

ABSTRACT
Spinal muscular atrophy (SMA) is a devastating neuromuscular disorder characterized by loss of motor neurons and muscle atrophy, generally presenting in childhood. SMA is caused by low levels of the survival motor neuron protein (SMN) due to inactivating mutations in the encoding gene SMN1. A second duplicated gene, SMN2, produces very little but sufficient functional protein for survival. Therapeutic strategies to increase SMN are in clinical trials, and the first SMN2-directed antisense oligonucleotide (ASO) therapy has recently been licensed. However, several factors suggest that complementary strategies may be needed for the long-term maintenance of neuromuscular and other functions in SMA patients. Pre-clinical SMA models demonstrate that the requirement for SMN protein is highest when the structural connections of the neuromuscular system are being established, from late fetal life throughout infancy. Augmenting SMN may not address the slow neurodegenerative process underlying progressive functional decline beyond childhood in less severe types of SMA. Furthermore, individuals receiving SMN-based treatments may be vulnerable to delayed symptoms if rescue of the neuromuscular system is incomplete. Finally, a large number of older patients living with SMA do not fulfill the present criteria for inclusion in gene therapy and ASO clinical trials, and may not benefit from SMN-inducing treatments. Therefore, a comprehensive whole-lifespan approach to SMA therapy is required that includes both SMN-dependent and SMN-independent strategies that treat the CNS and periphery. Here, we review the range of non-SMN pathways implicated in SMA pathophysiology and discuss how various model systems can serve as valuable tools for SMA drug discovery.
KEY WORDS: Animal models, Cellular models, Combinatorial therapies, Skeletal muscle, Spinal muscular atrophy, Survival motor neuron
Summary: Translational research for spinal muscular atrophy (SMA) should address the development of non-CNS and survival motor neuron (SMN)-independent therapeutic approaches to complement and enhance the benefits of CNS-directed and SMN-dependent therapies.
Introduction
Spinal muscular atrophy (SMA) is the most common genetic disease resulting in death in infancy, affecting approximately 1 in 6000 to 1 in 10,000 births (Crawford and Pardo, 1996). This autosomal recessive disorder, resulting from the loss-of-function of the survival motor neuron 1 (SMN1) gene, is characterized by loss of spinal cord motor neurons, muscular atrophy, neuromuscular junction (NMJ) denervation and paralysis (Crawford and Pardo, 1996; Kariya et al., 2008; Kong et al., 2009; Lefebvre et al., 1995; Murray et al., 2008). SMN1 is highly conserved and present as a single copy in the genome of all eukaryotic organisms (Bergin et al., 1997; Miguel-Aliaga et al., 1999, 2000; Paushkin et al., 2000). In humans, however, a genomic duplication has given rise to a second gene, SMN2 (Lefebvre et al., 1995; Rochette et al., 2001). A crucial C-to-T substitution at position 6 of exon 7 in SMN2, which occurs in all individuals, leads to the aberrant splicing of exon 7 and the subsequent production of an unstable SMNΔ7 protein (Lorson et al., 1999; Monani et al., 2000). An important sequence in intron 7 of SMN2, termed intron splicing silence N1 (ISS-N1) has been demonstrated to further favour the exclusion of exon 7 in the transcript (Singh et al., 2006). Thus, the telomeric SMN1 copy gives rise to the full-length (FL) SMN protein while the centromeric SMN2 copy predominantly produces the SMNΔ7 protein. However, the SMN2 gene always generates a small amount of functional protein, which maintains viability, as homozygous deletion of SMN1 is uniformly lethal (Gennarelli et al., 1995; Lefebvre et al., 1995). Deletions or intragenic mutations in SMN1 are found in all forms of SMA, with SMN2 acting to modulate the disease severity through variation in its copy number (Gennarelli et al., 1995; Lefebvre et al., 1995) (Fig. 1). As the number of copies of SMN2 increases, so does the quantity of stable FL-SMN protein produced. Thus, the variation in clinical severity seen in SMA is mostly explained by the total level of residual SMN protein.
Fig. 1.
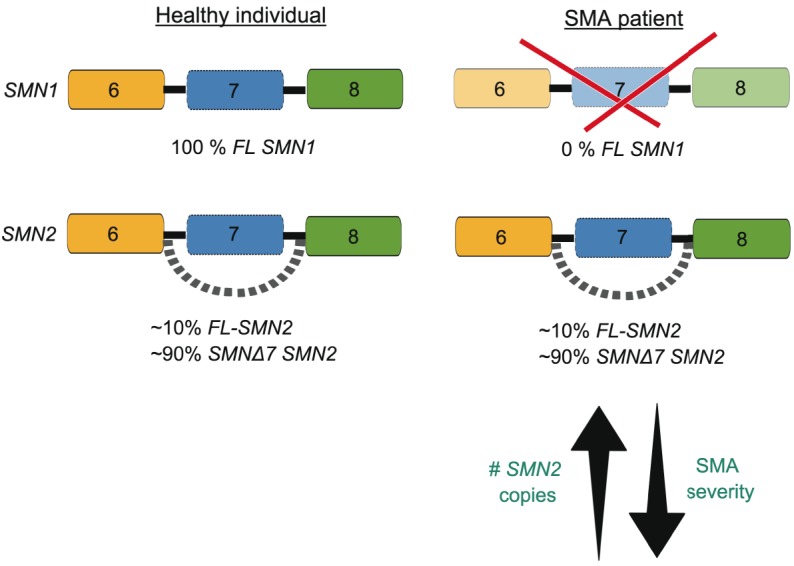
SMN1 and SMN2 contribute to spinal muscular atrophy (SMA). In healthy individuals, the survival motor neuron 1 (SMN1) gene produces 100% full length (FL) SMN protein while the SMN2 gene produces ∼10% FL SMN and ∼90% of a non-functional product that lacks exon 7 (SMNΔ7) due to aberrant alternative splicing. In SMA patients, the SMN1 gene is lost due to mutations or deletions. SMN2 remains and the small amount of FL SMN is sufficient for survival. The number of SMN2 copies correlates with disease severity, with a lower copy number being linked to more severe types of SMA.
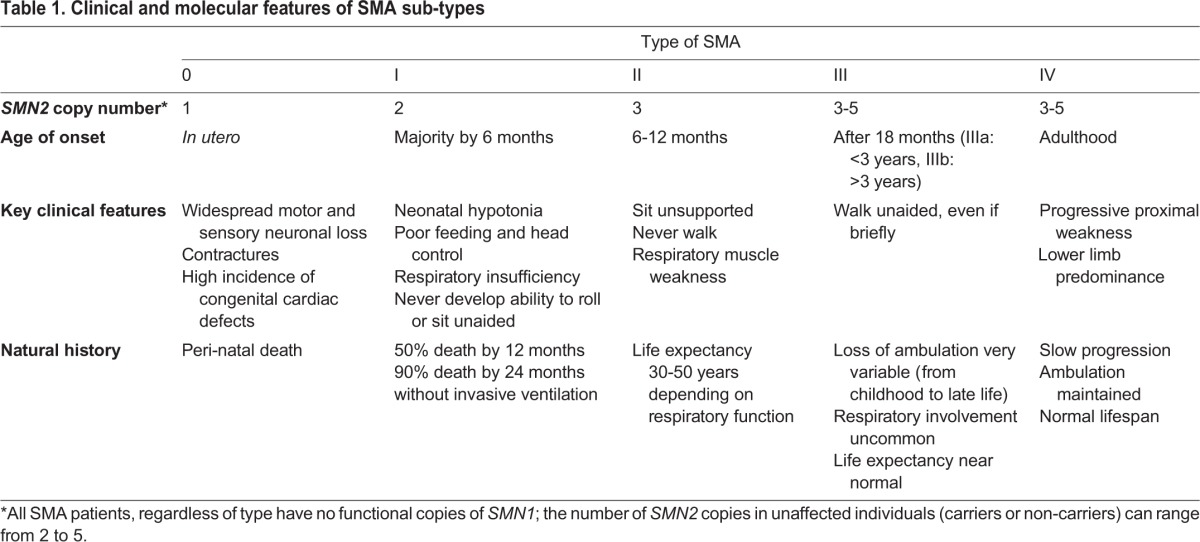
SMA is clinically heterogeneous and has been categorized into five types (0-IV) based on age of onset, severity of motor decline and life expectancy. The term ‘Type 0’, in which the minimal complement of one SMN2 gene is present, describes SMA with a clear in utero onset, arthrygryphosis (limited joint contracture) and complex motor and sensory nerve deficits, and death before or just after birth. Type I SMA patients display the most severe symptoms, with death in infancy if invasive ventilation is not implemented. Types II and III have a later childhood onset and are associated with survival into adulthood, and the potential for a normal lifespan, albeit with considerable physical disability (Munsat and Davies, 1992; Pearn, 1980). The clinical course of Type II and III patients living with SMA is characterized by long periods of relative stability with superimposed periods of accelerated functional decline, for example during the pubertal growth spurt, and a subsequent long period of slowly progressive age-related loss of motor function (Kaufmann et al., 2012). Advances in respiratory, musculoskeletal and nutritional care mean that greater numbers of patients with Type I SMA are surviving beyond infancy. Most SMA Type II patients are living full lives into adulthood and Type III SMA is, in the majority of cases, associated with a normal lifespan (Rudnik-Schöneborn et al., 2001). The clinical and molecular features of the five types of SMA are presented in Table 1. Given the range of severity and ages of onset, it will be necessary for any therapeutic strategy to address the needs of all individuals affected by SMA, from infancy to adulthood.
Table 1.
Clinical and molecular features of SMA sub-types
The SMN protein is ubiquitously expressed and is localized in the cytoplasm (Liu and Dreyfuss, 1996), neuronal growth cones (Fan and Simard, 2002), neuronal extensions (Fallini et al., 2010), the nucleolus (Charroux et al., 2000; Wehner et al., 2002) and in punctate nuclear structures called Gemini of coiled bodies (Gems) and Cajal bodies (Carvalho et al., 1999; Liu and Dreyfuss, 1996). The SMN protein has thus been attributed several key regulatory cellular functions in neuronal cells, including roles in RNA metabolism [specifically small nuclear ribonucleoproteins (snRNPs)] (Li et al., 2014), actin cytoskeleton dynamics (Hensel and Claus, 2017), mRNA transport (Donlin-Asp et al., 2016), ubiquitin homeostasis (Groen and Gillingwater, 2015), bioenergetics pathways (Boyd et al., 2017) and synaptic vesicle release (Kong et al., 2009) (Fig. 2). Importantly, to date, none of these roles has been identified as being solely responsible for SMA pathophysiology.
