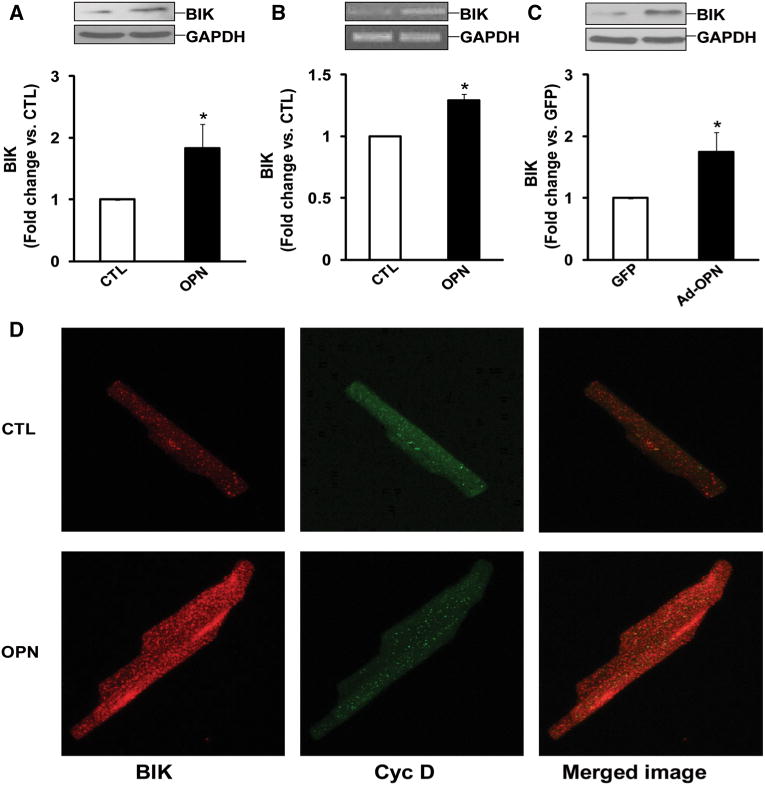
OPN increases BIK expression. a ARVMs were treated with OPN for 24 h. Total cell lysates were analyzed by Western blot using anti-BIK antibodies. The lower panel exhibits the mean data normalized to GAPDH, *p < 0.05 versus CTL; n = 9. b ARVMs were treated with OPN for 24 h. Total RNA were reverse transcribed and resulting cDNAs were subjected for amplification of BIK and GAPDH genes, *p < 0.05 versus CTL; n = 4. c ARVMs were infected with adenoviruses expressing OPN or GFP for 48 h. Cell lysates were analyzed by Western blot using anti-BIK antibodies. The lower panel exhibits the mean data normalized to GAPDH, *p < 0.05 versus CTL; n = 5. d ARVMs were treated with OPN for 24 h and immunostained using anti-BIK and anti-cyclophilin D antibodies. Cells were visualized using confocal microscope. The figure depicts representative images from three independent experiments
To further confirm increased BIK expression in response to OPN, cells were immunostained using anti-BIK antibodies. Dual immunostaining with cyclophilin D (a vital component of mitochondrial permeability transition pore; MPTP) was used to define localization of BIK to the mitochondria. Analysis of cells using confocal microscopy revealed an increased overall staining for BIK in OPN-treated cells. Dual immunostaining showed only a few co-localized spots of BIK with cyclophilin D, suggesting that majority of BIK localizes in cellular compartments other than mitochondria (Fig. 6d).
Role of BIK in OPN-stimulated apoptosis
To investigate the involvement of BIK in OPN-stimulated apoptosis, ARVMs were infected with adenoviruses expressing dominant-negative BIK (Mut BIK) or GFP for 24 h followed by treatment with OPN for 24 h. In Mut BIK, the conserved leucine−61 in the BH3 domain is changed to glycine, thereby making it inactive [22]. Analysis of apoptosis using TUNEL-assay indicated that expression of Mut BIK significantly inhibits OPN-stimulated apoptosis (GFP, 8.00 ± 1.32; OPN, 17.30 ± 1.34*; Mut BIK + OPN, 8.87 ± 1.71; Mut BIK, 10.00 ± 0.76; *p < 0.05 vs. CTL; $p < 0.05 vs. OPN, n = 5, Fig. 7).
Fig. 7.
