

Abstract
Macromolecular structure is governed by the strict rules of stereochemistry. Several approaches to the validation of the correctness of the interpretation of crystallographic and NMR data that underlie the models deposited in the PDB are utilized in practice. The stereochemical rules applicable to macromolecular structures are discussed in this chapter. Practical, computer-based methods and tools of verification of how well the models adhere to those established structural principles to assure their quality are summarized.
Keywords: Crystal structure, NMR structure, Ramachandran plot, bond lengths, bond angles, quality check, geometrical criteria, Protein Data Bank (PDB)
1 Introduction
Biological macromolecules (proteins, as well as other biopolymers, such as nucleic acids and carbohydrates) are composed of atoms belonging to a very limited number of elements (primarily carbon, nitrogen, oxygen and hydrogen, and to a lesser extent sulfur and phosphorus). A few other elements, for example selenium, are occasionally present in the covalent structure of biologically relevant macromolecules, whereas other elements, primarily metal cations, are frequently coordinated to atoms belonging to the macromolecule, or are located within functional groups that may be either coordinated or covalently attached to their macromolecular targets. The rules of chemical bonding derived from the accurate crystal structures of small organic and organometallic molecules (mainly from over 800,000 crystal structures currently present in the Cambridge Structural Database (CSD) [1]) must apply equally to the macromolecular structures as well. A significant difference between the interpretation of small-molecule and macromolecular structures is due to very different ratios of experimental observations, such as the number of reflection intensities, to the number of model parameters, such as atomic coordinates and displacement parameters (ADPs, formerly known as temperature factors). Except when atomic-resolution crystallographic data (defined as having dmin < 1.2 Å; [2]) are available, the vast majority of macromolecular structures (currently ~97.5% of crystal structures and 100% of NMR- and electron microscopy-derived structures in the Protein Data Bank (PDB [3]) could not be properly refined on the basis of the experimental data alone. Thus, as described elsewhere in this volume [Jaskolski, this volume], stereochemical restraints are almost always applied in some form during the refinement of macromolecules.
Some stereochemical properties of proteins had been elucidated before the first protein structures were experimentally determined. For example, the bond lengths, angles, and the planar nature of the peptide bond had been known since early 1950s. Pauling et al. [4] estimated that deviation of the peptide bond from planarity by 10° should destabilize it by about 1 kcal.mole−1. In the same paper, they described the stereochemical parameters of the α-helix (not yet named as such), by postulating a twist of the polypeptide with a non-integral number of residues (3.7, later revised to 3.6) per turn. Another classical secondary structure element defined at that time, was the β-sheet [5]. Properties of these secondary structures, together with those of other structural elements, were summarized in detail two years later [6]. These types of structures were subsequently found in experimentally determined structures of proteins [7–9]. Correct knowledge of the stereochemical properties of nucleic acid polymers (together with other evidence, such as X-ray fiber diffraction) led to the proposed model of the double-helical structure of DNA [10], which changed our understanding of fields even very indirectly related to structure, such as genetics.
2 Restraining bonds and angles
Whereas the lengths and angles of the peptide bond were known quite accurately early on [6], knowledge of the corresponding parameters in the side chains of amino acids (and in the nucleic acids and carbohydrates) came gradually, first from only the structures of the individual components of these polymeric compounds, and later from high resolution structures of macromolecules. Bond and angle information was subsequently compiled into stereochemical restraint libraries. Except for the few structures for which diffraction data could be collected to extremely high resolution (0.8 Å or better), all macromolecular refinement procedures utilize such standard stereochemical information [11]. X-ray- and neutron-diffraction structures of individual amino acids were initially used for the construction of restraint libraries for programs such as PROLSQ [12,13], but the restraints were subsequently improved on the basis of large databases. Almost universally, the currently used refinement programs, such as CNS [14], SHELXL [15], REFMAC5 [16], or PHENIX [17] use the parameters compiled 25 years ago by Engh and Huber [18] and subsequently updated [19] ten years later. These parameters were obtained by careful analysis of the CSD [1], and were later slightly modified through the analysis of highest-resolution macromolecular structures [20]. It was also pointed out that the values of bond lengths and angles depend, to a certain extent, on the secondary structure and they were modified accordingly to correct for such effects [21]. Other details, such as protonation-dependent variations of the geometry of the imidazole ring of histidine, were analyzed more recently [22]. Some adjustments to the parameters used in the refinement of nucleic acids [23] was proposed on the basis of an ultrahigh-resolution (0.55 Å) crystal structure of Z-DNA [24].