Fig. 2.

Localization of the survival motor neuron (SMN) protein in neuronal cells and associated general cellular functions. SMN regulates small nuclear ribonucleoprotein (snRNP) biogenesis, maturation and recycling in Gemini of coiled bodies (Gems) and Cajal bodies; ribosome biogenesis in nucleolus; snRNP biogenesis and actin dynamics in the cytoplasm; mRNA transport in axons; actin dynamics and vesicle release in the synpase.
The most advanced therapies currently in clinical trials for SMA are aimed at increasing FL SMN either by exogenously expressing SMN1 or upregulating FL SMN2 production (d'Ydewalle and Sumner, 2015). Unless these current SMN-dependent approaches can be given pre-symptomatically, when motor neuron dysfunction may still be reversible, and delivered with a very high level of efficiency to drastically induce SMN levels in spinal cord motor neurons, it is likely that the progressive neurodegenerative process will not be completely abrogated but simply slowed down. Thus, treated SMA patients may be vulnerable to a delayed deterioration of the neuromuscular system. There are also a large number of older children and adults living with SMA who do not fulfill the present criteria for inclusion (on the grounds of age and various clinical parameters) in the ongoing clinical trials and for whom it is not currently clear that SMN-inducing treatments will be beneficial because of the significant and irreversible SMN-related neuromuscular decline that is already established. Furthermore, it has become evident that SMA pathophysiology extends beyond the neuromuscular system, whereby numerous peripheral organs and tissues demonstrate pathological changes in pre-clinical models and patients (Hamilton and Gillingwater, 2013). Therapies that improve neuromuscular function as well as maintain lifelong general health of people living with SMA are therefore a major priority and an unmet clinical need.
As the first successful SMN-targeted therapeutic approaches are emerging into the clinical arena (Finkel et al., 2016; Gillingwater, 2016), we review here how to best move forward in the development of combinatorial therapeutic approaches for SMA that, ideally, would target the CNS and the periphery, operating via SMN-dependent and SMN-independent processes. We will firstly consider the various existing and alternative experimental models that could be used to identify novel SMA therapeutic targets. We will then discuss how the current SMN-specific compounds presently in clinical trials inform the potential development of treatments aimed at non-SMN targets in non-CNS tissues. Finally, we will expand on the idea of developing drug discovery and delivery approaches that enable systemic delivery of therapies.
Advantages and limitations of current animal and cell models
Animal models
A range of in vivo model systems have been developed to aid understanding of the pathogenesis of SMA and to test the efficacy of therapies. The similarities in anatomy and physiology to the human neuromuscular system, coupled with the ease of genetic manipulation, mean that the mouse has been an extremely valuable model for exploring the basic pathogenesis and evaluating potential treatments for SMA. Various mouse models have been developed over the years, displaying differing ranges of disease severity. While the complete knockout (Smn−/−) is embryonic lethal (Schrank et al., 1997), the heterozygous animals (Smn+/−) do not develop a typical SMA phenotype (Bowerman et al., 2014; Schrank et al., 1997), which is in accordance with data demonstrating that loss of ∼85% of normal Smn levels is required to reflect an SMA phenotype in mice (Bowerman et al., 2012a). Genetic modifications were thus integrated in the Smn–/– mice to allow their survival, whilst still retaining the criteria for an SMA model. The severely afflicted Smn–/–;SMN2 mice (Hsieh-Li et al., 2000; Monani et al., 2000) harbor a human SMN2 transgene that produces the typical ∼15% FL SMN2 transcript, whereas the Smn–/–;SMN2;SMNΔ7/Δ7 transgenic mice (Le et al., 2005) additionally express the partially functional human SMNΔ7 protein, which confers an increase in survival. Both these severe SMA mouse models typically do not survive past the first two post-natal weeks and therefore do not reflect the chronic phase of the disease, making them suitable for modelling only severe infantile SMA. This limitation means that these mice are also unsuitable for the evaluation of non-SMN therapies that may benefit some aspects of SMA pathology. In addition, severe SMA mouse models do not allow for the long-term evaluation of extra-CNS defects that might emerge over time in people living with Type II, III and IV SMA. To address this, animals that have a significantly longer asymptomatic phase have been developed. Smn2B/– mice (Bowerman et al., 2012a; Hammond et al., 2010) are an intermediate model with an average lifespan of 30 days. These mice carry an endogenous mutation within the murine Smn gene that mimics the human SMN2 gene by principally producing SMNΔ7 transcripts. Recently, longer-lived intermediate models were generated by administration of sub-optimal doses of exon 7 inclusion-promoting ISS-N1 antisense oligonucleotides (ASOs, Box 1) to human SMN2-carrying SMA mice (to promote an insufficient increase of FL-SMN for complete rescue) (Hosseinibarkooie et al., 2016; Zhou et al., 2015). Mouse models thus remain an important system for augmenting our understanding of the SMA disease process and in the evaluation of potential therapeutic approaches.
Box 1. Glossary.
AAV: adeno-associated virus. A small DNA virus that infects humans and other primate species without causing disease.
ASO: antisense oligonucleotide. A synthetic polymer, typically 15-20 nucleotides long and complementary to the sense sequence of a target mRNA.
BBB: blood-brain barrier. A highly selective semi-permeable endothelial cell barrier separating the circulating blood from the brain in the central nervous system.
CPP: cell-penetrating peptide. A short peptide that facilitates cellular uptake of molecules.
Discordant: in disagreement.
FDA: Food and Drug Administration of the United States Department of Health and Human Services; responsible for protecting and promoting public health.
Hypomorphic: genetic mutation causing partial loss of function.
Intracerebroventricular: direct injection into the ventricular system (i.e. into the cerebral ventricles) of the brain.
Intrathecal: direct injection into the sub-arachnoid space so that a drug reaches the cerebrospinal fluid.
NMJ: neuromuscular junction. The chemical synapse between a motor neuron and a muscle fibre.
SMN: survival motor neuron protein encoded by the human SMN1 and SMN2 genes that are ubiquitously expressed in most cells and tissues with a range of cell-specific and housekeeping functions.
More targeted hypotheses about the role of SMN and its interaction with specific proteins can be successfully explored in invertebrates, particularly Drosophila (fruit flies) (Chan et al., 2003; Chang et al., 2008) and Caenorhabditis elegans (Burt et al., 2006), and genetically tractable vertebrate model organisms that present a developmental phenotype, such as zebrafish (Hao et al., 2011; McWhorter et al., 2003). However, these models may display phenotypes that differ greatly from what is observed in mouse models and SMA patients. The locomotor and motility defects characterized in Drosophila larval SMA models (Praveen et al., 2012, 2014) can only have an indirect relationship to the disruptions in neuromuscular function that occur in patients. In zebrafish models of SMA generated by antisense morpholinos or maternal zygotic genetic mutations (Hao et al., 2013; McWhorter et al., 2003), developing motor axons and dendrites display outgrowth and branching defects, whereas in mouse and human, SMA motor axons correctly reach the target muscle and form the NMJ, followed by denervation of the muscle as the disease progresses (Ling et al., 2012). Nevertheless, such model systems are more efficient than mammalian models for high-throughput screening and are likely to have significant advantages if utilized efficiently and thoughtfully.
Finally, there have also been initiatives to develop large animal models for SMA, particularly for the evaluation of the delivery, benefits and toxicity of clinical-grade therapeutics. Specifically, endogenous and exogenous genetic modifications have been introduced in the pig to generate a porcine SMA model (Duque et al., 2015; Towne et al., 2010). More work is needed to evaluate whether the pig will become the pre-clinical model of choice for therapeutic assessment.
Cellular models
There are obvious limitations in the extent to which mouse models and the other in vivo models described above might be predictive of effects in humans, owing to inherent species-specific differences in cellular function. The inability to directly study neurons in individuals affected by a neurological disorder such as SMA has been a key impediment to understanding basic pathological mechanisms, particularly those occurring early in the disease process. Recent developments in stem cell technology have substantially expanded the range of cellular models available in motor neuron disease research by allowing the direct observation of pathological mechanisms in neurons derived from induced pluripotent stem cells (iPSCs) obtained from fibroblasts derived from affected individuals. SMA was the first neurological disorder in which a disease-relevant phenotype was demonstrated in iPSC-derived motor neurons (Ebert et al., 2009).
Beyond neurons, other cell types have been demonstrated to contribute to SMA pathology, as described below. iPSCs can be converted into pancreatic and cardiac cells, and thus represent an even more powerful tool, as these cell lines can be studied separately or in co-culture systems that mimic the dynamic interaction that exists in the human body (De Vos et al., 2016). Furthermore, iPSCs can also be used to generate endothelial cells (Lippmann et al., 2014) to model the blood-brain barrier (BBB; Box 1) that protects the CNS, but also limits the entry of therapeutic compounds (Abbott, 2013; Pardridge, 2005), thus allowing screening of novel and established drugs for the ability to cross the BBB as well as compare the properties of SMA and healthy endothelial cells.
iPSC-derived motor neurons can be generated from families in which genotypically matched individuals are discordant for the SMA phenotype, to serve as a tool to identify disease modifiers (Boza-Morán et al., 2015). Similarly, iPSCs can be generated from Type I, II and III patients, allowing the exploration of the impact of small changes in SMN expression at a cellular level. While iPSCs hold a lot of promise, experimental caveats include heterogeneity of the cell populations derived, as well as the limitations of studying cells in culture, isolated from other tissues within the context of the whole organism (Rouhani et al., 2014; De Filippis et al., 2017; Ebert et al., 2012). Nevertheless, iPSC-based models will give new insights into disease mechanisms and will also serve as a screening and validation tool for potential therapies identified in model organisms, especially as part of an experimental workflow designed to identify novel molecular targets and drugs and evaluate their combinatorial potential. Indeed, testing of candidate therapies across multiple platforms will most likely be key to the efficient and successful advancement of complementary therapeutic approaches (Fig. 3).
Fig. 3.

A model experimental plan to determine the therapeutic potential of a candidate non-survival motor neuron (SMN) molecular target or drug, alone and as a combinatorial therapy. Various in vitro and in vivo models such as an in vitro blood-brain barrier (BBB) model, Smn mutant zebrafish, hypomorphic SMA mouse models and induced pluripotent stem cell (iPSC)-dervied cells (e.g. motor neurons) could be used to evaluate different activity and efficiency parameters of the candidate target or drug. Each model system has a particular informative value depending on the candidate target or drug. Finally, the same experimental paradigm should be followed to assess the synergistic or additive value of combining the non-SMN treatment strategy with an SMN-dependent therapy.
