

Abstract
To identify genetic factors contributing to amyotrophic lateral sclerosis (ALS), we conducted whole-exome analyses of 1,022 index familial ALS (FALS) cases and 7,315 controls. In a new screening strategy, we performed gene-burden analyses trained with established ALS genes and identified a significant association between loss-of-function (LOF) NEK1 variants and FALS risk. Independently, autozygosity mapping for an isolated community in the Netherlands identified a NEK1 p.Arg261His variant as a candidate risk factor. Replication analyses of sporadic ALS (SALS) cases and independent control cohorts confirmed significant disease association for both p.Arg261His (10,589 samples analyzed) and NEK1 LOF variants (3,362 samples analyzed). In total, we observed NEK1 risk variants in nearly 3% of ALS cases. NEK1 has been linked to several cellular functions, including cilia formation, DNA-damage response, microtubule stability, neuronal morphology and axonal polarity. Our results provide new and important insights into ALS etiopathogenesis and genetic etiology.
In recent years, the combination of exome sequencing, segregation analysis and bioinformatic filtering has proven to be an effective strategy to rapidly identify new disease genes1. Unfortunately, this method can be difficult to apply to disorders such as ALS, for which late age of onset and low-to-modest variant penetrance make it difficult to obtain large informative multigenerational pedigrees. Owing to high genetic heterogeneity, ALS is also difficult to analyze using filtering methods designed to exploit unrelated patient groups2. Recently, we had demonstrated the utility of exome-wide rare variant burden (RVB) analysis as an alternate approach, identifying a replicable association between FALS risk and TUBA4A in a cohort of 363 cases3. In brief, RVB analysis is used to compare the combined frequency of rare variants in each gene in a case–control cohort. Candidate associations are identified by significant differences after multiple-test correction. Since this initial study, we extended our data set to include complete exome sequencing for 1,376 index FALS cases and 13,883 controls. Of these, 1,022 cases and 7,315 controls met all required data, inter-relatedness and ancestral quality control criteria (Supplementary Figs. 1 and 2, and Online Methods).
Successful detection of disease associations through RVB analysis can depend heavily on the appropriate setting of test parameters. As genetic loci often contain many alleles of no or low effect, prior filtering of variants based on minor allele frequency (MAF) and pathogenicity predictors can identify disease signatures otherwise masked by normal human variability. As appropriate MAF or pathogenicity predictor settings may not be obvious in advance, comprehensive assessment of all pursuable analysis strategies is desirable but can in turn introduce excessive multiple-test burden. To overcome these limitations, we performed 308 distinct RVB analyses of ten well-established ALS genes using 44 functional and 7 MAF filters (Fig. 1a). All tests included correction for gene coverage and ancestral covariates (Online Methods). In the final cohort, 72 cases and 0 controls harbored known ALS pathogenic mutations in these ten genes (Online Methods). An additional 26 cases harbored a repeat expansion in the C9orf72 gene. Tests differed in their capacity to detect individual known ALS genes (Supplementary Table 1), but we achieved the highest net sensitivity when we restricted analyses to variants with MAF < 0.001 and functional classifications of either nonsense, splice-altering4 or deemed deleterious by functional analysis through hidden Markov models (FATHMM)5. Under these settings, four genes exhibited disease association at exome-wide (Bonferroni-corrected P < 2.5 × 10−6) significance (SOD1, TARDBP, UBQLN2 and FUS), three achieved near exome-wide significance (TUBA4A, TBK1 and VCP), and three displayed modest to marginal disease association (PFN1, VAPB and OPTN) (Fig. 1b). Genes exhibiting the strongest disease associations included those reported as major ALS genes in population-based studies, whereas those exhibiting weaker associations are believed to constitute rarer causes of disease.
Figure 1.
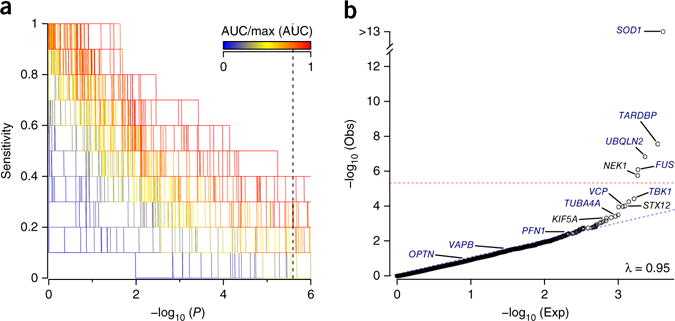
RVB analysis of FALS exomes. (a) RVB analyses of 1,022 index FALS cases and 7,315 controls for 10 known ALS genes, to assess 308 different combinations of MAF and functional prediction filters (Supplementary Table 1). The set of analysis parameters achieving the highest sensitivity for known ALS genes was identified as that achieving the highest area under the curve (AUC) in a plot of sensitivity (proportion of training genes achieving significance) across an increasing minimum P-value threshold. Dotted vertical line denotes Bonferroni-corrected P value for exome-wide significance. (b) Extension of the highest performing known gene-trained analysis to the entire exome. Threshold for exome-wide significance is denoted by the dotted red line. λ, observed genomic inflation factor. ‘Obs’ describes the P-value distribution for the observed data. ‘Exp’ describes the P-value distribution under null expectation.