Rms (root-mean-square) deviations from standard stereochemistry indicate how much a refined model departs from the geometrical targets present in the dictionaries. Different parameters can be evaluated by the rmsd criterion, but it is most common to use the values for bond lengths and angles when comparing different models. The allowed departure from the targets depends on the resolution of the diffraction data used in the refinement. Good-quality, medium-to-high resolution structures are expected to have rmsd(bond) values of about 0.02 Å (corresponding to the standard uncertainty of the targets themselves), although numbers half that size are also acceptable [20]. When this number becomes too high (above ~0.03 Å), this may indicate problems with the model. On the other hand, attempts to lower the rmsd further may lead to models that are more idealized, but less accurate. The common values for rmsd(angle) are between 0.5° and 2.0°. These levels of variations in the geometric parameters are in line with the rmsd values averaged for all classes of bonds and angles in polypeptides listed in the original Engh and Huber compilation (0.022 Å and 1.85° for bonds and angles, respectively) [18]. The default target values in different refinement programs are also in the same ranges [20].
3 Ramachandran plot and peptide planarity
One of the most useful tools for validation of protein structures is the Ramachandran plot [25], showing the mapping of pairs of φ/ψ torsion angles of the polypeptide backbone on the backdrop of the “allowed” or expected values. The allowed areas of the Ramachandran plot differ very significantly between glycines and the other amino acids, and to a lesser extent also between different amino acids [26]. The φ/ψ angles have a strong validation power because their values are usually not restrained in the refinement, unless a special torsion-angle-refinement method is used [14]. It was originally suggested that more than 90% of the φ/ψ pairs should be found in the most favored areas of the plots [27], although these areas were later redefined and the more recent estimate is that over 98% of the angles should be found in them [28]. On the other hand, the presence of φ/ψ conformations in the disallowed areas may indicate local problems with the structure. However, it is not unusual to occasionally find very strained torsion angles in some parts of proteins, particularly if the corresponding side chains are involved in multiple contacts and if the distortion has a functional significance. The correctness of the interpretation of such areas will ultimately rely on the appearance of the electron density maps.
The third main-chain conformational parameter of proteins, the peptide torsion angle ω, is expected to be close to 180° or exceptionally to 0° for cis-peptides (the latter seen more frequently than originally thought [29]). Cis-peptides are most commonly observed in the Xxx-Pro peptides, but are occasionally seen in peptide bonds connecting other types of amino acids as well (estimated at 1 cis bond per ~2000 residues [30]). However, due to their expected rarity such bonds are sometimes modeled as trans and the incorrect assignment is not detected during structure refinement. A recent global analysis of the structures deposited in the PDB found more than 4,000 instances of potential trans-cis flips that could be corrected based on stereochemical considerations alone [31]. Thus the assumption that all non-Xxx-Pro peptides should be necessarily in trans configuration should be applied with care. The opposite, however, may also be true, since another recent analysis of the PDB indicated a fairly high rate of the presence of non-Xxx-Pro cis peptides in structures refined at moderate-to-low resolution [32]. A majority of these non-standard peptides may represent fitting errors [32].
The peptide planes are usually under very tight stereochemical restraints, although there is growing evidence that deviations of ±20° from strict planarity should not be treated as abnormal if strongly supported by high-resolution electron density [20,33,34]). Whereas it was recently proposed that the ω angles could be more tightly restrained even in atomic-resolution structures without deteriorating the model [35], that proposal has been already refuted [36]. Unreasonably tight peptide planarity may lead to artificial distortions of the neighboring φ/ψ angles in the Ramachandran plot. On the other hand, some structures that have been deposited in the PDB quite recently exhibit deviations from peptide planarity exceeding 30° (for example, 4oiw, 3ja8, 2j6r, 4zkt, 3j9p, etc.). Models containing such violations should be regarded as highly suspicious, unless created on the basis of atomic-resolution data with absolutely clear electron density maps. For example, structure 4oiw includes several peptides in which the ω torsion angle deviates from planarity by as much as 40°. While some of such peptides are located in the loops with poor electron density and thus are clearly wrong, others are in quite good density, but do not represent a proper fit and may be due to the application of too weak planarity restraints. However, since such problems are highly localized, they may not be critical to the interpretation of the affected structures unless they are found in areas of high significance, such as the active sites of enzymes. On the other hand, the low-resolution structure 5dsv includes almost 200 ω torsion angles deviating from planarity by more than 30° (some by as much as 90°), thus it represents a clear case of improper use of planarity restraints during refinement, or outright wrong modeling.
