

ABSTRACT
The myotonic dystrophies are prototypic toxic RNA gain-of-function diseases. Myotonic dystrophy type 1 (DM1) and type 2 (DM2) are caused by different unstable, noncoding microsatellite repeat expansions – (CTG)DM1 in DMPK and (CCTG)DM2 in CNBP. Although transcription of mutant repeats into (CUG)DM1 or (CCUG)DM2 appears to be necessary and sufficient to cause disease, their pathomechanisms remain incompletely understood. To study the mechanisms of (CCUG)DM2 toxicity and develop a convenient model for drug screening, we generated a transgenic DM2 model in the fruit fly Drosophila melanogaster with (CCUG)n repeats of variable length (n=16 and 106). Expression of noncoding (CCUG)106, but not (CCUG)16, in muscle and retinal cells led to the formation of ribonuclear foci and mis-splicing of genes implicated in DM pathology. Mis-splicing could be rescued by co-expression of human MBNL1, but not by CUGBP1 (CELF1) complementation. Flies with (CCUG)106 displayed strong disruption of external eye morphology and of the underlying retina. Furthermore, expression of (CCUG)106 in developing retinae caused a strong apoptotic response. Inhibition of apoptosis rescued the retinal disruption in (CCUG)106 flies. Finally, we tested two chemical compounds that have shown therapeutic potential in DM1 models. Whereas treatment of (CCUG)106 flies with pentamidine had no effect, treatment with a PKR inhibitor blocked both the formation of RNA foci and apoptosis in retinae of (CCUG)106 flies. Our data indicate that expression of expanded (CCUG)DM2 repeats is toxic, causing inappropriate cell death in affected fly eyes. Our Drosophila DM2 model might provide a convenient tool for in vivo drug screening.
KEY WORDS: Myotonic dystrophy, DM2, RNA toxicity, Drosophila, Muscleblind, Apoptosis
Summary: A Drosophila model of myotonic dystrophy type 2 (DM2) recapitulates several features of the human disease, identifies apoptosis as a contributing factor to DM2, and is likely to provide a convenient tool for drug screening.
INTRODUCTION
Myotonic dystrophy (DM) is the most common adult-onset neuromuscular disorder (Harper, 2001). DM is characterized by myotonia, muscle weakness and wasting, as well as multi-systemic manifestations, including insulin resistance, gonadal atrophy, cataracts and neuropsychiatric symptoms (La Spada and Taylor, 2010; Udd and Krahe, 2012; Thornton, 2014). There are two genetically distinct types, DM1 and DM2, which are caused by similar noncoding repeat expansions in different genes: a (CTG)n expansion in the 3′ UTR of the DM1 protein kinase (DMPK) gene in DM1; and a (CCTG)n expansion in the first intron of the CCHC-type zinc finger nucleic acid binding protein (CNBP) gene [also known as zinc finger protein 9 (ZNF9)] in DM2 (La Spada and Taylor, 2010; Udd and Krahe, 2012; Timchenko, 2013; Thornton, 2014). Whereas expansion size generally correlates with disease severity in DM1 and is the basis for the observed pronounced anticipation, there does not appear to be a genotype/phenotype correlation in DM2 (Udd and Krahe, 2012). DM2 expansions up to 44 kb (11,000 CCTG) have been reported (Liquori et al., 2001; Day et al., 2003; Sallinen et al., 2004); the smallest expansions associated with clinically detectable manifestations are between 55 and 100 CCTG repeats (Liquori et al., 2001; Lucchiari et al., 2008; Bachinski et al., 2009).
The prevailing paradigm is that both DM1 and DM2 are toxic RNA-mediated spliceopathies, mediated by the mutant expansions of normally polymorphic (CTG)n or (CCTG)n repeats: transcription into (CUG)DM1 or (CCUG)DM2 RNA is necessary and sufficient to cause disease (Osborne and Thornton, 2006; Klein et al., 2011; Sicot and Gomes-Pereira, 2013). Mutant RNAs accumulate in ribonuclear foci and interfere with RNA splicing, transcription and/or translation of downstream effector genes, resulting in the characteristic pleiotropic phenotype (Schoser and Timchenko, 2010; Jones et al., 2011; Sicot et al., 2011; Udd and Krahe, 2012; Timchenko, 2013).
Mechanistically, (CUG)DM1 or (CCUG)DM2 RNA foci sequester Muscleblind-like (MBNL) proteins, which are zinc-finger RNA-binding proteins involved in alternative RNA splicing (Miller et al., 2000; Mankodi et al., 2001, 2003; Kanadia et al., 2003; Pascual et al., 2006; Lee and Cooper, 2009; Schoser and Timchenko, 2010; Jones et al., 2011; Meola et al., 2013). MBNL proteins are highly conserved from flies to humans. The fruit fly Drosophila melanogaster has a single MBNL gene, muscleblind (mbl), which is involved in muscle development and photoreceptor neuron differentiation in the eye (Begemann et al., 1997; Artero et al., 1998; Pascual et al., 2006). Loss of mbl causes muscle defects and blindness, hence the name of the gene (Begemann et al., 1997; Artero et al., 1998). Similarly, in DM1 and DM2 patients [humans have three MBNL homologous genes: MBNL1-3 (Fardaei et al., 2002)], the sequestration of MBNL proteins in RNA foci reduces the amount of functional MBNL proteins available for proper splicing, resulting in a shift from the normal adult splice pattern to an inappropriate embryonic/fetal pattern of target transcripts (Miller et al., 2000; Mankodi et al., 2001; Jiang et al., 2004; Kanadia et al., 2006; Holt et al., 2009).
More than 20 transcripts have been shown to be mis-spliced in DM (Jiang et al., 2004; Gatchel and Zoghbi, 2005; Botta et al., 2007; Du et al., 2010). For example, aberrant splicing of the muscle-specific chloride channel CLCN1 and the insulin receptor (INSR) accounts for myotonia in DM (Savkur et al., 2001, 2004; Mankodi et al., 2002; Wheeler et al., 2007; Tonevitsky and Trushkin, 2009; Tang et al., 2012; Santoro et al., 2013). Other mis-spliced genes in DM include the muscle contractile proteins cardiac troponin (TNNT2) and skeletal muscle troponin (TNNT3) (Philips et al., 1998; Yuan et al., 2007; Vihola et al., 2010).
In addition to the MBNL family of proteins, at least two other RNA-binding proteins have been implicated in DM1. Expanded CUG repeats increase the activities of CUG-binding protein (CUGBP1; also known as CELF1) and dsRNA-dependent protein kinase (PKR; also known as EIF2AK2) (Tian et al., 2000; Timchenko et al., 2001a,b; Mankodi et al., 2003; Ward et al., 2010; Jones et al., 2011). Whether these factors are involved in DM2 is unclear.
There is currently no cure for DM. Most efforts to identify therapeutic modes of intervention are focused on the reversal of RNA toxicity. To develop a convenient model for drug screening, we generated a DM2 model in the fruit fly Drosophila melanogaster. We obtained transgenes that express noncoding transcripts of variable size, with the largest at 106 CCUG repeats (DM2-106). Transgenic DM2-106 flies recapitulate many features observed in the human disease condition. They form RNA foci in muscles and retinal cells and affect RNA splicing of splicing reporter genes. Although we did not observe muscle atrophy in DM2-106 flies, they displayed strong disruption in the external morphology of the eye and underlying retina. Expression of MBNL1, but not CUGBP1, was able to rescue the eye phenotype of DM2-106 flies. Furthermore, DM2-106 flies exhibited a strong apoptotic response in developing retinae, and inhibition of apoptosis rescued the retinal disruption. Finally, we tested two chemical compounds with therapeutic potential in DM1. Whereas treatment of DM2-106 flies with pentamidine had no effect, treatment with a PKR inhibitor blocked both the formation of RNA foci and apoptosis in retinae of DM2-106 flies. These data suggest that the Drosophila DM2 model described here may provide a suitable tool for drug screening.
RESULTS
Transcripts with expanded (CCUG)n repeats form RNA foci
The smallest reported DM2 expansions associated with clinically detectable manifestations are between 55 and 100 CCTG repeats (Liquori et al., 2001; Lucchiari et al., 2008; Bachinski et al., 2009). To generate a DM2 model in Drosophila, we prepared two transgenes: a control transgene expressing a noncoding transcript with 16 CCUG repeats in the normal range (referred to as N-16), and an experimental transgene expressing a noncoding RNA with 106 CCUG repeats (DM2-106) (Fig. 1A). Because the (CCTG)DM2 expansion is part of a complex polymorphic motif (Bachinski et al., 2003, 2009) of the form (TG)n(TCTG)n(CCTG)>26 and the (TG)n(TCTG)n polymorphic repeats have been shown to affect DNA structure (Edwards et al., 2009), we included a (TG)n(TCTG)n tract in our (CCTG)DM2 constructs. Both control and DM2 transgenes contained the polymorphic (TG)n(TCTG)n repeats upstream of the (CCTG)n tract: the N-16 allele had a (TG)20(TCTG)12(CCTG)16 motif, while the DM2-106 allele had a (TG)22(TCTG)2(CCTG)106 motif (Fig. 1A). These transgenes are under the control of a UAS promoter (Brand and Perrimon, 1993) and expression can be induced using convenient Gal4 drivers, such as muscle-specific Mhc-Gal4 and eye-specific GMR-Gal4.