Extension of the optimal known ALS gene parameters to all protein-coding genes identified one new gene displaying exome-wide significant disease association (Fig. 1b). The gene, NEK1 (odds ratio (OR) = 8.2, P = 1.7 × 10−6), encodes the serine/threonine kinase NIMA (never in mitosis gene-A)-related kinase. Retesting NEK1 under alternate analysis parameters identified strong disease associations across most analysis strategies, particularly where we included LOF (nonsense and predicted splice-altering) variants (Supplementary Table 2 and Supplementary Fig. 3). We observed no evidence for systematic genomic inflation (λ = 0.95), confounding related to sample ascertainment (Supplementary Fig. 4) or case–control biases in NEK1 gene coverage (Supplementary Fig. 5). Removal of samples carrying rare variants of known ALS genes did not influence the association (OR = 8.9, P = 7.3 × 10−7).
In an independent line of research, we performed whole-genome sequencing for four ALS patients from an isolated community in the Netherlands (population < 25,000). We observed high inbreeding coefficients for each of the four patients, confirming their high degree of relatedness and supporting a restricted genetic lineage (Supplementary Fig. 6). Autozygosity mapping, allowing for genetic heterogeneity, identified four candidate disease variants occurring in detectable runs of homozygosity (ROH) (Supplementary Fig. 7). These variants included a p.Arg261His variant of NEK1. Two of the four SALS cases were homozygous for p.Arg261His and two were heterozygous, raising the possibility that even a single copy of the allele may increase disease risk. Clinical evaluation of the four cases did not find any overt differences in disease phenotype. None of the other three candidate variants exhibited homozygosity in multiple patients or occurred at all in more than two patients. Analysis of the region identified a shared p.Arg261His haplotype spanning 3 Mb in all four samples (Supplementary Table 3).
To validate the risk effects of p.Arg261His, we tested for disease association among 6,172 SALS cases and 4,417 matched controls from eight countries (Supplementary Figs. 8 and 9, and Online Methods). We genotyped this cohort using the Illumina exome chip or by whole-genome sequencing, allowing for checking of any overlap or detectable relatedness to the FALS case–control cohort, which was not present. Meta-analysis of all independent population strata identified a clear minor allele excess in cases with a combined significance of P = 4.8 × 10−5 and OR = 2.4 (Fig. 2). We also observed disease association in the FALS case–control data (OR = 2.7, P = 1.5 × 10−3) and a meta-analysis of FALS, SALS and all controls combined (OR = 2.4, P = 1.2 × 10−7).
Figure 2.
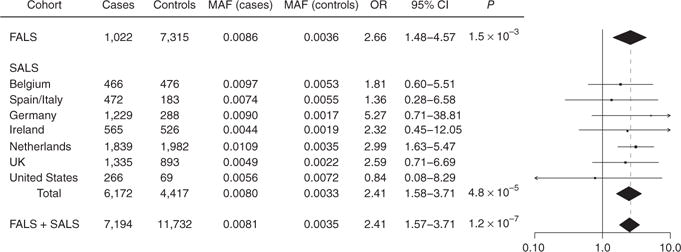
Replication analysis of NEK1 p.Arg261His. NEK1 p.Arg261His genotypes were ascertained for 1,022 FALS samples, 6,172 SALS samples and 11,732 controls. The SALS cohort was divided into seven geographically based case–control strata. Logistic regression was used to conduct tests of allelic association for all subcohorts and was followed by a fixed-effects meta-analysis. In the distribution of OR estimates across study cohorts (right), vertical dotted line denotes OR estimated under meta-analysis. CI, confidence interval.
DNA availability facilitated segregation analysis of only one NEK1 LOF variant, a p.Arg550* variant, which we also detected in the affected mother of the identified proband. To validate the effect of LOF variants observed in FALS and assess any potential contribution to sporadic disease, we analyzed full sequencing data of the NEK1 coding region for 2,303 SALS cases and 1,059 controls (Supplementary Fig. 3 and Online Methods). RVB analysis confirmed a significant excess of LOF variants in cases (23/2,303 SALS samples versus 0/1,059 controls, OR = 22.2, P = 1.5 × 10−4; Supplementary Table 2). Meta-analysis of discovery and replication LOF analyses yielded a combined significance of P = 3.4 × 10−8 and OR = 8.8.
In total, we detected 120 predicted nonsynonymous NEK1 variants in FALS samples, SALS samples and controls. These were distributed throughout the gene including in the sequence encoding protein kinase domain (PKD) and six coiled-coil domains thought to be involved in mediating protein–protein interactions (Supplementary Fig. 3). After conditioning for LOF variants and p.Arg261His, we observed tentative excesses of case variants in analyses of rarer variant categories, but larger sample sizes will be required to confirm the pathogenicity beyond p.Arg261His and LOF variants (Supplementary Table 4). Analysis of other members of the NEK gene family (NEK2–NEK11) identified no associations in the FALS data set meeting multiple-test criteria (Supplementary Table 5).
Although no other gene achieved discovery significance, ten candidate loci exhibited P < 1.0 × 10−3 in the FALS discovery analysis (Table 1). These included the gene encoding the SNARE (soluble NSF attachment protein receptor) complex protein synataxin 12 (STX12, OR = 33.1, P = 9.7 × 10−5). Analysis of the SALS replication cohort identified missense variants in 5/2,303 cases versus 0/1,059 in controls. However, the cohort was not sufficiently powered to assess events of this frequency, and larger sample sizes will be required to establish effects on ALS risk (Supplementary Table 6). Another identified candidate gene was the known hereditary spastic paraplegia gene KIF5A6 (OR = 7.1, P = 4.8 × 10−4); however, no observed elevations in patient variant frequencies within the SALS replication cohort reached statistical significance (Supplementary Table 7).
Table 1.