4 Tools for validation of macromolecular structures
The need to validate the correctness of macromolecular structures was pointed out soon after the progress in the development of computational hardware and software made refinement of large structures possible [37]. Some structure validation tools were built directly into computer programs such as FRODO [38] and O [39] that allowed manual or automated fitting of models to electron density. Programs such as WHAT IF [40], although originally written as general display and modeling tools, contained a number of routines that allowed assessment of the departure of geometrical parameters of macromolecular models from the expected values. A variant of WHAT IF, called WHAT CHECK [41,42], was specially designed to flag potential deviations of models from the expected geometry. Analysis of the geometry and stereochemical validation of the models is equally applicable to structures determined by either crystallography or NMR, whereas assessment of the agreement of the models with the primary experimental data is, of course, quite different for these two techniques, and is basically absent in the NMR field.
For a number of years the main structure validation tool was the program PROCHECK [27]. Its subroutines analyzed the geometry of protein structures and compared them to other well-refined structures determined at comparable resolution. In addition to analyzing the Ramachandran plots (see above), the programs analyzed the planarity of peptide bonds, bad non-bonded interactions, distortions of the geometry around the Cα atoms, energies of hydrogen bonds, and the departure of the side chain χ torsion angles from expected values. The graphical output of the program allowed its users to quickly identify the most problematic areas. Thus PROCHECK has been extensively used as a tool for guiding the process of structure refinement and rebuilding. An example of a Ramachandran plot for a comparatively large protein structure refined at atomic resolution of 1 Å is shown in Figure 1A. However, since the database serving this program suite was quite limited as compared to the current situation, PROCHECK is now considered to be obsolete. Nevertheless, since it was used in the past to verify many structures that still serve to explain biologically-relevant data, it is still useful to understand its advantages and limitations.
Figure 1.


Ramachandran plots for the tetrameric molecule of Erwinia chrysanthemi L-asparaginase [54]. The structure was refined with data extending to 1 Å resolution. A) A plot prepared with PROCHECK [27], showing that almost all φ/ψ torsion angles are found in the favored or additionally allowed regions. However, Thr204 in each subunit is marked as being in a disallowed region, and Asp296 in a generously allowed area. (The unusual conformation of these residues is supported by the original electron density.) B) An analogous plot prepared with MolProbity [45]. The two residues outside of the allowed region in PROCHECK are no longer considered to be outliers, whereas GlyB28 is now marked as such.
A newer approach to validation of macromolecular structures makes use of various versions of the program suite MolProbity [43–45], used as a web server, in a stand-alone mode, or as part of other program systems, such as Phenix [46]. In addition to analyzing the geometrical parameters discussed above, MolProbity relies very heavily on the analysis of interatomic clashes. For that purpose the program calculates the positions of the hydrogen atoms and adds them to the coordinate files (sometimes replacing the riding H atoms that might already be present there). The side chains of Asn, Gln, and His are subjects of special attention aimed at verification of the most likely orientation of their O/N side chains, or the proper orientation of the imidazole rings. Once these residues have been placed in their most likely orientations and the H atoms have been added, all-atom contacts are analyzed in detail. Close interatomic distances and clashes are shown graphically and in printouts, providing information useful for rebuilding the offending areas, or at least raising a red flag for the users of deposited structures. The program provides plots of the Ramachandran angles, using a much more extensive database than the one utilized by PROCHECK. The most favored areas are based on the analysis of quality-filtered data for as many as 100,000 residues, 98% of which are found therein, whereas the allowed regions encompass 99.95% of good reference data. This change of definitions of the allowed regions leads to some differences in the interpretation of the Ramachandran plots in comparison with PROCHECK (Figure 1B). The number of residues in the favored regions of the Ramachandran plot, as well as of the outliers, is listed by the program.
Another function of MolProbity, which is now based on a much larger database compared to PROCHECK, involves the analysis of the side-chain χ torsion angles. The preferred rotamers of the side chains are contoured by excluding 1% of high-quality data, and these definitions are periodically updated [45]. It was pointed out that unusual rotamers may often be found in the core of proteins due to inter-residue interactions, but it was also postulated that surface residues should not be modeled with unusual rotamers, especially when the electron density is not completely clear [45]. A typical problem leading to bad rotamers is fitting branched side chains (Thr/Val/Ile/Leu/Arg) backwards (by turning the chains around χ1 by ~180°) into unclear density and the output of MolProbity may help in taking remedial action. The summary of MolProbity analysis reports the percentage of poor rotamers and this number serves as another useful guide to assessing structure quality.
5 Protein Data Bank validation reports
Validation reports are extremely useful tools for both the depositors and the users of the PDB. The format of the reports has been under development for some time and will most likely change again in the future. The current standard is based on the recommendations of the wwPDB X-ray Validation Task Force [26]. Submission of a validation report is now required by a number of scientific journals as a companion piece to manuscripts that describe crystal structures. The availability of validation reports is expected to help reviewers in assessing whether the structure discussed in a manuscript is reliable and of sufficiently high quality. Of course, since the report had been made available to the depositors first, it should have been scrutinized prior to the final submission of the structure to the PDB. Sadly, as will be shown below, that seems not always to be the case.