Current SMN-targeted trials: early successes
At the forefront of SMA translational research are efforts now entering clinical trials for therapies that promote SMN2 exon 7 inclusion via ISS-N1 inhibition, that increase FL SMN2 transcription (small molecules) or that directly replace SMN1 (viral gene therapy) (Table 2) (d'Ydewalle and Sumner, 2015). Two decades of basic research characterizing the molecular basis of exon 7 splicing (Singh et al., 2017) have recently culminated in the first successful clinical trial of an ASO therapy (nusinersen, commercial name Spinraza) developed and commercialized by Ionis Pharmaceuticals and Biogen (Finkel et al., 2016; Gillingwater, 2016). Not only did intrathecal delivery of nusinersen improve some disease symptoms in Type I patients, but there was also evidence that levels of FL SMN were increased in spinal motor neurons of treated individuals. Although this was an open-label (unblinded) trial, treatment can be cautiously compared favourably to the devastating natural history of Type I SMA patients where the majority of children have died or become ventilator-dependent before the age of 12 months. Whilst the results of an ongoing Phase III study in a larger cohort of patients are awaited, the drug was approved in December 2016 by the United States Food and Drug Administration (US FDA) for all types of SMA based on the strength of the existing data. Likewise, nusinersen was recommended for European Union approval by the EMA in April 2017 and given marketing authorization in June 2017.
Table 2.
SMN-dependent therapies in clinical trials*
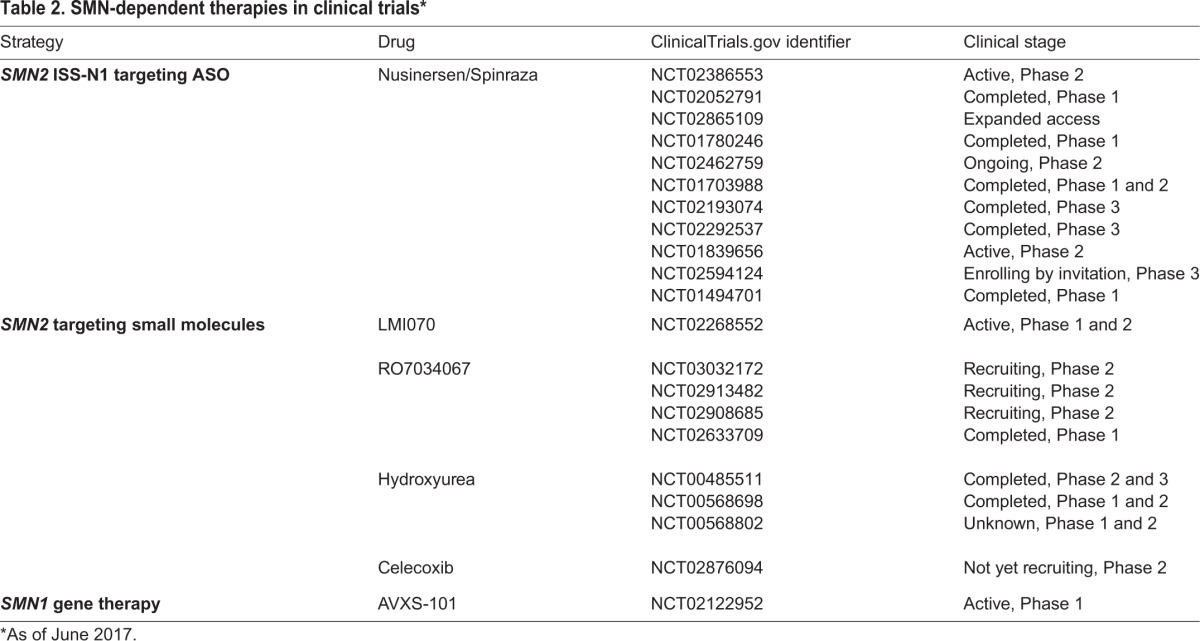
The ASO therapeutic approach, however, requires invasive intrathecal and intracerebroventricular administration for adequate delivery to the CNS, thus not addressing the issue of expression levels of FL SMN required in peripheral tissues, which are also potentially key contributors to SMA pathology (see below). A non-replicating adeno-associated virus (AAV9) vector has been developed by AveXis to deliver a functional copy of a human SMN1 gene. A potential advantage of this approach is that AAV9 crosses the perinatal BBB, allowing a single intravenous dose to ensure widespread systemic delivery. A Phase 1 clinical trial is now underway. Small molecules that increase FL SMN2 expression can also be administered systemically but their ability to cross the BBB may be limited and their exact mode of action needs to be deciphered to minimize potential off-target adverse effects. Small molecules targeting SMN2 developed by Novartis Pharmaceuticals and Hoffmann-La Roche are currently in clinical trials.
Given that nusinersen and other small molecules act on SMN2, the evaluation of their activity in pre-clinical studies was limited to severe SMN2-harboring mice and patient fibroblasts (Pinard et al., 2017; Ratni et al., 2016; Singh et al., 2017). Similarly, the efficiency of AAV9-SMN1 to increase SMN expression was demonstrated in mice with severe SMA (Armbruster et al., 2016; Passini et al., 2010). Whilst use of additional models such as Drosophila and zebrafish may not be relevant in these instances, assessing the activity of SMN-dependent approaches in iPSC-derived motor neurons, in vitro BBB models and other cell types may help shed light on cell-specific activity and efficiency of these various drugs, which could help in the optimization of their respective dosing regimens.
Expanding the repertoire of targets to non-neuronal tissues
Although SMA has generally been considered as an archetypal disorder of selective motor neuron vulnerability, there is now abundant evidence that other tissues and cells are either overtly or sub-clinically affected in SMA patients and animal models (Hamilton and Gillingwater, 2013), including skeletal muscle (Boyer et al., 2013; Martínez-Hernández et al., 2009; Mutsaers et al., 2011; Walker et al., 2008), pancreas (Bowerman et al., 2012c, 2014), liver (Bowerman et al., 2012c, 2014; Szunyogova et al., 2016), spleen (Deguise et al., 2017; Thomson et al., 2017), vasculature (Somers et al., 2012, 2016), heart (Bevan et al., 2010; Heier et al., 2010; Shababi et al., 2010) and Schwann cells (Aghamaleky Sarvestany et al., 2014; Hao et al., 2015; Hunter et al., 2014, 2016). The clinical implications of these observations is that treatment modalities that solely target the nervous system might be an inadequate long-term therapeutic strategy for SMA, especially in Type I patients where the consequences of low levels of SMN for the function of non-CNS tissues beyond infancy is unknown.
To date, a single clinical trial provides evidence for the potential benefit of non-SMN therapies in the chronic phase of SMA. Olesoxime is an orally active drug, which is predicted to have favourable bioenergetics effects as it acts on the outer mitochondrial membrane to modulate the permeability transition pore opening in response to oxidative stress. It has a neuroprotective effect in a range of in vitro and in vivo models (Bordet et al., 2010). A recent clinical trial in SMA Type II and III (non-ambulatory) patients did not achieve its primary endpoint, although it showed some evidence of disease-stabilising effects using secondary endpoints (Bertini et al., 2017).
Pharmacological compounds aimed at specifically targeting skeletal muscle are presently the only non-SMN, non-CNS drugs in clinical trials for SMA patients. In particular, Cytokinetics Inc. has developed a fast skeletal troponin activator (CK-2127107) that is currently in a Phase 2 study in SMA patients. Troponin mediates muscle contraction (Gresslien and Agewall, 2016), and is abnormally distributed in skeletal muscle of Type I-III SMA patients (Stevens et al., 2008). Interestingly, CK-2127107 has not been evaluated in either pre-clinical or Phase 1 clinical stages as a therapeutic strategy for SMA. Instead, the drug was investigated in a rat model of heart failure, where its oral administration resulted in an overall amelioration of muscle endurance and performance following exercise (Hwee et al., 2015). A first generation form of the compound (tirasemtiv or CK-2017357) was also assessed in rodent neuromuscular models for nemaline myopathy (Lee et al., 2013), myasthenia gravis (Russell et al., 2012) and amyotrophic lateral sclerosis (Hwee et al., 2014), demonstrating a significant improvement in muscle strength in each. Nevertheless, the FDA has recently granted the Orphan Drug designation to CK-2127107 for the treatment of SMA patients.
Another interesting muscle target is the myostatin-follistatin pathway, in which myostatin acts as a negative regulator of muscle growth (McPherron et al., 1997) and is itself inhibited by follistatin (Lee and McPherron, 2001). Several therapeutics strategies aimed at modulating this signaling cascade to promote muscle mass have therefore been evaluated in pathologies characterized by muscle atrophy, including SMA (Rodino-Klapac et al., 2009). Administration of recombinant follistatin to Smn–/–;SMN2;SMNΔ7/Δ7 mice resulted in significant improvement in muscle mass, gross motor function and lifespan. However, inhibition of myostatin by genetic (overexpression of follistatin) or pharmacological [soluble activin receptor IIB (ActRIIB-Fc) that inhibits the myostatin-promoting receptor] interventions had no obvious beneficial effects on the phenotype of the same mouse model (Sumner et al., 2009). Similar negative results were obtained upon genetically deleting myostatin in severe SMA mice (Rindt et al., 2012). Conversely, the recent intraperitoneal administration of a soluble form of ActRIIB encoded by an AAV2/8 viral vector in a milder model of SMA improved mass, contractile properties and size of SMN-depleted muscles (Liu et al., 2016). Results from SMA mouse studies have therefore been varied and the discrepancies may be due to the differing severities of the models used, the developmental timing of the approach and the specific targeting/delivery strategy utilized. Several strategies based on the myostatin-follistatin pathway are currently in clinical trials for muscle pathologies such as Duchenne muscular dystrophy (DMD), Becker muscular dystrophy and inclusion body myositis but have yet to be initiated for SMA. However, a recent report shows that serum and muscle biopsies from SMA patients display decreased expression of myostatin and increased levels of follistatin (Mariot et al., 2017), suggesting that additional mechanistic insight in the relevance of targeting this pathway for SMA therapy is required.
Taken together, these findings highlight an important need to better understand the intrinsic pathologies not only in SMA neurons but also in muscle and other non-CNS afflicted organs, so that cell- and tissue-specific treatments can be developed and eventually used in combination with SMN- and CNS-targeted strategies.