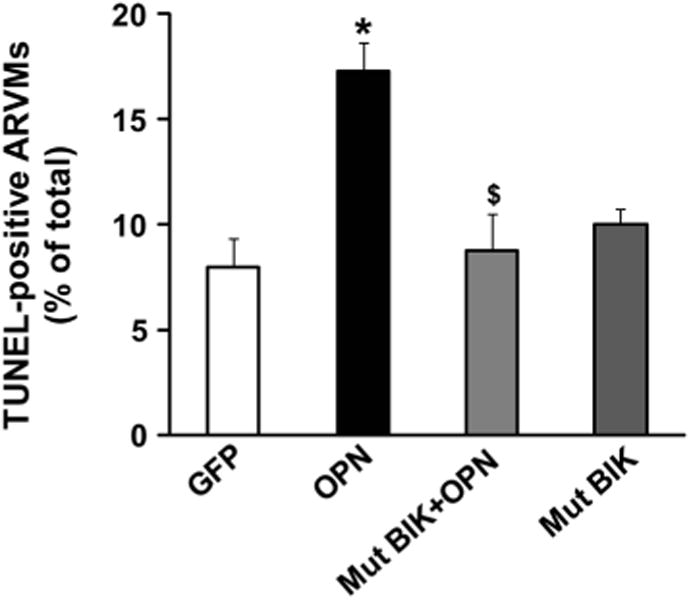
Adenoviral-mediated expression of mutant BIK decreases OPN-stimulated apoptosis. ARVMs were infected with adenoviruses expressing BIK or GFP for 24 h followed by treatment with OPN for 24 h. Apoptosis was measured using TUNEL-staining assay, *p < 0.05 versus CTL; $p < 0.05 versus OPN; n = 5
Myocyte-specific expression of OPN in adult heart increases oxidative stress, and expression of NOX-4 and BIK
Previously, we have shown that increased OPN expression in adult heart in myocyte-specific manner associates with increased myocyte apoptosis and myocardial dysfunction [12]. Analyses of LV lysates, 4 weeks after doxycycline withdrawal, using OxyBlot assay indicated a significant increase in protein carbonylation in OPN-expressing hearts (MHC-OPN) versus SHAM (Fig. 8a). Western blot analyses of LV lysates indicated a significant increase in NOX−4 and BIK protein levels in OPN-expressing hearts (Fig. 8b, c).
Fig. 8.
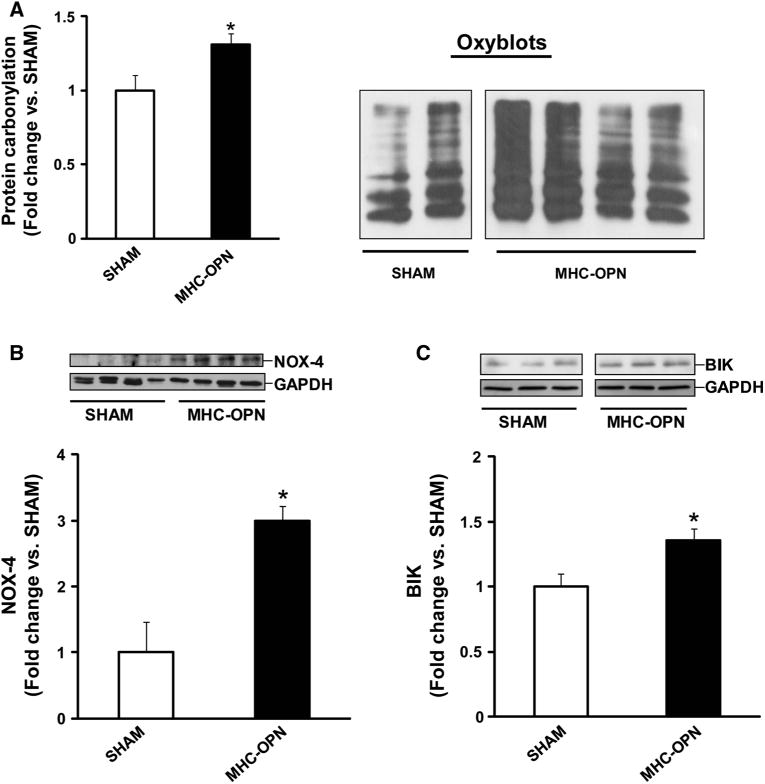
Increased OPN expression in the heart induces oxidative stress, and increases expression of NOX−4 and BIK Left ventricular lysates, prepared 4 weeks after doxycycline withdrawal, were analyzed by oxyblot assay (a), or by western blots using anti-NOX−4 (b) and anti-BIK (c) antibodies. a The right panel depicts oxyblot image obtained from SHAM and MHC-OPN samples, while the left panel depicts the mean data normalized to SYPRO ruby staining, *p < 0.05 versus SHAM; n = 3–4. The lower panels in b and c exhibit the mean data normalized to GAPDH, *p < 0.05 versus SHAM; n = 3–4
Discussion
Increased OPN expression, specifically in myocytes, associates with increased myocyte apoptosis in different models of heart disease [7–10]. Previously, we provided evidence that OPN stimulates cardiac myocyte apoptosis via the involvement of ER stress and mitochondrial death pathways [12]. A major finding of this study is that OPN increases intracellular ROS levels and induces oxidative stress in myocytes. Increased oxidative stress in response to OPN involves NOX, SOD−2 and mitoKATP. NOX may act upstream in activation of mitochondrial death pathway since inhibition of NOX decreases cytosolic cytochrome c levels and apoptosis. OPN induces mitochondrial depolarization and affects mitochondrial integrity. OPN increases expression of BIK, a pro-apoptotic protein serving as a crucial link between ER and mitochondria. Adenoviral-mediated expression of dominant-negative BIK decreases OPN-stimulated apoptosis. In vivo, increased OPN expression in myocyte-specific manner in adult heart associates with increased protein oxidation, and expression of NOX−4 and BIK.