This chapter will describe PDB validation reports as applied to crystal structures only – some aspects of the validation process are not applicable to models determined by NMR spectroscopy. The reports are based on the output of several programs and refer to the restraint libraries that are clearly identified in the output. Bond distances and angles in proteins are checked against the latest version of the Engh and Huber library [19], although - quite surprisingly - some refinement programs, such as REFMAC, have been for decades (sic!) using the obsolete early version [18]. However, the PDB validation report takes no account of the conformation-dependent libraries (CDL) [21]. For nucleic acids, the reference library is provided by Parkinson et al. [23] plus two other of the same vintage [47,48]. The geometrical parameters are checked principally by a recent version of MolProbity [45], whereas the agreement of the model with the diffraction data is monitored by procedures introduced by Kleywegt et al. [49] in their Uppsala Electron-Density Server (EDS; http://eds.bmc.uu.se/eds/).
The first page of the validation report (and the PDB web page for that structure) includes a graphical summary of the “quality indicators” of the structure. While helpful in providing a first-glance impression, this graph provides only fragmentary information about how the structure in question compares to other structures present in the PDB. Five different quality metrics are shown in colors ranging from deep red (poor) to blue (excellent). Black boxes indicate how the structure compares to all structures in the PDB, whereas open boxes provide a more meaningful comparison with structures at comparable resolution.
Examples of such graphs for three recently deposited medium-resolution structures are shown in Figure 2. Structure 5duy is considered to be quite acceptable, 4xok shows a number of problems, whereas 4ru7 (described in the original publication [50] but now made obsolete and replaced by 5fco) exhibits some grave problems that put its validity into question.
Figure 2.
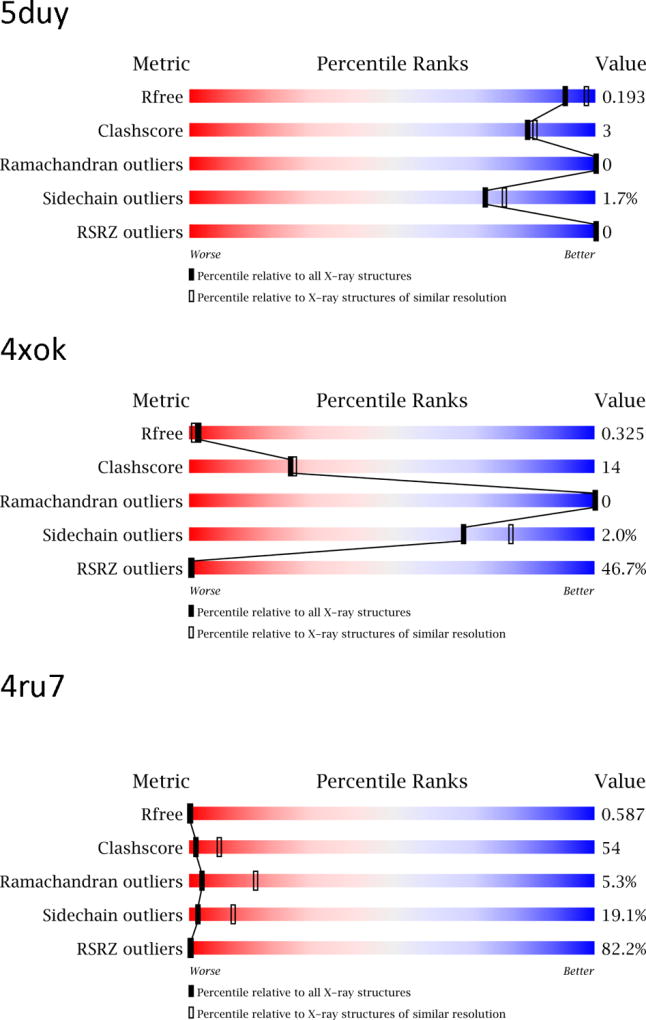
Summary graphs from the PDB validation reports for three medium-resolution structures, presented here only as examples. The deposit 4ru7 has by now been obsoleted and replaced, but is shown here since it corresponds directly to the original publication [50].