FALS discovery analysis identifies candidate genes
| Gene | ALS | ALS frequency | Control | Control frequency | OR | OR 95% CI | P |
|---|---|---|---|---|---|---|---|
| NEK1 | 12 | 0.0117 | 14 | 0.0019 | 8.2 | 3.7–18.0 | 1.7 × 10−6 |
| ATRN | 8 | 0.0078 | 7 | 0.0010 | 10.3 | 3.6–29.6 | 3.7 × 10−5 |
| STX12 | 4 | 0.0039 | 1 | 0.0001 | 33.1 | 5.8–339.0 | 9.7 × 10−5 |
| CREB3L2 | 4 | 0.0039 | 0 | 0.0000 | 64.9 | 6.6–8695.3 | 1.1 × 10−4 |
| DCC | 4 | 0.0039 | 2 | 0.0003 | 18.6 | 4.1–108.1 | 3.1 × 10−4 |
| WDR49 | 5 | 0.0049 | 2 | 0.0003 | 15.8 | 3.5–92.1 | 4.4 × 10−4 |
| KIF5A | 7 | 0.0068 | 8 | 0.0011 | 7.1 | 2.5–19.7 | 4.8 × 10−4 |
| C1QTNF7 | 12 | 0.0117 | 26 | 0.0036 | 3.6 | 1.8–7.1 | 6.7 × 10−4 |
| PEAK1 | 5 | 0.0049 | 3 | 0.0004 | 11.6 | 2.9–51.5 | 7.5 × 10−4 |
| BIRC6 | 10 | 0.0098 | 18 | 0.0025 | 4.3 | 1.9–9.3 | 8.4 × 10−4 |
| ZSCAN5B | 4 | 0.0039 | 2 | 0.0003 | 16.3 | 3.3–98.0 | 8.8 × 10−4 |
RVB analysis results for all genes exhibiting case association at P < 1 × 10−3 in FALS discovery cohort.
NEK1 has been previously described as a candidate gene for ALS7,8. Here our findings show that NEK1 in fact constitutes a major ALS-associated gene with risk variants present in ~3% of European and European-American ALS cases. We identified LOF variants in 1.2% of FALS samples (OR = 8.2) and 1.0% of SALS samples (OR = 22.2) versus 0.17% of controls, whereas we identified the p.Arg261His variant in 1.7% of FALS samples (OR = 2.7) and 1.6% of SALS samples (OR = 2.4) versus 0.69% of controls. We identified variants of unknown clinical importance (missense, MAF < 0.001) in a further 1.8% of FALS samples and 1.3% of SALS samples versus 1.2% of controls. In comparison, risk variants in previously established ALS genes occur at approximately the following percentages: C9orf72, <10%; SOD1, <2%; TARDBP, <1%; FUS, <1%; and others, <<1% or uncertain9–12. However, caution must be taken when comparing the frequency of variants or mutations that differ in penetrance (i.e., highly penetrant mutations to lower-penetrance risk variants). Furthermore, assessment of the true odds ratio for variants in a gene may be difficult because of the presence of neutral variants that dilute out the observed effect. The actual odds ratio may therefore be even higher for specific subsets of patient variants. The LOF variants in NEK1 displayed a higher odds ratio relative to p.Arg261His. The p.Arg261His variant occurs adjacent to the protein kinase domain and is classified as deleterious by most bioinformatic prediction algorithms (SIFT, PolyPhen, LRT, MutationTaster, Mutation Assessor, PROVEAN, CADD, GERP and SiPhy). One model to account for the difference in p.Arg261His and LOF variant toxicity could be a correlation between phenotypic expression and the predicted extent of NEK1 LOF. This model would also be consistent with previous findings that homozygosity for NEK1 LOF variants causes a severe developmental phenotype; short rib polydactyly syndrome type II (SRPS)13. In the current study, no individuals carried multiple LOF alleles. However, in SRPS, homozygous carriers of NEK1 LOF variants have been reported to exhibit a 64% reduction of NEK1 mRNA levels whereas unaffected heterozygous parents exhibit a 30–40% reduction13.
NEK1 represents one of 11 members of the highly conserved NIMA kinase family, which has conserved functions in cell-cycle progression and mitosis. In postmitotic cells, NEK1 is a primary regulator of the formation of nonmotile primary cilium14,15. Disruption in the structure or function of primary cilia has been linked to neurological defects such as brain dysgenesis, hydrocephalus and intellectual disability16,17, and abnormalities in cilia number, structure and microtubule state occur in fibroblasts derived from SRPS patients homozygous for NEK1 truncation variants13. In vitro disruption of the activity of other neuronally expressed NEK family members has similarly been shown to disrupt neuronal morphology, neurite outgrowth, microtubule stability and microtubule dynamics18,19. Microtubule integrity and kinesin and dynein intraflagellar transport are essential to maintain cilia structure and function. This is of particular relevance as disruption of the microtubule cytoskeleton has been associated to the development of ALS3, and mutations of the dynein subunit dynactin are associated with motor neuron degeneration20. Additionally, motor neurons derived from mice expressing human SOD1 G93A show a selective loss of cilia both in vitro and in vivo21. Besides its role in ciliogenesis, NEK1 is also known to regulate mitochondrial membrane permeability22 and DNA repair23. Both of these processes have been extensively investigated in relation to ALS, and have been postulated to explain the toxicity of ALS- associated mutations in SOD1 and FUS24,25. Mutations in DNA-repair genes cause several early-onset neurological phenotypes, and multiple lines of evidence suggest defective DNA repair may contribute to both late-onset neurodegeneration and brain aging in general26. For example, oxidative damage and DNA strand breaks have been observed to be elevated in ALS, Alzheimer’s disease and Parkinson’s disease cases27, and a recent large-scale genome-wide association study (GWAS) implicated DNA-repair genes as age-of-onset modifiers in Huntington’s disease28. The pathological importance of DNA damage in ALS, and whether modifier effects observed in Huntington’s disease may generalize to repeat-expansion disorders such as C9orf72-associated ALS, constitute important questions to be addressed. Finally, through its coiled-coil domain, NEK1 has been shown to interact with multiple other proteins of potential importance, including the ALS-associated proteins VAPB and ALS2 (ref. 7) and the axonal outgrowth regulator FEZ1 (ref. 29).
