

SUMMARY
Proper brain function requires a substantial energy supply, up to 20% of whole body energy in humans, and brain activation produces large dynamic variations in energy demand. While local increases in cerebral blood flow are well known, the cellular responses to energy demand are controversial. During brain excitation, glycolysis of glucose to lactate temporarily exceeds the rate of mitochondrial fuel oxidation; although the increased energy demand occurs mainly within neurons, some have suggested this glycolysis occurs mainly in astrocytes, which then shuttle lactate to neurons as their primary fuel. Using metabolic biosensors in acute hippocampal slices and brains of awake mice, we find that neuronal metabolic responses to stimulation do not depend on astrocytic stimulation by glutamate release, nor do they require neuronal uptake of lactate; instead they reflect increased direct glucose consumption by neurons. Neuronal glycolysis temporarily outstrips oxidative metabolism, and provides a rapid response to increased energy demand.
Graphical Abstract
INTRODUCTION
How the brain responds to the metabolic challenge of activation has been both intriguing and controversial for many decades. It is generally agreed that the “resting” brain, under normal metabolic circumstances, consumes glucose and metabolizes it nearly completely to CO2 (Clarke and Sokoloff, 1994; Madsen et al., 1999). However, with brain activation, glucose consumption increases much more than oxygen consumption (Fox and Raichle, 1986; Fox et al., 1988). This mismatch is typically explained as a temporary increase in “aerobic glycolysis”: conversion of glucose to pyruvate and then to lactate, instead of total mitochondrial oxidation of the pyruvate (Dienel, 2012).
Most controversial has been the cellular site of this increased aerobic glycolysis during brain stimulation. A popular hypothesis is the existence of an “astrocyte-neuron lactate shuttle” (ANLS), which proposes that astrocytes are stimulated by neuronal glutamate release to convert more glucose to lactate, which is then released by the astrocytes to serve as fuel for the neighboring neurons (Bélanger et al., 2011; Pellerin et al., 1998). Proponents of the ANLS have argued that neurons preferentially use this astrocyte-produced lactate rather than glucose (Magistretti and Pellerin, 1999). Most of the experimental support for the ANLS has come from separate experiments on neurons and astrocytes, both conducted in cell culture conditions (Pellerin et al., 2007). In vivo measurements to learn which cell type shows greater stimulated uptake of glucose analogs have given conflicting results (Chuquet et al., 2010; Lundgaard et al., 2015), perhaps because the fluorescent glucose derivatives used are substantially different from glucose in structure and size; their cell-specific uptake may reflect the different types of glucose transporters in neurons and astrocytes (or even other uptake pathways) rather than the different rates of glucose phosphorylation.
Here we have used genetically-encoded fluorescent biosensors to measure directly the metabolic responses of individual neurons to stimulation, in the context of brain tissue containing both neurons and astrocytes, in acute hippocampal slices and in the brains of awake mice. The behavior of metabolic transients in neurons contradicts the predictions of the ANLS hypothesis, and instead is consistent with direct neuronal metabolism of glucose.
RESULTS
Stimulation produces transient increases of cytosolic NADH:NAD+ in individual neurons
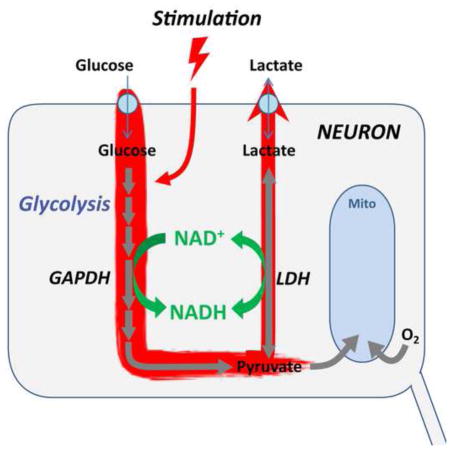
We first monitored stimulation-induced transients in the cytosolic NADH:NAD+ ratio, which responds to both glucose and lactate metabolism. Neuronal NADH:NAD+ ratio is controlled by three major processes (Fig. 1E): glycolysis, lactate dehydrogenase activity, and mitochondrial NADH shuttles (Salway, 2004; in neurons, mainly the malate-aspartate shuttle, McKenna et al., 2006); unbiased proteomics and transcriptomics surveys of central neurons reveal that these are by far the dominant NADH dehydrogenase enzymes in the cytosol (Sharma et al., 2015; Zeisel et al., 2015; Suppl. Fig. 1). NADHCYT can rise in response to either increased glycolysis (which converts NAD+ to NADH at the GAPDH step), to increased conversion of lactate to pyruvate, or to decreased activity of mitochondrial NADH shuttles. The Peredox sensor (Hung et al., 2011) was expressed in mouse hippocampus and studied by imaging in acute hippocampal slices using a two-photon microscope equipped for fluorescence lifetime imaging (2p-FLIM). The fluorescence lifetime (LT) of the sensor protein, measured in nanoseconds, gives a direct report of sensor occupancy and thus of the cytosolic NADH:NAD+ ratio (Mongeon et al., 2016). We also used the fluorescence LT response of the genetically-encoded red Ca2+ sensor RCaMP1h (Akerboom et al., 2013; Yellen and Mongeon, 2015) to monitor neuronal excitation.
Figure 1. NADHCYT transients with synaptic stimulation.

(A) Filmstrip of metabolic transients in dentate granule neurons. A sequence of acquisitions shows the brief increase in RCaMP lifetime reflecting an augmented Ca2+ concentration after synaptic stimulation. The peak in the Peredox lifetime elevation occurs approximately 1 min later, reflecting changes in the cytosolic NADH/NAD+ ratio (NADHCYT) toward a more reduced state. Images are pseudocolored according to sensor lifetimes. The color scale bar to the right shows the range of lifetime values for both RCaMP and Peredox sensors. For the latter, the color scale is accompanied by an axis converting lifetimes to calibrated NADH/NAD+ ratios. (B) Individual traces from a cell in the filmstrip illustrate the time course of synaptically stimulated transients in Peredox (top) and RCaMP1h lifetimes (bottom). (C) Average transient increases in Peredox lifetime after synaptic stimulation of 60 brief pulses delivered at 20 Hz. Traces represent the mean ± SEM (events=447, neurons=166, slices=27, mice=14; SEM calculated using number of neurons). (D) The magnitude of the NADHCYT increase correlates with the amplitude of the neuronal Ca2+ spike in a wide range of Ca2+ responses elicited by synaptic stimulation. The scatter plot summarizes data resulting of events elicited with different stimulation strengths (events=554, neurons=166, slices=27, mice=14). (E) Glycolysis and lactate utilization enhance neuronal NADH production. The cartoon summarizes the main pathways leading to an increased cytosolic NADH/NAD+ ratio in neurons. After passive uptake via GLUT transporters, glucose is phosphorylated and enters glycolysis, where NADH is produced by the enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Lactate import occurs through monocarboxylate transporters (MCT) and is then converted to pyruvate by the enzyme lactate dehydrogenase (LDH), also reducing NAD+ to NADH.
Without stimulation, the NADH:NAD+ ratio is quite low (oxidized) in dentate granule neurons (DGNs) in the hippocampal slice (Mongeon et al., 2016). Upon brief synaptic stimulation of the DGNs by electrical stimulation in the molecular layer of the dentate gyrus (60 brief pulses), there was a prominent and reliable elevation of the cytosolic NADH:NAD+ ratio for a period of about 3 minutes (Figure 1A–C). The 3 second long stimulation produced an immediate, brief rise in RCaMP1h lifetime (left hand panel of Fig. 1A, at t=0). This was followed by a slower and more prolonged rise in Peredox lifetime. The increase in cytosolic NADH:NAD+ ratio (hereafter referred to as NADHCYT) was quite substantial, with 60 stimuli typically producing a change from a baseline ratio of ~1:1000 up to a stimulated ratio of ~1:500, a ~2-fold change (corresponding to a Peredox LT of 1.73 ± 0.01 ns at baseline and 1.88 ± 0.01 ns when stimulated; mean ± SEM, n=166 neurons).
Recording from many neurons, we observed a wide range of both Ca2+ and NADHCYT responses, but the two were well-correlated even for the smallest responses (Fig. 1D), showing that the metabolic NADHCYT transient occurs not just for large neuronal responses.
Astrocyte stimulation by glutamate is neither sufficient nor necessary for neuronal NADH transients
The ANLS hypothesis would attribute the rise in neuronal NADHCYT to increased supply of lactate by astrocytes, which would then raise neuronal NADHCYT through the increased oxidation of lactate to pyruvate. Astrocyte release of lactate is thought to be stimulated by synaptic release of glutamate, which is then transported into astrocytes with Na+ ions, stimulating astrocytic glycolysis (Pellerin and Magistretti, 1994; Pellerin et al., 2007). We first tested for a possible role of astrocytes in this metabolic transient by leaving glutamate release intact but blocking the postsynaptic activation of the DGNs with inhibition of ionotropic glutamate receptors by NBQX and D-AP5. If synaptic stimulation of astrocytes is sufficient to produce release of lactate, the ANLS would predict that the rise in neuronal NADHCYT would occur even with the blockade of the postsynaptic response. The opposite was seen: blockade of the postsynaptic response (as judged from the disappearance of the postsynaptic Ca2+ transient, and the disappearance of the EPSP from an extracellular electrode; Fig 2A, B and Suppl. Fig. S3) was accompanied by disappearance of the NADHCYT transient (Fig 2B, C). Under these conditions, glutamate is still released from the presynaptic terminals and will be transported into astrocytes, and signaling through metabotropic glutamate receptors (GPCRs) is still intact, but these effects were not sufficient to produce an NADHCYT transient in the neurons. This result agrees with the conclusions of Ivanov and colleagues (2014), who found that glutamate release per se was not sufficient to produce NAD(P)H autofluorescence transients in brain slices (the NAD(P)H signal is addressed further in the Discussion).
Figure 2. Glutamate release is neither sufficient nor necessary for NADHCYT transients.

(A) Typical electrode placement for synaptic stimulation. The picture shows an infra-red differential interference contrast image of a horizontal slice from the hippocampus, where the stimulating electrode (s.e.) is placed in the molecular layer (m.l.) and an extracellular recording electrode (r.e.) is inserted in the layer of dentate granule neurons (DGNs, following the arrow direction). (B) Inhibition of ionotropic glutamate receptors abolishes the metabolic transients. A representative trace of a synaptically stimulated DGN (using a train of 60 pulses at 20 Hz) shows that the Peredox signal disappears after blocking synaptic transmission with a mixture of 5 μM NBQX and 25 μM D-AP5 in the ACSF. The complete blockade of synaptic transmission is corroborated by the absence of the typical RCaMP1h signal after stimulation. The effect of synaptic blockers was partially reversed after 5 min of washout in regular ACSF, and both Ca2+ and NADH transients were completely recovered after 10 min in ACSF. (C) Dataset for the effect of ionotropic glutamate receptor block. Data were compared by a paired Student’s t-test (neurons=46, slices=6, mice=3). (D) Typical electrode placement for antidromic (axonal) stimulation. In this experimental paradigm, the stimulating electrode (s.e.) is placed in the the hilus (hil) and an extracellular recording electrode (r.e.) is inserted in the layer of dentate granule neurons (DGNs, following the arrow direction). (E) Average transient increases in Peredox lifetime after antidromic stimulation of 100 brief pulses delivered at 50 Hz. Traces represent the mean ± SEM (events=179, neurons=84, slices=18, mice=11; SEM calculated using number of neurons). The lower panel shows the putative average Ca2+ spike in the cells, as a proxy for neuronal excitation. (F) Elevation in the NADH/NAD+ ratio is directly correlated with the neuronal Ca2+ increase after electrical antidromic stimulation in DGNs, similarly to synaptic stimulation. Scatter plots of changes in Peredox lifetime versus variations in RCaMP lifetime for events recorded after antidromic stimulation (open circles; events=205, neurons=87, slices=19, mice=12), overlap with data collected using synaptic stimulation (dark squares).
Alternatively, we asked if astrocyte stimulation by glutamate was necessary for the metabolic transient. We stimulated the neurons not through their synaptic inputs, but rather by direct stimulation of their axons (antidromic stimulation), using an electrode placed in the hilus of the dentate gyrus (Fig. 2D). This should remove nearly all of the glutamate release in the vicinity of the DGN cell bodies where we are observing the sensor responses. Consistent with this, there is little or no reduction in the antidromically-induced DGN calcium response with the application of synaptic blockers (Suppl. Fig. S3). Nevertheless, it is known that hilar stimulation triggers synaptic transmission between mossy fibers (DGNs axons) and CA3 pyramidal neurons, which send backprojections to the dentate gyrus (Scharfman, 2007). Although our recordings show that it is unlikely that this contributes to our DGN responses (Suppl. Fig. S3), a combination of synaptic blockers was continuously applied to minimize any contribution of this loop from CA3 back to the dentate gyrus. Despite the relative absence of glutamate release, we observed that antidromic stimulation produced NADHCYT transients in the DGN cell bodies very similar to those produced by synaptic stimulation (Fig 2E, F), showing that glutamate stimulation of astrocytes is not required for the transients.