Identifying non-SMN targets to develop combinatorial therapeutic approaches
SMN-dependent gene therapies will require administration as early as possible, even pre-symptomatically, to exert the maximum effect (Bevan et al., 2010; Foust et al., 2010; Valori et al., 2010), and at present can be expected to reduce disease severity rather than effect a complete cure. In the absence of a routine screening program for newborns, the potential benefit may also be limited by the delay in diagnosis in milder forms, which generally have an insidious onset. An important step forward would be to develop therapeutic approaches targeting pathways that reflect the chronic pathological process in SMA, facilitating treatments that are adjunctive to SMN replacement therapy to improve and maintain neuromuscular integrity and function throughout the life of the individual.
One of the first SMN-independent targets to prove beneficial with respect to potential SMA therapy is the RhoA-ROCK pathway. The small GTPase RhoA and its downstream effector, the Ser/Thr protein kinase ROCK, are key modulators of actin dynamics (Luo et al., 1997). It has been demonstrated that the RhoA-ROCK pathway is aberrantly upregulated in SMN-depleted rodent neuronal cells (Bowerman et al., 2007) and in the spinal cord and skeletal muscle of Smn2B/– mice (Bowerman et al., 2010, 2012b). Importantly, it was shown that pharmacological inhibition of ROCK significantly increases lifespan and muscle pathology of Smn2B/– SMA mice (Bowerman et al., 2010, 2012b). Additional investigators have further confirmed the contribution of the RhoA-ROCK pathway to SMA in neuronal cells (Hensel et al., 2014), patient fibroblasts (Nölle et al., 2011) and glial cells (Caraballo-Miralles et al., 2012). The tumour suppressor protein PTEN is a member of the protein tyrosine phosphatase family that can regulate cell migration, spreading and growth (Lachyankar et al., 2000; Li et al., 2003; Sano et al., 1999; Tamura et al., 1999). Interestingly, PTEN is phosphorylated by ROCK (Bermúdez Brito et al., 2015), thus leading to increased PTEN inhibitory activity on neuronal survival. While PTEN activity in SMA mice has yet to be investigated, we can hypothesize that the increased activity of the RhoA-ROCK pathway reported in SMA mice (Bowerman et al., 2010, 2012b) induces the increased phosphorylation of PTEN. Concordantly, it has been found that suppressing PTEN in SMA mice through a gene therapy approach led to improvements in NMJ pathology and a significant extension in lifespan (Little et al., 2015). Combined, these studies have highlighted actin modulators as potential targets for combinatorial therapeutic approaches for SMA.
Further regulators of actin dynamics have emerged as potential therapeutic targets for SMA. Plastin 3 (PLS3) is an actin-bundling protein that was identified as a modifier of disease severity in an investigation of discordant family members that carried the same SMN1 mutations (Oprea et al., 2008). Additional analyses of serum (Yanyan et al., 2014) and iPSC-derived motor neurons from SMA patients have further supported the influence of plastin 3 levels on disease progression in certain families but not others (Boza-Morán et al., 2015; Heesen et al., 2016). Indeed, overexpression of plastin 3 in a zebrafish model of SMA significantly rescues the axonal growth and branching defects caused by smn1 gene depletion (Oprea et al., 2008). Further analysis of smn1 mutant zebrafish reveals that reduced SMN levels lead to decreased plastin 3 protein expression, NMJ defects and aberrant motor function, and these effects can be corrected by plastin 3 overexpression (Hao et al., 2012). More recently, studies in mice have shown that increased expression of plastin 3 delays axonal degeneration and improves NMJ function (Ackermann et al., 2013) as well as ameliorates survival and neuromuscular phenotype (Kaifer et al., 2017), possibly through the modulation of endocytic pathways (Hosseinibarkooie et al., 2016). However, it must be noted that a number of animal and patient studies do not reflect the suggested modifying powers of plastin 3 on SMA pathogenesis (McGovern et al., 2015; Stratigopoulos et al., 2010), highlighting the complex relationship that may exist between SMN and plastin 3. The pathological relevance and therapeutic importance of non-SMN targets can be highly dependent on the severity of the disease (Kaifer et al., 2017) and as such, should be evaluated in hypomorphic models, whether transgenically or pharmacologically induced. Nonetheless, the studies on RhoA-ROCK, PTEN and plastin 3 have highlighted actin modulators as potential targets for combinatorial therapeutic approaches for SMA.
Subsequent work has also revealed other promising non-SMN targets, including chondrolectin. It has been shown that this transmembrane protein (encoded by Chodl) and its binding partners are potential modifiers of axonal integrity in SMA mice and that altered expression of Chodl is found in spinal motor neurons of SMA mice (Bäumer et al., 2009). Importantly, increasing the expression of chodl rescues motor neuron outgrowth defects in a zebrafish model of SMA (Sleigh et al., 2014). Experiments in mouse models are underway to fully evaluate the therapeutic potential of Chodl modulation.
As described above, one of the housekeeping functions of SMN is the regulation of RNA metabolism, particularly in the biogenesis of snRNPs (Li et al., 2014), essential components of the RNA splicing machinery (Will and Lührmann, 2001). It has been demonstrated that SMN depletion specifically impacts the activity of the spliceosome complex containing the U12 snRNP (Doktor et al., 2017; Gabanella et al., 2007) and that the Drosophila stasimon (stas) gene is a direct target of U12-dependent splicing (Lotti et al., 2012). Stasimon (also known as Tmem41b in mammals) plays a role in synaptic transmission in neuronal synapses and its expression is significantly reduced and splicing similarly altered in motor and sensory neurons of SMA mice (Lotti et al., 2012). Importantly, overexpression of stas restored neurotransmitter release in Drosophila smn mutants and rescued motor axon growth and branching defects in an SMA zebrafish model (Lotti et al., 2012).
The activity of cyclin-dependent kinase 5 (Cdk5) has been reported to be upregulated in SMA mice and patient iPSC-derived motor neurons (Miller et al., 2015). The increased abundance of Cdk5 is responsible for the pathological hyperphosphorylation of the tau protein in SMN-depleted neuronal cells (Miller et al., 2015). A transgenic approach was used to completely knockout Cdk5 expression in SMA mice, which led to significant rescue of motor neuron synaptic stripping, motor neuron death and NMJ denervation (Miller et al., 2015). Interestingly, a recent unbiased RNA-sequencing-based assessment of global gene changes in SMN-depleted mouse tissues confirmed the specific missplicing of U12 snRNP-dependent genes, several of which are Ca2+ channel genes and may be upstream regulators of Cdk5 activity (Doktor et al., 2017), thus further highlighting Cdk5 as a potential pathological effector in SMA.
Finally, the ubiquitin-like modifier activating enzyme Uba1 (Groen and Gillingwater, 2015) and its downstream effectors [including the Wnt signaling effector β-catenin (Ctnnb1)], have been identified as major targets acting downstream of SMN to regulate neuromuscular and systemic pathology in SMA. Reduced levels of Uba1 were reported in all tissues and organs investigated from SMA mouse models (Aghamaleky Sarvestany et al., 2014; Wishart et al., 2014). Furthermore, β-catenin, which accumulates in neuromuscular tissues in SMA, has been uncovered as a key downstream target of Uba1 deficiencies in SMA. Importantly, pharmacological inhibition of β-catenin dramatically ameliorated neuromuscular pathology in zebrafish, Drosophila and mouse models of SMA (Wishart et al., 2014) while systemic Uba1 gene therapy increased survival and improved neuromuscular and peripheral pathology of SMA mice (Powis et al., 2016).
Thus, a range of pathways are already candidates for non-SMN therapy approaches. As the list of molecular effectors grows, drug-screening approaches to identify pharmacological compounds that can modulate them will be essential. In addition, identifying novel targets should combine proteomics and transcriptomics studies with genome-based network analysis and drug-repositioning strategies. Combinatorial experimental paradigms should then be put in place to evaluate the therapeutic potential of a ‘cocktail treatment’ comprising SMN gene therapy and a non-SMN-targeting drug, optimally making use of the multiple in vitro and in vivo models discussed above (Fig. 3). While several non-SMN pathways and molecular targets have been highlighted as being aberrantly regulated in SMA models and display therapeutic potential, these studies remain, for the most part, in the pre-clinical discovery phase, in contrast to SMN-dependent strategies that are quickly dominating the clinical trial landscape. For non-SMN treatments to become a practical reality in the combinatorial approach paradigm, an efficient strategic plan needs to be established to facilitate their transition to the clinic.
Improving systemic delivery of drugs to target CNS and non-CNS tissues
While motor neurons are undoubtedly the primary cellular target in SMA (Powis and Gillingwater, 2016), cumulative evidence highlights the role of other cells and tissues that may be clinically or sub-clinically affected. However, most of these studies have investigated these tissues or cells independently of the others. The hierarchal contribution of each to Type 0-IV SMA therefore remains unclear. While current gene therapies in clinical trials are promising, nusinersen (Ionis Pharmaceuticals/Biogen) delivery circumvents the peripheral tissues and organs by being injected directly to the CNS, and, although AAV9-SMN1 gene therapy (AveXis) can be delivered systemically, multiple rounds of administration might not be possible due to immunogenicity (Basner-Tschakarjan and Mingozzi, 2014). In both of these cases, this raises the risk of incomplete rescue of SMN deficiency in peripheral organs and the potential for development of non-CNS pathologies later in life.
Development of novel therapeutic approaches targeting non-SMN targets should therefore include careful consideration of both CNS and systemic delivery methods. The optimal dosing regimen for a pharmacological compound should balance its ability to target all relevant tissues with the need to make therapy as non-invasive as possible. An option for systemic delivery of molecules is to conjugate them with a vehicle that can transport them across the membrane of multiple cell types. This has proven efficient for the delivery of ASOs under the neutrally charged chemistry of phosphorodiamidate morpholino (PMO) (Douglas and Wood, 2013). Cell-penetrating peptides (CPPs) have previously been shown to cross both plasma and endosomal membranes (Mitchell et al., 2000). One such peptide-conjugated PMO has been developed, termed peptide nucleic acids/PMO internalization peptide 6a (Pip6a)-PMO, that efficiently modulates splicing in various tissues of a DMD mouse model (Betts et al., 2012). Importantly, it is delivered via a single intravenous (IV) injection. Recently, it has been reported that conjugation of Pip6a to the SMN2 ISS-N1 PMO results in dramatic improvements in survival and neuromuscular phenotype associated with increased FL-SMN levels in both CNS and peripheral tissues (Hammond et al., 2016). CPPs therefore have significant potential to facilitate targeting of SMN to the whole body as well the eventual delivery of therapeutic non-SMN targets or drugs in a similar fashion.