OPN is suggested to act as a promoter or inhibitor of oxidative stress. In angiotensin II-induced renal damage, lack of OPN associated with increased expression of NOX subunits such as NOX−2, gp47phox and NOX−4 in kidney cortex [27] implicating that deficiency of OPN promotes oxidative stress by upregulating NOX subtypes. In contrast, lack of OPN associated with decreased mRNA levels of NOX subunits (p47Phox, p67Phox, and gp91Phox) and oxidative stress in aldosterone-induced renal damage [26] suggesting that OPN deficiency protects against oxidative stress. In aortic mesenchymal cells, OPN treatment increased superoxide and the oxylipid product 8-iso-prostaglandin F2 α-isoprostane levels, and expression of NOX−2 [32]. Aortic adventitial myofibroblasts isolated from mice lacking OPN exhibited decreased expression of NOX subunits (NOX−2, NOX−1, p67, and p47) [32]. These studies point towards the possibility that oxidative stress induced by OPN may be tissue- and disease-specific. In cardiac myocytes, OPN-induced oxidative stress as evidenced by increased protein carbonylation, number of ROS-positive cells and H2DCFDA staining. Furthermore, scavenging ROS using antioxidants inhibited OPN-stimulated increases in oxidative stress, cytosolic cytochrome c (a biomarker of mitochondrial death pathway) and apoptosis.
NOX−4 subtype is expressed primarily in the mitochondria of cardiac myocytes. It is suggested to be a major source of mitochondrial oxidative stress mediating mitochondrial and cardiac dysfunction in response to pressure overload [16, 19]. Here, OPN treatment increased expression of NOX−4 in myocytes. Expression of OPN in myocyte-specific manner in the heart also associated with increased NOX−4 expression. These data suggest a role for NOX−4 in OPN-induced oxidative stress in myocytes. Apocynin, a specific inhibitor of NOX, decreased cytosolic cytochrome c levels and apoptosis. Intracellular antioxidant enzymes help the cells to recover from oxidative stress. However, decreased expression of antioxidant enzymes and prolonged oxidative stress may induce cell death. Our studies provide evidence that OPN induces mitochondrial depolarization and inhibits expression of SOD−2 (mitochondrial antioxidant). Pretreatment of cells with SOD mimetic, MnTBAP, inhibited OPN-stimulated increases in oxidative stress, cytosolic cytochrome c and apoptosis. Collectively, these studies suggest that mitochondria may serve as a major source of oxidative stress in ARVMs in response to OPN. NOX may act upstream in mitochondrial ROS production and apoptosis.