Fig. 1.

A Drosophila DM2 model forms nuclear CCUG foci. (A) Schematic (not to scale) of the noncoding CCTG repeat constructs used in this study. The control contains (CCTG)16 repeats (N-16), which is non-toxic in humans. The mutant construct contains (CCTG)106 repeats (DM2-106). Both constructs are preceded by polymorphic (TG)n(TCTG)n repeats, as indicated, that are also part of the complex human repeat motif. These constructs are under control of the UAS promotor. (B) In situ hybridization using a locked nucleic acid (LNA) probe was performed on 15 μm cryosections of thoracic muscles of flies expressing DM2-106 and control repeats using the myosin Mhc-Gal4 driver. DM2-106 expression is associated with the presence of ribonuclear foci (red) in DAPI-stained nuclei (blue), whereas no foci are detected in controls using the same Gal4 driver. Two representative foci are indicated (arrows). (C) Quantification of nuclei with ribonuclear foci in control and DM2-106 muscle cells using Mhc-Gal4. Error bars indicate s.d.
Because myotonia and muscle wasting are associated with human DM2, we first expressed the control and disease transgenes using Mhc-Gal4 and analyzed the morphology of the indirect flight muscle (IFM). As nuclear retention of RNA-protein aggregates (foci) is a hallmark of DM2 (Mankodi et al., 2003; Jones et al., 2011; Udd and Krahe, 2012; Meola et al., 2013), we first determined that DM2-106 flies mirror this disease-linked trait and performed FISH analysis to detect foci in the nucleus of IFM cells of DM2-106 flies. No foci were detected in control IFM, whereas more than 50% of the cells analyzed had nuclear foci in DM2-106 flies (Fig. 1B,C), demonstrating that 106 CCUG repeats are sufficient to cause biochemical changes. The average fraction of nuclei with ribonuclear foci in DM2-106 muscle cells is similar to that observed in a DM1 fly model expressing 480 CTG repeats (García-Alcover et al., 2014).
Expression of DM2-106 in Drosophila muscles causes mis-splicing
In order to evaluate DM2-106 flies as a suitable DM2 model, we examined mis-splicing events in transgenic flies expressing the 106 CCUG repeats in IFM. We studied alternative splicing of the endogenous Fhos gene (Fig. 2A), which showed aberrant splicing regulation in DM1 flies expressing a (CTG)480 tract (Garcia-Lopez et al., 2008) (see also Fig. 2B). For this analysis, we used two different transgenes for control and DM2-106 constructs, located on chromosomes 2 and 3. Expression of both DM2-106 transgenes increased the frequency at which exon 24 was aberrantly included (Fig. 2B): quantification revealed an increase from ∼30% in N-16 control flies to >70% in DM2-106 flies (Fig. 2C), similar to DM1.
Fig. 2.

DM2-106 expression in muscle causes mis-splicing of MBNL1-dependent transcripts. (A) Outline of the intron/exon structure of Fhos (CG42610) showing the exons implicated in the splicing event studied. Wild-type flies mainly skipped exon 24 (solid line), whereas DM2-106 expression in IFM led to aberrant inclusion of exon 24 (dotted lines). Arrows indicate primers used for semi-quantitative PCR analysis. (B,C) Agarose gel and quantification of Fhos RT-PCR products from IFM expressing control (N-16) and DM2-106 transgenes located on chromosomes 2 and 3. These transgenes were driven by Mhc-Gal4. Flies that only contain the Mhc-Gal4 driver without a UAS transgene show an average frequency of exon 24 inclusion of ∼30%. Compared with this control, expression of normal repeat length (CCUG)16 does not significantly alter Fhos splicing, whereas in the (CCUG)106 repeat-expressing cells exon 24 is retained at ∼70%, levels similar to those of DM1 flies expressing an interrupted 480 CUG repeat sequence (iCUG)480. (D,E) Agarose gel and quantification of Fhos RT-PCR products from flies expressing the indicated transgenes with the Mhc-Gal4 driver. Simultaneous expression of human MBNL1 and DM2-106 induces exon 24 exclusion, restoring wild-type levels (Mhc-Gal4 only). Error bars represent s.d. and each experiment was repeated at least twice in adults of 0-5 days of age. (F-H) Luminescence levels of Mhc-Gal4>UAS-minigene,DM2-106 normalized to the levels of Mhc-Gal4>UAS-minigene,UAS-GFP. Relative luminescence decreased from 100% in control flies to 78% for the human INSR reporter minigene (F), 38% for TNNT2 (cTNT) (G) and 68% for mouse Tnnt3 (H). RLU, relative light units. **P<0.005, ***P<0.001 (Student's t-test). (F′) RT-PCR analysis of the INSR spliceosensor in N-16 and DM2-106 background. The percentage is the average of two experiments.
The MBNL proteins are sequestered in (CCUG)DM2 foci and have been implicated as important mediators of DM2-associated spliceopathy. To validate an involvement of MBNL factors in DM2 flies, we co-expressed the human MBNL1 gene and DM2-106 in Drosophila IFM. As shown in Fig. 2D, MBNL1 expression rescued exon 24 inclusion levels in IFM in the presence of (CCUG)106, unlike GFP protein, which was used as a negative control in this co-expression experiment. The frequency of disease-linked exon 24 inclusion was reduced from 73% (DM2-106+GFP) to 48% (DM2-106+MBNL1), close to control levels in the non-disease situation (38%) (Fig. 2E).
The suitability of our Drosophila DM2 system as a disease model was further demonstrated by the observation that different spliceosensor luciferase reporters, which express specific mammalian reporter mini-genes for identified mis-splicing events in DM1 and DM2 (human INSR exon 11 and mouse Tnnt3 fetal exon) (Savkur et al., 2004; Vihola et al., 2010; García-Alcover et al., 2014), were also responsive to the presence of expanded CCUG repeats (Fig. 2F,H). In addition, we tested a TNNT2 exon 5 spliceosensor reporter that shows mis-splicing in DM1 (Philips et al., 1998). All three spliceosensor reporters revealed alternative splicing aberrations, resulting in reduced luciferase luminescence, when (CCUG)106 repeats were expressed in the IFM (Fig. 2F-H). To verify that the significant changes in luciferase luminescence were due to mis-splicing, we examined the splicing pattern of the INSR spliceosensor reporter directly by RT-PCR in our DM2 fly model. In the Drosophila DM1 model, two splice isoforms of the INSR spliceosensor were detectable due to the inclusion (isoform B) or exclusion (isoform A) of an alternative exon between exons 11 and 12 (García-Alcover et al., 2014). Isoform A is preferentially observed in the disease state (García-Alcover et al., 2014). In the N-16 controls expressing (CCUG)16, isoform A was present at 42.5% (Fig. 2F′), consistent with the previous report by García-Alcover et al. (2014). By contrast, in the presence of expanded (CCUG)106 repeats (DM2-106), the relative amount of isoform A increased to 65% (Fig. 2F′), which correlated with the decreased luciferase luminescence observed in Fig. 2F. These results demonstrated that DM2-106 transgenic flies display a spliceopathy phenotype similar to that seen in human DM2 patients and thus validate it as a suitable DM2 model.
Expression of (CCUG)106 in Drosophila IFM does not cause muscle atrophy
To study the extent of (CCUG)DM2 toxicity in our DM2 model, we analyzed IFM samples expressing control or expanded (CCUG)n (N-16 or DM2-106) for morphological defects similar to those described in patients (Vihola et al., 2003; Bassez et al., 2008). However, in contrast to the phenotypic alterations of the IFM in DM1 models (Fig. 3C), no significant differences were observed between the IFMs of control and expanded (CCUG)n-expressing flies (Fig. 3A,B, quantified in Fig. 3D). DM2-106 flies appeared to be able to fly normally and even aged flies (40 days) did not display any obvious flight defects. These results demonstrated that although (CCUG)106 repeats are sufficient to cause biochemical abnormalities, they are not sufficient to cause morphological and behavioral phenotypes in the IFM.
Fig. 3.