Concluding remarks
Gene therapy and ASO approaches to increase SMN levels are now entering the clinical arena. In the severest form (Type I) of SMA, promising preliminary results must be balanced with a full appreciation of the potential limitations of such strategies. The value of SMN-based therapies in older Type II and III patients is unclear and it may be some time before these can be accurately evaluated. Translational research should therefore address the development of non-CNS and SMN-independent therapeutic approaches to complement and enhance the benefits of CNS-directed and SMN-dependent therapies, taking into account the need to maintain the neuromuscular system of an SMA patient through childhood and puberty, when there is maximal growth of the axial skeleton, and into adult life when the process of age-related attrition of motor units is likely to contribute to progressive loss of motor function (Fig. 4).
Fig. 4.

Overview of the natural history of spinal muscular atrophy (SMA), major developmental milestones and treatment strategies. Although the precise details of SMN expression in the developing human nervous system are difficult to study, evidence from animal models suggests that SMN levels peak in the period of maximum neuromuscular development and then decline to a stable low level. This means that there are different windows of opportunity for the various types of proposed therapies to be effectively employed. Whether combinatorial therapies might be particularly applicable in the more chronic phase of SMA or from the outset is a priority area for research. ASO therapy in utero will be dependent on an optimal route of delivery, which at present remains unclear.
There remains a need for the use of various in vitro and in vivo models as well as molecular high-throughput approaches for the rapid identification of new targets and drugs. It will be crucial to develop tools to evaluate the effects of combination pharmacological therapies at different disease stages. It is therefore of utmost importance that the SMA research and clinical community, as well as those living with SMA, recognise the need to develop and test combinatorial therapeutic approaches that can be effectively delivered systemically and target both SMN and non-SMN molecular effectors. This will allow for a better understanding of the tissue requirements for SMN and non-SMN treatments and, ultimately, provide the best therapeutic strategy for SMA. As with most chronic progressive neurodegenerative disorders, it is likely that, once loss of neuronal integrity has been initiated, combinatorial approaches to therapy will be required to maintain neuromuscular health throughout life.
Acknowledgements
The work of the UK SMA Research Consortium is funded by The SMA Trust. For details of members of the UK SMA Research Consortium, see www.smatrust.org/research/uk-sma-research-consortium/meet-the-uk-sma-research-consortium-team/. We thank Lynn Ossher for graphical contributions. M.B. is an SMA Trust Career Development Fellow.
Footnotes
This article is part of a special subject collection ‘Neurodegeneration: from Models to Mechanisms to Therapies’, which was launched in a dedicated issue guest edited by Aaron Gitler and James Shorter. See related articles in this collection at http://dmm.biologists.org/collection/neurodegenerative-disorders.
Competing interests
The authors declare no competing or financial interests.
Funding
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
References
- Abbott N. J. (2013). Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 36, 437-449. 10.1007/s10545-013-9608-0 [DOI] [PubMed] [Google Scholar]
- Ackermann B., Kröber S., Torres-Benito L., Borgmann A., Peters M., Hosseini Barkooie S. M., Tejero R., Jakubik M., Schreml J., Milbradt J. et al. (2013). Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 22, 1328-1347. 10.1093/hmg/dds540 [DOI] [PubMed] [Google Scholar]
- Aghamaleky Sarvestany A., Hunter G., Tavendale A., Lamont D. J., Llavero Hurtado M., Graham L. C., Wishart T. M. and Gillingwater T. H. (2014). Label-free quantitative proteomic profiling identifies disruption of ubiquitin homeostasis as a key driver of Schwann cell defects in spinal muscular atrophy. J. Proteome Res. 13, 4546-4557. 10.1021/pr500492j [DOI] [PubMed] [Google Scholar]
- Armbruster N., Lattanzi A., Jeavons M., Van Wittenberghe L., Gjata B., Marais T., Martin S., Vignaud A., Voit T., Mavilio F. et al. (2016). Efficacy and biodistribution analysis of intracerebroventricular administration of an optimized scAAV9-SMN1 vector in a mouse model of spinal muscular atrophy. Mol. Ther. Methods Clin. Dev. 3, 16060 10.1038/mtm.2016.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basner-Tschakarjan E. and Mingozzi F. (2014). Cell-mediated immunity to AAV vectors, evolving concepts and potential solutions. Front. Immunol. 5, 350 10.3389/fimmu.2014.00350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumer D., Lee S., Nicholson G., Davies J. L., Parkinson N. J., Murray L. M., Gillingwater T. H., Ansorge O., Davies K. E. and Talbot K. (2009). Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 5, e1000773 10.1371/journal.pgen.1000773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergin A., Kim G., Price D. L., Sisodia S. S., Lee M. K. and Rabin B. A. (1997). Identification and characterization of a mouse homologue of the spinal muscular atrophy-determining gene, survival motor neuron. Gene 204, 47-53. 10.1016/S0378-1119(97)00510-6 [DOI] [PubMed] [Google Scholar]
- Bermúdez Brito M., Goulielmaki E. and Papakonstanti E. A. (2015). Focus on PTEN regulation. Front. Oncol. 5, 166 10.3389/fonc.2015.00166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini E., Dessaud E., Mercuri E., Muntoni F., Kirschner J., Reid C., Lusakowska A., Comi G. P., Cuisset J.-M., Abitbol J.-L. et al. (2017). Safety and efficacy of olesoxime in patients with type 2 or non-ambulatory type 3 spinal muscular atrophy: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 16, 513-522. 10.1016/S1474-4422(17)30085-6 [DOI] [PubMed] [Google Scholar]
- Betts C., Saleh A. F., Arzumanov A. A., Hammond S. M., Godfrey C., Coursindel T., Gait M. J. and Wood M. J. A. (2012). Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Mol. Ther. Nucleic Acids 1, e38 10.1038/mtna.2012.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan A. K., Hutchinson K. R., Foust K. D., Braun L., McGovern V. L., Schmelzer L., Ward J. G., Petruska J. C., Lucchesi P. A., Burghes A. H. M. et al. (2010). Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum. Mol. Genet. 19, 3895-3905. 10.1093/hmg/ddq300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordet T., Berna P., Abitbol J.-L. and Pruss R. M. (2010). Olesoxime (TRO19622): a novel mitochondrial-targeted neuroprotective compound. Pharm. Basel Switz. 3, 345-368. 10.3390/ph3020345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman M., Shafey D. and Kothary R. (2007). Smn depletion alters profilin II expression and leads to upregulation of the RhoA/ROCK pathway and defects in neuronal integrity. J. Mol. Neurosci. MN 32, 120-131. 10.1007/s12031-007-0024-5 [DOI] [PubMed] [Google Scholar]
- Bowerman M., Beauvais A., Anderson C. L. and Kothary R. (2010). Rho-kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum. Mol. Genet. 19, 1468-1478. 10.1093/hmg/ddq021 [DOI] [PubMed] [Google Scholar]
- Bowerman M., Murray L. M., Beauvais A., Pinheiro B. and Kothary R. (2012a). A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscul. Disord. NMD 22, 263-276. 10.1016/j.nmd.2011.09.007 [DOI] [PubMed] [Google Scholar]
- Bowerman M., Murray L. M., Boyer J. G., Anderson C. L. and Kothary R. (2012b). Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Med. 10, 24 10.1186/1741-7015-10-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman M., Swoboda K. J., Michalski J.-P., Wang G.-S., Reeks C., Beauvais A., Murphy K., Woulfe J., Screaton R. A., Scott F. W. et al. (2012c). Glucose metabolism and pancreatic defects in spinal muscular atrophy. Ann. Neurol. 72, 256-268. 10.1002/ana.23582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman M., Michalski J.-P., Beauvais A., Murray L. M., DeRepentigny Y. and Kothary R. (2014). Defects in pancreatic development and glucose metabolism in SMN-depleted mice independent of canonical spinal muscular atrophy neuromuscular pathology. Hum. Mol. Genet. 23, 3432-3444. 10.1093/hmg/ddu052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd P. J., Tu W.-Y., Shorrock H. K., Groen E. J. N., Carter R. N., Powis R. A., Thomson S. R., Thomson D., Graham L. C., Motyl A. A. L. et al. (2017). Bioenergetic status modulates motor neuron vulnerability and pathogenesis in a zebrafish model of spinal muscular atrophy. PLoS Genet. 13, e1006744 10.1371/journal.pgen.1006744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer J. G., Murray L. M., Scott K., De Repentigny Y., Renaud J.-M. and Kothary R. (2013). Early onset muscle weakness and disruption of muscle proteins in mouse models of spinal muscular atrophy. Skelet. Muscle 3, 24 10.1186/2044-5040-3-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boza-Morán M. G., Martínez-Hernández R., Bernal S., Wanisch K., Also-Rallo E., Le Heron A., Alías L., Denis C., Girard M., Yee J.-K. et al. (2015). Decay in survival motor neuron and plastin 3 levels during differentiation of iPSC-derived human motor neurons. Sci. Rep. 5, 11696 10.1038/srep11696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt E. C., Towers P. R. and Sattelle D. B. (2006). Caenorhabditis elegans in the study of SMN-interacting proteins: a role for SMI-1, an orthologue of human Gemin2 and the identification of novel components of the SMN complex. Invert. Neurosci. 6, 145-159. 10.1007/s10158-006-0027-x [DOI] [PubMed] [Google Scholar]
- Caraballo-Miralles V., Cardona-Rossinyol A., Garcera A., Villalonga P., Soler R. M., Olmos G. and Lladó J. (2012). SMN deficiency attenuates migration of U87MG astroglioma cells through the activation of RhoA. Mol. Cell. Neurosci. 49, 282-289. 10.1016/j.mcn.2011.12.003 [DOI] [PubMed] [Google Scholar]