Cardiac myocytes express sarcolemmal KATP [33] and mitoKATP. The mitoKATP are present in the inner mitochondrial membraneand modulate mitochondrial membrane potential, ion homeostasis and matrix volume [34]. MitoKATP are suggested to play an important role in modulation of ischemic preconditioning as selective antagonism of mitoKATP inhibits the protective effects of ischemic preconditioning [35], while selective mitoKATP agonist mimic the protective effects of ischemic preconditioning with respect to infarct size and myocyte apoptosis [36]. Blocking of mitoKATP using GLIB is shown to inhibit aldosterone-induced ROS production in the heart [37]. Reconstitution of myocardial mitoKATP into the lipid bilayers in vitro suggest that these channels are activated by superoxides [38]. Here OPN-stimulated increases in oxidative stress, cytosolic cytochrome c, and apoptosis were inhibited by the selective inhibition of mitoKATP using GLIB. These data further support a role for mitochondria, specifically mitoKATP, in OPN-stimulated myocyte apoptosis.
Opening of the MPTP occurs upon induction of apoptosis. The mitochondrial network fragments and cytochrome-c are released activating downstream caspases [20]. A large influx of calcium into mitochondria from the ER promotes apoptosis. This influx of calcium is regulated by the pro-apoptotic members of BCL−2 family proteins such as BIK and BAX. Under normal conditions, BAX is a cytosolic protein. During apoptosis, BAX undergoes conformational change and insertion into the outer mitochondrial membrane, resulting in cytochrome c release and apoptosis [39]. BAX is also suggested to translocate to the ER and initiate a parallel ER stress pathway of activation of caspases and apoptosis [40]. BIK localizes mainly in the ER [22]. Evidence has been provided for the localization of BIK to the mitochondria and nucleus [23, 41]. BIK is suggested to induce remodeling of the mitochondrial cristae and apoptosis by mobilizing calcium influx from ER to the mitochondria [24, 42]. Besides affecting their cellular localization, apoptotic stimuli generally enhance the expression of pro-apoptotic proteins. Previously, we have shown that OPN increases expression of BAX in ARVMs and the heart [12]. Here we provide evidence for increased expression of BIK in response to OPN. OPN treatment-induced mitochondrial depolarization and remodeling. Expression of a mutant BIK decreased OPN-stimulated increases in myocyte apoptosis. Collectively, these data suggest a role for BIK in OPN-stimulated abnormal mitochondrial morphology and apoptosis.
Expression of OPN increases in the heart under a variety of pathophysiological conditions of the heart in animal models and in humans. Angiotensin II, endothelin, norepinephrine, cytokines, and glucocorticoid hormone are suggested to induce OPN expression in the heart [2, 3]. The data presented here suggest that OPN induces oxidative stress in myocytes via the involvement of NOX−4, SOD−2, and mitoKATP. OPN affects expression of apoptotic proteins and induces morphological changes in the mitochondria. Further studies aimed at identifying the molecular signals leading to oxidative stress and expression of apoptosis-related proteins in response to OPN may have important implications for the treatment of myocardial dysfunction.
Acknowledgments
The authors appreciate the technical help received from Dr. Dennis Defoe, Dr. Robert V. Schoborg, Mr. Rolf Fritz, Ms. Jennifer Kintner, and Ms. Barbara A. Connelly.
Funding: This work was supported by Merit Review awards (BX002332 and BX000640) from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development, National Institutes of Health (R15HL129140), and funds from Institutional Research and Improvement account (to KS).
Abbreviations
- OPN
Osteopontin
- ARVMs
Adult rat ventricular myocytes
- ROS
Reactive oxygen species
- NADPH oxidase
Nicotinamide adenine dinucleotide phosphate-oxidase
- NOX
NADPH oxidase
- NAC
N-acetyl-l-cysteine
- SOD-2
Mitochondrial superoxide dismutase-2
- DMEM
Dulbecco's modified eagle medium
- APO
Apocynin
- GLIB
Glibenclamide
- 5-HD
5-hydroxydecanoate
- H2DCFDA
2′,7′-dichlorohydrofluorescein diacetate
- mitoKATP
Mitochondrial KATP channels
- MnTBAP
Manganese (III) tetrakis (4-benzoic acid) porphyrin
- CTL
Control
- MAM
Mitochondrial-associated endoplasmic reticulum membrane
- MHC-OPN
OPN transgenic mice
- Mut BIK
Mutant BIK
- ER
Endoplasmic reticulum
Footnotes
Compliance with ethical standards: Conflicts of interest: No conflicts of interest, financial or otherwise, are declared by the author(s).