DM2-106 expression does not cause morphological defects in Drosophila musculature. (A-C) IFM transverse sections from control flies (Mhc-Gal4/+) or flies expressing either (CCUG)106 (DM2-106) or (CUG)480 (DM1). (CUG)480 expression leads to vacuolization and muscle disorganization (Garcia-Lopez et al., 2008), whereas (CCUG)106 expression was not disruptive to muscle fiber morphology. (D) Relative muscle areas of at least six independent thoraces of each genotype were calculated after binarization using ImageJ and statistically analyzed using a two-tailed, non-paired t-test (P=0.118). Error bars indicate s.d.
Expanded (CCUG)106 repeats cause severe disorganization of eye morphology, which is modified by loss or gain of MBNL proteins
Because (CCUG)106-expressing flies did not show significant phenotypic alterations in the IFM, we turned our focus to a different phenotype commonly observed in DM2 pathogenesis, namely ocular manifestations. For that purpose, we expressed control (N-16) and DM2-106 transgenes using GMR-Gal4 in the posterior half of developing eye imaginal discs, which form the retina during late larval and pupal stages. Adult flies expressing the (CCUG)106 transgenes developed eyes of severely disorganized morphology (Fig. 4C). By contrast, control GMR-Gal4>N-16 flies displayed only mildly rough eyes, as compared with GMR-Gal4-only eyes (Fig. 4A,B). Both transgenes are expressed at similar levels (Fig. 4H). Thus, in contrast to the IFM, expression of expanded (CCUG)106 repeats caused severe phenotypic abnormalities in the fly eye. A similar observation has recently been reported for a different DM2 fly model (Yu et al., 2015).
Fig. 4.

Expression of expanded (CCUG)n causes severe disruption of eye morphology, which can be modified by loss or gain of MBNL. (A) The eye of a GMR-Gal4-only fly. (B) Expression of the control N-16 transgene under the GMR-Gal4 driver shows a very mild eye roughening, which is caused by GMR-Gal4. (C) Disruption of the external eye morphology of a fly that expresses DM2-106 using GMR-Gal4. (D) Expression of a UAS control transgene, UAS-GFP, does not rescue the eye phenotype of GMR-Gal4>DM2-106 flies. (E) Heterozygosity for a null allele of muscleblind (mblKG08885), resulting in functional hemizygosity, severely enhances the DM2-106 eye phenotype owing to the 50% reduction in MBL protein levels. (F) Expression of human MBNL1 rescues the eye phenotype of DM2-106 flies under GMR-Gal4 control. (G) Co-expression of a CUGBP1 transgene using GMR-Gal4 does not rescue or provides only very little rescue of the external eye morphology of DM2-106 flies. (H) Expression levels of control (N-16) and experimental (DM2-106) transgenes under GMR-Gal4 control in eye imaginal discs. Statistical analysis was by two-tailed, non-paired t-test (P=0.222). (I-I″) MBNL1 protein (green) accumulates in CCUG foci (red) in GMR-Gal4>DM2-106 eye imaginal discs (arrows). Interestingly, MBNL1 also aggregates in foci independently of CCUG repeats (arrowheads). Nuclei are labeled with DAPI (blue).
As described above, MBNL proteins are thought to be involved in DM2 pathogenesis. To confirm this in our DM2 fly model, we examined whether alterations to the gene dose of Drosophila mbl would modify the DM2-106 phenotype. In a heterozygous mbl+/− background, the eye phenotype of adult DM2-106 flies is severely enhanced (Fig. 4E). The eyes are rougher and more disorganized, and often also reduced in size. Phenotypic rescue is observed when the human MBNL1 protein is overexpressed in DM2-106 flies, which suppressed the DM2-106 eye phenotype (Fig. 4F). The eyes appear almost normal and ommatidial integrity is visible. Expression of a control UAS transgene, UAS-GFP, does not affect the DM2-106 eye phenotype (Fig. 4D), suggesting that the rescue by MBNL1 expression is not due to the additional UAS transgene. Immunolocalization reveals that MBNL1 protein is localized in CCUG foci (Fig. 4I-I″, arrows), but we also observe aggregates of MBNL1 protein outside of CCUG foci (Fig. 4I,I″, arrowheads).
Another protein implicated in the pathology of DM1 is CUGBP1 (Timchenko et al., 2001a,b; de Haro et al., 2006; Jones et al., 2011; Timchenko, 2013). However, in contrast to MBNL1, expression of human CUGBP1 has little or no effect on the morphology of DM2-106 eyes (compare Fig. 4G with 4C).
DM2-106 causes severe disruption of retinal organization
To further characterize the eye phenotype of GMR>DM2-106 flies, we examined the underlying retinal morphology. The developing retina is fully differentiated at 42 h after puparium formation (APF). In wild-type retinae at that stage, photoreceptor neurons, cone cells and pigment cells are highly organized in a stereotypical pattern to form the individual ommatidia. Each ommatidium contains four concentrically aligned cone cells surrounded by pigment cells (Fig. 5A″). In GMR-Gal4>N-16 control flies, this pattern is not significantly disturbed (Fig. 5A-A″). By contrast, in GMR>DM2-106 retinae the precise cellular arrangement is severely disrupted, with photoreceptor neurons, cone cells and pigment cells irregularly positioned and numbered (Fig. 5B-B″). Ommatidia were fused and ommatidial identity was not observed. Thus, expression of (CCUG)106 RNA caused severe disruption of retinal morphology.
Fig. 5.
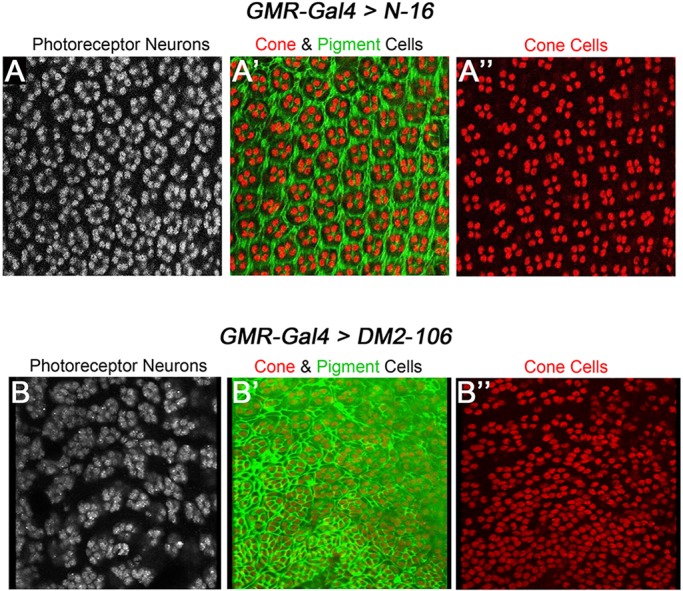
DM2-106 causes severe disruption of retinal morphology. Fully differentiated retinae of pupal eye imaginal discs at 42 h APF labeled with antibodies against ELAV (a marker for photoreceptor neurons; A,B), Cut (a marker of cone cells; A′,A″,B′,B″) and Dlg (to visualize cell outline and thus reveal pigment cells; A′,B′). (A′,B′) Double labeling for Cut and Dlg. (A-A″) GMR>N-16 control retina showing the regular pattern of photoreceptors (A), cone (A′,A″) and pigment cells (A′). (B-B″) Retina expressing DM2-106 under GMR-Gal4 control shows irregularities of photoreceptor neurons (B), cone and pigment cells (B′,B″).
Apoptosis induced by DM2-106 causes retinal disruption and disorganization
Recently, apoptosis has been implicated in muscle degeneration in a Drosophila DM1 model (Bargiela et al., 2015). Therefore, we examined whether apoptosis contributes to the retinal phenotype in GMR>DM2-106 transgenic flies. In GMR-Gal4>N-16 control flies, no or very little apoptosis occurs in eye imaginal discs, the larval precursors of the adult eyes (Fig. 6A). However, in GMR>DM2-106 eye imaginal discs, apoptosis is strongly induced in the GMR domain in the posterior half of the larval disc (Fig. 6B, arrow), suggesting that (CCUG)106 RNA triggers apoptosis. P35 is a potent inhibitor of apoptosis, and specifically inhibits effector caspases in flies (Hay et al., 1994; Hawkins et al., 2000; Meier et al., 2000). Co-expression of p35 together with DM2-106 under GMR-Gal4 control suppressed the apoptotic phenotype in larval eye imaginal discs (Fig. 6C). Because P35 potently suppressed apoptosis in DM2-106 flies, we were able to dissect the relative contribution of apoptosis to the DM2-106 retinal and eye morphology phenotypes. Co-expression of p35 in the DM2-106 model normalized the external eye morphology of adult flies (Fig. 6D, compare with Fig. 4C). Furthermore, co-expression of p35 suppressed the misalignment of photoreceptor neurons and cone cells, restoring ommatidial integrity (Fig. 6E,F, compare with Fig. 5B). These data illustrate that expression of the DM2-106 transcript caused apoptosis that resulted in retinal disruption and disorganization.