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
FALS discovery cohort
The FALS discovery cohort included 1,376 FALS patients and 13,883 non-ALS controls analyzed by exome sequencing. Patients were recruited at specialist clinics in Ireland (n = 18), Italy (n = 143), Spain (n = 49), the UK (n = 219), the United States (n = 511), the Netherlands (n = 50), Canada (n = 34), Belgium (n = 12), Germany (n = 202), Turkey (n = 47) and Australia (n = 91). Variants occurring at very low frequency in the general population (ExAC MAF <0.0001), which have been both previously reported as ALS-associated and annotated as either ‘pathogenic’ or ‘likely pathogenic’ by ClinVar within the ten genes, were considered to be pathogenic mutations. The breakdown of the 72 mutations observed in the final cohort included the following: SOD1 (28), TARDBP (12), FUS (9), PFN1 (6), TBK1 (1), TUBA4A (4), UBQLN2 (4), VAPB (2) and VCP (6). An additional 26 cases harbored a repeat expansion in the C9orf72 gene. Controls included 29 internal samples and samples obtained from dbGAP30. Sequencing obtained from dbGAP was generated under the following projects: Genetic Epidemiology of chronic obstructive pulmonary disease (COPD) (COPDGene) phs000179; NHLBI Grand Opportunity Exome Sequencing Project (GO-ESP): Lung Cohorts Exome Sequencing Project (cystic fibrosis) phs000254; NHLBI GO-ESP: Women’s Health Initiative Exome Sequencing Project (WHI)-WHISP phs000281; NHLBI GO-ESP: Lung Cohorts Exome Sequencing Project (pulmonary arterial hypertension) phs000290; NHLBI GO-ESP: Lung Cohorts Exome Sequencing Project (Lung Health Study of Chronic Obstructive Pulmonary Disease) phs000291; NHLBI GO-ESP: Lung Cohorts Exome Sequencing Project (COPDGene) phs000296; NHLBI Framingham Heart Study Allelic Spectrum Project phs000307; NHLBI GO-ESP: Family Studies (Thoracic aortic aneurysms leading to acute aortic dissections) phs000347; NHLBI GO-ESP Family Studies: pulmonary arterial hypertension phs000354; NHLBI GO-ESP: Family Studies: (familial atrial fibrillation) phs000362; NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (ARIC) phs000398; NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (CHS) phs000400; NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (FHS) phs000401; NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (JHS) phs000402; NHLBI GO-ESP: Heart Cohorts Exome Sequencing Project (MESA) phs000403; NHLBI GO-ESP: Lung Cohorts Exome Sequencing Project (asthma) phs000422; Jackson Heart Study Allelic Spectrum Project phs000498; NHLBI GO-ESP Family Studies: Idiopathic Bronchiectasis phs000518; Alzheimer’s Disease Sequencing Project (ADSP) phs000572; NHLBI GO-ESP: Family Studies (Hematological Cancers) phs000632; Building on GWAS for NHLBI diseases: the US CHARGE consortium (CHARGE-S): FHS phs000651; Building on GWAS for NHLBI diseases: The US CHARGE Consortium (CHARGE-S): CHS phs000667; Building on GWAS for NHLBI Diseases: the US CHARGE Consortium (CHARGE-S): ARIC phs000668; NIH Exome Sequencing of FALS Project phs000101.v4.p1. Familial history was considered positive for ALS if the proband had at least one affected relative within three generations. We received approval for this study from the institutional review boards of the participating centers, and written informed consent was obtained from all patients (consent for research).
SALS replication cohort
The SALS replication cohort included 2,387 SALS cases and 1,093 controls analyzed by whole-genome sequencing, and 5,834 SALS cases and 4,117 controls analyzed by exome chip. All individuals were recruited at specialist clinics in Ireland, Italy, Spain, the UK, the United States, the Netherlands and Belgium. Details of sample contributions per country are shown in Figure 2. Evaluation of C9orf72 status was performed in 2,387 SALS cases and 166 (7%) displayed a repeat expansion. We received approval for this study from the institutional review boards of the participating centers, and written informed consent was obtained from all patients (consent for research).
Exome sequencing
Exome sequencing of patients was performed as previously described3. Raw sequence data for controls was obtained from dbGaP. Sequence reads were aligned to human reference GRCh37 using Burrows–Wheeler aligner (BWA) and processed according to recommended best practices31. Variant detection and genotyping were performed using the GATK HaplotypeCaller. Variant quality control was performed using the GATK variant quality score recalibration method, with a VQSLOD cutoff of 2.27 (truth set sensitivity of 99%). A minimum variant quality by depth (QD) score of 2 was also imposed and all genotypes associated with genotype quality (GQ) < 20 were reset to missing. Variants were also excluded in the event of case or control call rates < 70% (post genotype QC). Exome sequencing data was not used to infer the presence or absence of indels due to the limited sensitivity and comparatively high false positive rates associated with available calling algorithms32.