Lactate import does not contribute to neuronal NADHCYT transients
It remains possible that astrocytes depend on other stimuli (e.g., K+ release from neurons) for strong stimulation of lactate release (Peng et al., 1994; Bittner et al., 2011). We therefore used a different test of the role of lactate import in producing the neuronal NADHCYT transients: we blocked the plasma membrane monocarboxylate transporters MCT1 and MCT2 with a high affinity blocker, AR-C155858 (Ovens et al., 2010a, 2010b). These two transporters are used both for lactate export from astrocytes and for lactate import into neurons. The prediction of the ANLS hypothesis is clear: when the monocarboxylate transporters (MCTs) are inhibited, the uptake of lactate by neurons and the consequent rise in NADHCYT should be abolished.
To test the effectiveness of MCT inhibition by AR-C155858, we confirmed that it was able to block a rise in neuronal NADHCYT by direct application of exogenous lactate. In the experiment of Figure 3A, we first applied synaptic stimulation and observed the transient rise in NADHCYT, as described earlier. Then we applied 10 mM Na-lactate in the solution bathing the hippocampal slice. This produced a large and prompt increase in NADHCYT, likely mediated by the rapid action of lactate dehydrogenase (LDH), which converts NAD+ to NADH in parallel with the conversion of lactate to pyruvate (Williamson et al., 1967; Hung et al., 2011). Washout of lactate returned the NADHCYT level to baseline. Next we applied the MCT inhibitor AR-C155858 (2 μM), and observed a small decline in the resting NADHCYT (as discussed below, this is consistent with some astrocyte-to-neuron lactate shuttling at rest, as previously suspected; Mongeon et al., 2016). In the presence of the MCT inhibitor, the NADHCYT response to direct application of exogenous lactate was much smaller (27% ± 2% of the original response, mean ± SEM, n=36 neurons), as expected if lactate uptake is mostly blocked. This blockade of lactate uptake did not, however, abolish the NADHCYT response to synaptic stimulation. In fact, the NADHCYT response was significantly larger than the original response in the absence of the MCT inhibitor (Figures 3A, C; the 95% confidence interval of the increase in ΔPeredox-LT/ΔRCaMP-LT was 1.23-fold to 1.34-fold, using a paired comparison, when the second stimulation was performed 5 min after MCT inhibitor application).
Figure 3. Neuronal lactate import is not required for the NADHCYT transients.

(A) Monocarboxylate transporter (MCT) inhibition abolishes NADH transients induced by the application of external lactate but enhances the NADHCYT rise after synaptic stimulation. The MCT inhibitor AR-C155858 (2 μM, 5 min) prevents the increase in the NADH/NAD+ ratio in response to external lactate (10 mM). However, the endogenous NADH transients in response to synaptic stimulation (60 pulses at 20 Hz) were bigger in the presence of the MCT inhibitor (MCT inhib). (B) Lactate-induced changes in Peredox lifetime were significantly reduced in the presence of the MCT inhibitor. Data were compared by a paired Student’s t-test (neurons=36, slices=5, mice=5). (C) Scatter plots of changes in Peredox lifetime versus changes in RCaMP lifetime, show that acute (5 min) or long-lasting (>10 min; as in A) exposure to the MCT inhibitor increases the size of NADHCYT transients for similar Ca2+ transients, making the slope of the positive correlation steeper. All comparisons were made using a paired Student’s t-test. Control data points are open symbols while symbols for treatments are filled, using circles or squares for acute (neurons=39, slices=7, mice=5) and long-lasting (neurons=36, slices=5, mice=5) application of the MCT inhibitor respectively. The dashed lines and surrounding shaded zones show the linear fit and 95% confidence interval for the (unpaired) data with (black) and without (gray) MCT inhibitor; the lower dashed line shows the prediction of the ANLS that the stimulation-induced transients should be reduced comparably to the lactate-induced transients in B. (D) Lactate dehydrogenase (LDH) inhibition also decreases the NADHCYT transients induced by the application of external lactate and increases the NADH/NAD+ ratio after synaptic stimulation. (E) The LDH inhibitor GSK-2837808A (2 μM, 30 min) diminishes the change of the cytosolic NADH/NAD+ ratio in response to external lactate (10 mM). Data were compared by a Wilcoxon signed-rank test (neurons=54, slices=5, mice=5). (F) Scatter plots of changes in Peredox lifetime versus changes in RCaMP lifetime, show that sustained (>30 min; as in A) exposure to the LDH inhibitor increases the size of NADHCYT transients for similar Ca2+ transients, making the slope of the positive correlation steeper. Control data points are open circles (events=179, neurons=54, slices=5, mice=5) while black squares correspond to LDH inhibition (events=54, neurons=54, slices=5, mice=5). The dashed lines and surrounding shaded zones show the linear fit and 95% confidence interval for the (unpaired) data with (black) and without (gray) LDH inhibitor; the lower dashed line shows the prediction of the ANLS that the stimulation-induced transients should be reduced comparably to the lactate-induced transients in E. (G) Left. Representative traces of changes in CFP/YFP fluorescence emission ratio for Laconic before and after the application of the MCT inhibitor (2 μM, 5 min). Right. The relative increase in the CFP/YFP ratio was unaffected by MCT inhibition (neurons=41, slices=5, mice=4; Wilcoxon Signed Rank Test, p>0.05).
The failure of the MCT inhibitor to abolish (or even diminish) the stimulated rise in neuronal NADHCYT, despite its ability to nearly abolish the direct response to exogenous lactate, demonstrates conclusively that the neuronal transients in NADHCYT are not produced by lactate import. The fact that the transients are actually larger when the MCTs are inhibited may indicate that neurons normally export lactate when their metabolism is activated by stimulation, as discussed below.
We also tested the effect of the MCT inhibitor on lactate levels in the neurons. The FRET-based biosensor Laconic changes its CFP/YFP fluorescence emission ratio upon the binding of lactate (San Martín et al., 2013). We saw reproducible increases in the Laconic signal of DGNs with synaptic stimulation, corresponding to an increase in neuronal intracellular [lactate] (Fig. 3G, left). The response to lactate has a very shallow concentration dependence (~25% change in the FRET ratio over 4 logs of [lactate]); the small pH-dependence of the FRET signal thus translates into a significant uncertainty in the lactate concentration (an alkalinization of 0.1 pH unit produces approximately a 2-fold increase in the apparent [lactate], according to the values in Fig. 2C of San Martín et al., 2013). However, changes in pH with stimulation were small, on the order of a 0.1 pH unit acidification with strong stimulation (Suppl. Fig. S5C), measured using the red fluorescence lifetime biosensor pHRed (Tantama et al., 2011). This small acidification during stimulation would have given an opposite response (an apparent decrease in [lactate]). With application of the MCT inhibitor, there was no significant change in these neuronal intracellular [lactate] transients (Fig. 3G), again arguing against the idea that astrocytes contribute significantly to neuronal lactate levels during the stimulation-induced transients.
Also consistent with the hypothesis that the neuronal transients in NADHCYT are derived from increased NADH produced directly in neurons rather than from lactate import, the neuronal NADHCYT transients were elevated rather than abolished upon inhibition of lactate dehydrogenase by the high-affinity inhibitor GSK-2837808A (2 μM; Fig. 3D, F). As expected, application of the LDH inhibitor reduced the NADHCYT response to direct application of exogenous lactate (by 78% ± 3% of the original response, mean ± SEM, n=54 neurons; Fig. 3E). But there was no diminution of the stimulation-induced NADHCYT response, as would have been seen if the elevated NADHCYT depended on LDH-dependent production of NADH from imported lactate. Instead, the stimulation-induced NADHCYT responses were larger (by 37% ± 5%; n=54 neurons; Fig. 3F), as expected for production of NADH within the neuron when LDH-dependent recycling to NAD+ is inhibited.
Cytosolic NADH transients reflect increased neuronal glycolysis upon stimulation
The main alternative explanations for the neuronal NADHCYT transients are increased production of NADH by neuronal glycolysis, or decreased recycling of NADH to NAD+ by diminished flux of the mitochondrial NADH shuttles (see Fig 1E). We tested for a contribution of neuronal glycolysis to the NADH transients by inhibiting glycolysis at the GAPDH step by brief treatments with iodoacetic acid (IAA). We supplied 2 mM exogenous lactate as an alternative fuel to glucose, to keep cells and mitochondria reasonably well-fed despite the inhibition of glycolysis. This amount of lactate is consistent with the upper end of the estimated extracellular concentration in brain (Langemann et al., 2001), and did not raise neuronal cytosolic NADH:NAD+ so high that it obscured the normal metabolic transients seen with stimulation.
Even with lactate present, rundown of slices occurred fairly quickly after glycolysis was inhibited (as evaluated from higher baseline Ca2+ reported by RCaMP1h, and from neuronal swelling). We therefore shifted to a cooler temperature (~27–28°C) to perform the IAA treatment, which allowed better control and better slice health when we shifted back to the elevated temperature (34°C). We used antidromic stimulation of DGNs to avoid complications from changes in synaptic Ca2+ buffering.
Addition of 2 mM lactate produced a modest increase in baseline NADHCYT, as expected, but NADHCYT transients were still readily observed (Figure 4A). The shift to cooler temperatures produced a reversible increase in fluorescence LT of both sensors, as expected (Hung et al., 2011). IAA treatment diminished the baseline (consistent with a contribution of glycolysis to the resting NADHCYT level), and upon return to the experimental temperature, the stimulation-induced NADHCYT transient was much smaller, despite a slightly larger Ca2+ transient in response to stimulation. Resting Ca2+ did not rise in most experiments (Suppl. Fig. S4), consistent with maintenance of cellular integrity.
Figure 4. NADHCYT transients most likely arise from neuronal glycolysis.

(A) Treatment with the GAPDH inhibitor iodoacetic acid (IAA) reduces the NADHCYT transients in response to antidromic stimulation. Representative trace of NADHCYT transients elicited by antidromic stimulation (same conditions as Fig. 2), before and after IAA treatment. The bars indicate the times of application of both lactate (2 mM) and IAA (0.5 mM). IAA inhibits glycolysis irreversibly; the temperature was lowered (shaded zone) before and during IAA application to minimize damage to the slice’s health. (Temperature affects sensor lifetime directly, which is why there are large changes in the sensors’ response during the temperature changes.) After this treatment, the cells were stimulated again, leading to a much smaller NADHCYT transient than before IAA treatment. (B) IAA decreased the ability of neurons to produce NADH in response to excitation, as observed in the reduction of the Δ(Peredox LT)/Δ(RCaMP LT) ratio (neurons=18, slices=5, mice=5). This ratio normalizes the NADHCYT change to the size of the Ca2+ transients, as they are correlated (Figs. 1, 2) and the Ca2+ transients are somewhat affected by IAA treatment. Figure S4 shows additional controls and analysis.
These results suggest that the neuronal NADHCYT transient may be produced by a transient increase in the rate of neuronal glycolysis. We sought further evidence for this idea by monitoring the effect of stimulation on the intracellular glucose concentration in dentate granule neurons, using a genetically-encoded glucose sensor with a fluorescence lifetime change (based on the Sweetie sensor; Keller et al., 2014, Suppl. Fig. S5). We observed small transient drops (dips) in neuronal intracellular [glucose] upon synaptic stimulation that had a very similar time course to the NADHCYT transients (Fig. 5A). We hypothesized that these dips were due to increased glucose utilization with an approximately constant (but rapid) rate of glucose transport, rather than due to reduced glucose transport. When we deliberately reduced glucose transport rate chronically, by partial inhibition with cytochalasin B, the baseline [glucose]i was predictably lower, but the stimulation-induced dips in [glucose]i actually became larger (Fig 5A, B), consistent with our hypothesis. This result is opposite from what would be expected if the dips had been caused by a dip in the extracellular glucose concentration, secondary to increased astrocyte glucose utilization; in such a case, partial blockade of glucose transport should have diminished the size of the dips.
Figure 5. Stimulation induces transient dips of neuronal intracellular glucose, consistent with increased neuronal glycolysis.

(A) Representative trace of glucose dips elicited by synaptic stimulation of 60 pulses at 50 Hz. Glucose transporter inhibition by 0.2 μM Cytochalasin B (10 min) increased the size of the glucose dips. Figure S5 shows the properties of the SweetieTS sensor. (B) The structural analog Cytochalasin D (0.2 μM, 10 min) does not affect the glucose dips. This experiment serves as a negative control for the possible effects of CytoB on the cytoskeleton. The effect sizes for CytoB (neurons=22, slices=4, mice=4) and CytoD (neurons=17, slices=4, mice=3) were compared using an unpaired Student’s t-test. (C) Representative recording of glucose dips in the continuous presence of 0.2 μM CytoB (to enhance the glucose dips), before and after the application of MCT inhibitor (2 μM). (D) The acute application of 2 μM MCT inhibitor (5 min) reduced the glucose dips. Comparisons were done using the ratio between two consecutive control pulses and the ratio between the dip in the presence of MCT inhibitor and the previous control pulse (neurons=28, slices=7, mice=4). The nonparametric Wilcoxon signed-rank test was used because at least one of the distributions was not Gaussian.