- Carvalho T., Almeida F., Calapez A., Lafarga M., Berciano M. T. and Carmo-Fonseca M. (1999). The spinal muscular atrophy disease gene product, SMN: A link between snRNP biogenesis and the Cajal (coiled) body. J. Cell Biol. 147, 715-728. 10.1083/jcb.147.4.715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan Y. B., Miguel-Aliaga I., Franks C., Thomas N., Trülzsch B., Sattelle D. B., Davies K. E. and van den Heuvel M. (2003). Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum. Mol. Genet. 12, 1367-1376. 10.1093/hmg/ddg157 [DOI] [PubMed] [Google Scholar]
- Chang H. C.-H., Dimlich D. N., Yokokura T., Mukherjee A., Kankel M. W., Sen A., Sridhar V., Fulga T. A., Hart A. C., Van Vactor D. et al. (2008). Modeling spinal muscular atrophy in Drosophila. PLoS ONE 3, e3209 10.1371/journal.pone.0003209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charroux B., Pellizzoni L., Perkinson R. A., Yong J., Shevchenko A., Mann M. and Dreyfuss G. (2000). Gemin4. A novel component of the SMN complex that is found in both gems and nucleoli. J. Cell Biol. 148, 1177-1186. 10.1083/jcb.148.6.1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford T. O. and Pardo C. A. (1996). The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 3, 97-110. 10.1006/nbdi.1996.0010 [DOI] [PubMed] [Google Scholar]
- De Filippis L., Zalfa C. and Ferrari D. (2017). Neural Stem Cells and Human induced pluripotent stem cells to model rare CNS diseases. CNS Neurol. Disord. Drug Targets doi:10.2174/1871527316666170615121753 [Epub ahead of print] 10.2174/1871527316666170615121753 [DOI] [PubMed] [Google Scholar]
- Deguise M.-O., De Repentigny Y., McFall E., Auclair N., Sad S. and Kothary R. (2017). Immune dysregulation may contribute to disease pathogenesis in spinal muscular atrophy mice. Hum. Mol. Genet 26, 801-819. 10.1093/hmg/ddw434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos J., Bouckenheimer J., Sansac C., Lemaître J.-M. and Assou S. (2016). Human induced pluripotent stem cells: a disruptive innovation. Curr. Res. Transl. Med. 64, 91-96. 10.1016/j.retram.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Doktor T. K., Hua Y., Andersen H. S., Brøner S., Liu Y. H., Wieckowska A., Dembic M., Bruun G. H., Krainer A. R. and Andresen B. S. (2017). RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns. Nucleic Acids Res. 45, 395-416. 10.1093/nar/gkw731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlin-Asp P. G., Bassell G. J. and Rossoll W. (2016). A role for the survival of motor neuron protein in mRNP assembly and transport. Curr. Opin. Neurobiol. 39, 53-61. 10.1016/j.conb.2016.04.004 [DOI] [PubMed] [Google Scholar]
- Douglas A. G. L. and Wood M. J. A. (2013). Splicing therapy for neuromuscular disease. Mol. Cell. Neurosci. 56, 169-185. 10.1016/j.mcn.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duque S. I., Arnold W. D., Odermatt P., Li X., Porensky P. N., Schmelzer L., Meyer K., Kolb S. J., Schümperli D., Kaspar B. K. et al. (2015). A large animal model of Spinal Muscular Atrophy and correction of phenotype. Ann. Neurol. 77, 399-414. 10.1002/ana.24332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Ydewalle C. and Sumner C. J. (2015). Spinal muscular atrophy therapeutics: where do we stand? Neurother. J. Am. Soc. Exp. Neurother. 12, 303-316. 10.1007/s13311-015-0337-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert A. D., Yu J., Rose F. F., Mattis V. B., Lorson C. L., Thomson J. A. and Svendsen C. N. (2009). Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277-280. 10.1038/nature07677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert A. D., Liang P. and Wu J. C. (2012). Induced pluripotent stem cells as a disease modeling and drug screening platform. J. Cardiovasc. Pharmacol. 60, 408-416. 10.1097/FJC.0b013e318247f642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C., Bassell G. J. and Rossoll W. (2010). High-efficiency transfection of cultured primary motor neurons to study protein localization, trafficking, and function. Mol. Neurodegener. 5, 17 10.1186/1750-1326-5-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L. and Simard L. R. (2002). Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum. Mol. Genet. 11, 1605-1614. 10.1093/hmg/11.14.1605 [DOI] [PubMed] [Google Scholar]
- Finkel R. S., Chiriboga C. A., Vajsar J., Day J. W., Montes J., De Vivo D. C., Yamashita M., Rigo F., Hung G., Schneider E. et al. (2016). Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet Lond. Engl. 388, 3017-3026. 10.1016/S0140-6736(16)31408-8 [DOI] [PubMed] [Google Scholar]
- Foust K. D., Wang X., McGovern V. L., Braun L., Bevan A. K., Haidet A. M., Le T. T., Morales P. R., Rich M. M., Burghes A. H. M. et al. (2010). Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 28, 271-274. 10.1038/nbt.1610 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gabanella F., Butchbach M. E. R., Saieva L., Carissimi C., Burghes A. H. M. and Pellizzoni L. (2007). Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE 2, e921 10.1371/journal.pone.0000921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarelli M., Lucarelli M., Capon F., Pizzuti A., Merlini L., Angelini C., Novelli G. and Dallapiccola B. (1995). Survival motor-neuron gene transcript analysis in muscles from spinal muscular-atrophy patients. Biochem. Biophys. Res. Commun. 213, 342-348. 10.1006/bbrc.1995.2135 [DOI] [PubMed] [Google Scholar]
- Gillingwater T. H. (2016). Dawn of a new therapeutic era for spinal muscular atrophy. Lancet Lond. Engl. 388, 2964-2965. 10.1016/S0140-6736(16)32390-X [DOI] [PubMed] [Google Scholar]
- Gresslien T. and Agewall S. (2016). Troponin and exercise. Int. J. Cardiol. 221, 609-621. 10.1016/j.ijcard.2016.06.243 [DOI] [PubMed] [Google Scholar]
- Groen E. J. N. and Gillingwater T. H. (2015). UBA1: at the crossroads of ubiquitin homeostasis and neurodegeneration. Trends Mol. Med. 21, 622-632. 10.1016/j.molmed.2015.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton G. and Gillingwater T. H. (2013). Spinal muscular atrophy: going beyond the motor neuron. Trends Mol. Med. 19, 40-50. 10.1016/j.molmed.2012.11.002 [DOI] [PubMed] [Google Scholar]
- Hammond S. M., Gogliotti R. G., Rao V., Beauvais A., Kothary R. and DiDonato C. J. (2010). Mouse survival motor neuron alleles that mimic SMN2 splicing and are inducible rescue embryonic lethality early in development but not late. PLoS ONE 5, e15887 10.1371/journal.pone.0015887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond S. M., Hazell G., Shabanpoor F., Saleh A. F., Bowerman M., Sleigh J. N., Meijboom K. E., Zhou H., Muntoni F., Talbot K. et al. (2016). Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 113, 10962-10967. 10.1073/pnas.1605731113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L. T., Burghes A. H. M. and Beattie C. E. (2011). Generation and Characterization of a genetic zebrafish model of SMA carrying the human SMN2 gene. Mol. Neurodegener. 6, 24 10.1186/1750-1326-6-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L. T., Wolman M., Granato M. and Beattie C. E. (2012). Survival motor neuron affects plastin 3 protein levels leading to motor defects. J. Neurosci. Off. J. Soc. Neurosci. 32, 5074-5084. 10.1523/JNEUROSCI.5808-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L. T., Duy P. Q., Jontes J. D., Wolman M., Granato M. and Beattie C. E. (2013). Temporal requirement for SMN in motoneuron development. Hum. Mol. Genet. 22, 2612-2625. 10.1093/hmg/ddt110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L. T., Duy P. Q., Jontes J. D. and Beattie C. E. (2015). Motoneuron development influences dorsal root ganglia survival and Schwann cell development in a vertebrate model of spinal muscular atrophy. Hum. Mol. Genet. 24, 346-360. 10.1093/hmg/ddu447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heesen L., Peitz M., Torres-Benito L., Hölker I., Hupperich K., Dobrindt K., Jungverdorben J., Ritzenhofen S., Weykopf B., Eckert D. et al. (2016). Plastin 3 is upregulated in iPSC-derived motoneurons from asymptomatic SMN1-deleted individuals. Cell. Mol. Life Sci. CMLS 73, 2089-2104. 10.1007/s00018-015-2084-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heier C. R., Satta R., Lutz C. and DiDonato C. J. (2010). Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum. Mol. Genet. 19, 3906-3918. 10.1093/hmg/ddq330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensel N. and Claus P. (2017). The actin cytoskeleton in SMA and ALS: how does it contribute to motoneuron degeneration? Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry. doi: 10.1177/1073858417705059 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Hensel N., Stockbrügger I., Rademacher S., Broughton N., Brinkmann H., Grothe C. and Claus P. (2014). Bilateral crosstalk of rho- and extracellular-signal-regulated-kinase (ERK) pathways is confined to an unidirectional mode in spinal muscular atrophy (SMA). Cell. Signal. 26, 540-548. 10.1016/j.cellsig.2013.11.027 [DOI] [PubMed] [Google Scholar]
- Hosseinibarkooie S., Peters M., Torres-Benito L., Rastetter R. H., Hupperich K., Hoffmann A., Mendoza-Ferreira N., Kaczmarek A., Janzen E., Milbradt J. et al. (2016). The power of human protective modifiers: PLS3 and CORO1C unravel impaired endocytosis in spinal muscular atrophy and rescue SMA phenotype. Am. J. Hum. Genet. 99, 647-665. 10.1016/j.ajhg.2016.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh-Li H. M., Chang J.-G., Jong Y.-J., Wu M.-H., Wang N.-M., Tsai C. H. and Li H. (2000). A mouse model for spinal muscular atrophy. Nat. Genet. 24, 66-70. 10.1038/71709 [DOI] [PubMed] [Google Scholar]
- Hunter G., Aghamaleky Sarvestany A., Roche S. L., Symes R. C. and Gillingwater T. H. (2014). SMN-dependent intrinsic defects in Schwann cells in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 23, 2235-2250. 10.1093/hmg/ddt612 [DOI] [PubMed] [Google Scholar]