Genome sequencing
Whole-genome sequencing of 2,387 SALS samples and 1,093 controls was performed with Illumina’s FastTrack services using PCR free library preparation and paired-end (100 bp or 150 bp) sequencing on the HiSeq 2500 or Hiseq X platform (Illumina) to yield 35× coverage at minimum. BWA was used to align sequencing reads to genome build hg19, and the Isaac variant caller was used to call single-nucleotide variants (SNVs), insertions and deletions (indels)33. Both the aligned and unaligned reads were delivered in binary sequence alignment/map format (BAM) together with variant call format (VCF) files containing the SNVs and indels. gVCF files were generated per individual, and variants that failed the Isaac-based quality filter were excluded.
Exome chip
A total of 5,815 ALS patients and 4,614 healthy controls from the Netherlands, Belgium, Germany, Ireland, Italy, Spain and the UK were included. Genotyping was conducted using Illumina HumanExome-12v1 BeadChips in accordance with the manufacturer’s recommendations. The GenTrain 2.0 clustering algorithm was used for genotype calling, as implemented in the Illumina GenomeStudio software package. Initial genotype calls were made based on the HumanExome clusterfile provided by Illumina. More accurate cluster boundaries were determined based on the actual study data, after the exclusion of samples with a GenCall quality score in the lower 10th percentile of the distribution across all variants genotyped (p10GC) < 0.38 or call rate < 0.99. Subsequently, the excluded samples were added back into the data set, and new genotypes calls were made using the previously obtained cluster boundaries.
Sample filtering
Samples from the FALS discovery and SALS replication cohorts were excluded from analysis in the event of failing to meet genotype call rate, heterozygosity, gender concordance, duplication, relatedness or population stratification filters as summarized in Supplementary Figures 1 and 7. All samples from the FALS cohort were required to exhibit filtered exome-wide call rates > 70%. For both the FALS and SALS cohorts, PLINK (v1.07)34 was used to define an LD-pruned (r2 < 0.5, window size = 50, step = 5) set of autosomal markers with MAF > 0.01 and P > 0.001 for deviation from Hardy–Weinberg equilibrium. These marker sets were then used to calculate inbreeding coefficients for use in heterozygosity filtering, identify study duplicates, conduct relatedness filtering, perform tests of pairwise population concordance for stratification filtering, conduct PCA for a second round of stratification filtering and conduct PCA to generate covariates for stratification correction in RVB analysis and single-variant analysis of filtered cohorts. Samples from the SALS replication cohort were required to exhibit no relatedness/duplication with samples from the FALS discovery cohort. PLINK was used to calculate inbreeding coefficients, test for discordance in reported and SNV predicted gender and conduct tests of pairwise population concordance. Identification of sample duplicates and sample relatedness was performed using KING35. PCA was conducted using genome-wide complex trait analysis (GCTA)36. Details of results from population stratification analysis are provided in Supplementary Figures 2 and 8.
Statistical analyses
RVB analyses were performed by logistic regression of case–control status to number of minor alleles observed per sample per gene3,37. Results from underpowered tests (≤3 observations in combined case–control cohort) were excluded and did not contribute to assessments of genomic inflation. Variants were included for RVB analyses on the basis of MAF within the combined case–control cohort, MAF within the 1000 Genomes project38, and pathogenicity predictions generated using snpEFF (single nucleotide polymorphism effect)39, PolyPhen2 (polymorphism phenotyping version 2)40, SIFT (sorting intolerant from tolerant)41, LRT (likelihood ratio test)42, MutationTaster43, MutationAssessor44, FATHMM (functional analysis through hidden Markov models)5, CADD (combined annotation dependent depletion)45, PROVEAN (protein variation effect analyzer)46, GERP (genomic evolutionary rate profiling)47, phyloP (phylogenetic P value)48, SiPhy (SiPhylogenic)49, dbNSFP (database nonsynonymous SNP functional prediction)50 and dbscSNV (database of splice site consequences of single nucleotide variants)4 as described in Supplementary Table 1. All RVB analyses were conditioned for a missing variant MAF-weighted measure of sample gene call rate and the first four PCs derived from common variant profiles. Homozygosity mapping was performed using HomozygosityMapper51 allowing for genetic heterogeneity. ROH were selected as all loci achieving a homozygosity score ≥ 8,483 (0.6 × max). Single variant analyses were allele-count-based, conducted using PLINK, and also included correction for the first four PCs derived from common variant profiles. Meta-analyses were conducted using METAL52 under a fixed-effect model with weighting by inverted effect size standard error. All statistical tests were two-sided.