These dips in glucose concentration also appeared to be tightly coupled to NADH production in the cell. When lactate export is blocked by MCT inhibition, NAD+ recycling should be impaired, and the rate of glycolysis should be slower. Indeed, we found that application of MCT inhibitor to the slice did inhibit the stimulation-induced dips reported by the glucose sensor (Fig 5C, D). Thus there is bidirectional coupling between glucose and NADHCYT: the NADHCYT transients were diminished when glycolysis was inhibited, and glucose dips were diminished when NAD+ recycling was inhibited (by blocking lactate export). This argues that the two effects are mechanistically connected, and thus it argues against the idea that the glucose dips result from a reported activity-induced decrease in glucose transport (Porras et al., 2004). Such a decrease in glucose transport would presumably lead to a transient decrease in NADHCYT (rather than the observed increase), and it would also impair the ability of a neuron to respond to metabolic challenge.
Transients in cytosolic NADH do not simply reflect mitochondrial NAD(P)H transients
Many studies of neurons and brain slices have used changes in “NAD(P)H” autofluorescence to monitor metabolic changes (Chance et al., 1962; Duchen, 1992; Schuchmann et al., 2001; Shuttleworth, 2010); these small fluorescence changes are generally observed using widefield fluorescence microscopy and are thought to reflect mainly changes in mitochondrial NADH, which constitutes the bulk of cellular NADH. (The cofactor NADPH, which exhibits identical fluorescence properties to NADH, also contributes to the autofluorescence, as indicated by the designation NAD(P)H.) After neuronal stimulation, a rapid (tens of seconds) “dip” in the autofluorescence is followed by an “overshoot” that lasts several minutes. The general consensus is that the dip represents mitochondrial NADH recycling to restore the mitochondrial membrane potential (due to challenge from Ca2+ uptake and ATP synthesis), and the overshoot represents stimulation of NADH production by Krebs cycle enzymes that are activated by increased matrix calcium (Shuttleworth, 2010).
The NADHCYT transients reported by Peredox are in principle compartmentally separated from the changes in mitochondrial NADH, and NADH does not pass through the mitochondrial membranes. The NADH:NAD+ ratio is substantially higher in mitochondria than in cytosol, but uptake of reducing equivalents from cytosol to mitochondria is produced by the “malate-aspartate shuttle” (MAS), a system that involves multiple enzymes and transporters and whose activity is coupled to the proton electrochemical gradient across the mitochondrial membrane. This normally drives the uphill transport of reducing equivalents from cytosol to mitochondria. Nevertheless, a slowdown or even reversal of the MAS could contribute to the NADHCYT transients that we observed.
We tested the role of the MAS by blocking the key enzyme aspartate aminotransferase with a low-affinity inhibitor, aminooxyacetate (AOA). Application of AOA (10 mM) to hippocampal slices produced a prompt, large increase in NADHCYT (Fig. 6A) as expected from inhibition of a major contributor to cytosolic NADH recycling. The large increase in baseline NADHCYT would prevent us from observing transients, and also starves mitochondria of pyruvate by driving cytosolic pyruvate to be converted to lactate. We reversed both of these problems by adding exogenous pyruvate (1.5 mM), which restores baseline NADHCYT to near-normal levels (Fig. 6A, B).
Figure 6. NADHCYT transients occur independently of the mitochondrial malate-aspartate shuttle.

(A) Inhibition of the malate-aspartate shuttle (MAS) by aminooxyacetate (AOA) increases the cytosolic NADH/NAD+ ratio. The right panel shows representative traces of Peredox lifetime in two neurons, which increases after the application of 10 mM AOA in the ACSF. In the control cell, Peredox LT remains high in the continuous presence of 10 mM AOA, whereas the exposure of the second cell to 1.5 mM pyruvate (Pyr) decreased the Peredox LT to baseline levels, even in the presence of the shuttle inhibitor. The right panel summarizes the effect of AOA and pyruvate (as well as their combination) on Peredox lifetime (neurons=40, slices=5, mice=5). Values are significantly different from each other with a p<0.01, except for the conditions 1.5Pyr and 1.5Pyr+10 AOA. (B) Representative traces of activity-induced changes in Peredox lifetime of a neuron before and after AOA application, both obtained in the presence of 1.5 mM pyruvate in the ACSF. Antidromic stimulation was delivered in the presence of picrotoxin (100 μM) in addition to the ionotropic glutamate receptor blockers. This demonstrates that neuronal activation still produces a rise in the cytosolic NADH/NAD+ ratio, in spite of MAS inhibition. (C) The size of the NADHCYT transient is marginally affected by MAS blockade. Left. The repetitive exposure to pyruvate increases the NADHCYT transients around 11% in slices that were not treated with AOA (neurons=33, slices=3, mice=3). By contrast, the transients obtained in the simultaneous presence of AOA and pyruvate are ~8% smaller than the initial transients in the presence of pyruvate alone (neurons=40, slices=5, mice=5; Wilcoxon matched-pairs signed rank test, *p<0.05, **p<0.01). Right. For comparisons, we took into account the hysteresis effect by dividing the Peredox LT/RCaMP LT ratio at the final condition (second exposure to pyruvate in controls or AOA plus pyruvate in treated neurons) by the initial pulse in the presence of pyruvate alone. NADHCYT transients in the presence of AOA plus pyruvate, are approximately 19% smaller than expected (Mann-Whitney test, **p<0.01).
We then compared stimulation-induced NADHCYT transients in the presence of 1.5 mM pyruvate alone with those in 1.5 mM pyruvate and 10 mM AOA combined (Fig. 6B, C). There was only a small reduction (~10%) in the transients in the presence of AOA, relative to the slight increase in parallel controls with pyruvate (Fig. 6C, Suppl. Fig. S6). This small decrease could be due to a small role for the MAS in producing the cytosolic NADHCYT transients, or alternatively to an effect of AOA on the alanine transaminase that converts cytosolic pyruvate to alanine (blocking the transaminase could cause more accumulation of pyruvate, which would decrease the transients). In either case, the contribution of the MAS to the NADHCYT transients appears to be minor.
Similar neuronal cytosolic NADH transients occur in vivo with normal sensory stimulation
Stimulation-induced transients in NADHCYT were not unique to the in vitro hippocampal slice preparation, but were also observed in a more natural in vivo situation, in the brains of awake, unanesthetized mice. The Peredox (nuclear localized) and RCaMP1h sensors were expressed in layer 2/3 neurons of the whisker-sensing barrel region of primary somatosensory cortex (area S1-BF), and a cranial window was surgically implanted to permit imaging of the sensors (Holtmaat et al., 2009). A subset of sensor-expressing neurons – typically 1–3 neurons per mouse – responded to a 10-second whisker stimulation with a rapid transient of intracellular [Ca2+], as indicated by the transient increase in RCaMP1h lifetime (Figure 7A). Practically all of these responsive neurons exhibited an approximately 4–5 minute long elevation in NADHCYT (Figure 7A, B), while neighboring non-responsive neurons showed no significant change. As for the in vitro case, the size of the NADHCYT transients was well-correlated with the Ca2+ response even for small responses (Figure 7C). Thus, similar NADHCYT transients can occur in vivo in response to the neuronal activity produced by naturalistic sensory stimulation.
Figure 7. NADHCYT transients in vivowith sensory stimulation.

(A) Representative pseudo-colored lifetime image of neurons expressing RCaMP1h (left panels) and nuclear-localized Peredox (right panels) in layers II/III of primary somatosensory cortex of an awake mouse. Scale bar = 10 μm. The images were acquired 0.33 minutes before (top panels), during (middle panels), and 1 minute after (bottom panels) whisker stimulation. Stimulation resulted in a brief increase in RCaMP lifetime in the responsive neuron, reflecting an augmented Ca2+ concentration (middle left panel). This was followed approximately 1 minute later by an increase in Peredox lifetime for the responsive neuron, reflecting changes in the cytosolic NADH/NAD+ ratio (NADHCYT) toward a more reduced state (bottom right panel). The color scale bar to the right shows the range of lifetime values for both RCaMP1h and Peredox sensors. (B) Average transient increases, within neuronal regions of interest, in cytosolic NADH/NAD+ ratio (top panel) and Ca2+ concentration (bottom panel) following whisker stimulation (red arrow) consisting of 10 seconds of mechanical stimulation at 5 Hz. Traces represent the mean ± SEM; n=19 neurons, N= 9 mice (3 males, 6 females). (C) Elevation in the NADH/NAD+ ratio is directly correlated with the increase in intracellular Ca2+ following whisker stimulation. (D) Comparison of lactate-induced Δ(Peredox LT) (50mM lactate) before and after MCT inhibition (50 μM AR-C155858) in vivo using a paired Student’s t-test. n= 59 neurons, N= 6 mice (3 females, 3 males). (E) Representative experiment on the effect of MCT inhibition on neuronal NADHCYT transients elicited by whisker stimulation. In vivo application of MCT inhibitor AR-C155858 (50μM, 20 minutes) did not reduce the endogenous NADH transients in response to whisker stimulation (10 sec of mechanical stimulation at 5 Hz). (F) The scatter plot of Δ(Peredox LT)/Δ(RCaMP LT) shows that in vivo exposure to MCT inhibitor does not reduce the size of the NADH/NAD+ transients and the slope of the relationship. The ratio of stimulation-induced Δ(Peredox LT)/Δ(RCaMP LT) in the absence of MCT inhibitor was 0.13 with a 95%CI: 0.10–0.16. In the presence of MCT inhibitor, the ratio of stimulation-induced Δ(Peredox LT)/Δ(RCaMP LT) was 0.15 with a 95%CI: 0.11–0.20. n=4 neurons, N=3 mice (2 females, 1 male). The dashed line shows the prediction of the ANLS that the stimulation-induced transients should be reduced comparably to the lactate-induced transients in D (based on adjusting the slope for the linear fit of the control measurements).
In order to test the pharmacological sensitivity of the in vivo stimulation-induced NADHCYT transients, we devised a “permeable window” method to allow application of metabolites and drugs. Two weeks after normal placement of a glass coverslip cranial window, the glass was replaced with a transparent, porous filter (details in the Methods section). Although the dura is surgically removed when the permeable window is placed, solution changes above the filter do require somewhat higher concentrations and longer times to reach the imaged cells than for a superfused brain slice.
Exogenous lactate (applied at 50 mM rather than the 10 mM used in slice) reliably produced increases in neuronal NADHCYT observed by Peredox signals in layer 2/3 neurons (Fig. 7D, left). Application of the MCT inhibitor AR-C155858 (at 50 μM rather than the 2 μM used for slice) effectively reduced the size of the lactate-induced increase (by 85.3% ± 6.3%, mean ± SEM, n=59 neurons in 6 animals; Fig. 7D). Nevertheless, neuronal NADHCYT transients induced by whisker stimulation were not diminished by the MCT inhibitor (Fig. 7E, F). Because only a minority of imaged neurons responded to whisker stimulation with RCaMP-monitored calcium transients, and the calcium transients are quite variable in size, we compared the ratio of NADHCYT to calcium signals (ΔPeredox-LT/ΔRCaMP-LT) before and after MCT inhibitor application, and found that these were not significantly different; all transients were well above the level suggested by the degree of MCT inhibition, as judged from the responses to exogenous lactate (Fig. 7D, F). The failure of MCT inhibitor to abolish the stimulation-induced transients in vivo supports the conclusion that such metabolic transients are endogenous to the neurons and do not rely on lactate import from the extracellular space or from astrocytes.
DISCUSSION
The NADH biosensor reports metabolic transients specifically in neuronal cytosol
Using the Peredox biosensor, we observe reliable transient increases in fluorescence lifetime when neurons are stimulated. The Peredox biosensor mainly reports the ratio of free NADH to free NAD+, although there is some effect of the absolute concentration of NADH as well (Hung et al., 2011). The biosensor is expressed in cytosol and not targeted to mitochondria; it therefore reports cytosolic NADH: NAD+ ratio. In dentate granule neurons in a hippocampal slice, the typical increase in this ratio is substantial - approximately a 2-fold increase above the resting level. In neurons of barrel cortex observed in vivo, the typical increase is smaller (about a 33% increase over the resting level), but still substantial.