- Hunter G., Powis R. A., Jones R. A., Groen E. J. N., Shorrock H. K., Lane F. M., Zheng Y., Sherman D. L., Brophy P. J. and Gillingwater T. H. (2016). Restoration of SMN in Schwann cells reverses myelination defects and improves neuromuscular function in spinal muscular atrophy. Hum. Mol. Genet. 25, 2853-2861. 10.1093/hmg/ddw141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwee D. T., Kennedy A., Ryans J., Russell A. J., Jia Z., Hinken A. C., Morgans D. J., Malik F. I. and Jasper J. R. (2014). Fast skeletal muscle troponin activator tirasemtiv increases muscle function and performance in the B6SJL-SOD1G93A ALS mouse model. PLoS ONE 9, e96921 10.1371/journal.pone.0096921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwee D. T., Kennedy A. R., Hartman J. J., Ryans J., Durham N., Malik F. I. and Jasper J. R. (2015). The small-molecule fast skeletal troponin activator, CK-2127107, improves exercise tolerance in a rat model of heart failure. J. Pharmacol. Exp. Ther. 353, 159-168. 10.1124/jpet.114.222224 [DOI] [PubMed] [Google Scholar]
- Kaifer K. A., Villalón E., Osman E. Y., Glascock J. J., Arnold L. L., Cornelison D. D. W. and Lorson C. L. (2017). Plastin-3 extends survival and reduces severity in mouse models of spinal muscular atrophy. JCI Insight 2, e89970 10.1172/jci.insight.89970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya S., Park G.-H., Maeno-Hikichi Y., Leykekhman O., Lutz C., Arkovitz M. S., Landmesser L. T. and Monani U. R. (2008). Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 17, 2552-2569. 10.1093/hmg/ddn156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann P., McDermott M. P., Darras B. T., Finkel R. S., Sproule D. M., Kang P. B., Oskoui M., Constantinescu A., Gooch C. L., Foley A. R. et al. (2012). Prospective cohort study of spinal muscular atrophy types 2 and 3. Neurology 79, 1889-1897. 10.1212/WNL.0b013e318271f7e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L., Wang X., Choe D. W., Polley M., Burnett B. G., Bosch-Marcé M., Griffin J. W., Rich M. M. and Sumner C. J. (2009). Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. Off. J. Soc. Neurosci. 29, 842-851. 10.1523/JNEUROSCI.4434-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachyankar M. B., Sultana N., Schonhoff C. M., Mitra P., Poluha W., Lambert S., Quesenberry P. J., Litofsky N. S., Recht L. D., Nabi R. et al. (2000). A role for nuclear PTEN in neuronal differentiation. J. Neurosci. Off. J. Soc. Neurosci. 20, 1404-1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le T. T., Pham L. T., Butchbach M. E. R., Zhang H. L., Monani U. R., Coovert D. D., Gavrilina T. O., Xing L., Bassell G. J. and Burghes A. H. M. (2005). SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 14, 845-857. 10.1093/hmg/ddi078 [DOI] [PubMed] [Google Scholar]
- Lee S.-J. and McPherron A. C. (2001). Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 98, 9306-9311. 10.1073/pnas.151270098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E.-J., De Winter J. M., Buck D., Jasper J. R., Malik F. I., Labeit S., Ottenheijm C. A. and Granzier H. (2013). Fast skeletal muscle troponin activation increases force of mouse fast skeletal muscle and ameliorates weakness due to nebulin-deficiency. PLoS ONE 8, e55861 10.1371/journal.pone.0055861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S., Bürglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P. and Zeviani M. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155-165. 10.1016/0092-8674(95)90460-3 [DOI] [PubMed] [Google Scholar]
- Li L., Liu F. and Ross A. H. (2003). PTEN regulation of neural development and CNS stem cells. J. Cell. Biochem. 88, 24-28. 10.1002/jcb.10312 [DOI] [PubMed] [Google Scholar]
- Li D. K., Tisdale S., Lotti F. and Pellizzoni L. (2014). SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin. Cell Dev. Biol. 32, 22-29. 10.1016/j.semcdb.2014.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling K. K. Y., Gibbs R. M., Feng Z. and Ko C.-P. (2012). Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 21, 185-195. 10.1093/hmg/ddr453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann E. S., Al-Ahmad A., Azarin S. M., Palecek S. P. and Shusta E. V. (2014). A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci. Rep. 4, 4160 10.1038/srep04160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little D., Valori C. F., Mutsaers C. A., Bennett E. J., Wyles M., Sharrack B., Shaw P. J., Gillingwater T. H., Azzouz M. and Ning K. (2015). PTEN depletion decreases disease severity and modestly prolongs survival in a mouse model of spinal muscular atrophy. Mol. Ther. J. Am. Soc. Gene Ther. 23, 270-277. 10.1038/mt.2014.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. and Dreyfuss G. (1996). A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 15, 3555-3565. [PMC free article] [PubMed] [Google Scholar]
- Liu M., Hammers D. W., Barton E. R. and Sweeney H. L. (2016). Activin receptor type IIB inhibition improves muscle phenotype and function in a mouse model of spinal muscular atrophy. PLoS ONE 11, e0166803 10.1371/journal.pone.0166803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson C. L., Hahnen E., Androphy E. J. and Wirth B. (1999). A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 96, 6307-6311. 10.1073/pnas.96.11.6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti F., Imlach W. L., Saieva L., Beck E. S., Hao L. T., Li D. K., Jiao W., Mentis G. Z., Beattie C. E., McCabe B. D. et al. (2012). An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151, 440-454. 10.1016/j.cell.2012.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L., Jan L. Y. and Jan Y.-N. (1997). Rho family small GTP-binding proteins in growth cone signalling. Curr. Opin. Neurobiol. 7, 81-86. 10.1016/S0959-4388(97)80124-9 [DOI] [PubMed] [Google Scholar]
- Mariot V., Joubert R., Hourdé C., Servais L., Hanna M. G., Maisonobe T., Muntoni F., Féasson L., Panse R. L., Benvensite O. et al. (2017). Myostatin inhibition for neuromuscular disorders: defining the good candidate. Neuromuscul. Disord. 27, S8 10.1016/S0960-8966(17)30240-7 [DOI] [Google Scholar]
- Martínez-Hernández R., Soler-Botija C., Also E., Alias L., Caselles L., Gich I., Bernal S. and Tizzano E. F. (2009). The developmental pattern of myotubes in spinal muscular atrophy indicates prenatal delay of muscle maturation. J. Neuropathol. Exp. Neurol. 68, 474-481. 10.1097/NEN.0b013e3181a10ea1 [DOI] [PubMed] [Google Scholar]
- McGovern V. L., Massoni-Laporte A., Wang X., Le T. T., Le H. T., Beattie C. E., Rich M. M. and Burghes A. H. M. (2015). Plastin 3 expression does not modify spinal muscular atrophy severity in the Δ7 SMA mouse. PLoS ONE 10, e0132364 10.1371/journal.pone.0132364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron A. C., Lawler A. M. and Lee S.-J. (1997). Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387, 83-90. 10.1038/387083a0 [DOI] [PubMed] [Google Scholar]
- McWhorter M. L., Monani U. R., Burghes A. H. M. and Beattie C. E. (2003). Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 162, 919-931. 10.1083/jcb.200303168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel-Aliaga I., Culetto E., Walker D. S., Baylis H. A., Sattelle D. B. and Davies K. E. (1999). The Caenorhabditis elegans orthologue of the human gene responsible for spinal muscular atrophy is a maternal product critical for germline maturation and embryonic viability. Hum. Mol. Genet. 8, 2133-2143. 10.1093/hmg/8.12.2133 [DOI] [PubMed] [Google Scholar]
- Miguel-Aliaga I., Chan Y. B., Davies K. E. and van den Heuvel M. (2000). Disruption of SMN function by ectopic expression of the human SMN gene in Drosophila. FEBS Lett. 486, 99-102. 10.1016/S0014-5793(00)02243-2 [DOI] [PubMed] [Google Scholar]
- Miller N., Feng Z., Edens B. M., Yang B., Shi H., Sze C. C., Hong B. T., Su S. C., Cantu J. A., Topczewski J. et al. (2015). Non-aggregating tau phosphorylation by cyclin-dependent kinase 5 contributes to motor neuron degeneration in spinal muscular atrophy. J. Neurosci. Off. J. Soc. Neurosci. 35, 6038-6050. 10.1523/JNEUROSCI.3716-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell D. J., Kim D. T., Steinman L., Fathman C. G. and Rothbard J. B. (2000). Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. Off. J. Am. Pept. Soc. 56, 318-325. 10.1034/j.1399-3011.2000.00723.x [DOI] [PubMed] [Google Scholar]
- Monani U. R., Sendtner M., Coovert D. D., Parsons D. W., Andreassi C., Le T. T., Jablonka S., Schrank B., Rossoll W., Rossol W. et al. (2000). The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 9, 333-339. 10.1093/hmg/9.3.333 [DOI] [PubMed] [Google Scholar]
- Munsat T. L. and Davies K. E. (1992). International SMA consortium meeting. (26-28 June 1992, Bonn, Germany). Neuromuscul. Disord. NMD 2, 423-428. 10.1016/S0960-8966(06)80015-5 [DOI] [PubMed] [Google Scholar]
- Murray L. M., Comley L. H., Thomson D., Parkinson N., Talbot K. and Gillingwater T. H. (2008). Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 17, 949-962. 10.1093/hmg/ddm367 [DOI] [PubMed] [Google Scholar]
- Mutsaers C. A., Wishart T. M., Lamont D. J., Riessland M., Schreml J., Comley L. H., Murray L. M., Parson S. H., Lochmüller H., Wirth B. et al. (2011). Reversible molecular pathology of skeletal muscle in spinal muscular atrophy. Hum. Mol. Genet. 20, 4334-4344. 10.1093/hmg/ddr360 [DOI] [PubMed] [Google Scholar]
- Nölle A., Zeug A., van Bergeijk J., Tönges L., Gerhard R., Brinkmann H., Al Rayes S., Hensel N., Schill Y., Apkhazava D. et al. (2011). The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profilin. Hum. Mol. Genet. 20, 4865-4878. 10.1093/hmg/ddr425 [DOI] [PubMed] [Google Scholar]
- Oprea G. E., Kröber S., McWhorter M. L., Rossoll W., Müller S., Krawczak M., Bassell G. J., Beattie C. E. and Wirth B. (2008). Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science 320, 524-527. 10.1126/science.1155085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge W. M. (2005). The blood-brain barrier: bottleneck in brain drug development. NeuroRx J. Am. Soc. Exp. Neurother. 2, 3-14. 10.1602/neurorx.2.1.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini M. A., Bu J., Roskelley E. M., Richards A. M., Sardi S. P., O'Riordan C. R., Klinger K. W., Shihabuddin L. S. and Cheng S. H. (2010). CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Invest. 120, 1253-1264. 10.1172/JCI41615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paushkin S., Charroux B., Abel L., Perkinson R. A., Pellizzoni L. and Dreyfuss G. (2000). The survival motor neuron protein of Schizosacharomyces pombe. Conservation of survival motor neuron interaction domains in divergent organisms. J. Biol. Chem. 275, 23841-23846. 10.1074/jbc.M001441200 [DOI] [PubMed] [Google Scholar]