Supplementary Material
Acknowledgments
We acknowledge all of the study participants and our collaborators for enabling this study by graciously providing samples for this study. Funding was provided by US National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) (R01NS073873, J.E.L.), the American ALS Association (N.T., V.S., C.E.S., J.E.L. and R.H.B.Jr.), the Motor Neuron Disease (MND) Association (N.T., V.S., C.E.S. and J.E.L.), the Angel Fund (R.H.B.Jr.), Project ALS/P2ALS (R.H.B.Jr.), the ALS Therapy Alliance (R.H.B.Jr. and J.E.L.), The Netherlands ALS Foundation (Project MinE; J.H.V. and L.H.v.d.B.), ALS liga Belgium (P.V.D. and W.Ro.), Suna and Inan Kirac Foundation (N.A.B.). Computer resources for this study were provided by the Green High Performance Computing Center at the University of Massachusetts Medical School. L.H.v.d.B. received grants from the Netherlands Organization for Health Research and Development (Vici Scheme; the SOPHIA and STRENGTH projects through the EU Joint Programme – Neurodegenerative Disease Research, JPND). I.P.B. received grant funding from the National Health and Medical Research Council (NHMRC) of Australia (1095215, 1107644). P.C.S. was supported through the auspices of H. Robert Horvitz (Massachusetts Institute of Technology), an Investigator of the Howard Hughes Medical Institute. M.A.v.E. received a grant from the Netherlands Organization for Health Research and Development (Veni scheme) and travel grants from Baxter. This is an EU Joint Programme – Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organizations under the aegis of JPND (United Kingdom, Medical Research Council; Netherlands, ZonMW; Italy, Ministero dell’Istruzione, dell’Università e della Ricerca; Belgium, Fonds Wetenschappelijk Onderzoek; Germany, Bundesministerium für Bildung und Forschung). C.E.S. and A.A.-C. receive salary support from the National Institute for Health Research (NIHR) Dementia Biomedical Research Unit at South London and Maudsley NHS Foundation Trust and King’s College London. The work leading up to this publication was funded by the European Community’s Health Seventh Framework Programme (FP7/2007–2013; grant agreement number 259867). Samples used in this research were in part obtained from the UK National DNA Bank for MND Research, funded by the MND Association and the Wellcome Trust. I.R.C.C.S. Istituto Auxologico Italiano; AriSLA – Fondazione Italiana di Ricerca per la SLA co-financed with support of “5×1000” – Healthcare Research of the Italian Ministry of Health (grants EXOMEFALS 2009 and NOVALS 2012 (N.T., C.T., C.G., V.S. and J.E.L.)), (grant RepeatALS 2013 (S.D.T. and L.C.)), Italian Ministry of Health (grant GR-2011-02347820 – IRisALS (N.T., C.T. and D.C.)). This work was supported by a grant from the Flemish agency for Innovation by Science and Technology (IWT, Project MinE), the Interuniversity Attraction Poles (IUAP) program P7/16 of the Belgian Federal Science Policy Office, by the FWO-Vlaanderen under the frame of E-RARE-2, the ERA-Net for Research on Rare Diseases (PYRAMID), by a EU JPND project (STRENGTH). P.V.D. is supported by FWO Vlaanderen and the Belgian ALS liga. In Australia, this work was supported by a Leadership Grant to I.P.B. from MND Australia and an NHMRC fellowship (1092023) to K.L.W. G.A.R. is funded by the Canadian Institute of Health Research (CIHR), Genome-wide exon capture for targeted resequencing in patients with FALS (#208973) the Muscular Dystrophy Association, and Whole exome sequencing in patients with FALS (#153959). We thank JMB Vianney de Jong for collection of clinical data. C.S.L. is recipient of Tim E. Noël fellowship from ALS society of Canada. W.Ro. is supported through the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders, the Laevers Fund for ALS Research, the ALS Liga België, the fund ‘Een Hart voor ALS’ and the fund ‘Opening the Future’. The research leading to these results has received funding from the European Research Council und the European’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement 340429 and from the Geneeskundige Stichting Koningin Elisabeth (G.S.K.E.). M.S. and C.D. were supported by the Deutsche Forschungsgemeinschaft, SE697/4-1, BMBF EnergI and the Deutsche Gesellschaft für Muskelkranke, Project He 2/2. This work was supported in whole or in parts by a grant from the German Federal Ministry of Education and Research (JPND STRENGTH consortium; German network for ALS research MND-NET), the Charcot Foundation for ALS Research, the virtual Helmholtz Institute “RNA-Dysmetabolismus in ALS and FTD” and the DFG-funded Swabian ALS Registry. A.Ch. is funded in part by Italian Ministry of Health (Ricerca Sanitaria Finalizzata 2010, grant RF-2010-2309849, project EXPALS), the European Community’s Health Seventh Framework Programme (FP7/2007-2013 under grant agreements 259867), the Joint Programme – Neurodegenerative Disease Research (Italian Ministry of Education and University) (Sophia, and Strength Projects), A.C. is funded in part by Italian Ministry of Health (Ricerca Sanitaria Finalizzata 2010, grant GR-2010-2320550, project EXTRALS) and Fondazione Vialli e Mauro per la Ricerca sulla SLA onlus (grant #4). FUNDELA – Spanish Foundation to the development of ALS research, ISCIII – Carlos III Institute / Fondo de Investigación Sanitaria of Spain (PI10/00092; PI14/00088), ADELA – ALS Spanish Association. Part of this work was carried out on the Dutch national e-infrastructure with the support of SURF Foundation. The Alzheimer’s Disease Sequencing Project (ADSP), phs000572.v7.p4, is comprised of two Alzheimer’s disease (AD) genetics consortia and three National Human Genome Research Institute (NHGRI) funded Large Scale Sequencing Centers (LSSC). The two AD genetics consortia are the Alzheimer’s Disease Genetics Consortium (ADGC) funded by the National Institute on Aging (NIA) (U01 AG032984), and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) funded