References
- 1.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635–688. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh M, Foster CR, Dalal S, Singh K. Osteopontin: role in extracellular matrix deposition and myocardial remodeling post-MI. J Mol Cell Cardiol. 2010;48:538–543. doi: 10.1016/j.yjmcc.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh M, Dalal S, Singh K. Osteopontin: at the cross-roads of myocyte survival and myocardial function. Life Sci. 2014;118:1–6. doi: 10.1016/j.lfs.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang KX, Denhardt DT. Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev. 2008;19:333–345. doi: 10.1016/j.cytogfr.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Denhardt DT, Noda M, O'Regan AW, et al. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest. 2001;107:1055–1061. doi: 10.1172/JCI12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kahles F, Findeisen HM, Bruemmer D. Osteopontin: a novel regulator at the cross roads of inflammation, obesity and diabetes. Mol Metab. 2014;3:384–393. doi: 10.1016/j.molmet.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsui Y, Jia N, Okamoto H, et al. Role of osteopontin in cardiac fibrosis and remodeling in angiotensin II-induced cardiac hypertrophy. Hypertension. 2004;43:1195–1201. doi: 10.1161/01.HYP.0000128621.68160.dd. [DOI] [PubMed] [Google Scholar]
- 8.Satoh M, Nakamura M, Akatsu T, et al. Myocardial osteopontin expression is associated with collagen fibrillogenesis in human dilated cardiomyopathy. Eur J Heart Fail. 2005;7:755–762. doi: 10.1016/j.ejheart.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 9.Subramanian V, Krishnamurthy P, Singh K, Singh M. Lack of osteopontin improves cardiac function in streptozotocin-induced diabetic mice. Am J Physiol Heart Circ Physiol. 2007;292:H673–H683. doi: 10.1152/ajpheart.00569.2006. [DOI] [PubMed] [Google Scholar]
- 10.Sam F, Xie Z, Ooi H, et al. Mice lacking osteopontin exhibit increased left ventricular dilation and reduced fibrosis after aldosterone infusion. Am J Hypertens. 2004;17:188–193. doi: 10.1016/j.amjhyper.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 11.Renault MA, Robbesyn F, Reant P, et al. Osteopontin expression in cardiomyocytes induces dilated cardiomyopathy. Circ Heart Fail. 2010;3:431–439. doi: 10.1161/CIRCHEARTFAILURE.109.898114. [DOI] [PubMed] [Google Scholar]
- 12.Dalal S, Zha Q, Daniels CR, et al. Osteopontin stimulates apoptosis in adult cardiac myocytes via the involvement of CD44 receptors, mitochondrial death pathway, and endoplasmic reticulum stress. Am J Physiol Heart Circ Physiol. 2014;306:H1182–H1191. doi: 10.1152/ajpheart.00954.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugamura K, Keaney JF. Reactive oxygen species in cardiovascular disease. Free Radic Biol Med. 2011;51:978–992. doi: 10.1016/j.freeradbiomed.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L, Fan H, Yang L, et al. Tyrosol prevents ischemia/reperfusion-induced cardiac injury in H9c2 cells: involvement of ROS, Hsp70, JNK and ERK, and apoptosis. Molecules. 2015;20:3758–3775. doi: 10.3390/molecules20033758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vichova T, Motovska Z. Oxidative stress: predictive marker for coronary artery disease. Exp Clin Cardiol. 2013;18:e88–e91. [PMC free article] [PubMed] [Google Scholar]
- 16.Ago T, Kuroda J, Pain J, et al. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss JN, Korge P, Honda HM, Ping P. Role of the mitochondrial permeability transition in myocardial disease. Circ Res. 2003;93:292–301. doi: 10.1161/01.RES.0000087542.26971.D4. [DOI] [PubMed] [Google Scholar]
- 18.Baines CP, Kaiser RA, Purcell NH, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 19.Kuroda J, Ago T, Matsushima S, et al. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bravo-Sagua R, Rodriguez AE, Kuzmicic J, et al. Cell death and survival through the endoplasmic reticulum-mitochondrial axis. Curr Mol Med. 2013;13:317–329. doi: 10.2174/156652413804810781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005;24:1546–1556. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Germain M, Mathai JP, Shore GC. BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J Biol Chem. 2002;277:18053–18060. doi: 10.1074/jbc.M201235200. [DOI] [PubMed] [Google Scholar]