These NADHCYT signals are distinct from the autofluorescence signal referred to as NAD(P)H, which arises from the total fluorescence of the two functionally distinct species NADH and NADPH. The NAD(P)H autofluorescence is almost certainly dominated by mitochondrial NADH (Brennan et al., 2006; Shuttleworth, 2010; Berndt et al., 2015). It shows a rapid decline (dip) over the initial ~10 seconds, which is thought to arise from a rapid depolarization of the mitochondrial membrane potential (due to Ca2+ influx and to ATP synthase activity), which then allows the electron transport chain to oxidize mitochondrial NADH using molecular O2. The dip in NAD(P)H autofluorescence is then followed by an overshoot above its initial value. While some have attributed this overshoot to glycolytic NADH production in the cytosol of astrocytes (Kasischke et al., 2004), it is almost certainly due instead to replenishment of mitochondrial NADH in neurons, by multiple means (Brennan et al., 2006; Shuttleworth, 2010; Berndt et al., 2015): stimulation of TCA cycle dehydrogenases by calcium as well as increased shuttling of NADH from the cytosol (which may itself be secondary to production of NADH by cytosolic glycolysis; Ivanov et al., 2014). The maximal changes in NAD(P)H autofluorescence are typically much smaller (5–10%) than the changes we observe here using Peredox, employing comparable stimuli. In addition, the NAD(P)H transients have a signal-to-noise ratio that is too small to permit unambiguous assignment of the signal to specific cells or cell types.
The Peredox signals observed here are instead direct reports of neuronal cytosolic NADH:NAD+ ratio (NADHCYT), which reflects changes in glycolysis and lactate utilization. Our experiments were designed to determine which of these factors actually contribute to the metabolic transients provoked by stimulation.
Astrocyte-to-neuron lactate shuttling may occur at rest, but not during neuronal stimulation
The astrocyte-neuron lactate shuttle (ANLS) hypothesis holds that lactate exported by astrocytes becomes an important fuel for neurons, particularly during stimulation. It postulates that the temporal separation between glycolysis (which temporarily outstrips oxidative metabolism and results in net lactate production during excitation) is also a spatial separation: astrocytes are responsible for the glycolytic production of lactate, which is then consumed by neurons. The ANLS hypothesis continues to generate controversy in the literature (Hertz, 2004; Chuquet et al., 2010; Patel et al., 2014; Lundgaard et al., 2015). Although there are strong arguments against the logic of the ANLS on its face (Dienel, 2017), there has been little conclusive evidence at the cellular level.
There is indeed evidence that astrocytes are “more glycolytic” than neurons, both from experiments on cultured cells and from measurements in brain tissue. Cultured astrocytes produce lactate, and cultured neurons can consume it (Pellerin et al., 1998); and astrocytes have higher levels of the regulatory PFK2 enzyme that can stimulate glycolysis (Herrero-Mendez et al., 2009). In anesthetized mouse brain, astrocytes have been inferred to have higher resting levels of lactate than neurons, suggestive of a net flux from astrocytes to neurons (Mächler et al., 2016). In resting hippocampal slices, the cytosolic NADH: NAD+ ratio is higher in astrocytes than neurons (Mongeon et al., 2016). These observations under resting conditions are compatible with higher lactate production by astrocytes and a redox gradient that would favor lactate transfer to neurons (see also Cerdán et al., 2006).
Our results support the possibility of an ANLS that functions at rest but not during neuronal stimulation. Blockade of MCTs, which interrupts the movement of lactate into and out of neurons, does produce a decline in the resting level of neuronal NADHCYT, consistent with an ANLS that functions at rest. But during stimulation, MCT blockade did not reduce the large transient increases in neuronal NADHCYT, despite its clear ability to suppress neuronal responses to direct elevation of extracellular lactate both in acute brain slices and in vivo (Fig. 3A–C, Fig. 7D–F). Similarly, blockade of LDH eliminates the ability of exogenous lactate to influence neuronal NADHCYT, but clearly augments the stimulation-induced NADHCYT transients (Fig. 3D–F). These experimental results are incompatible with the hypothesis of lactate shuttling from astrocytes to neurons during stimulation. Because our experiments were all done with optical signals from the cell bodies of neurons, we cannot exclude the possibility of ANLS activity at synaptic terminals, but recent results on metabolism at synaptic terminals of cultured rat neurons, and in C. elegans, support the idea that they too rely on direct neuronal metabolism of glucose (Ashrafi et al., 2017; Jang et al., 2016).
Coordinated dips in neuronal [glucose] and increases in NADHCYT point to increased glycolysis after stimulation
Given that neuronal metabolic responses to stimulation do not rely on lactate import, it seems very likely that neurons turn instead to increased glucose metabolism for their increased energy needs. Consistent with this, the intracellular glucose concentration is seen to dip after stimulation, during the same period that NADHCYT is elevated (Figure 5). We attribute this dip to an increased rate of neuronal glucose consumption that exceeds any increase in glucose uptake. The dip in glucose concentration is not attributable to a simple dip in extracellular [glucose], which could be produced, for instance, by increased glucose uptake by astrocytes: such a change would be blunted by a glucose uptake inhibitor (cytochalasin B), whereas the observed dip actually increases with partial inhibition of glucose uptake.
The conclusion that the NADHCYT transients are produced by a transient increase in glycolysis is also supported by the substantial inhibition of the transients by IAA treatment, which inhibits glycolysis (Figure 4). These experiments were done in the presence of 2 mM lactate in the bath, comparable to measured extracellular lactate in the brain (Langemann et al., 2001), to provide continued support of mitochondrial oxidation, which is clearly important for sustained neuronal health. These experiments also demonstrate that the NADHCYT transients are not mainly a consequence of mitochondrial responses: either reduced mitochondrial shuttle activity (which would elevate NADHCYT by decreased recycling of NADH to NAD+) or increased pyruvate consumption (which would elevate NADHCYT by redistribution of the LDH equilibrium to produce more NADH). In the presence of extracellular lactate, both of these processes would have continued normally, yet the transients became much smaller when glycolysis was inhibited. While we cannot exclude some contribution of the mitochondrial NADH shuttles to producing the NADHCYT transients, it appears that glycolysis plays a dominant role.
Neuronal use of glucose is not simply an adaptation to MCT inhibition that allows the NADHCYT transients to persist when lactate uptake is abolished. If neuronal glycolysis occurs mainly when neurons are deprived of lactate import, the glucose dips should become larger when MCTs are inhibited, but this is the opposite of the experimental result. Instead, glucose dips are somewhat smaller when MCTs are inhibited, while the NADHCYT transients are larger.
Transient aerobic glycolysis may provide a rapid first response to energy resupply, followed by the higher capacity response by oxidative phosphorylation
The large stimulation-induced increase in NADHCYT that we observe using the Peredox sensor indicates that the production of NADH from glycolysis exceeds the rate of NADH shuttling to mitochondria. Moreover, the rise in NADH is almost certainly accompanied by an increase in the lactate/pyruvate ratio, due to the rapid action of lactate dehydrogenase that serves to recycle NAD+ for use in further glycolysis. This likely rapid depletion of pyruvate suggests that there is at least some mismatch between the rate of pyruvate production by glycolysis and the rate of pyruvate oxidation by mitochondria - in other words, that there is at least some net “aerobic glycolysis” of glucose to lactate in the neurons. This picture is bolstered by our observation that NADHCYT actually rises more when the movement of lactate across the plasma membrane (via MCTs) is blocked -- suggesting that conversion of pyruvate to lactate and export of lactate are normally required to recycle NAD+ and permit a continued high rate of glycolysis. This also accounts for the diminished glucose dip when MCTs are blocked.
We hypothesize that this transient of neuronal glycolysis acts as a metabolic “first responder” to meet the increased energy needs of the stimulated neuron. The ability to perform glycolysis – to recycle NAD+ by producing and exporting lactate – allows uncoupling between the rates of cytosolic glucose metabolism and mitochondrial metabolism. This “elasticity” between the two rates may allow for faster resupply of ATP. Although mitochondrial metabolism has much higher capacity than glycolysis to produce ATP, it involves many more steps, including transport of ADP and phosphate into the matrix and transport of ATP back to the cytosol. These additional steps may limit the ability to respond to a sudden increase in energy demand.
Measurements of oxygen levels in brain slices indicate that oxygen consumption does increase promptly in response to stimulation (Hall et al., 2012; Ivanov et al., 2014), suggesting a coordinated response of both glycolysis and oxidative phosphorylation to meet increased demand. The physiological coordination of both glycolysis and oxidative metabolism in response to neuronal activity may be choreographed by calcium influx, which, as shown in Figs. 1D and 7C, is tightly correlated to the cytosolic NADH transients. However, it remains to be learned whether this correlation is produced by a direct modulation of glycolytic rate by cytosolic calcium (e.g. Singh et al., 2004; Kim et al., 2016; Nicholson-Fish et al., 2016; Hu et al., 2016), by indirect effects due to Ca2+ uptake by mitochondria (Duchen, 1992; Satrústegui and Bak, 2015), or simply by the ATP burden produced by the demands of Ca2+ extrusion.
Despite the fact that both glycolysis and oxidative metabolism are elevated after stimulation, there is ample evidence that immediately after brain activation, oxygen consumption does not rise commensurate with the increases in blood flow and glucose consumption. In fact, this mismatch between blood flow and oxygen consumption is the source of the BOLD signal that is commonly used to detect brain activation in functional imaging experiments (Kwong et al., 1992): blood oxygenation level is somewhat paradoxically increased in activated regions, despite the increased metabolic demand. PET studies also show that blood flow and glucose consumption are dramatically increased by brain activation, but oxygen consumption barely increases (Fox and Raichle, 1986; Fox et al., 1988). These observations support the idea that increased glucose consumption is not accompanied by an equivalent increase in mitochondrial pyruvate oxidation, and that instead glycolysis – the net conversion of glucose to lactate –becomes a substantial component of the transient response to activation. Our results argue that neurons are themselves a site of this increased glycolysis, and that during activation they are likely to be exporters of glycolytic lactate rather than consumers of lactate from astrocytes. The ultimate fate of the exported lactate may be a combination of “catch-up” oxidation by many neighboring brain cells (and by the stimulated neurons themselves) as well as some net lactate excretion from the brain (Dienel, 2012; Glenn et al., 2015), particularly when activation is strong and synchronous, as in epilepsy (Collins et al., 1970; During et al., 1994)
We do not yet know the relative contributions of glycolysis and oxidative phosphorylation to the immediate energy demands of the neuron. However, it is known that in the evolution of skeletal muscle, the muscle fibers with the most dynamic energy requirements -- the fast-twitch fibers -- tend to rely on glycolysis for prompt energy production (Crow and Kushmerick, 1982; Spriet, 1989). Like muscle, the brain has no doubt evolved to meet the large instant-to-instant changes in energy demands with a choreographed regulation of the core metabolic pathways, to ensure not only brain cell health but organismal survival.
STAR★Methods
Contact for Reagent and Resource Sharing
Dr. Gary Yellen (gary_yellen@hms.harvard.edu) is the Lead Contact for reagent and resource sharing. All published reagents will be shared on an unrestricted basis; reagent requests should be directed to the lead author.
Experimental Models and Subject Details
Mice
Wild-type C57BL/6NCrl mice of both genders were bred in house and used in all acute brain slice and in vivo experiments, and mice were randomly allocated to experimental groups. For in vivo imaging experiments, mice were imaged at 10–13 weeks of age.
Animals were housed in a barrier facility in individually ventilated cages with ad libitum access to food (Picolab Rodent Diet 5053) and water, with a controlled cage temperature of 24°C and relative humidity of 53%. All experiments were performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals and Animal Welfare Act. Specific protocols were approved by the Harvard Medical Area Standing Committee on Animals.
Hippocampal brain slices from mice
Hippocampal Slice Preparation
Mice between 14 and 24 days-old were anesthetized with isoflurane, decapitated, and the brain was placed in ice-cold slicing solution containing (in mM): 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.25 CaCl2, 7 MgCl2, 75 sucrose and 25 D-glucose (335–340 mOsm/kg). Brains were glued by the dorsal side in a chamber containing the same slicing-solution and horizontal slices were cut at a thickness of 275 μm using a vibrating slicer (7000smz-2, Campden Instruments, Loughborough, England).
Slices were immediately transferred to a chamber filled with artificial cerebrospinal fluid (ACSF) at physiologic temperature (37°C), containing (in mM): 120 NaCl, 2.5 KCl, 1 NaH2PO4, 26 NaHCO3, 2 CaCl2, 1 MgCl2 and 10 D-glucose (~290 mOsm/kg). All solutions were continuously bubbled with a mix of 95% O2 and 5% CO2, which provides adequate oxygenation and stabilizes the pH around 7.4. Slices were incubated at 37°C for 35 min and then at room temperature for at least 30 min before the experiments, which were executed in the next 4 hours after the preparation.