- Pearn J. (1980). Classification of spinal muscular atrophies. Lancet Lond. Engl. 1, 919-922. 10.1016/S0140-6736(80)90847-8 [DOI] [PubMed] [Google Scholar]
- Pinard E., Green L., Reutlinger M., Weetall M., Naryshkin N. A., Baird J., Chen K. S., Paushkin S. V., Metzger F. and Ratni H. (2017). Discovery of a novel class of survival motor neuron 2 splicing modifiers for the treatment of spinal muscular atrophy. J. Med. Chem. 60, 4444-4457. 10.1021/acs.jmedchem.7b00406 [DOI] [PubMed] [Google Scholar]
- Powis R. A. and Gillingwater T. H. (2016). Selective loss of alpha motor neurons with sparing of gamma motor neurons and spinal cord cholinergic neurons in a mouse model of spinal muscular atrophy. J. Anat. 228, 443-451. 10.1111/joa.12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis R. A., Karyka E., Boyd P., Côme J., Jones R. A., Zheng Y., Szunyogova E., Groen E. J. N., Hunter G., Thomson D. et al. (2016). Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight 1, e87908 10.1172/jci.insight.87908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praveen K., Wen Y. and Matera A. G. (2012). A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. Cell Rep. 1, 624-631. 10.1016/j.celrep.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praveen K., Wen Y., Gray K. M., Noto J. J., Patlolla A. R., Van Duyne G. D. and Matera A. G. (2014). SMA-causing missense mutations in survival motor neuron (Smn) display a wide range of phenotypes when modeled in drosophila. PLoS Genet. 10, e1004489 10.1371/journal.pgen.1004489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratni H., Karp G. M., Weetall M., Naryshkin N. A., Paushkin S. V., Chen K. S., McCarthy K. D., Qi H., Turpoff A., Woll M. G. et al. (2016). Specific correction of alternative survival motor neuron 2 splicing by small molecules: discovery of a potential novel medicine to treat spinal muscular atrophy. J. Med. Chem. 59, 6086-6100. 10.1021/acs.jmedchem.6b00459 [DOI] [PubMed] [Google Scholar]
- Rindt H., Buckley D. M., Vale S. M., Krogman M., Rose F. F., Garcia M. L. and Lorson C. L. (2012). Transgenic inactivation of murine myostatin does not decrease the severity of disease in a model of Spinal Muscular Atrophy. Neuromuscul. Disord. NMD 22, 277-285. 10.1016/j.nmd.2011.10.012 [DOI] [PubMed] [Google Scholar]
- Rochette C. F., Gilbert N. and Simard L. R. (2001). SMN gene duplication and the emergence of the SMN2 gene occurred in distinct hominids: SMN2 is unique to Homo sapiens. Hum. Genet. 108, 255-266. 10.1007/s004390100473 [DOI] [PubMed] [Google Scholar]
- Rodino-Klapac L. R., Haidet A. M., Kota J., Handy C., Kaspar B. K. and Mendell J. R. (2009). Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve 39, 283-296. 10.1002/mus.21244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouhani F., Kumasaka N., de Brito M. C., Bradley A., Vallier L. and Gaffney D. (2014). Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 10, e1004432 10.1371/journal.pgen.1004432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnik-Schöneborn S., Hausmanowa-Petrusewicz I., Borkowska J. and Zerres K. (2001). The predictive value of achieved motor milestones assessed in 441 patients with infantile spinal muscular atrophy types II and III. Eur. Neurol. 45, 174-181. 10.1159/000052118 [DOI] [PubMed] [Google Scholar]
- Russell A. J., Hartman J. J., Hinken A. C., Muci A. R., Kawas R., Driscoll L., Godinez G., Lee K. H., Marquez D., Browne W. F. et al. (2012). Activation of fast skeletal muscle troponin as a potential therapeutic approach for treating neuromuscular diseases. Nat. Med. 18, 452-455. 10.1038/nm.2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano T., Lin H., Chen X., Langford L. A., Koul D., Bondy M. L., Hess K. R., Myers J. N., Hong Y. K., Yung W. K. et al. (1999). Differential expression of MMAC/PTEN in glioblastoma multiforme: relationship to localization and prognosis. Cancer Res. 59, 1820-1824. [PubMed] [Google Scholar]
- Schrank B., Götz R., Gunnersen J. M., Ure J. M., Toyka K. V., Smith A. G. and Sendtner M. (1997). Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc. Natl. Acad. Sci. USA 94, 9920-9925. 10.1073/pnas.94.18.9920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shababi M., Habibi J., Yang H. T., Vale S. M., Sewell W. A. and Lorson C. L. (2010). Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum. Mol. Genet. 19, 4059-4071. 10.1093/hmg/ddq329 [DOI] [PubMed] [Google Scholar]
- Singh N. K., Singh N. N., Androphy E. J. and Singh R. N. (2006). Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 26, 1333-1346. doi: 10.1038/gt.2017.34 [Epub ahead of print]. 10.1128/MCB.26.4.1333-1346.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N. N., Howell M. D., Androphy E. J. and Singh R. N. (2017). How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy. Gene Ther. doi: 10.1038/gt.2017.34 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh J. N., Barreiro-Iglesias A., Oliver P. L., Biba A., Becker T., Davies K. E., Becker C. G. and Talbot K. (2014). Chondrolectin affects cell survival and neuronal outgrowth in in vitro and in vivo models of spinal muscular atrophy. Hum. Mol. Genet. 23, 855-869. 10.1093/hmg/ddt477 [DOI] [PubMed] [Google Scholar]
- Somers E., Stencel Z., Wishart T. M., Gillingwater T. H. and Parson S. H. (2012). Density, calibre and ramification of muscle capillaries are altered in a mouse model of severe spinal muscular atrophy. Neuromuscul. Disord. NMD 22, 435-442. 10.1016/j.nmd.2011.10.021 [DOI] [PubMed] [Google Scholar]
- Somers E., Lees R. D., Hoban K., Sleigh J. N., Zhou H., Muntoni F., Talbot K., Gillingwater T. H. and Parson S. H. (2016). Vascular defects and spinal cord hypoxia in spinal muscular atrophy. Ann. Neurol. 79, 217-230. 10.1002/ana.24549 [DOI] [PubMed] [Google Scholar]
- Stevens L., Bastide B., Maurage C. A., Dupont E., Montel V., Cieniewski-Bernard C., Cuisset J. M., Vallée L. and Mounier Y. (2008). Childhood spinal muscular atrophy induces alterations in contractile and regulatory protein isoform expressions. Neuropathol. Appl. Neurobiol. 34, 659-670. 10.1111/j.1365-2990.2008.00950.x [DOI] [PubMed] [Google Scholar]
- Stratigopoulos G., Lanzano P., Deng L., Guo J., Kaufmann P., Darras B., Finkel R., Tawil R., McDermott M. P., Martens W. et al. (2010). Association of plastin 3 expression with disease severity in spinal muscular atrophy only in postpubertal females. Arch. Neurol. 67, 1252-1256. 10.1001/archneurol.2010.239 [DOI] [PubMed] [Google Scholar]
- Sumner C. J., Wee C. D., Warsing L. C., Choe D. W., Ng A. S., Lutz C. and Wagner K. R. (2009). Inhibition of myostatin does not ameliorate disease features of severe spinal muscular atrophy mice. Hum. Mol. Genet. 18, 3145-3152. 10.1093/hmg/ddp253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szunyogova E., Zhou H., Maxwell G. K., Powis R. A., Francesco M., Gillingwater T. H. and Parson S. H. (2016). Survival motor neuron (SMN) protein is required for normal mouse liver development. Sci. Rep. 6, 34635 10.1038/srep34635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M., Gu J., Danen E. H. J., Takino T., Miyamoto S. and Yamada K. M. (1999). PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway. J. Biol. Chem. 274, 20693-20703. 10.1074/jbc.274.29.20693 [DOI] [PubMed] [Google Scholar]
- Thomson A. K., Somers E., Powis R. A., Shorrock H. K., Murphy K., Swoboda K. J., Gillingwater T. H. and Parson S. H. (2017). Survival of motor neurone protein is required for normal postnatal development of the spleen. J. Anat. 230, 337-346. 10.1111/joa.12546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towne C., Schneider B. L., Kieran D., Redmond D. E. and Aebischer P. (2010). Efficient transduction of non-human primate motor neurons after intramuscular delivery of recombinant AAV serotype 6. Gene Ther. 17, 141-146. 10.1038/gt.2009.119 [DOI] [PubMed] [Google Scholar]
- Valori C. F., Ning K., Wyles M., Mead R. J., Grierson A. J., Shaw P. J. and Azzouz M. (2010). Systemic delivery of scAAV9 expressing SMN prolongs survival in a model of spinal muscular atrophy. Sci. Transl. Med. 2, 35ra42 10.1126/scitranslmed.3000830 [DOI] [PubMed] [Google Scholar]
- Walker M. P., Rajendra T. K., Saieva L., Fuentes J. L., Pellizzoni L. and Matera A. G. (2008). SMN complex localizes to the sarcomeric Z-disc and is a proteolytic target of calpain. Hum. Mol. Genet. 17, 3399-3410. 10.1093/hmg/ddn234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner K. A., Ayala L., Kim Y., Young P. J., Hosler B. A., Lorson C. L., Baserga S. J. and Francis J. W. (2002). Survival motor neuron protein in the nucleolus of mammalian neurons. Brain Res. 945, 160-173. 10.1016/S0006-8993(02)02750-6 [DOI] [PubMed] [Google Scholar]
- Will C. L. and Lührmann R. (2001). Spliceosomal UsnRNP biogenesis, structure and function. Curr. Opin. Cell Biol. 13, 290-301. 10.1016/S0955-0674(00)00211-8 [DOI] [PubMed] [Google Scholar]
- Wishart T. M., Mutsaers C. A., Riessland M., Reimer M. M., Hunter G., Hannam M. L., Eaton S. L., Fuller H. R., Roche S. L., Somers E. et al. (2014). Dysregulation of ubiquitin homeostasis and β-catenin signaling promote spinal muscular atrophy. J. Clin. Invest. 124, 1821-1834. 10.1172/JCI71318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanyan C., Yujin Q., Jinli B., Yuwei J., Hong W. and Fang S. (2014). Correlation of PLS3 expression with disease severity in children with spinal muscular atrophy. J. Hum. Genet. 59, 24-27. 10.1038/jhg.2013.111 [DOI] [PubMed] [Google Scholar]
- Zhou H., Meng J., Marrosu E., Janghra N., Morgan J. and Muntoni F. (2015). Repeated low doses of morpholino antisense oligomer: an intermediate mouse model of spinal muscular atrophy to explore the window of therapeutic response. Hum. Mol. Genet. 24, 6265-6277. 10.1093/hmg/ddv329 [DOI] [PMC free article] [PubMed] [Google Scholar]