Fig. 6.

Induction of apoptosis results in disruption of the photoreceptor neuron pattern in DM2-106 retinae. (A-C) TUNEL labeling as a marker for apoptosis of N-16 (A), DM2-106 (B) and DM2-106+p35 (C) eye imaginal discs from third instar larvae under GMR-Gal4 control. The extent of the GMR expression domain in the larval eye disc is indicated. The arrow (B) highlights the induced apoptosis in the posterior part of the larval eye disc where GMR-Gal4 is expressed. (D) Rescue of the external eye morphology of adult DM2-106 flies expressing the caspase inhibitor p35 under GMR-Gal4 control. (E,F) GMR>DM2-106+p35 pupal retinae at 42 h APF labeled for the photoreceptor marker ELAV (E) and the cone cell marker Cut (F). Inhibition of apoptosis by co-expression of the caspase inhibitor P35 normalizes the photoreceptor and cone cell pattern in GMR>DM2-106 retinae (compare with Fig. 5B).
Feasibility of the DM2-106 model for chemical screening
The data presented here suggest that expression of DM2-106 in the Drosophila retina mimics pathological manifestations seen in the human condition, including the formation of toxic CCUG foci, as well as retinal disorganization and degeneration. Therefore, DM2-106 might provide a suitable and convenient model for drug screening and identification of lead compounds (García-Alcover et al., 2013). To assess the feasibility of our DM2-106 model for drug screening, we tested two compounds that have previously been shown to have therapeutic potential in DM1 models. Pentamidine is a dsRNA-intercalating drug that was found to disrupt the MBNL1-CUG repeat complex in DM1 (Warf et al., 2009). It was recently reported that pentamidine treatment can also rescue cardiac dysfunction in a Drosophila DM1 model (Chakraborty et al., 2015). The second drug, the oxindole/imidazole derivative C16, is an inhibitor of the dsRNA-dependent protein kinase PKR (PKR-I), which is activated by expanded CUG repeats in DM1 (Tian et al., 2000, 2005; Huichalaf et al., 2010; Wojciechowska et al., 2014). As an assay for drug treatment, we examined the ability of the selected inhibitors to block the formation of toxic CCUG foci in the DM2-106-expressing retina (Fig. 7B). Interestingly, these foci were not only nuclear, but could also be observed in the cytoplasm (Fig. 7B, arrows). Pentamidine treatment up to 350 μM, a concentration that has been shown to be effective in DM1 (Warf et al., 2009), has no visible effect on RNA foci formation in the DM2-106 model (Fig. 7C-E). By contrast, treatment with PKR-I showed a pronounced decrease in the abundance of CCUG RNA foci in a concentration-dependent manner: 4 μM PKR-I caused a significant reduction of RNA foci, and 7 μM completely disrupted foci formation (Fig. 7F-H), resembling wild-type retinae (Fig. 7A). Consistently, loss of CCUG foci by PKR-I treatment correlated with reduction and loss of apoptosis (Fig. 7I-L). These examples illustrate that the DM2-106 retina might provide a convenient model for drug screening in flies.
Fig. 7.
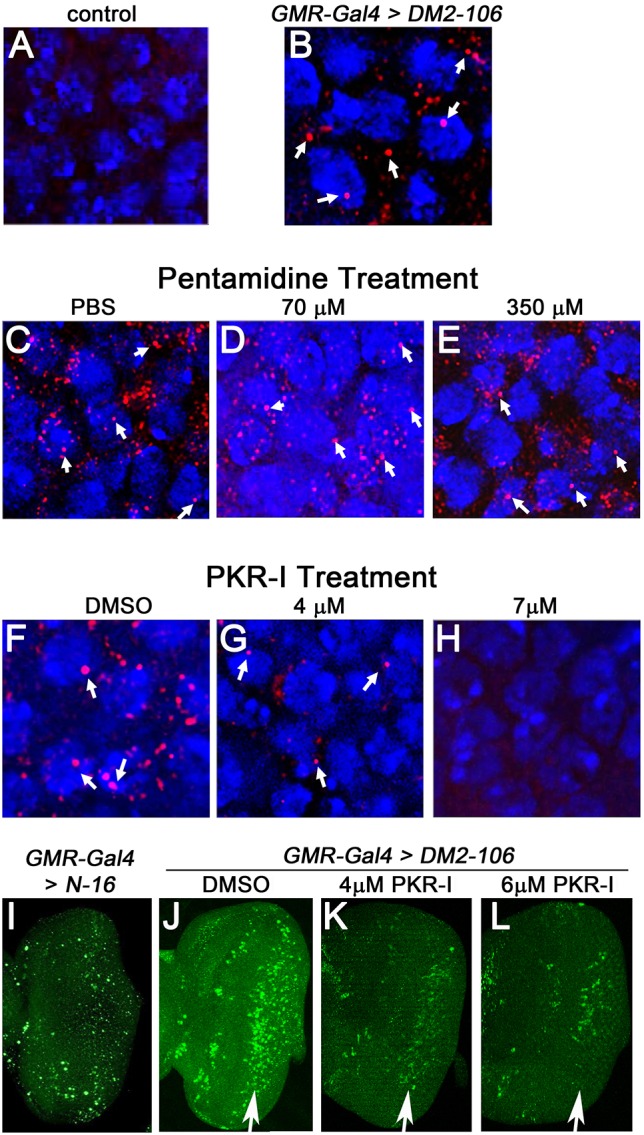
Treatment with PKR inhibitor, but not pentamidine, blocks foci formation and apoptosis in DM2-106 retinae. (A-H) Shown are 42 h APF retinae from control (A), untreated GMR>DM2-106 (B) and GMR>DM2-106 treated with various buffers and drugs (C-H) as indicated. These retinae were labeled for CCUG foci (red) and nuclei (blue). Arrows indicate example foci, both nuclear and cytoplasmic. Whereas pentamidine treatment did not block foci formation up to a concentration of 350 μM, treatment with PKR inhibitor (oxindole/imidazole derivative C16, PKR-I) suppressed foci formation in a concentration-dependent manner. (I-L) Eye imaginal discs from control (I), untreated GMR>DM2-106 (J) and GMR>DM2-106 treated with the indicated concentrations of PKR-I (K,L). Eye discs were obtained from third instar larvae and were labeled by TUNEL as an apoptotic marker. Arrows indicate apoptosis in the GMR-expression area.
The suppression of foci formation and apoptosis by PKR-I suggests that PKR activity is increased in the retinae of DM2-106 expressing pathogenic CCUG repeats. Activation of PKR by expanded CUG repeats in DM1 has been reported (Tian et al., 2000; Huichalaf et al., 2010; Wojciechowska et al., 2014). It is currently unknown whether PKR is also activated in DM2. To examine this possibility, we tested a known PKR phosphorylation target, eukaryotic translation initiation factor 2 alpha (eIF2α) (Proud, 2005). In humans, PKR phosphorylates and inactivates eIF2α on Ser51 (Proud, 2005). This phosphorylation site is conserved in Drosophila eIF2α, and phospho-specific eIF2α (P-Ser51) antibodies cross-react with phosphorylated Drosophila eIF2α (Williams et al., 2001; Farny et al., 2009). These antibodies detect a strong increase in eIF2α phosphorylation on Ser51 in DM2-106 retinae as compared with the N-16 control (Fig. 8A-C). Importantly, this strong increase in Ser51 phosphorylation in DM2-106 retinae was significantly reduced upon dietary administration of 7 μM PKR-I (Fig. 8D). These data suggest that PKR activity is strongly induced in the presence of 106 CCUG repeats in the retina.
Fig. 8.

Expression of pathogenic (CCUG)106 increases PKR activity in retinae of GMR>DM2-106 flies. The posterior portions of eye imaginal discs from third instar larvae of the indicated genotype labeled with phospho-specific eIF2α (P-Ser51) antibody to detect PKR activity. GMR-Gal4>UAS-N-16 (A,B) or GMR-Gal4>UAS-DM2-106 (C,D) discs were treated (B,D) or not (A,C) with 7 μM PKR-I.
DISCUSSION
The goal of this work was to develop a fly model that can be used for drug screening to identify therapeutic compounds for potential treatment of DM2 patients. The rationale was that many cell biological processes, including apoptosis, alternative RNA splicing and the genes/proteins involved, are highly conserved between flies and humans. Furthermore, the genetic tools available in Drosophila allow for rapid characterization of the underlying phenotypes. Finally, experimentation with flies is relatively inexpensive and the short generation time enables rapid genetic and chemical screening.