Method Details
Reagents
All reagents were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise specified. The synaptic blockers NBQX and D-AP5 were obtained from Toronto Research Chemicals (Toronto, ON) and Abcam (Cambridge, MA), respectively. The MCT inhibitor AR-C155858 was purchased from Tocris (Bristol, UK); it is highly specific to MCT1 and MCT2 (Ovens et al., 2010a), unlike the commonly used MCT inhibitor α-cyano-4-hydroxycinnamate (4-CIN), which blocks mitochondrial pyruvate uptake with higher affinity than it blocks MCTs (Halestrap and Denton, 1974).
Viral vectors
Custom-made adeno-associated vectors (AAV; obtained either from the Penn Vector Core, University of Pennsylvania, PA or the Viral Core Facility from Children Hospital in Boston, MA) were used for biosensor expression in brain tissue. For the expression of Peredox and the glucose sensor SweetieTS in hippocampus, we used the AAV2/8 serotype and the universal promoter CAG (Mongeon et al., 2016). For expression of the lactate sensor Laconic in hippocampus, we used the AAV2/9 serotype and the CAG promoter. For expression of Peredox in layers 2/3 of the primary somatosensory cortex, we used a nuclear-localized version of the sensor with the AAV2/9 serotype under the control of neuron-specific synapsin promoter. For expression of the Ca2+ sensor RCaMP1h, the AAV2/9 serotype and the synapsin promoter were used.
Biosensor expression in hippocampus
For sensor expression in the hippocampus, mice at postnatal day 1 were anesthetized using cryoanesthesia. Following confirmation of anesthesia, pups were injected intracranially with 300nl of either a 1:1 mixture of AAV2/8.CAG.Peredox and AAV2/9.hSyn.RCaMP1h or a 1:1 mixture of AAV2/8.CAG.SweetieTS and AAV2/9.hSyn.RCaMP1h. Pups received dual hemisphere viral injections at the following coordinates with respect to lambda: 0 μm in the anterior-posterior direction, ±2 μm in the medial-lateral axis, and −2.3 μm in the dorsal-ventral direction.
Although in principle it is possible that expression of the Peredox sensor, an NADH binding protein, could buffer increases in free [NADH], we saw no evidence of this: the average change in sensor LT was uncorrelated with the level of sensor protein expression, judged from fluorescence intensity (Suppl. Fig. S2). Most likely the changes in free [NADH] are already buffered by high levels of cytosolic dehydrogenases that bind NADH (Zhang et al., 2002), and the additional expression of sensor makes only a negligible change in the buffering capacity.
Electrophysiology
The brain slice was attached to a poly-lysine coated coverslip and the recordings were performed in a “single-perfusion” chamber with a continuous supply of oxygenated ACSF. Local field potentials were recorded in the current clamp configuration. Borosilicate pipettes (Warner Instruments, Hamden, CT) were used with tip resistances of ~ 1–3 MΩ and inserted in the dentate gyrus at a depth of 50–100 μm. The pipette solution consisted of (mM): 150 NaCl, 2.5 KCl, 10 HEPES, 2 CaCl2 and 1 MgCl2 (~300 mOsm/l; pH 7.3).
During the experiments, the slices were perfused with ACSF at a flow rate of 5 ml/min and maintained at a near-physiologic temperature of 34°C using inline heaters (Warner Instruments, Hamden, CT). The solutions were preheated (~38°C) by maintaining solution bottles in a waterless bead bath (Cole-Parmer, Vernon Hills, IL) to prevent out-gassing.
Dentate granule neurons were stimulated with a concentric bipolar electrode (FHC, Bowdoin, ME) placed in the molecular layer or the hilus for synaptic and antidromic stimulation respectively. Stimulation was delivered in trains of 20 to 100 brief (100 μsec) pulses at a frequency of 20 or 50 Hz, using an A360 stimulus isolation unit (WPI, Sarasota, FL). Stimulation intensity was maintained at the minimum level required to reliably evoke spike activity, which typically range from 100–500 μA for synaptic, and 250–1500 μA for antidromic stimulation.
Signals were measured with a Multiclamp 700B (Molecular Devices, Sunnyvale, CA). The recordings were filtered at 2.8 kHz and sampled at 10 kHz. Signals were digitized using a PCIe-6353 digitizer (National Instruments, Austin, TX) and acquired using custom-made software running on MATLAB.
Two-photon Fluorescence Lifetime Imaging Microscopy
Lifetime imaging data were acquired with a modified Thorlabs Bergamo II microscope (Thorlabs Imaging Systems, Sterling, VA), with hybrid photodetectors R11322U-40 (Hamamatsu Photonics, Shizuoka, Japan); the light source was a Chameleon Vision-S tunable Ti-Sapphire mode-locked laser (80 MHz, ~75 fs; Coherent, Santa Clara, CA). The objective lens used for brain slice imaging was an Olympus LUMPLFLN 60x/W (NA 1.0), and for in vivo imaging, an Olympus XLUMPLFLN 20x/W (NA 1.0). An excitation wavelength of 790 nm was used for the Peredox, SweetieTS, and RCaMP sensors; for the pHRed sensor, excitation was at 850 nm. Fluorescence emission light was split with an FF562-Di03 dichroic mirror and bandpass filtered for green (FF01-525/50) and red (FF01-641/75) channels (all filter optics from Semrock, Rochester, NY). For the Laconic sensor, excitation was at 850 nm, and emission light was split with an FF506-Di03 dichroic mirror and bandpass filtered for CFP (FF01-475/35) and YFP (FF01-542/27) channels. The photodetector signals and laser sync signals were preamplified and then digitized at 1.25 gigasamples per second using a field-programmable gate array board (PC720 with FMC125 and FMC122 modules, 4DSP, Austin, TX).
Laboratory-built firmware and software performed time-correlated single photon counting to determine the arrival time of each photon relative to the laser pulse; the distribution of these arrival times indicates the fluorescence lifetime (Yellen and Mongeon, 2015; Mongeon et al., 2016). Lifetime histograms were fitted using nonlinear least-squares fitting in MATLAB (Mathworks, Natick, MA), with a two-exponential decay convolved with a Gaussian for the impulse response function (Yasuda et al., 2006). Microscope control and image acquisition were performed by a modified version of the ScanImage software written in MATLAB (Pologruto et al., 2003) (provided by B. Sabatini and modified by G.Y.).
Cranial window surgery with AAV injections
Mice (postnatal day ~60) were first administered dexamethasone sodium phosphate (0.03 ml at 4 mg/ml) by an intramuscular injection to the quadriceps 2 hours before surgery. Animals were anesthetized (isoflurane; induction: 2–5%, maintenance: 1.4%), the stereotactic coordinates overlying the primary somatosensory (barrel field) cortex were marked (1.3 mm posterior and 3.5 mm lateral to bregma) and a titanium headplate was attached to the skull using opaque C&B-Metabond (Parkell Inc., Edgewood, NY). A 3 mm round craniotomy with the marked spot at its center was made using a hand-held drill. Following exposure of the brain, the tissue was irrigated with sterile saline and any bleeding stopped using sterile absorbable gelatin sponges. Four injections of a solution containing 1:1 mixture of AAV2/9.hSyn.Peredox-NLS and AAV2/9.hSyn.RCaMP1h (~100 nl per injection, ~7 min per injection) were made near the center of the craniotomy ~200 μm apart from each other at a depth of ~200 μm below the dural surface using a beveled glass micropipette attached to a UMP3-1 UltraMicroPump (World Precision Instruments, Sarasota, FL). The surface of the exposed tissue was then covered with a thin layer of Kwik-Sil (to reduce animal movement-induced brain motion that could interfere with imaging; World Precision Instruments, Sarasota, FL) and a glass plug (consisted of a 3 mm diameter coverslip glued to a 5 mm diameter coverslip (both from Warner Instruments, Hamden, CT) using UV-curable optical adhesive (NOA 68; Norland Products, Cranbury, NJ)). The glass plug was then cemented to the skull using opaque C&B-Metabond. After the window implantation surgery, the mice typically woke up after ~10 min and were walking around the cage within ~15 min. Following 5 days of post-operative recovery, mice were habituated to head-post restraint over a 5-day period. Imaging experiments began approximately 10 days post-surgery to allow proper recovery and also steady-state expression of sensors.
Permeable window placement surgery
Two weeks post-cranial window surgery and head-restraint habituation, some mice underwent a second surgical procedure to replace the glass window with a custom-made permeable window for drug delivery during in vivo imaging. Animals were anesthetized (isoflurane; induction: 2–5%, maintenance: 1.4%), and a hand-held drill was used to remove the dental cement holding the previously implanted glass window in place. Following exposure of the brain, the tissue was irrigated with ice-cold sterile sucrose-based saline solution (in mM: 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.25 CaCl2, 7 MgCl2, 75 sucrose and 25 D-glucose (335–340 mOsm/kg)) and any bleeding stopped using sterile absorbable gelatin sponges. Once bleeding stopped, a durotomy was carefully performed using a pair of fine-tipped bent tweezers (500234; World Precision Instruments, Sarasota, FL); the tissue was covered in ice-cold sucrose-based saline solution throughout the durotomy. The exposed brain was covered with a thin layer of 2% agarose dissolved in ACSF (in mM: 120 NaCl, 2.5 KCl, 1 NaH2PO4, 26 NaHCO3, 2 CaCl2, 1 MgCl2) and a custom-made permeable window. The permeable window consists of a 4mm square of a bilayer polymeric microporous filter with 8μm pores and 25μm-thick grid structural support (Precision Membranes, Provo, UT) super-glued to a round polyimide plastic washer (91435A160; McMaster Carr, Princeton, NJ) and cut to size. The border of the permeable window was then attached to the skull using super glue gel (LOC1255800; WB Mason, Boston, MA) and opaque C&B Metabond. The membrane was kept moist at all times with sucrose-based saline solution. An imaging well consisting of a size 014 Buna-N O-Ring (7855-412; Ace Glass, Vineland, NJ) was cemented to the previously implanted titanium headplate using C&B Metabond. The imaging well was then filled with ACSF, and the imaging area was sealed with a custom-3D printed cap and a thin layer of Kwik-Sil. Mice were allowed to recover from surgery for 1–2 hours prior to imaging session.
In vivo awake imaging
In vivo imaging experiments were conducted with awake, unanesthetized mice, as it has been previously demonstrated that anesthesia increases the apparent oxygen concentration of intact brain (Lyons et al., 2016). Imaging experiments began approximately 10 days post-surgery to allow proper recovery and also steady-state expression of sensors. During imaging, mice were placed on a 6-inch foam ball that could spin noiselessly on ball bearings. Their head was restrained using the previously implanted headplate. Prior to imaging, mice were habituated to head restraint and the foam ball over a 5-day period. Sessions of head restraint increased in duration over this period from 5 minutes to 1 hour.
Imaging sessions were 1–2 hours in duration. Viral expression of Peredox and RCaMP1h permitted recording from different neurons in the same mice on different days (Andermann et al., 2010; O’Connor et al., 2010). When imaging from the same cortical region on multiple days, previously imaged neurons were re-identified using the pattern of the surface vasculature as guidance, and then an adjacent volume was selected to ensure that all neurons in the sample were unique.
Neuronal responses in the primary somatosensory area (barrel cortex) were obtained by delivering a 10-second 5 Hz mechanical stimulation using a custom-built foam pad to brush past the whiskers of the mouse. The foam-pad was attached to a servo-motor and controlled by an Arduino microcontroller (both from Sparkfun, Niwot, CO). This stimulation was presented 3 times in a given imaging session with a 20 minute interval between stimulations.
Drug delivery during in vivo awake imaging
At the start of the imaging session, the custom-made cap covering the permeable window was removed and the solution covering the membrane was replaced with ACSF at physiologic temperature (37°C), containing (in mM): 120 NaCl, 2.5 KCl, 1 NaH2PO4, 26 NaHCO3, 2 CaCl2, 1 MgCl2. During imaging, solutions were exchanged by removing the previous solution manually while washing in the new solution into the imaging well. The solution exchange was repeated 3x prior to continuation of imaging. At the end of the experiment, the imaging well was washed with fresh ACSF and the custom-made cap was replaced to protect the permeable window.
Quantification and Statistical Analyses
Image analysis was performed using MATLAB software developed in our laboratory. Regions of interest (ROIs) were defined around individual cells, and photon statistics were calculated for all pixels within the ROI. Typical ROIs encompassed 100–900 image pixels, in images of 128×128 or 256×256 pixels acquired at a scanning rate of ~2 ms per line. Most data points in the time series plots of lifetimes are for the mean value of 20 sequentially acquired frames, except that for RCaMP1h (calcium) data in the 10 seconds after stimulation, the data points represent individual frames.