Free R factor, in use for practically all structures since its introduction into crystallographic practice in 1992 [51], depicts the relative deviation of the observed and calculated structure factors (in analogy to the conventional R-factor) for a subset of (5–10% or ~1000) reflections never used in structure refinement. Rfree should be higher (by ~4–7 percent points) than R. If it is much higher, it indicates serious problems with the model (without pointing out the errors, however), or overinterpretation of the experimental data by too many (unjustified) model parameters. If the Rfree-R gap is too small, it strongly suggests a compromised test data set (e.g. used, deliberately or not, at some stage in the refinement) and questions the validity of the Rfree test in such a case. The expected value of Rfree depends very much on the resolution of the data sets and its interpretation was discussed in detail previously [52]. In exceptionally good cases Rfree can be as low as 10% for the best-refined structures using ultrahigh-resolution data; typically it is about 20–25% for medium-resolution structures, but should not exceed 30% even for structures refined against low-resolution data. Thus the value of 19.3% for 5duy (refined at 2.12 Å resolution) is in the expected range, it is rather high (32.5%) for 4xok (2.2 Å), and is absolutely random (58.7%) for 4ru7 (2.97 Å; now obsoleted and replaced by 5fco), suggesting that in the latter case the deposited structure factors might not correspond to the coordinates.
Rfree is a better (even if not ideal) measure of model quality than the standard crystallographic R (residual) factor, calculated as R = 100·Σ | |Fo| − |Fc| | / Σ |Fo|. The problem with R is that it has poor statistical properties and will essentially go down on any, even unreasonable model expansion. A much better metric would be the weighted (note the use of weights, w) wR2 factor (wR2 = 100·[Σ (|Fo| − |Fc|)2 / Σ |Fo|2]1/2) but it is seldom used in macromolecular crystallography.
The second line of the “quality”graph summarizes a parameter called clashscore, introduced in MolProbity. This score is related to the number of interatomic distances that are shorter by more than 0.4 Å than the sum of the van der Waals radii and is expressed as the number of such close contacts per 1000 atoms in the structure. Of course, interatomic clashes do not have any physical meaning (after all, atoms cannot interpenetrate), but since the models cannot provide an error-free description of the structure, some (small) number of such violations is inevitable. The clashscore of 3 for 5duy is almost exactly in the typical range (which seems to be almost independent of resolution). For 4xok this parameter is definitely much worse than the average, and the clashscore of 54 for 4ru7 is certainly unacceptable.
The next bar in the summary picture shows the number of Ramachandran outliers. There are no such violations in 5duy and 4xok, whereas the presence of 5.3% outliers in the case of 4ru7 indicates very poor geometry of the main chain of the model. This number represents the percentage of all residues in the structure that are found in the disallowed areas of the plot (Figure 1B). It needs to be stressed that whereas the presence of violations (as in 4ru7) is suggestive of problems with the structure, their absence (as in 4xok) does not necessarily prove that the model is of high quality.
Side-chain outliers are defined as the percentage of side chains with a combination of χ torsion angles that are not similar to any combination preferred for that given residue type, calculated in the same way as for the Ramachandran violations. Clearly, some residues will have unusual torsion angles due to packing constraints, but there is no real justification for having outliers in the surface areas where the electron density is relatively weak; thus only proper rotamers should be assumed to be present there. The percentage of side-chain outliers lower or equal to 2% in 5duy and 4xok indicates that the residues were generally modeled with their preferred rotamers and were not distorted during the refinement process. However, more than 19% of outliers found in 4ru7 is a clear indication that the model was allowed to depart very far from a typical conformation, reiterating a real question about the quality of this model.
The final line of the summary plot is related to the number of residues that do not fit well the corresponding electron density. RSR (real space R factor), calculated for each residue separately, is a measure of the quality of fit between the coordinates of a residue and the corresponding electron density. The RSR Z score (RSRZ) compares the fit to electron density for each residue with the mean and standard deviation of the fit for all such residues in a similar resolution bin. A residue is considered to be an RSRZ outlier if its RSR is more than 2 standard deviations worse than the average for this residue type, and the plotted score corresponds to the percentage of residues that are considered to be outliers based on this criterion. A well-refined protein structure may show no RSRZ outliers at all, whereas the presence of poorly defined regions in the structure (for example, due to local disorder that was still modeled) will lead to an increase of this parameter. In practice, the number of RSRZ outliers is often correlated with higher-than-average Rfree. This can be seen in the comparison of the structures in Figure 2, where 5duy has no RSRZ outliers, whereas almost half of the residues in 4xok do not seem to fit the electron density. Very few residues in 4ru7 appear to fit the electron density, again pointing to serious problems with the structure factor file.
The next summary graph contains two lines for each polypeptide (or polynucleotide) chain present in the coordinate file. The lower bar is colored green, yellow, orange, or red to denote deviation of a particular residue from 0–3 (or more) stereochemical quality standards (see below). The top line, if present and colored red, indicates that this segment of the chain exhibits poor fit to the electron density. This plot is followed by a table showing similar outliers for non-polymeric components (such as buffer molecules) of the structure.