by NIA (R01 AG033193), the National Heart, Lung, and Blood Institute (NHLBI), other NIH institutes and other foreign governmental and nongovernmental organizations. The Discovery Phase analysis of sequence data is supported through UF1AG047133 to G.D. Schellenberg, L.A. Farrer, M.A. Pericak-Vance, R. Mayeux and J.L. Haines; U01AG049505 to S. Seshadri; U01AG049506 to E. Boerwinkle; U01AG049507 to E. Wijsman; and U01AG049508 to A.M.Goate. The ADGC cohorts include: Adult Changes in Thought (ACT), the Alzheimer’s Disease Centers (ADC), the Chicago Health and Aging Project (CHAP), the Memory and Aging Project (MAP), Mayo Clinic, Mayo Parkinson’s Disease controls, University of Miami, the Multi-Institutional Research in Alzheimer’s Genetic Epidemiology Study (MIRAGE), the National Cell Repository for Alzheimer’s disease (NCRAD), the National Institute on Aging Late Onset Alzheimer’s Disease Family Study (NIA-LOAD), the Religious Orders Study (ROS), the Texas Alzheimer’s Research and Care Consortium (TARC), Vanderbilt University/Case Western Reserve University (VAN/CWRU), the Washington Heights-Inwood Columbia Aging Project (WHICAP) and the Washington University Sequencing Project (WUSP), the Columbia University Hispanic-Estudio Familiar de Influencia Genetica de Alzheimer (EFIGA), the University of Toronto, and Genetic Differences (GD). The CHARGE cohorts, with funding provided by 5RC2HL102419 and HL105756, include the following: Atherosclerosis Risk in Communities (ARIC) Study which is carried out as a collaborative study supported by NHLBI contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C and HHSN268201100012C), Austrian Stroke Prevention Study (ASPS), Cardiovascular Health Study (CHS), Erasmus Rucphen Family Study (ERF), Framingham Heart Study (FHS), and Rotterdam Study (RS). The three LSSC are: the Human Genome Sequencing Center at the Baylor College of Medicine (U54 HG003273), the Broad Institute Genome Center (U54HG003067), and the Washington University Genome Institute (U54HG003079). Biological samples and associated phenotypic data used in primary data analyses were stored at Study Investigators institutions, and at the National Cell Repository for Alzheimer’s Disease (NCRAD, U24AG021886) at Indiana University funded by NIA. Associated phenotypic data used in primary and secondary data analyses were provided by Study Investigators, the NIA-funded Alzheimer’s Disease Centers (ADCs), and the National Alzheimer’s Coordinating Center (NACC, U01AG016976) and the National Institute on Aging Alzheimer’s Disease Data Storage Site (NIAGADS, U24AG041689) at the University of Pennsylvania, funded by NIA, and at the Database for Genotypes and Phenotypes (dbGaP) funded by NIH. Contributors to the Genetic Analysis Data included Study Investigators on projects that were individually funded by NIA, and other NIH institutes, and by private US organizations, or foreign governmental or nongovernmental organizations. We thank people with MND, their families and control individuals for their participation in this project.
Footnotes
Data access. Full details of variants identified in ALS patients are publicly available through the ALS Variant Server at http://als.umassmed.edu/.
URLs. Exome Variant Server, NHLBI Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/; Exome Aggregation Consortium (ExAC), http://exac.broadinstitute.org; ALS Variant Server http://als.umassmed.edu/.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
Sample collection, preparation and clinical evaluation: P.T.C.v.D., A.M.D., N.T., F.P.D., W.v.R., K.R.v.E., A.R.J., P.K., A.S., W.S., B.N.S., M.A.v.E., S.D.T., A. Kenna, J.W.M., C. Tiloca, R.L.M., C.V., C. Troakes, C. Colombrita, G.M., A. Calvo, F.V., S.A.-S., A. King, D.C., J.d.B., F.B., A.J.v.d.K., M.d.V., A.L.M.A.t.A., P.C.S., D.M.-Y., M.P., S.A., J.L.M.-B., T.M.S., T.M., K.E.M., S.D’A., L.M., G.P.C., R.D.B., M.C., S.G., G.Q., C.B., V.P., B.C., S.C., C. Cereda, L.C., G.S., G.L., K.L.W., P.N.L., G.A.N., I.P.B., C.S.L., P.A.D., G.A.R., H.P., P.J.S., M.R.T., K.T., F.T., K.B.B., M.V.B., R.R., J.E.-P., A.G.-R., P.V.D., W.R., A. Chio, C.G., C.D., M.S., A.R., J.D.G., J.S.M., N.A.B., O.H., A.C.L., P.M.A., J.H.W., R.H.B., A.A.-C., V.S., C.E.S., L.H.v.d.B., J.H.V. and J.E.L. Experiments and data analysis: K.P.K., P.T.C.v.D., A.M.D., N.T., B.J.K., F.P.D., W.v.R., K.R.v.E., A.R.J., P.K., A.S., W.S., B.N.S., M.A.v.E., S.D.T., A. Kenna, J.W.M., C.F., C.T., R.L.M., C.V., C. Troakes, C. Colombrita, G.M., A. Calvo, F.V., S.A.-S., A. King, D.C., P.C.S., D.M.-Y., K.L.W., C.S.L., P.A.D., M.v.B., R.R., J.E.-P., A.G.-R., P.v.D., W.R., A. Chio, C.G., C.D., M.S., A.R., J.D.G., J.S.M., N.A.B., O.H., A.C.L., P.M.A., J.H.W., R.H.B.Jr, A.A.-C., V.S., C.E.S., L.H.v.d.B., J.H.V. and J.E.L. Scientific planning and direction: K.P.K., P.T.C.v.D., A.M.D., N.T., B.J.K., C.F., I.P.B., C.S.L., P.A.D., G.A.R., H.P., P.J.S., M.R.T., K.T., F.T., K.B.B., M.v.B., R.R., J.E.-P., A.G.-R., P.v.D., W.R., A. Chio, C.G., C.D., M.S., A.R., J.D.G., J.S.M., N.A.B., O.H., A.C.L., P.M.A., J.H.W., R.H.B.Jr, A.A.-C., V.S., C.E.S., L.H.v.d.B., J.H.V. and J.E.L. Initial manuscript preparation: K.P.K., P.T.C.v.D., A.M.D., N.T., A.A.-C., V.S., C.E.S., L.H.v.d.B., J.H.V. and J.E.L.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking Mendelian disease using exome sequencing. Genome Biol. 2011;12:228. doi: 10.1186/gb-2011-12-9-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ng SB, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith BN, et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014;84:324–331. doi: 10.1016/j.neuron.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014;42:13534–13544. doi: 10.1093/nar/gku1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shihab HA, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65. doi: 10.1002/humu.22225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reid E, et al. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10) Am J Hum Genet. 2002;71:1189–1194. doi: 10.1086/344210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cirulli ET, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenner D, et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain. 2016;139:e28. doi: 10.1093/brain/aww033. [DOI] [PubMed] [Google Scholar]