Bleedthrough of green Peredox fluorescence into the red RCaMP optical channel was corrected using the ratio of measured red to green fluorescence observed when only Peredox was expressed (Suppl. Fig. S2), approximately 6.0%. Likewise, green glucose sensor fluorescence produced a bleedthrough of ~5.9% to the red channel. Similarly, RCaMP1h expression alone led to some signal in the green optical channel, probably due to an immature fluorophore, in direct experiments approximately 4.4% of the red fluorescence intensity, and having a lifetime of ~0.71 ns; these values were used to correct the baseline in dual expression experiments. Even with this correction, the effect of some green signal from RCaMP1h still appears to produce some shift of the baseline lifetime values for Peredox, compared to those seen in the absence of RCaMP1h (Mongeon et al., 2016). For this reason, we mostly measured the net lifetime change for the transients, relative to baseline (ΔLT). To estimate the actual NADH:NAD+ ratios, we use the baseline values determined previously in the absence of RCaMP and add the transient ΔLT to calculate the change according to the previous calibration.
Working data were tested for normality by a Kolmogorov-Smirnov test. For data fulfilling normality, a paired or unpaired Student’s t-test was used as appropriate. Equivalent non-parametric tests (Mann-Whitney or Wilcoxon tests) were used if one of the groups did not comply with normality. Correlations were performed using the non-parametric Spearman r method. The statistical analysis was done using the software GraphPad Prism version 7 (San Diego, CA). Data are reported as mean ± SEM (unless indicated otherwise) and p < 0.05 was considered statistically significant. Graphics were constructed using Origin 2015 (OriginLab, Northampton, MA). The statistical parameters can be found in the figures and figure legends; in general, SEM’s were calculated using the number of neurons studied.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bacterial and Virus Strains | ||
| AAV9.Syn.RCaMP1h.WPRE.SV40 | Penn Vector Core | Discontinued |
| AAV9.Syn.jRCaMP1b-NES.WPRE.SV40 | Penn Vector Core | Cat#AV-9-PV3852 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| NBQX (6-Nitro-7-sulfamoylbenzo[f]quinoxaline-2,3-dione, Disodium Salt) | Toronto Research Chemicals | Cat#N550005; CAS:479347-86-9 |
| D-AP5 (D-(-)-2-Amino-5-phosphonopentanoic acid) | Abcam | Cat#ab120003; CAS:79055-68-8 |
| AR-C155858 | Tocris Bioscience | Cat#4960; CAS:496791-37-8 |
| Picrotoxin | Sigma-Aldrich | Cat#P1675; CAS:124-87-8 |
| Cytochalasin B | Tocris Bioscience | Cat#5474; CAS:14930-96-2 |
| Cytochalasin D | Tocris Bioscience | Cat#1233; CAS:22144-77-0 |
| GSK-2837808A | Tocris Bioscience | Cat#5189; CAS:1445879-21-9 |
| Aminooxyacetate (O-(carboxymethyl)hydroxylamine hemihydrate) | Sigma-Aldrich | Cat#C13408; CAS:2921-14-4 |
| Sodium L-lactate | Sigma-Aldrich | Cat#71718; CAS:867-56-1 |
| Sodium pyruvate | Sigma-Aldrich | Cat#P8574; CAS:113-24-6 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| C57BL/6NCrl mice | Charles River | RRID:IMSR_CRL:27 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| AAV.CAG.Peredox.WPRE.SV40 | Mongeon et al., 2016 | Addgene #73807 |
| AAV.hSyn.Peredox-NLS.WPRE.SV40 | This paper | Derived from Addgene #32384 |
| AAV.CAG.SweetieTS.WPRE.SV40 | This paper | |
| AAV.CAG.Laconic.WPRE.SV40 | San Martín et al., 2013 | Derived from Addgene #44238 |
| AAV.CAG.pHRed.WPRE.SV40 | Tantama et al., 2011 | Derived from Addgene #31472 |
| Software and Algorithms | ||
| GraphPad Prism Version 7 | GraphPad | |
| Origin 2015 | OriginLab | |
| ScanImage | Pologruto et al., 2003 | |
| Other | ||
| Polymeric microporous filters with 8μm pores and 25 μm thick grid structural support | Precision Membranes, Provo, UT | N/A |
Supplementary Material
Figure S1. Relative abundance of cytosolic NADH-specific dehydrogenases in neurons. Related to Figure 1.
The graph shows the most highly abundant cytosolic NADH-specific dehydrogenases in mouse neurons, from unbiased proteomic analysis of cultured neurons (black bars, left axis; Sharma et al. 2015) and from unbiased transcriptomic analysis (single-cell RNA-seq) on CA1 pyramidal neurons (grey bars, right axis; Zeisel et al. 2015). The x-axis shows the gene names.
By far the four dominant dehydrogenases are GAPDH, LDHA/LDHB, and MDH1, corresponding to the three main NADHCYT-controlling processes in Figure 1E. The second mitochondrial NADH shuttle, the glycerol phosphate shuttle, uses the GPD1 dehydrogenase, expressed a much lower levels than MDH1.
Figure S2. Bleedthrough correction and buffering effects of sensor expression. Related to Figure 1.
(A) Bleedthrough correction was performed in DGNs exclusively expressing one sensor. The spectral contamination of photons emitted by the green-fluorescent proteins SweetieTS and Peredox into the red channel was calculated as the slope from the linear fit of the relationship between the photon count in the red channel (left Y-axis) versus the photon count in the green channel (X-axis). For the fitting, at least ten points within the 80th–100th percentiles of photon count in the green channel were used because of a better signal to noise ratio in this range. Bleedthrough from RCaMP fluorescence to the green channel was analyzed in a similar way, although it must be taken into account that it does not reflect the spectral contamination of photons emitted by the red proteins, but the emission of immature RCaMP proteins that emit in the green spectrum. The values of bleedthrough correction for each sensor (slope) appear embedded in the graphs.
(B) Peredox expression does not buffer NADH or NAD+ concentrations. No correlation was observed between the photon count of Peredox fluorescence (used as a proxy for Peredox expression in DGNs) and the Peredox lifetimes at baseline. Likewise, the magnitude of the change in Peredox lifetime in response to antidromic stimulation was not correlated with Peredox expression in DGNs (events=132, neurons=49, slices=9, mice=6).
Figure S3. Effects of ionotropic glutamate receptor blockers on synaptically and antidromically stimulated DGNs. Related to Figure 2.
(A) Representative traces of excitatory local field potentials (LFP) recorded in dentate granule neurons after synaptic stimulation. LFPs recorded in regular ACSF (Control) were abolished by the application of ionotropic glutamate receptor blockers (5 μM NBQX + 25 μM D-AP5). LFPs were recovered after the washout of synaptic blockers from the ACSF. The trace shows a couple of LFPs at the beginning and the end in a typical stimulation train. The stimulation artifacts are blanked for better visualization of the signals.
(B) Representative trace of NADHCYT and Ca2+ transients elicited by antidromic stimulation, before and after washout (10 min) of synaptic blockers in the ACSF.
(C) As expected, the removal of synaptic blockers from the ACSF does not affect the changes in RCaMP LT induced by antidromic stimulation. Data were compared by a paired Student’s t-test (neurons=22, slices=4, mice=2) and differences between both conditions were not significant (n.s.).
Figure S4. Further analysis of IAA and temperature shifts. Related to Figure 4.
(A) Peredox lifetime at baseline increases with the addition of 2 mM lactate (2Lac), as expected from the conversion of lactate to pyruvate mediated by the enzyme LDH. IAA treatment reduces the Peredox LT at levels even lower than the initial values without lactate supplementation, indicating the occurrence of neuronal glycolysis at baseline. Data were compared by a paired Student’s t-test (neurons=18, slices=5, mice=5).
(B) Absolute Peredox lifetime changes (ΔPeredox LT) upon stimulation are slightly but significantly enhanced by lactate supplementation in the ACSF, but IAA treatment drastically reduces the metabolic NADHCYT responses, indicating that glycolysis is a major contributor for these transients.
(C) The intracellular Ca2+ concentration at rest is not affected by our protocol for IAA treatment. This means that the slice health was not compromised at baseline.
(D) Ca2+ transients are enhanced after IAA treatment, suggesting that the cells are more excitable or less effective in extruding (or buffering) this ion upon stimulation.
(E) Representative trace of NADHCYT transients obtained under the same conditions as in Fig 4, but without IAA treatment. The bar indicates the time of application of lactate (2 mM) and the temperature was lowered (shaded zone) to mimic the protocol for IAA application. (Temperature affects sensor lifetime directly, which is why there are large changes in the sensors’ response during the temperature changes.) After returning to near-physiologic temperature, Ca2+ spikes and NADHCYT transients remained unaltered, indicating that the reduction in NADHCYT transients observed after IAA treatment is attributable to GAPDH inhibition and not because of a collateral effect of manipulating the temperature of the slices.
(F) Addition of 2 mM lactate slightly increases the magnitude of neuronal NADHCYT transients with respect to the extent of neuronal excitation, measured as the change in RCaMP LT. Moreover, the proportional NADHCYT response to Ca2+ transients (ΔPeredox LT/ΔRCaMP LT) remained unchanged after transiently cooling the slices. Data were compared by a paired Student’s t-test (neurons=19, slices=4, mice=4).
(G) Peredox lifetime at baseline is not affected by changes in temperature during the protocol used in Fig 4.
(H) Absolute Peredox lifetime changes (ΔPeredox LT) remain the same throughout the experiments.
(I) The intracellular Ca2+ concentration at rest is not affected by the experimental paradigm. This means that the slice health was not compromised at baseline because of temperature changes.
(J) Ca2+ responses to stimulation also remained unaltered, indicating no changes in neuronal excitability or Ca2+ handling after the transient change in temperature.
Figure S5. Properties of the SweetieTS glucose sensor, and direction of pH responses with stimulation. Related to Figure 5.
(A) Calibration of the purified glucose sensor SweetieTS at near-physiologic temperature (34°C). Data represent the average of three independent experiments. The curve resulted from averaging individual fittings of each experimental dataset to a Hill equation, yielding a Km= 3.4 ± 0.7 mM for glucose and a Hill coefficient of n=1.28 ± 0.03 (mean ± SD).
(B) pH dependence of SweetieTS. pH dose response curves were constructed at three different glucose concentrations (0.5mM, 1mM, 3mM) for two independent purified protein samples at 34°C. For the 3 glucose concentrations tested, the lifetime of SweetieTS increased with acidification – neuronal stimulation produces a small acidification (see C below), and any acidification-induced change in SweetieTS lifetime is in the opposite direction from the observed dips in sensor lifetime (interpreted as dips in [glucose]). Measurements were normalized to SweetieTS lifetime at pH7.4 for each glucose concentration. Data are mean ± SEM.
(C) DGNs show a small acidification with stimulation. The pH sensor pHRed (Tantama et al., 2011) was expressed in DGNs under the universal promoter CAG using an AAV2/8 vector. Neuronal activity was detected using the Ca2+ sensor jRCaMP1b (Dana et al., 2016), which was co-expressed by infecting DGNs with AAV2/9 viruses using the neuron-specific promoter synapsin. As both sensors emit in the red spectrum, cross-contamination between their signals was minimized by targeting pHRed and jRCaMP1b to the nucleus and cytoplasm respectively. Signals from both sensors can be also distinguished by their baseline lifetime values, which in our experimental conditions were pHRed LT= 2.14 ± 0.10 ns and jRCaMP1b= 1.45 ± 0.25 ns (mean ± SD from events=23, neurons=10, slices=5, mice=3). The average pH response (mean ± SEM) to synaptic stimulation (60 pulses, 100 μs of duration and 400–500 μA of intensity, delivered at 20Hz) shows a small transient intracellular acidification, as calculated using the calibration curve previously reported for pHRed lifetimes (Tantama et al., 2011). Sensors were imaged using a 2p excitation wavelength of 850 nm.
(D) Monocarboxylate inhibition (MCT inhib) by 2 μM AR-C155858 does not affect the magnitude of the synaptically- stimulated acidification in the intracellular pH of DGNs. Data for the effect of MCT inhibition were compared by a paired Student’s t-test (neurons=6, slices=4, mice=3).
(E) Amino acid sequence of the SweetieTS sensor. The sequence of the circularly-permuted variant of TSapphire is highlighted.
Figure S6. Inhibition of the mitochondrial malate-aspartate shuttle. Related to Fig. 6.
Representative trace of an experiment for MAS inhibition. After obtaining stable NADHCYT transients, a transient in the presence of 1.5 mM pyruvate was obtained as a control response for the test pulse after MAS inhibition. Washout of pyruvate was confirmed after the recovery of the baseline and also by another stimulation, which exhibited a response with a similar size to the initial transients. AOA was then applied until the effect of MAS blockade was evident as a rise in Peredox lifetime at baseline, which reached a value near saturation after 10 min. At this point, 1.5 mM pyruvate were added to the circulating ACSF+AOA and the baseline decreased and stabilized after 20 min. Antidromic stimulation produces NADHCYT transients in DGNs, still in the presence of AOA plus pyruvate.
The success of this experiment relied heavily on the timing of exposure to the different test conditions. To minimize delays because of perfusion exchange, the test compounds were added directly to the flask containing the ACSF recirculating in the chamber. Stock solutions were freshly prepared in ACSF the day of experiments, at 200X the concentration needed (i.e. 0.3 M for Na-Pyruvate). An aliquot equivalent to 0.5% of the recirculating volume was added to achieve the desired working concentrations. Picrotoxin (100 μM) was added to the ACSF (in addition to the iGluR blockers) to prevent potential effects due to AOA inhibition of GABA transaminase (McKenna et al., 2006).