As discussed above, a quick glance at the structure quality diagram may be sufficient for the first impression regarding the quality of a PDB deposit, but clean plots are not necessarily sufficient to affirm that the model is excellent. A graph showing large departures from the expected average values, however, should immediately alert the user of such a file (and especially its depositor!) of potentially serious problems.
The next section of the validation report deals with the composition of the entry and is useful for checking the sequence of the macromolecule against relevant databases, what kinds of expression tags are present, and how many atoms are modeled with zero occupancy and/or in alternate conformations.
The third part of the validation report provides the details behind the chain quality plot found on the summary page. It includes residue-by-residue plots colored as defined above for the presence of geometric outliers, with red dots denoting poor fit to electron density (RSRZ>2). Residues to be present in the sample but not included in the model (most likely because of disorder and/or poor/absent electron density) are marked in gray. Stretches of residues with no apparent problems are marked by a green line.
A table of data and refinement statistics, included in the fourth part of the report, provides selected statistics derived directly from the deposit, or recalculated by the PDB. This table is worth attention, especially if the numbers claimed by the authors are significantly different from the ones recalculated during the validation process. Differences in the resolution limit may be due to the inclusion in the deposited structure factor files of shells that were not actually used in refinement (quite common at low resolution), but very large deviations (such as 1.41 Å computed with EDS vs. 2.98 Å claimed by the depositors of 4ru7) are certainly worth close examination. The values of Rmerge or Rsym (if cited by the depositors) give some indication of the internal consistency of the diffraction data, whereas <I/σ(I)>, computed by the program Xtriage, indicates whether statistically significant observations were present in the outermost data shell. If the value of <I/σ(I)> is much less than ~2.0, this indicates that the claimed resolution limits may have been overly optimistic.
The values of the refinement R and Rfree factors are listed as claimed by the depositors, and as recalculated during validation. Some differences, such as R of 13.9% claimed by depositors of 5duy and 15.5% calculated during validation, are not unexpected, due to different assumptions used by different computer programs, e.g. Phenix or REFMAC. The corresponding numbers for 4xok, 30.3% and 30.1%, were most likely calculated by the same software. However, the difference between 21.0% and 57.1% found in the validation report of 4ru7 is a clear indication that the structure factor file does not correspond to the coordinates present in this deposit.
Other parameters listed in that table are helpful in deciding whether the diffraction data could have been twinned or if translational non-crystallographic symmetry is present, and show how the mean B factor for all atoms compares with the Wilson B factor for the diffraction data.
The fifth section of the report is particularly relevant to the subject of this chapter, since it provides a detailed description of the geometric parameters of the modeled coordinates. The four stereochemical criteria are bond lengths, bond angles, chirality (where present), and planarity (where present). All bond lengths and angles with individual Z scores larger than 5 are listed, together with the RMSZ scores for the whole chain. Chiral center volumes (signed volume of the tetrahedron formed by the four substituents of an sp3 atom) that differ significantly from the expected values are tagged, and potentially planar groups which appear to be non-planar are similarly marked. Close contacts are evaluated for all-atom models that include hydrogen atoms (either as already present in the deposit or added computationally) and the ones for which the distance is shorter than the sum of their van der Waals radii minus 0.4 Å are highlighted. Additional analysis presents the deviations of the Ramachandran main-chain torsion angles and χ side chain angles from the allowed (or most likely) targets, and summarizes the percentile values with respect to all crystal structures, or structures at similar resolution. A percentile value of 100% means that there are no outliers, whereas a lower number indicates the percentage of PDB structures with more problems (the value of 0% would portend the worst value of that parameter in the entire PDB). The Ramachandran and non-rotameric outliers are explicitly identified, together with the candidate Asp/Gln/His residues that might need flipping. The latter information might be particularly useful if these residues are expected to be important for the function of the protein.
The last section of the validation report deals with the agreement between the coordinates and the electron density maps. The RSRZ values are computed individually for each chain and the number of RSRZ outliers (RSRZ>2) is given, together with the percentage scores relative to all crystal structures, as well as structures at similar resolution. Thus for 5duy, with no outliers, the percentage is 100%, whereas it is 1% for all entries and 0% for entries at similar resolution for 4xok. The numbers are all 0% for 4ru7.