Here, we describe a DM2 fly model that expresses 106 CCUG repeats in a non-coding transcript (DM2-106). Several features of DM2-106 flies indicate that expression of expanded (CCUG)106 RNA elicits molecular and cellular phenotypes similar to those associated with DM2 pathology in human patients. DM2-106 transcripts aggregate in RNA foci that are predominantly nuclear, but can also be observed in the cytoplasm, at least in retinal cells. Cytoplasmic (CCUG)DM2 foci have also recently been described in human HeLa cells (Jones et al., 2015). These RNA foci sequester MBNL proteins, which causes mis-splicing in muscles similar to that seen in human DM2 patients. Although we did not observe muscle atrophy in DM2-106 flies, the retinae and eyes of these flies were severely disrupted. Functional complementation by overexpression of human MBNL1 protein in GMR>DM2-106 rescued the retinal degeneration. Furthermore, inhibition of apoptosis restored the retinal pattern and eye morphology, suggesting that expression of (CCUG)106 in DM2-106 flies induced apoptosis as the underlying cause of the retinal degeneration. The involvement of apoptosis in retinal degeneration is consistent with the recent finding that apoptosis also contributes to muscle degeneration in a Drosophila DM1 model (Bargiela et al., 2015). In a pilot drug screening experiment in DM2-106 eyes, we found that an inhibitor of PKR activity efficiently blocked formation of RNA foci and apoptosis, whereas pentamidine failed to inhibit foci formation. Finally, we show that pathogenic (CCUG)n DM2 repeat expansions activate the dsRNA-dependent protein kinase PKR, similar to previous reports in DM1 (Tian et al., 2000; Huichalaf et al., 2010; Wojciechowska et al., 2014). Taken together, these data suggest that our DM2-106 fly model provides a convenient tool for drug screening.
While this work was under way, another group published a different DM2 model in Drosophila (Yu et al., 2015). These authors were able to express more than 700 CCUG repeats in flies. Consistent with our observations, expression of (CCUG)700 repeats caused retinal and eye disruption. Interestingly, muscle atrophy was also not reported (Yu et al., 2015), suggesting that in Drosophila the eye is perhaps more sensitive to RNA perturbations than skeletal muscle. In this context, it is worth noting that muscle weakness and atrophy are generally weaker in DM2 patients than in DM1 (Udd and Krahe, 2012). Alternatively, it is possible that there is an expansion threshold that underlies tissue-specific manifestations of the overall DM2 phenotype and that the expression of (CCUG)106 repeats is insufficient in itself to induce muscle phenotypes due to mis-splicing, but could involve the recently identified RAN (repeat-associated non-ATG) translation as another pathomechanism (Zu et al., 2011).
Despite the fact that DM1 and DM2 share many pathological manifestations, they are not identical diseases (Udd and Krahe, 2012). For example, they affect different types of muscle and the neurological symptoms in DM2 are generally less severe (Thornton, 2014; Ulane et al., 2014). We also observed differences between the DM1 and DM2 models in Drosophila. Whereas expression of CUGBP1 enhanced the DM1 phenotype (de Haro et al., 2006), it had no obvious effect on the eye phenotype of GMR>DM2-106 flies. Furthermore, pentamidine treatment, which was shown to be effective in DM1 (Chakraborty et al., 2015), had no effect on foci formation in DM2-106. Therefore, comparative analysis of DM1 and DM2 fly models might reveal additional differences that underlie the two diseases and thereby provide important insights into the etiology of the human phenotypes.
Our pilot drug screen revealed that the DM2-106 Drosophila model is well suited for drug screening. Treatment of DM2-106 flies with increasing concentrations of a PKR inhibitor disrupted CCUG RNA foci formation and apoptosis in eye imaginal discs, the larval precursor tissue of adult retinae and eyes. PKR encodes a dsRNA-dependent protein kinase, which was found to be activated in DM1. Our data suggest that PKR activity is also induced by expanded (CCUG)n DM2 repeats. Unfortunately, although PKR-I feeding of larvae disrupted CCUG foci formation in GMR>DM2-106 eye imaginal discs, the resulting eye phenotype of adult flies was not rescued (Fig. S1). A possible explanation for this observation is that flies stop feeding after the larval stage, so that during pupal stages the eye phenotype can still develop. Nevertheless, we are confident that modeling of DM2 in Drosophila will further contribute to our understanding of the pathology of DM2 and provide an excellent platform for genetic and chemical (drug) screening.
MATERIALS AND METHODS
Generation of control and expanded (CCUG)n repeat expression clones
The (CCTG)DM2 expansion is part of a complex polymorphic motif (Bachinski et al., 2003, 2009) of the form (TG)12-26(TCTG)7-12(CCTG)3-9(G/TCTG)0-4(CCTG)4-15. DM2 expansions can be as large as 40 kb with the CCTG motif uninterrupted (Liquori et al., 2001; Bachinski et al., 2003; Sallinen et al., 2004). Reported normal alleles have repeat tract lengths of up to 26 CCTG motifs with one or more interruptions (Bachinski et al., 2009). The smallest reported DM2 expansions associated with clinically detectable manifestations are between 55 and 100 CCTG repeats (Liquori et al., 2001; Lucchiari et al., 2008; Bachinski et al., 2009). Because this complex polymorphic repeat motif has been shown to have an effect on DNA structure (Edwards et al., 2009), we included the (TG)n(TCTG)n tracts in the (CCTG)DM2 constructs. We took advantage of the repeat-primed PCR (RP-PCR) assay developed in our laboratory (R.K.) for the diagnostic detection of the DM2 expansions (Sallinen et al., 2004; Bachinski et al., 2009). Using this approach, we amplified repeats from a clinically affected, genetically confirmed DM2 patient to produce (TG)n(TCTG)n(CCTG)n repeats with 16 to 189 pure (CCTG)n motifs. Cloned repeats were verified by sequencing to ensure purity of the expanded (CCTG)n repeat tract. In order to express the (CCTG)n repeats in Drosophila, mutant fragments containing 106 repeats with the upstream region were recovered from the TOPO vector and cloned into pUAST (Brand and Perrimon, 1993). The same cloning procedure was used with genomic DNA from a normal individual to generate the control vector containing a normal (CCTG)16 allele. The presence and the length of the (CCTG)n repeats in the pUAST vector were confirmed by sequencing in both directions: DM2-106, (TG)22(TCTG)2(CCTG)106; and N-16, (TG)20(TCTG)12(CCTG)16.
Generation of the MBNL1 expression clone
The human MBNL1 clone was obtained from OriGene (TrueClone accession number NM_021038.3). The plasmid was digested with NotI, to separate the insert from the vector, and with SpeI, to decrease the vector size and distinguish it from the insert. The NotI insert with the entire coding sequence for MBNL1 was then cloned into the NotI site of the pUAST vector. Proper orientation was confirmed by restriction enzyme digestion and sequence analysis from both ends.
Generation of the CUGBP1 expression clone
The human CUGBP1 clone was obtained from OriGene (TrueClone accession number NM_006560.2). This variant is the predominant transcript and encodes isoform 1. To generate the expression clone, we used the same procedure as for the MBNL1 expression clone, except for the SpeI digestion, since insert and vector were readily distinguishable by size in gel electrophoresis.
Fly husbandry
Flies were raised on normal corn agar and crosses were incubated at 25°C. The following mutants and transgenic stocks were used: UAS-[CCTG]16 (control); UAS-[CCTG]106 (DM2-106); UAS-MBNL1; UAS-CUGBP1; UAS-p35; Mhc-Gal4; GMR-Gal4; mblKG08885. Generation and management of DM1 spliceosensor flies was as described (García-Alcover et al., 2014). To simplify crosses, DM2-106 transgenes on chromosome 2 or 3 were recombined with GMR-Gal4 on the same chromosome to yield GMR>DM2-106 on chromosome 2 or 3. Fly eyes were photographed using a Zeiss Axio Imager Z1 compound microscope.
Drug treatment
Fly food was supplemented with drugs at the final concentrations indicated in Figs 7, 8 and Fig. S1. Pentamidine was obtained from Sigma-Aldrich (439843) and PKR-I from Calbiochem (527451). Because PKR-I needs to be dissolved in 100% DMSO, a DMSO-only control was also performed. The same volume of DMSO-containing solutions was mixed into the food.