- 23.Hegde R, Srinivasula SM, Ahmad M, et al. Blk, a BH3-containing mouse protein that interacts with Bcl-2 and Bcl-xL, is a potent death agonist. J Biol Chem. 1998;273:7783–7786. doi: 10.1074/jbc.273.14.7783. [DOI] [PubMed] [Google Scholar]
- 24.Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005;24:1546–1556. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coultas L, Bouillet P, Stanley EG, et al. Proapoptotic BH3-only Bcl-2 family member Bik/Blk/Nbk is expressed in hemopoietic and endothelial cells but is redundant for their programmed death. Mol Cell Biol. 2004;24:1570–1581. doi: 10.1128/MCB.24.4.1570-1581.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Irita J, Okura T, Jotoku M, et al. Osteopontin deficiency protects against aldosterone-induced inflammation, oxidative stress, and interstitial fibrosis in the kidney. Am J Physiol Ren Physiol. 2011;301:F833–F844. doi: 10.1152/ajprenal.00557.2010. [DOI] [PubMed] [Google Scholar]
- 27.Wolak T, Kim H, Ren Y, et al. Osteopontin modulates angiotensin II-induced inflammation, oxidative stress, and fibrosis of the kidney. Kidney Int. 2009;76:32–43. doi: 10.1038/ki.2009.90. [DOI] [PubMed] [Google Scholar]
- 28.Deka S, Vanover J, Sun J, et al. An early event in the herpes simplex virus type-2 replication cycle is sufficient to induce Chlamydia trachomatis persistence. Cell Microbiol. 2007;9:725–737. doi: 10.1111/j.1462-5822.2006.00823.x. [DOI] [PubMed] [Google Scholar]
- 29.Dalle-Donne I, Aldini G, Carini M, et al. Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luk E, Carroll M, Baker M, Culotta VC. Manganese activation of superoxide dismutase 2 in Saccharomyces cerevisiae requires MTM1, a member of the mitochondrial carrier family. Proc Natl Acad Sci USA. 2003;100:10353–10357. doi: 10.1073/pnas.1632471100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 32.Lai CF, Seshadri V, Huang K, et al. An osteopontin-NADPH oxidase signaling cascade promotes pro-matrix metalloproteinase 9 activation in aortic mesenchymal cells. Circ Res. 2006;98:1479–1489. doi: 10.1161/01.RES.0000227550.00426.60. [DOI] [PubMed] [Google Scholar]
- 33.Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- 34.Holmuhamedov EL, Jovanović S, Dzeja PP, et al. Mitochondrial ATP-sensitive K+ channels modulate cardiac mitochondrial function. Am J Physiol. 1998;275:H1567–H1576. doi: 10.1152/ajpheart.1998.275.5.H1567. [DOI] [PubMed] [Google Scholar]
- 35.Hu H, Sato T, Seharaseyon J, et al. Pharmacological and histochemical distinctions between molecularly defined sarcolemmal KATP channels and native cardiac mitochondrial KATP channels. Mol Pharmacol. 1999;55:1000–1005. [PubMed] [Google Scholar]
- 36.Garlid KD, Paucek P, Yarov-Yarovoy V, et al. The mitochondrial KATP channel as a receptor for potassium channel openers. J Biol Chem. 1996;271:8796–8799. doi: 10.1074/jbc.271.15.8796. [DOI] [PubMed] [Google Scholar]
- 37.Nolly MB, Caldiz CI, Yeves AM, et al. The signaling pathway for aldosterone-induced mitochondrial production of superoxide anion in the myocardium. J Mol Cell Cardiol. 2014;67:60–68. doi: 10.1016/j.yjmcc.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 38.Zhang DX, Chen YF, Campbell WB, et al. Characteristics and superoxide-induced activation of reconstituted myocardial mitochondrial ATP-sensitive potassium channels. Circ Res. 2001;89:1177–1183. doi: 10.1161/hh2401.101752. [DOI] [PubMed] [Google Scholar]
- 39.Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20:929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zong WX, Li C, Hatzivassiliou G, et al. Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol. 2003;162:59–69. doi: 10.1083/jcb.200302084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han J, Sabbatini P, White E. Induction of apoptosis by human Nbk/Bik, a BH3-containing protein that interacts with E1B 19K. Mol Cell Biol. 1996;16:5857–5864. doi: 10.1128/mcb.16.10.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chinnadurai G, Vijayalingam S, Rashmi R. BIK, the founding member of the BH3-only family proteins: mechanisms of cell death and role in cancer and pathogenic processes. Oncogene. 2008;27(Suppl 1):S20–S29. doi: 10.1038/onc.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]