This section of the report also contains an analysis of any non-standard residues in proteins, polynucleotides and carbohydrates, as well as for their ligands. An important parameter for the assessment of ligand quality is LLDF. It compares the electron density of the ligand with the electron density of the neighboring atoms of the macromolecule. Values of LLDF that exceed 2.0 indicate potential problems, thus the conformation of such ligands (or even their presence) should be carefully scrutinized. Since the PDB validation report does not provide information which would indicate whether the coordination of metal ions (if present) is plausible, it is worthwhile to obtain such information from a dedicated server CheckMyMetal (http://csgid.org/csgid/metal_sites/) [53].
6 Summary and Conclusions
The stereochemistry of properly determined macromolecular structures cannot deviate very much from standard values of bond lengths and angles, as well as from acceptable torsion angles. Validation is crucial in assuring good quality of the models and such tests should be routinely run during structure refinement. The standard arbiter should be the official PDB validation report. It is recommended that parameters such as the number of residues deviating from favored or allowed regions in the Ramachandran plot should be quoted according to that report. Different programs may report such parameters slightly differently but deferring to validation by the PDB will assure some level of conformity. Of course, depositors should pay real attention to the output of the validation reports; as shown here, this is not always the case. The users of macromolecular coordinates might also benefit from taking a look at such reports before using the data; it is always worthwhile to know how reliable a given structures is, and to adjust the level of confidence accordingly, especially if the user is interested in the atomic details of some specific areas, such as enzyme active sites or intermolecular interfaces. While not perfect, validation reports do contain a lot of useful and crucial information that should never be disregarded. Ultimately, if there is any doubt or controversy, the electron density map should be the final arbiter.
References
- 1.Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. 2002;B58:380–388. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 2.Sheldrick GM. Phase annealing in SHELX-90: direct methods for larger structures. Acta Crystallogr. 1990;A46:467–473. [Google Scholar]
- 3.Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pauling L, Corey RB, Branson HR. The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci USA. 1951;37:205–211. doi: 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pauling L, Corey RB. The pleated sheet, a new layer configuration of polypeptide chains. Proc Natl Acad Sci U S A. 1951;37:251–256. doi: 10.1073/pnas.37.5.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pauling L, Corey RB. Stable configurations of polypeptide chains. Proc R Soc Lond B Biol Sci. 1953;141:21–33. doi: 10.1098/rspb.1953.0012. [DOI] [PubMed] [Google Scholar]
- 7.Kendrew JC, Bodo G, Dintzis HM, et al. A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature. 1958;181:662–666. doi: 10.1038/181662a0. [DOI] [PubMed] [Google Scholar]
- 8.Perutz MF, Rossmann MG, Cullis AF, et al. Structure of Haemoglobin: A three-dimensional Fourier synthesis at 5.5-Å resolution, obtained by X-ray analysis. Nature. 1960;185:416–421. doi: 10.1038/185416a0. [DOI] [PubMed] [Google Scholar]
- 9.Blake CC, Fenn RH, North AC, et al. Structure of lysozyme. A Fourier map of the electron density at 6 Å resolution obtained by x-ray diffraction. Nature. 1962;196:1173–1176. doi: 10.1038/1961173a0. [DOI] [PubMed] [Google Scholar]
- 10.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 11.Evans PR. An introduction to stereochemical restraints. Acta Crystallogr. 2007;D63:58–61. doi: 10.1107/S090744490604604X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wlodawer A, Hendrickson WA. A procedure for joint refinement of macromolecular structures with X-ray and neutron diffraction data from single crystals. Acta Crystallogr. 1982;A38:239–247. [Google Scholar]
- 13.Hendrickson WA. Stereochemically restrained refinement of macromolecular structures. Methods Enzymol. 1985;115:252–270. doi: 10.1016/0076-6879(85)15021-4. [DOI] [PubMed] [Google Scholar]
- 14.Brünger AT, Adams PD, Clore GM, et al. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 15.Sheldrick GM, Schneider TR. SHELXL: High-Resolution Refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 16.Murshudov GN, Skubak P, Lebedev AA, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. 2011;D67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams PD, Grosse-Kunstleve RW, Hung LW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 18.Engh R, Huber R. Accurate bond and angle parameters for X-ray protein-structure refinement. Acta Crystallogr. 1991;A47:392–400. [Google Scholar]