- 9.Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kenna KP, et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet. 2013;50:776–783. doi: 10.1136/jmedgenet-2013-101795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lattante S, et al. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology. 2012;79:66–72. doi: 10.1212/WNL.0b013e31825dceca. [DOI] [PubMed] [Google Scholar]
- 12.Chiò A, et al. Extensive genetics of ALS: a population-based study in Italy. Neurology. 2012;79:1983–1989. doi: 10.1212/WNL.0b013e3182735d36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thiel C, et al. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am J Hum Genet. 2011;88:106–114. doi: 10.1016/j.ajhg.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shalom O, Shalva N, Altschuler Y, Motro B. The mammalian Nek1 kinase is involved in primary cilium formation. FEBS Lett. 2008;582:1465–1470. doi: 10.1016/j.febslet.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 15.White MC, Quarmby LM. The NIMA-family kinase, Nek1 affects the stability of centrosomes and ciliogenesis. BMC Cell Biol. 2008;9:29. doi: 10.1186/1471-2121-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JH, Gleeson JG. The role of primary cilia in neuronal function. Neurobiol Dis. 2010;38:167–172. doi: 10.1016/j.nbd.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee L. Riding the wave of ependymal cilia: genetic susceptibility to hydrocephalus in primary ciliary dyskinesia. J Neurosci Res. 2013;91:1117–1132. doi: 10.1002/jnr.23238. [DOI] [PubMed] [Google Scholar]
- 18.Cohen S, Aizer A, Shav-Tal Y, Yanai A, Motro B. Nek7 kinase accelerates microtubule dynamic instability. Biochim Biophys Acta. 2013;1833:1104–1113. doi: 10.1016/j.bbamcr.2012.12.021. [DOI] [PubMed] [Google Scholar]
- 19.Chang J, Baloh RH, Milbrandt J. The NIMA-family kinase Nek3 regulates microtubule acetylation in neurons. J Cell Sci. 2009;122:2274–2282. doi: 10.1242/jcs.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puls I, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 21.Ma X, Peterson R, Turnbull J. Adenylyl cyclase type 3, a marker of primary cilia, is reduced in primary cell culture and in lumbar spinal cord in situ in G93A SOD1 mice. BMC Neurosci. 2011;12:71. doi: 10.1186/1471-2202-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Craigen WJ, Riley DJ. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle. 2009;8:257–267. doi: 10.4161/cc.8.2.7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pelegrini AL, et al. Nek1 silencing slows down DNA repair and blocks DNA damage-induced cell cycle arrest. Mutagenesis. 2010;25:447–454. doi: 10.1093/mutage/geq026. [DOI] [PubMed] [Google Scholar]
- 24.Sama RR, Ward CL, Bosco DA. Functions of FUS/TLS from DNA repair to stress response: implications for ALS. ASN Neuro. 2014;6 doi: 10.1177/1759091414544472. 1759091414544472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tafuri F, Ronchi D, Magri F, Comi GP, Corti S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front Cell Neurosci. 2015;9:336. doi: 10.3389/fncel.2015.00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madabhushi R, Pan L, Tsai LH. DNA damage and its links to neurodegeneration. Neuron. 2014;83:266–282. doi: 10.1016/j.neuron.2014.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coppedè F, Migliore L. DNA damage in neurodegenerative diseases. Mutat Res. 2015;776:84–97. doi: 10.1016/j.mrfmmm.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 28.Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. 2015;162:516–526. doi: 10.1016/j.cell.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Surpili MJ, Delben TM, Kobarg J. Identification of proteins that interact with the central coiled-coil region of the human protein kinase NEK1. Biochemistry. 2003;42:15369–15376. doi: 10.1021/bi034575v. [DOI] [PubMed] [Google Scholar]
- 30.Tryka KA, et al. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2014;42:D975–D979. doi: 10.1093/nar/gkt1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang H, et al. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 2014;6:89. doi: 10.1186/s13073-014-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raczy C, et al. Isaac: ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics. 2013;29:2041–2043. doi: 10.1093/bioinformatics/btt314. [DOI] [PubMed] [Google Scholar]
- 34.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manichaikul A, et al. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heinze G, Schemper M. A solution to the problem of separation in logistic regression. Stat Med. 2002;21:2409–2419. doi: 10.1002/sim.1047. [DOI] [PubMed] [Google Scholar]
- 38.1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;7:20. doi: 10.1002/0471142905.hg0720s76. Chapter 7, Unit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 42.Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–1561. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 44.Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davydov EV, et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper GM, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garber M, et al. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics. 2009;25:i54–i62. doi: 10.1093/bioinformatics/btp190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human non-synonymous and splice site SNVs. Hum Mutat. 2016;37:235–241. doi: 10.1002/humu.22932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seelow D, Schuelke M, Hildebrandt F, Nürnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37:W593–W599. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.