Reverse transcription PCR (RT-PCR) analysis
For Fhos and INSR splicing assays, total RNA was extracted from ∼50 adult flies with Tri Reagent (Sigma) following the manufacturer's instructions. Contaminating DNA was degraded by RNase-free DNase I (Thermo Scientific). Reverse transcription was performed with SuperScript II reverse transcriptase (Invitrogen) following the manufacturer's guidelines. GoTaq polymerase (Promega) was used for PCR amplification with primers (5′-3′) Fhos-F (GTCATGGAGTCGAGCAGTGA) and Fhos-R (TGTGATGCGGGTATCTACGA), or with primers INSR-F (ACGTTTGAGGATTACCTGCACAA) and INSR-R (GAGATGGCCTGGAACGACAG), in each case for 29 cycles, with an annealing temperature of 60°C (García-Alcover et al., 2014). Band intensity was quantified using ImageJ (NIH).
For quantification of N-16 and DM2-106 transcript levels (Fig. 4H), total RNA was extracted from Drosophila eye imaginal discs using TRIzol reagent (Invitrogen). cDNA conversion was performed using the SuperScript II RNase H-Reverse Transcriptase Kit (Invitrogen). Quantitative PCR (qPCR) was performed using cDNA template and SYBR Green Power Mix (Applied Biosystems). Three sets of primers flanking the CCUG repeats were designed in the pUAST vector used to clone the transgenes: primer set #1, Fwd GTGGTGGAATGCCTTTAAT and Rev GGAGGAGTAGAATGTTGAGA; primer set #2, Fwd AAAGAAGAGAAAGGTAGAAGAC and Rev AGCAAAGCAAGCAAGAG; primer set #3, Fwd CTAGTGATGATGATGAGGCTACT and Rev TAGCAATTCTGAAGGAAAGTC. Transcript levels of Ribosomal protein 49 (Rp49; also known as RpL32) were used for normalization across samples, using primers Fwd ACCAGCTTCAAGATGACCATCC and Rev CTTGTTCGATCCGTAACCGATG.
Fluorescence in situ hybridization (FISH)
FISH analysis was performed as described (Salisbury et al., 2009), except that Drosophila tissue was used. Imaginal discs were imaged by confocal microscopy. RNA-FISH analysis of drug-treated retinae was performed with a (CUGG)10 probe.
For muscle preparation, thoraces of 0- to 5-day-old MHC-Gal4>UAS-(CCTG)106 or MHC-Gal4>UAS-(CCTG)16 females were dissected, embedded in OCT (Fisher HealthCare), frozen in liquid nitrogen and stored at −80°C until processed. At least five 40× magnification images of different focal planes along the z-axis were taken using a Leica DM2500 microscope for DAPI (UV channel) and Cy3 (green channel). The z-planes were stacked using Photoshop (Adobe) and the number of nuclei with foci counted with ImageJ software. At least 50 cells from each individual were counted and at least three individuals were analyzed for each compound. The percentage of cells with foci was compared between MHC-Gal4>UAS-(CCTG)106 and MHC-Gal4>UAS-(CCTG)16.
Luciferase readout
Three 0- to 5-day-old adult flies were placed in each well of a flat-bottom 96-well plate (Daslab, Barcelona, Spain) and homogenized in 150 μl 1× reporter lysis buffer (Promega). Then, 50 μl of the homogenate was transferred to a new white 96-well plate (Sterilin). Lysate luminescence was measured with an Envision plate reader (PerkinElmer) after dispensing 15 μl Luciferase Assay Reagent (Promega) with the Envision injector. At least 60 wells were analyzed for each genotype studied.
Muscle histology
Drosophila thoraces (7-12 days old) were embedded in Epon for semi-thin transverse sectioning as previously described (Tomlinson and Ready, 1987). Relative muscle areas of at least six different thoraces were calculated as described (Garcia-Lopez et al., 2011).
Immunohistochemistry
At least 20 imaginal discs per experiment were dissected from late third instar larvae and pupal retinae from 42-h-old pupae. They were fixed and stained using standard protocols (Fogarty and Bergmann, 2014). TUNEL was performed using a TUNEL assay kit (Roche Life Sciences) according to the manufacturer's instructions. Antibodies to the following primary antigens were used: ELAV [rat; 1:50; Developmental Studies Hybridoma Bank (DSHB)]; Cut (mouse; 1:50; DSHB); Dlg (rabbit; 1:100; from Kwang-Wook Choi, Korea Advanced Institute of Science and Technology, Daejeon, South Korea); MBNL1 (rabbit; 1:2000; from Charles Thornton, University of Rochester Medical Center, Rochester, NY, USA); cleaved Caspase 3 (rabbit; 1:200; Cell Signaling Technology, 9661); and eIF2α (P-Ser51) (rabbit; 1:100; Cell Signaling Technology, 3597). Secondary antibodies were donkey Fab fragments from Jackson ImmunoResearch (715-166-151, 711-096-152, 712-606-153, 711-166-152; all at 1:600). Nuclei were visualized by Hoechst and DAPI staining. Fluorescent images were taken with an Olympus Optical FV500 confocal microscope.
Acknowledgements
We thank Linda Bachinski, Juan Botas, Kwang-Wook Choi, Marek Mlodzik, Bruce Hay, Thomas Cooper, Maurice Swanson, Nicholas Webster, Eric Olson, Charles Thornton, Jillian Lindblad, the Bloomington Drosophila Stock Center and the Developmental Studies Hybridoma Bank, Iowa City, IA, USA, for providing plasmids, fly stocks, antibodies and other reagents. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: R.K., A.B.; Methodology: M.S., T.C.; Formal analysis: V.B.Y., A.A., A.L.C., R.K., A.B.; Investigation: V.B.Y., M.S., A.A., J.C.-B., I.G.-A., M.W., C.B., Z.C., A.L.C.; Writing - original draft: A.B.; Writing - review & editing: A.L.C., R.K., A.B.; Supervision: A.L.C., R.K., A.B.; Funding acquisition: R.K., A.B.
Funding
This work was supported by the National Institute of General Medical Sciences (NIGMS) under award number R35 GM118330 to A.B., and by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) under award number AR48171, the Muscular Dystrophy Association (RG 4321) and the Alice Kleberg Reynolds Foundation for Genetics to R.K. Supported in part by the National Institutes of Health/National Cancer Institute under award number P30 CA016672 and used for the Sequencing and Microarray Facility. Deposited in PMC for immediate release.