Acknowledgments
We thank the members of the Yellen lab for helpful discussions and comments on the manuscript, and to Binsen Li, Veronica Burnham, and Nidhi Nathwani for expert technical assistance. Thanks also to Andrew Lutas for technical contributions to the awake mouse setup. We also are very grateful to Bernardo Sabatini for helping us get started with two-photon microscopy, and to Ryohei Yasuda for helping us get started with FLIM. We thank the Viral Core of Boston Children’s Hospital and the U. Penn. Vector Core for packaging of AAVs; the GENIE project of HHMI Janelia Farms and Loren Looger and Douglas Kim for the RCaMP sensors; Loren Looger, Jonny Martin, and Jacob Keller for the original Sweetie sensor; and L. Felipe Barros for the Laconic sensor. This work was supported by NIH grants R01 NS055031 and DP1 EB016985 (to G.Y.); NIH fellowships F32 NS080455 (to R.M.), F32 NS093784 (to C.L.) and F32 NS100331 (to C.M.D-G).; a David Mahoney Fellowship (to R.M.); and a Fix Fund Postdoctoral Fellowship (to C.M.D-G.).
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, G.Y., R.M., C.M.D-G., C.L.; methodology, G.Y., R.M., C.L.; software, G.Y.; formal analysis and investigation, C.M.D-G., R.M., C.L., H.Z., D.K.; writing and visualization, G.Y., C.M.D-G., C.L., R.M., H.Z., D.K.; supervision and project administration, G.Y.; funding acquisition, G.Y., R.M., C.L., C.M.D-G.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akerboom J, Carreras Calderón N, Tian L, Wabnig S, Prigge M, Tolö J, Gordus A, Orger MB, Severi KE, Macklin JJ, et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci. 2013;6:2. doi: 10.3389/fnmol.2013.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann ML, Kerlin AM, Reid RC. Chronic cellular imaging of mouse visual cortex during operant behavior and passive viewing. Front Cell Neurosci. 2010;4:3. doi: 10.3389/fncel.2010.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Wu Z, Farrell RJ, Ryan TA. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron. 2017;93:606–615. e3. doi: 10.1016/j.neuron.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti PJ. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metabolism. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Berndt N, Kann O, Holzhütter HG. Physiology-based kinetic modeling of neuronal energy metabolism unravels the molecular basis of NAD(P)H fluorescence transients. J Cereb Blood Flow Metab. 2015;35:1494–1506. doi: 10.1038/jcbfm.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner CX, Valdebenito R, Ruminot I, Loaiza A, Larenas V, Sotelo-Hitschfeld T, Moldenhauer H, San Martín A, Gutiérrez R, Zambrano M, et al. Fast and reversible stimulation of astrocytic glycolysis by K+ and a delayed and persistent effect of glutamate. J Neurosci. 2011;31:4709–4713. doi: 10.1523/JNEUROSCI.5311-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AM, Connor JA, Shuttleworth CW. NAD(P)H fluorescence transients after synaptic activity in brain slices: predominant role of mitochondrial function. Journal of Cerebral Blood Flow & Metabolism. 2006;26:1389–1406. doi: 10.1038/sj.jcbfm.9600292. [DOI] [PubMed] [Google Scholar]
- Cerdán S, Rodrigues TB, Sierra A, Benito M, Fonseca LL, Fonseca CP, García-Martín ML. The redox switch/redox coupling hypothesis. Neurochem Int. 2006;48:523–530. doi: 10.1016/j.neuint.2005.12.036. [DOI] [PubMed] [Google Scholar]
- Chance B, Cohen P, Jobsis F, Schoener B. Intracellular oxidation-reduction states in vivo. Science. 1962;137:499–508. doi: 10.1126/science.137.3529.499. [DOI] [PubMed] [Google Scholar]
- Chuquet J, Quilichini P, Nimchinsky EA, Buzsáki G. Predominant enhancement of glucose uptake in astrocytes versus neurons during activation of the somatosensory cortex. J Neurosci. 2010;30:15298–15303. doi: 10.1523/JNEUROSCI.0762-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke DD, Sokoloff L. Circulation and energy metabolism of the brain. In: Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD, editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. Philadelphia: Lippincott, Williams, and Wilkins; 1994. pp. 637–669. [Google Scholar]
- Collins RC, Posner JB, Plum F. Cerebral energy metabolism during electroshock seizures in mice. Am J Physiol. 1970;218:943–950. doi: 10.1152/ajplegacy.1970.218.4.943. [DOI] [PubMed] [Google Scholar]
- Crow MT, Kushmerick MJ. Chemical energetics of slow- and fast-twitch muscles of the mouse. J Gen Physiol. 1982;79:147–166. doi: 10.1085/jgp.79.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D, et al. Sensitive red protein calcium indicators for imaging neural activity. Elife. 2016;5 doi: 10.7554/eLife.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA. Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab. 2012;32:1107–1138. doi: 10.1038/jcbfm.2011.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J Neurosci Res. 2017 doi: 10.1002/jnr.24015. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J. 1992;283(Pt 1):41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During MJ, Fried I, Leone P, Katz A, Spencer DD. Direct measurement of extracellular lactate in the human hippocampus during spontaneous seizures. J Neurochem. 1994;62:2356–2361. doi: 10.1046/j.1471-4159.1994.62062356.x. [DOI] [PubMed] [Google Scholar]
- Fox PT, Raichle ME. Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proceedings of the National Academy of Sciences. 1986;83:1140–1144. doi: 10.1073/pnas.83.4.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PT, Raichle ME, Mintun MA, Dence C. Nonoxidative glucose consumption during focal physiologic neural activity. Science. 1988;241:462–464. doi: 10.1126/science.3260686. [DOI] [PubMed] [Google Scholar]
- Glenn TC, Martin NA, Horning MA, McArthur DL, Hovda DA, Vespa P, Brooks GA. Lactate: brain fuel in human traumatic brain injury: a comparison with normal healthy control subjects. J Neurotrauma. 2015;32:820–832. doi: 10.1089/neu.2014.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Denton RM. Specific inhibition of pyruvate transport in rat liver mitochondria and human erythrocytes by α-cyano-4-hydroxycinnamate (Short Communication) Biochemical Journal. 1974;138:313–316. doi: 10.1042/bj1380313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Klein-Flügge MC, Howarth C, Attwell D. Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J Neurosci. 2012;32:8940–8951. doi: 10.1523/JNEUROSCI.0026-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. 2009;11:747–752. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- Hertz L. The astrocyte-neuron lactate shuttle: a challenge of a challenge. J Cereb Blood Flow Metab. 2004;24:1241–1248. doi: 10.1097/00004647-200411000-00008. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WCA, et al. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 2009;4:1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Juvekar A, Lyssiotis CA, Lien EC, Albeck JG, Oh D, Varma G, Hung YP, Ullas S, Lauring J, et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell. 2016;164:433–446. doi: 10.1016/j.cell.2015.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung YP, Albeck JG, Tantama M, Yellen G. Imaging cytosolic NADH-NAD+ redox state with a genetically encoded fluorescent biosensor. Cell Metab. 2011;14:545–554. doi: 10.1016/j.cmet.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AI, Malkov AE, Waseem T, Mukhtarov M, Buldakova S, Gubkina O, Zilberter M, Zilberter Y. Glycolysis and oxidative phosphorylation in neurons and astrocytes during network activity in hippocampal slices. J Cereb Blood Flow Metab. 2014;34:397–407. doi: 10.1038/jcbfm.2013.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, Nelson JC, Bend EG, Rodríguez-Laureano L, Tueros FG, Cartagenova L, Underwood K, Jorgensen EM, Colón-Ramos DA. Glycolytic enzymes localize to synapses under energy stress to support synaptic function. Neuron. 2016;90:278–291. doi: 10.1016/j.neuron.2016.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- Keller JP, Marvin JS, Schreiter E, Looger LL. Sweetie: An optimized genetically-encoded fluorescent sensor for glucose. Soc for Neurosci Abstr. 2014:658.14/VV17. [Google Scholar]
- Kim N, Lee JO, Lee HJ, Lee YW, Kim HI, Kim SJ, Park SH, Lee CS, Ryoo SW, Hwang GS, et al. AMPK, a metabolic sensor, is involved in isoeugenol-induced glucose uptake in muscle cells. J Endocrinol. 2016;228:105–114. doi: 10.1530/JOE-15-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong KK, Belliveau JW, Chesler DA, Goldberg IE, Weisskoff RM, Poncelet BP, Kennedy DN, Hoppel BE, Cohen MS, Turner R, Cheng HM, Brady TJ, Rosen BR. Dynamic magnetic resonance imaging of human brain activity during primary sensory stimulation. Proc Natl Acad Sci U S A. 1992;89:5675–9. doi: 10.1073/pnas.89.12.5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langemann H, Alessandri B, Mendelowitsch A, Feuerstein T, Landolt H, Gratzl O. Extracellular levels of glucose and lactate measured by quantitative microdialysis in the human brain. Neurol Res. 2001;23:531–536. doi: 10.1179/016164101101198785. [DOI] [PubMed] [Google Scholar]
- Lundgaard I, Li B, Xie L, Kang H, Sanggaard S, Haswell JDR, Sun W, Goldman S, Blekot S, Nielsen M, et al. Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat Commun. 2015;6:6807. doi: 10.1038/ncomms7807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons DG, Parpaleix A, Roche M, Charpak S. Mapping oxygen concentration in the awake mouse brain. Elife. 2016:5. doi: 10.7554/eLife.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mächler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, Kaelin V, Zuend M, San Martín A, Romero-Gómez I, et al. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 2016;23:94–102. doi: 10.1016/j.cmet.2015.10.010. [DOI] [PubMed] [Google Scholar]
- Madsen PL, Cruz NF, Sokoloff L, Dienel GA. Cerebral oxygen/glucose ratio is low during sensory stimulation and rises above normal during recovery: excess glucose consumption during stimulation is not accounted for by lactate efflux from or accumulation in brain tissue. J Cereb Blood Flow Metab. 1999;19:393–400. doi: 10.1097/00004647-199904000-00005. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L. Astrocytes couple synaptic activity to glucose utilization in the brain. Physiology. 1999;14:177–182. doi: 10.1152/physiologyonline.1999.14.5.177. [DOI] [PubMed] [Google Scholar]
- McKenna MC, Waagepetersen HS, Schousboe A, Sonnewald U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: Current evidence and pharmacological tools. Biochemical Pharmacology. 2006;71:399–407. doi: 10.1016/j.bcp.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Mongeon R, Venkatachalam V, Yellen G. Cytosolic NADH-NAD+ redox visualized in brain slices by two-photon fluorescence lifetime biosensor imaging. Antioxid Redox Signal. 2016;25:553–563. doi: 10.1089/ars.2015.6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson-Fish JC, Cousin MA, Smillie KJ. Phosphatidylinositol 3-kinase couples localised calcium influx to activation of Akt in central nerve terminals. Neurochem Res. 2016;41:534–543. doi: 10.1007/s11064-015-1663-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor DH, Peron SP, Huber D, Svoboda K. Neural activity in barrel cortex underlying vibrissa-based object localization in mice. Neuron. 2010;67:1048–1061. doi: 10.1016/j.neuron.2010.08.026. [DOI] [PubMed] [Google Scholar]
- Ovens MJ, Davies AJ, Wilson MC, Murray CM, Halestrap AP. AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7–10. Biochem J. 2010a;425:523–530. doi: 10.1042/BJ20091515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovens MJ, Manoharan C, Wilson MC, Murray CM, Halestrap AP. The inhibition of monocarboxylate transporter 2 (MCT2) by AR-C155858 is modulated by the associated ancillary protein. Biochem J. 2010b;431:217–225. doi: 10.1042/BJ20100890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, Lai JCK, Chowdhury GMI, Hyder F, Rothman DL, Shulman RG, Behar KL. Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc Natl Acad Sci USA. 2014;111:5385–5390. doi: 10.1073/pnas.1403576111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Pellegri G, Bittar PG, Charnay Y, Bouras C, Martin JL, Stella N, Magistretti PJ. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev Neurosci. 1998;20:291–299. doi: 10.1159/000017324. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55:1251–1262. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- Peng L, Zhang X, Hertz L. High extracellular potassium concentrations stimulate oxidative metabolism in a glutamatergic neuronal culture and glycolysis in cultured astrocytes but have no stimulatory effect in a GABAergic neuronal culture. Brain Res. 1994;663:168–172. doi: 10.1016/0006-8993(94)90475-8. [DOI] [PubMed] [Google Scholar]
- Pologruto TA, Sabatini BL, Svoboda K. ScanImage: Flexible software for operating laser scanning microscopes. BioMedical Engineering OnLine. 2003;2:13. doi: 10.1186/1475-925X-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras OH, Loaiza A, Barros LF. Glutamate mediates acute glucose transport inhibition in hippocampal neurons. J Neurosci. 2004;24:9669–9673. doi: 10.1523/JNEUROSCI.1882-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salway JG. Metabolism at a glance. Wiley-Blackwell; 2004. [Google Scholar]
- San Martín A, Ceballo S, Ruminot I, Lerchundi R, Frommer WB, Barros LF. A genetically encoded FRET lactate sensor and its use to detect the Warburg effect in single cancer cells. PLoS ONE. 2013;8:e57712. doi: 10.1371/journal.pone.0057712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satrústegui J, Bak LK. fluctuations in cytosolic calcium regulate the neuronal malate aspartate NADH shuttle: Implications for neuronal energy metabolism. Neurochemical Research. 2015;40:2425–2430. doi: 10.1007/s11064-015-1652-8. [DOI] [PubMed] [Google Scholar]
- Scharfman HE. The CA3 “backprojection” to the dentate gyrus. Prog Brain Res. 2007;163:627–637. doi: 10.1016/S0079-6123(07)63034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuchmann S, Kovacs R, Kann O, Heinemann U, Buchheim K. Monitoring NAD(P)H autofluorescence to assess mitochondrial metabolic functions in rat hippocampal-entorhinal cortex slices. Brain Res Brain Res Protoc. 2001;7:267–276. doi: 10.1016/s1385-299x(01)00080-0. [DOI] [PubMed] [Google Scholar]
- Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, et al. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015;18:1819–1831. doi: 10.1038/nn.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuttleworth CW. Use of NAD(P)H and flavoprotein autofluorescence transients to probe neuron and astrocyte responses to synaptic activation. Neurochemistry International. 2010;56:379–386. doi: 10.1016/j.neuint.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Salih M, Leddy JJ, Tuana BS. The muscle-specific calmodulin-dependent protein kinase assembles with the glycolytic enzyme complex at the sarcoplasmic reticulum and modulates the activity of glyceraldehyde-3-phosphate dehydrogenase in a Ca2+/calmodulin-dependent manner. J Biol Chem. 2004;279:35176–35182. doi: 10.1074/jbc.M402282200. [DOI] [PubMed] [Google Scholar]
- Spriet LL. ATP utilization and provision in fast-twitch skeletal muscle during tetanic contractions. Am J Physiol. 1989;257:E595–605. doi: 10.1152/ajpendo.1989.257.4.E595. [DOI] [PubMed] [Google Scholar]
- Tantama M, Hung YP, Yellen G. Imaging intracellular pH in live cells with a genetically encoded red fluorescent protein sensor. J Am Chem Soc. 2011;133:10034–10037. doi: 10.1021/ja202902d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J. 1967;103:514–527. doi: 10.1042/bj1030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R, Harvey CD, Zhong H, Sobczyk A, van Aelst L, Svoboda K. Supersensitive Ras activation in dendrites and spines revealed by two-photon fluorescence lifetime imaging. Nat Neurosci. 2006;9:283–291. doi: 10.1038/nn1635. [DOI] [PubMed] [Google Scholar]
- Yellen G, Mongeon R. Quantitative two-photon imaging of fluorescent biosensors. Curr Opin Chem Biol. 2015;27:24–30. doi: 10.1016/j.cbpa.2015.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L, Betsholtz C, et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science. 2002;295:1895–1897. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Relative abundance of cytosolic NADH-specific dehydrogenases in neurons. Related to Figure 1.