Supplementary information
Supplementary information available online at http://dmm.biologists.org/lookup/doi/10.1242/dmm.026179.supplemental
References
- Artero R., Prokop A., Paricio N., Begemann G., Pueyo I., Mlodzik M., Perez-Alonso M. and Baylies M. K. (1998). The muscle blind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev. Biol. 195, 131-143. 10.1006/dbio.1997.8833 [DOI] [PubMed] [Google Scholar]
- Bachinski L. L., Udd B., Meola G., Sansone V., Bassez G., Eymard B., Thornton C. A., Moxley R. T., Harper P. S., Rogers M. T. et al. (2003). Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am. J. Hum. Genet. 73, 835-848. 10.1086/378566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachinski L. L., Czernuszewicz T., Ramagli L. S., Suominen T., Shriver M. D., Udd B., Siciliano M. J. and Krahe R. (2009). Premutation allele pool in myotonic dystrophy type 2. Neurology 72, 490-497. 10.1212/01.wnl.0000333665.01888.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargiela A., Cerro-Herreros E., Fernandez-Costa J. M., Vilchez J. J., Llamusi B. and Artero R. (2015). Increased autophagy and apoptosis contribute to muscle atrophy in a myotonic dystrophy type 1 Drosophila model. Dis. Model. Mech. 8, 679-690. 10.1242/dmm.018127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassez G., Chapoy E., Bastuji-Garin S., Radvanyi-Hoffman H., Authier F.-J., Pellissier J. F., Eymard B. and Gherardi R. K. (2008). Type 2 myotonic dystrophy can be predicted by the combination of type 2 muscle fiber central nucleation and scattered atrophy. J. Neuropathol. Exp. Neurol. 67, 319-325. 10.1097/NEN.0b013e31816b4acc [DOI] [PubMed] [Google Scholar]
- Begemann G., Paricio N., Artero R., Kiss I., Perez-Alonso M. and Mlodzik M. (1997). muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development 124, 4321-4331. [DOI] [PubMed] [Google Scholar]
- Botta A., Vallo L., Rinaldi F., Bonifazi E., Amati F., Biancolella M., Gambardella S., Mancinelli E., Angelini C., Meola G. et al. (2007). Gene expression analysis in myotonic dystrophy: indications for a common molecular pathogenic pathway in DM1 and DM2. Gene Expr. 13, 339-351. 10.3727/000000006781510705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A. H. and Perrimon N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401-415. [DOI] [PubMed] [Google Scholar]
- Chakraborty M., Selma-Soriano E., Magny E., Couso J. P., Perez-Alonso M., Charlet-Berguerand N., Artero R. and Llamusi B. (2015). Pentamidine rescues contractility and rhythmicity in a Drosophila model of myotonic dystrophy heart dysfunction. Dis. Model. Mech. 8, 1569-1578. 10.1242/dmm.021428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day J. W., Ricker K., Jacobsen J. F., Rasmussen L. J., Dick K. A., Kress W., Schneider C., Koch M. C., Beilman G. J., Harrison A. R. et al. (2003). Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology 60, 657-664. 10.1212/01.WNL.0000054481.84978.F9 [DOI] [PubMed] [Google Scholar]
- de Haro M., Al-Ramahi I., De Gouyon B., Ukani L., Rosa A., Faustino N. A., Ashizawa T., Cooper T. A. and Botas J. (2006). MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Hum. Mol. Genet. 15, 2138-2145. 10.1093/hmg/ddl137 [DOI] [PubMed] [Google Scholar]
- Du H., Cline M. S., Osborne R. J., Tuttle D. L., Clark T. A., Donohue J. P., Hall M. P., Shiue L., Swanson M. S., Thornton C. A. et al. (2010). Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 17, 187-193. 10.1038/nsmb.1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S. F., Sirito M., Krahe R. and Sinden R. R. (2009). A Z-DNA sequence reduces slipped-strand structure formation in the myotonic dystrophy type 2 (CCTG) x (CAGG) repeat. Proc. Natl. Acad. Sci. USA 106, 3270-3275. 10.1073/pnas.0807699106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardaei M., Rogers M. T., Thorpe H. M., Larkin K., Hamshere M. G., Harper P. S. and Brook J. D. (2002). Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 11, 805-814. 10.1093/hmg/11.7.805 [DOI] [PubMed] [Google Scholar]
- Farny N. G., Kedersha N. L. and Silver P. A. (2009). Metazoan stress granule assembly is mediated by P-eIF2alpha-dependent and -independent mechanisms. RNA 15, 1814-1821. 10.1261/rna.1684009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty C. E. and Bergmann A. (2014). Detecting caspase activity in Drosophila larval imaginal discs. Methods Mol. Biol. 1133, 109-117. 10.1007/978-1-4939-0357-3_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Alcover I., Lopez Castel A., Perez-Alonso M. and Artero R. (2013). In vivo strategies for drug discovery in myotonic dystrophy disorders. Drug Discov. Today Technol. 10, e97-e102. 10.1016/j.ddtec.2012.02.001 [DOI] [PubMed] [Google Scholar]
- García-Alcover I., Colonques-Bellmunt J., Garijo R., Tormo J. R., Artero R., Álvarez-Abril M. C., López Castel A. and Pérez-Alonso M. (2014). Development of a Drosophila melanogaster spliceosensor system for in vivo high-throughput screening in myotonic dystrophy type 1. Dis. Model. Mech. 7: 1297-1306. 10.1242/dmm.016592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Lopez A., Monferrer L., Garcia-Alcover I., Vicente-Crespo M., Alvarez-Abril M. C. and Artero R. D. (2008). Genetic and chemical modifiers of a CUG toxicity model in Drosophila. PLoS ONE 3, e1595 10.1371/journal.pone.0001595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Lopez A., Llamusi B., Orzaez M., Perez-Paya E. and Artero R. D. (2011). In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc. Natl. Acad. Sci. USA 108, 11866-11871. 10.1073/pnas.1018213108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatchel J. R. and Zoghbi H. Y. (2005). Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 6, 743-755. 10.1038/nrg1691 [DOI] [PubMed] [Google Scholar]
- Harper P. S. (2001). Myotonic Dystrophy, 3rd edn. London, UK: W.B. Saunders. [Google Scholar]
- Hawkins C. J., Yoo S. J., Peterson E. P., Wang S. L., Vernooy S. Y. and Hay B. A. (2000). The Drosophila caspase DRONC cleaves following glutamate or aspartate and is regulated by DIAP1, HID, and GRIM. J. Biol. Chem. 275, 27084-27093. [DOI] [PubMed] [Google Scholar]
- Hay B. A., Wolff T. and Rubin G. M. (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 120, 2121-2129. [DOI] [PubMed] [Google Scholar]
- Holt I., Jacquemin V., Fardaei M., Sewry C. A., Butler-Browne G. S., Furling D., Brook J. D. and Morris G. E. (2009). Muscleblind-like proteins: similarities and differences in normal and myotonic dystrophy muscle. Am. J. Pathol. 174, 216-227. 10.2353/ajpath.2009.080520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huichalaf C., Sakai K., Jin B., Jones K., Wang G.-L., Schoser B., Schneider-Gold C., Sarkar P., Pereira-Smith O. M., Timchenko N. et al. (2010). Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J. 24, 3706-3719. 10.1096/fj.09-151159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H., Mankodi A., Swanson M. S., Moxley R. T. and Thornton C. A. (2004). Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 13, 3079-3088. 10.1093/hmg/ddh327 [DOI] [PubMed] [Google Scholar]
- Jones K., Jin B., Iakova P., Huichalaf C., Sarkar P., Schneider-Gold C., Schoser B., Meola G., Shyu A.-B., Timchenko N. et al. (2011). RNA Foci, CUGBP1, and ZNF9 are the primary targets of the mutant CUG and CCUG repeats expanded in myotonic dystrophies type 1 and type 2. Am. J. Pathol. 179, 2475-2489. 10.1016/j.ajpath.2011.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K., Wei C., Schoser B., Meola G., Timchenko N. and Timchenko L. (2015). Reduction of toxic RNAs in myotonic dystrophies type 1 and type 2 by the RNA helicase p68/DDX5. Proc. Natl. Acad. Sci. USA 112, 8041-8045. 10.1073/pnas.1422273112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia R. N., Johnstone K. A., Mankodi A., Lungu C., Thornton C. A., Esson D., Timmers A. M., Hauswirth W. W. and Swanson M. S. (2003). A muscleblind knockout model for myotonic dystrophy. Science 302, 1978-1980. 10.1126/science.1088583 [DOI] [PubMed] [Google Scholar]
- Kanadia R. N., Shin J., Yuan Y., Beattie S. G., Wheeler T. M., Thornton C. A. and Swanson M. S. (2006). Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA 103, 11748-11753. 10.1073/pnas.0604970103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A. F., Gasnier E. and Furling D. (2011). Gain of RNA function in pathological cases: Focus on myotonic dystrophy. Biochimie 93, 2006-2012. 10.1016/j.biochi.2011.06.028 [DOI] [PubMed] [Google Scholar]
- La Spada A. R. and Taylor J. P. (2010). Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 11, 247-258. 10.1038/nrg2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. E. and Cooper T. A. (2009). Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 37, 1281-1286. 10.1042/BST0371281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori C. L., Ricker K., Moseley M. L., Jacobsen J. F., Kress W., Naylor S. L., Day J. W. and Ranum L. P. (2001). Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293, 864-867. 10.1126/science.1062125 [DOI] [PubMed] [Google Scholar]
- Lucchiari S., Pagliarani S., Corti S., Mancinelli E., Servida M., Fruguglietti E., Sansone V., Moggio M., Bresolin N., Comi G. P. et al. (2008). Colocalization of ribonuclear inclusions with muscle blind like-proteins in a family with myotonic dystrophy type 2 associated with a short CCTG expansion. J. Neurol. Sci. 275, 159-163. 10.1016/j.jns.2008.08.007 [DOI] [PubMed] [Google Scholar]
- Mankodi A., Urbinati C. R., Yuan Q.-P., Moxley R. T., Sansone V., Krym M., Henderson D., Schalling M., Swanson M. S. and Thornton C. A. (2001). Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 10, 2165-2170. 10.1093/hmg/10.19.2165 [DOI] [PubMed] [Google Scholar]
- Mankodi A., Takahashi M. P., Jiang H., Beck C. L., Bowers W. J., Moxley R. T., Cannon S. C. and Thornton C. A. (2002). Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 10, 35-44. 10.1016/S1097-2765(02)00563-4 [DOI] [PubMed] [Google Scholar]
- Mankodi A., Teng-Umnuay P., Krym M., Henderson D., Swanson M. and Thornton C. A. (2003). Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann. Neurol. 54, 760-768. 10.1002/ana.10763 [DOI] [PubMed] [Google Scholar]
- Meier P., Silke J., Leevers S. J. and Evan G. I. (2000). The Drosophila caspase DRONC is regulated by DIAP1. EMBO J. 19, 598-611. 10.1093/emboj/19.4.598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meola G., Jones K., Wei C. and Timchenko L. T. (2013). Dysfunction of protein homeostasis in myotonic dystrophies. Histol. Histopathol. 28, 1089-1098. 10.14670/HH-28.1089 [DOI] [PubMed] [Google Scholar]
- Miller J. W., Urbinati C. R., Teng-Umnuay P., Stenberg M. G., Byrne B. J., Thornton C. A. and Swanson M. S. (2000). Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 19, 4439-4448. 10.1093/emboj/19.17.4439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne R. J. and Thornton C. A. (2006). RNA-dominant diseases. Hum. Mol. Genet. 15, R162-R169. 10.1093/hmg/ddl181 [DOI] [PubMed] [Google Scholar]
- Pascual M., Vicente M., Monferrer L. and Artero R. (2006). The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation 74, 65-80. 10.1111/j.1432-0436.2006.00060.x [DOI] [PubMed] [Google Scholar]
- Philips A. V., Timchenko L. T. and Cooper T. A. (1998). Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280, 737-741. 10.1126/science.280.5364.737 [DOI] [PubMed] [Google Scholar]
- Proud C. G. (2005). eIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 16, 3-12. 10.1016/j.semcdb.2004.11.004 [DOI] [PubMed] [Google Scholar]
- Salisbury E., Schoser B., Schneider-Gold C., Wang G.-L., Huichalaf C., Jin B., Sirito M., Sarkar P., Krahe R., Timchenko N. A. et al. (2009). Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am. J. Pathol. 175, 748-762. 10.2353/ajpath.2009.090047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallinen R., Vihola A., Bachinski L. L., Huoponen K., Haapasalo H., Hackman P., Zhang S., Sirito M., Kalimo H., Meola G. et al. (2004). New methods for molecular diagnosis and demonstration of the (CCTG)n mutation in myotonic dystrophy type 2 (DM2). Neuromuscul. Disord. 14, 274-283. 10.1016/j.nmd.2004.01.002 [DOI] [PubMed] [Google Scholar]
- Santoro M., Masciullo M., Bonvissuto D., Bianchi M. L. E., Michetti F. and Silvestri G. (2013). Alternative splicing of human insulin receptor gene (INSR) in type I and type II skeletal muscle fibers of patients with myotonic dystrophy type 1 and type 2. Mol. Cell. Biochem. 380, 259-265. 10.1007/s11010-013-1681-z [DOI] [PubMed] [Google Scholar]
- Savkur R. S., Philips A. V. and Cooper T. A. (2001). Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 29, 40-47. 10.1038/ng704 [DOI] [PubMed] [Google Scholar]
- Savkur R. S., Philips A. V., Cooper T. A., Dalton J. C., Moseley M. L., Ranum L. P. W. and Day J. W. (2004). Insulin receptor splicing alteration in myotonic dystrophy type 2. Am. J. Hum. Genet. 74, 1309-1313. 10.1086/421528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoser B. and Timchenko L. (2010). Myotonic dystrophies 1 and 2: complex diseases with complex mechanisms. Curr. Genomics 11, 77-90. 10.2174/138920210790886844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicot G. and Gomes-Pereira M. (2013). RNA toxicity in human disease and animal models: from the uncovering of a new mechanism to the development of promising therapies. Biochim. Biophys. Acta 1832, 1390-1409. 10.1016/j.bbadis.2013.03.002 [DOI] [PubMed] [Google Scholar]
- Sicot G., Gourdon G. and Gomes-Pereira M. (2011). Myotonic dystrophy, when simple repeats reveal complex pathogenic entities: new findings and future challenges. Hum. Mol. Genet. 20, R116-R123. 10.1093/hmg/ddr343 [DOI] [PubMed] [Google Scholar]
- Tang Z. Z., Yarotskyy V., Wei L., Sobczak K., Nakamori M., Eichinger K., Moxley R. T., Dirksen R. T. and Thornton C. A. (2012). Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum. Mol. Genet. 21, 1312-1324. 10.1093/hmg/ddr568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton C. A. (2014). Myotonic dystrophy. Neurol. Clin. 32, 705-719, viii 10.1016/j.ncl.2014.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B., White R. J., Xia T., Welle S., Turner D. H., Mathews M. B. and Thornton C. A. (2000). Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA 6, 79-87. 10.1017/S1355838200991544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B., Mukhopadhyay R. and Mathews M. B. (2005). Polymorphic CUG repeats in human mRNAs and their effects on gene expression. RNA Biol. 2, 149-156. 10.4161/rna.2.4.2446 [DOI] [PubMed] [Google Scholar]
- Timchenko L. (2013). Molecular mechanisms of muscle atrophy in myotonic dystrophies. Int. J. Biochem. Cell Biol. 45, 2280-2287. 10.1016/j.biocel.2013.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko N. A., Cai Z.-J., Welm A. L., Reddy S., Ashizawa T. and Timchenko L. T. (2001a). RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J. Biol. Chem. 276, 7820-7826. 10.1074/jbc.M005960200 [DOI] [PubMed] [Google Scholar]
- Timchenko N. A., Iakova P., Cai Z.-J., Smith J. R. and Timchenko L. T. (2001b). Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol. Cell. Biol. 21, 6927-6938. 10.1128/MCB.21.20.6927-6938.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson A. and Ready D. F. (1987). Cell fate in the Drosophila ommatidium. Dev. Biol. 123, 264-275. 10.1016/0012-1606(87)90448-9 [DOI] [PubMed] [Google Scholar]
- Tonevitsky E. A. and Trushkin E. V. (2009). Model for alternative splicing of insulin receptor in myotonic dystrophy type 1. Bull. Exp. Biol. Med. 147, 772-776. 10.1007/s10517-009-0611-2 [DOI] [PubMed] [Google Scholar]
- Udd B. and Krahe R. (2012). The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 11, 891-905. 10.1016/S1474-4422(12)70204-1 [DOI] [PubMed] [Google Scholar]
- Ulane C. M., Teed S. and Sampson J. (2014). Recent advances in myotonic dystrophy type 2. Curr. Neurol. Neurosci. Rep. 14, 429 10.1007/s11910-013-0429-1 [DOI] [PubMed] [Google Scholar]
- Vihola A., Bassez G., Meola G., Zhang S., Haapasalo H., Paetau A., Mancinelli E., Rouche A., Hogrel J. Y., Laforet P. et al. (2003). Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology 60, 1854-1857. 10.1212/01.WNL.0000065898.61358.09 [DOI] [PubMed] [Google Scholar]
- Vihola A., Bachinski L. L., Sirito M., Olufemi S.-E., Hajibashi S., Baggerly K. A., Raheem O., Haapasalo H., Suominen T., Holmlund-Hampf J. et al. (2010). Differences in aberrant expression and splicing of sarcomeric proteins in the myotonic dystrophies DM1 and DM2. Acta Neuropathol. 119, 465-479. 10.1007/s00401-010-0637-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A. J., Rimer M., Killian J. M., Dowling J. J. and Cooper T. A. (2010). CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum. Mol. Genet. 19, 3614-3622. 10.1093/hmg/ddq277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf M. B., Nakamori M., Matthys C. M., Thornton C. A. and Berglund J. A. (2009). Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. USA 106, 18551-18556. 10.1073/pnas.0903234106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler T. M., Lueck J. D., Swanson M. S., Dirksen R. T. and Thornton C. A. (2007). Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J. Clin. Invest. 117, 3952-3957. 10.1172/jci33355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D. D., Pavitt G. D. and Proud C. G. (2001). Characterization of the initiation factor eIF2B and its regulation in Drosophila melanogaster. J. Biol. Chem. 276, 3733-3742. 10.1074/jbc.M008041200 [DOI] [PubMed] [Google Scholar]
- Wojciechowska M., Taylor K., Sobczak K., Napierala M. and Krzyzosiak W. J. (2014). Small molecule kinase inhibitors alleviate different molecular features of myotonic dystrophy type 1. RNA Biol. 11, 742-754. 10.4161/rna.28799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z., Goodman L. D., Shieh S.-Y., Min M., Teng X., Zhu Y. and Bonini N. M. (2015). A fly model for the CCUG-repeat expansion of myotonic dystrophy type 2 reveals a novel interaction with MBNL1. Hum. Mol. Genet. 24, 954-962. 10.1093/hmg/ddu507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Compton S. A., Sobczak K., Stenberg M. G., Thornton C. A., Griffith J. D. and Swanson M. S. (2007). Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 35, 5474-5486. 10.1093/nar/gkm601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T., Gibbens B., Doty N. S., Gomes-Pereira M., Huguet A., Stone M. D., Margolis J., Peterson M., Markowski T. W., Ingram M. A. C. et al. (2011). Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 108, 260-265. 10.1073/pnas.1013343108 [DOI] [PMC free article] [PubMed] [Google Scholar]