The graph shows the most highly abundant cytosolic NADH-specific dehydrogenases in mouse neurons, from unbiased proteomic analysis of cultured neurons (black bars, left axis; Sharma et al. 2015) and from unbiased transcriptomic analysis (single-cell RNA-seq) on CA1 pyramidal neurons (grey bars, right axis; Zeisel et al. 2015). The x-axis shows the gene names.
By far the four dominant dehydrogenases are GAPDH, LDHA/LDHB, and MDH1, corresponding to the three main NADHCYT-controlling processes in Figure 1E. The second mitochondrial NADH shuttle, the glycerol phosphate shuttle, uses the GPD1 dehydrogenase, expressed a much lower levels than MDH1.
Figure S2. Bleedthrough correction and buffering effects of sensor expression. Related to Figure 1.
(A) Bleedthrough correction was performed in DGNs exclusively expressing one sensor. The spectral contamination of photons emitted by the green-fluorescent proteins SweetieTS and Peredox into the red channel was calculated as the slope from the linear fit of the relationship between the photon count in the red channel (left Y-axis) versus the photon count in the green channel (X-axis). For the fitting, at least ten points within the 80th–100th percentiles of photon count in the green channel were used because of a better signal to noise ratio in this range. Bleedthrough from RCaMP fluorescence to the green channel was analyzed in a similar way, although it must be taken into account that it does not reflect the spectral contamination of photons emitted by the red proteins, but the emission of immature RCaMP proteins that emit in the green spectrum. The values of bleedthrough correction for each sensor (slope) appear embedded in the graphs.
(B) Peredox expression does not buffer NADH or NAD+ concentrations. No correlation was observed between the photon count of Peredox fluorescence (used as a proxy for Peredox expression in DGNs) and the Peredox lifetimes at baseline. Likewise, the magnitude of the change in Peredox lifetime in response to antidromic stimulation was not correlated with Peredox expression in DGNs (events=132, neurons=49, slices=9, mice=6).
Figure S3. Effects of ionotropic glutamate receptor blockers on synaptically and antidromically stimulated DGNs. Related to Figure 2.
(A) Representative traces of excitatory local field potentials (LFP) recorded in dentate granule neurons after synaptic stimulation. LFPs recorded in regular ACSF (Control) were abolished by the application of ionotropic glutamate receptor blockers (5 μM NBQX + 25 μM D-AP5). LFPs were recovered after the washout of synaptic blockers from the ACSF. The trace shows a couple of LFPs at the beginning and the end in a typical stimulation train. The stimulation artifacts are blanked for better visualization of the signals.
(B) Representative trace of NADHCYT and Ca2+ transients elicited by antidromic stimulation, before and after washout (10 min) of synaptic blockers in the ACSF.
(C) As expected, the removal of synaptic blockers from the ACSF does not affect the changes in RCaMP LT induced by antidromic stimulation. Data were compared by a paired Student’s t-test (neurons=22, slices=4, mice=2) and differences between both conditions were not significant (n.s.).
Figure S4. Further analysis of IAA and temperature shifts. Related to Figure 4.
(A) Peredox lifetime at baseline increases with the addition of 2 mM lactate (2Lac), as expected from the conversion of lactate to pyruvate mediated by the enzyme LDH. IAA treatment reduces the Peredox LT at levels even lower than the initial values without lactate supplementation, indicating the occurrence of neuronal glycolysis at baseline. Data were compared by a paired Student’s t-test (neurons=18, slices=5, mice=5).
(B) Absolute Peredox lifetime changes (ΔPeredox LT) upon stimulation are slightly but significantly enhanced by lactate supplementation in the ACSF, but IAA treatment drastically reduces the metabolic NADHCYT responses, indicating that glycolysis is a major contributor for these transients.
(C) The intracellular Ca2+ concentration at rest is not affected by our protocol for IAA treatment. This means that the slice health was not compromised at baseline.
(D) Ca2+ transients are enhanced after IAA treatment, suggesting that the cells are more excitable or less effective in extruding (or buffering) this ion upon stimulation.
(E) Representative trace of NADHCYT transients obtained under the same conditions as in Fig 4, but without IAA treatment. The bar indicates the time of application of lactate (2 mM) and the temperature was lowered (shaded zone) to mimic the protocol for IAA application. (Temperature affects sensor lifetime directly, which is why there are large changes in the sensors’ response during the temperature changes.) After returning to near-physiologic temperature, Ca2+ spikes and NADHCYT transients remained unaltered, indicating that the reduction in NADHCYT transients observed after IAA treatment is attributable to GAPDH inhibition and not because of a collateral effect of manipulating the temperature of the slices.
(F) Addition of 2 mM lactate slightly increases the magnitude of neuronal NADHCYT transients with respect to the extent of neuronal excitation, measured as the change in RCaMP LT. Moreover, the proportional NADHCYT response to Ca2+ transients (ΔPeredox LT/ΔRCaMP LT) remained unchanged after transiently cooling the slices. Data were compared by a paired Student’s t-test (neurons=19, slices=4, mice=4).
(G) Peredox lifetime at baseline is not affected by changes in temperature during the protocol used in Fig 4.
(H) Absolute Peredox lifetime changes (ΔPeredox LT) remain the same throughout the experiments.
(I) The intracellular Ca2+ concentration at rest is not affected by the experimental paradigm. This means that the slice health was not compromised at baseline because of temperature changes.
(J) Ca2+ responses to stimulation also remained unaltered, indicating no changes in neuronal excitability or Ca2+ handling after the transient change in temperature.
Figure S5. Properties of the SweetieTS glucose sensor, and direction of pH responses with stimulation. Related to Figure 5.
(A) Calibration of the purified glucose sensor SweetieTS at near-physiologic temperature (34°C). Data represent the average of three independent experiments. The curve resulted from averaging individual fittings of each experimental dataset to a Hill equation, yielding a Km= 3.4 ± 0.7 mM for glucose and a Hill coefficient of n=1.28 ± 0.03 (mean ± SD).
(B) pH dependence of SweetieTS. pH dose response curves were constructed at three different glucose concentrations (0.5mM, 1mM, 3mM) for two independent purified protein samples at 34°C. For the 3 glucose concentrations tested, the lifetime of SweetieTS increased with acidification – neuronal stimulation produces a small acidification (see C below), and any acidification-induced change in SweetieTS lifetime is in the opposite direction from the observed dips in sensor lifetime (interpreted as dips in [glucose]). Measurements were normalized to SweetieTS lifetime at pH7.4 for each glucose concentration. Data are mean ± SEM.
(C) DGNs show a small acidification with stimulation. The pH sensor pHRed (Tantama et al., 2011) was expressed in DGNs under the universal promoter CAG using an AAV2/8 vector. Neuronal activity was detected using the Ca2+ sensor jRCaMP1b (Dana et al., 2016), which was co-expressed by infecting DGNs with AAV2/9 viruses using the neuron-specific promoter synapsin. As both sensors emit in the red spectrum, cross-contamination between their signals was minimized by targeting pHRed and jRCaMP1b to the nucleus and cytoplasm respectively. Signals from both sensors can be also distinguished by their baseline lifetime values, which in our experimental conditions were pHRed LT= 2.14 ± 0.10 ns and jRCaMP1b= 1.45 ± 0.25 ns (mean ± SD from events=23, neurons=10, slices=5, mice=3). The average pH response (mean ± SEM) to synaptic stimulation (60 pulses, 100 μs of duration and 400–500 μA of intensity, delivered at 20Hz) shows a small transient intracellular acidification, as calculated using the calibration curve previously reported for pHRed lifetimes (Tantama et al., 2011). Sensors were imaged using a 2p excitation wavelength of 850 nm.
(D) Monocarboxylate inhibition (MCT inhib) by 2 μM AR-C155858 does not affect the magnitude of the synaptically- stimulated acidification in the intracellular pH of DGNs. Data for the effect of MCT inhibition were compared by a paired Student’s t-test (neurons=6, slices=4, mice=3).
(E) Amino acid sequence of the SweetieTS sensor. The sequence of the circularly-permuted variant of TSapphire is highlighted.
Figure S6. Inhibition of the mitochondrial malate-aspartate shuttle. Related to Fig. 6.
Representative trace of an experiment for MAS inhibition. After obtaining stable NADHCYT transients, a transient in the presence of 1.5 mM pyruvate was obtained as a control response for the test pulse after MAS inhibition. Washout of pyruvate was confirmed after the recovery of the baseline and also by another stimulation, which exhibited a response with a similar size to the initial transients. AOA was then applied until the effect of MAS blockade was evident as a rise in Peredox lifetime at baseline, which reached a value near saturation after 10 min. At this point, 1.5 mM pyruvate were added to the circulating ACSF+AOA and the baseline decreased and stabilized after 20 min. Antidromic stimulation produces NADHCYT transients in DGNs, still in the presence of AOA plus pyruvate.
The success of this experiment relied heavily on the timing of exposure to the different test conditions. To minimize delays because of perfusion exchange, the test compounds were added directly to the flask containing the ACSF recirculating in the chamber. Stock solutions were freshly prepared in ACSF the day of experiments, at 200X the concentration needed (i.e. 0.3 M for Na-Pyruvate). An aliquot equivalent to 0.5% of the recirculating volume was added to achieve the desired working concentrations. Picrotoxin (100 μM) was added to the ACSF (in addition to the iGluR blockers) to prevent potential effects due to AOA inhibition of GABA transaminase (McKenna et al., 2006).