- 19.Engh RA, Huber R. International Tables for Crystallography. Kluwer Academic Publishers; Dordrecht: 2001. pp. 382–392. [Google Scholar]
- 20.Jaskolski M, Gilski M, Dauter Z, Wlodawer A. Stereochemical restraints revisited: how accurate are refinement targets and how much should protein structures be allowed to deviate from them? Acta Crystallogr. 2007;D63:611–620. doi: 10.1107/S090744490700978X. [DOI] [PubMed] [Google Scholar]
- 21.Tronrud DE, Karplus PA. A conformation-dependent stereochemical library improves crystallographic refinement even at atomic resolution. Acta Crystallogr. 2011;D67:699–706. doi: 10.1107/S090744491102292X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malinska M, Dauter M, Kowiel M, et al. Protonation and geometry of histidine rings. Acta Crystallogr D Biol Crystallogr. 2015;71:1444–1454. doi: 10.1107/S1399004715007816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkinson G, Vojtechovsky J, Clowney L, et al. New parameters for the refinement of nucleic acid-containing structures. Acta Crystallogr D Biol Crystallogr. 1996;52:57–64. doi: 10.1107/S0907444995011115. [DOI] [PubMed] [Google Scholar]
- 24.Brzezinski K, Brzuszkiewicz A, Dauter M, et al. High regularity of Z-DNA revealed by ultra high-resolution crystal structure at 0.55 Å. Nucleic Acids Res. 2011;39:6238–6248. doi: 10.1093/nar/gkr202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramakrishnan C, Ramachandran GN. Stereochemical criteria for polypeptide and protein chain conformations. II Allowed conformation for a pair of peptide units. Biophys J. 1965;5:909–933. doi: 10.1016/S0006-3495(65)86759-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Read RJ, Adams PD, Arendall WB, III, et al. A new generation of crystallographic validation tools for the protein data bank. Structure. 2011;19:1395–1412. doi: 10.1016/j.str.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laskowski RA, MacArthur MW, Moss DS, et al. PROCHECK: program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 28.Kleywegt GJ, Jones TA. Phi/psi-chology: Ramachandran revisited. Structure. 1996;4:1395–1400. doi: 10.1016/s0969-2126(96)00147-5. [DOI] [PubMed] [Google Scholar]
- 29.Weiss MS, Hilgenfeld R. On the use of the merging R factor as a quality indicator for X-ray data. J Appl Cryst. 1997;30:203–205. [Google Scholar]
- 30.Stewart DE, Sarkar A, Wampler JE. Occurrence and role of cis peptide bonds in protein structures. J Mol Biol. 1990;214:253–260. doi: 10.1016/0022-2836(90)90159-J. [DOI] [PubMed] [Google Scholar]
- 31.Touw WG, Joosten RP, Vriend G. Detection of trans-cis flips and peptide-plane flips in protein structures. Acta Crystallogr D Biol Crystallogr. 2015;71:1604–1614. doi: 10.1107/S1399004715008263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Croll TI. The rate of cis-trans conformation errors is increasing in low-resolution crystal structures. Acta Crystallogr D Biol Crystallogr. 2015;71:706–709. doi: 10.1107/S1399004715000826. [DOI] [PubMed] [Google Scholar]
- 33.EU 3-D Validation Network. Who checks the checkers? Four validation tools applied to eight atomic resolution structures. J Mol Biol. 1998;276:417–436. doi: 10.1006/jmbi.1997.1526. [DOI] [PubMed] [Google Scholar]
- 34.Addlagatta A, Krzywda S, Czapinska H, et al. Ultrahigh-resolution structure of a BPTI mutant. Acta Crystallogr. 2001;D57:649–663. doi: 10.1107/s0907444901003468. [DOI] [PubMed] [Google Scholar]
- 35.Chellapa GD, Rose GD. On interpretation of protein X-ray structures: Planarity of the peptide unit. Proteins. 2015;83:1687–1692. doi: 10.1002/prot.24854. [DOI] [PubMed] [Google Scholar]
- 36.Brereton AE, Karplus PA. On the reliability of peptide non-planarity seen in ultra-high resolution crystal structures. Protein Sci. 2016 doi: 10.1002/pro.2883. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brändén C-I, Jones TA. Between objectivity and subjectivity. Nature. 1990;343:687–689. [Google Scholar]
- 38.Jones TA. Interactive computer graphics: FRODO. Methods Enzymol. 1985;115:157–171. doi: 10.1016/0076-6879(85)15014-7. [DOI] [PubMed] [Google Scholar]
- 39.Jones TA, Zou JY, Cowan S, et al. Improved methods for building protein models in electron density maps and location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 40.Vriend G. WHAT IF: A molecular modelling and drug design program. J Mol Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 41.Hooft RW, Vriend G, Sander C, et al. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 42.Nabuurs S, Spronk C, Krieger E, et al. Computational Mechanical Chemistry for Drug Discovery. Marcel Dekker; New York and Basel: 2004. pp. 387–403. [Google Scholar]
- 43.Davis IW, Murray LW, Richardson JS, et al. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis IW, Leaver-Fay A, Chen VB, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen VB, Arendall WB, III, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clowney L, Jain SC, Srinivasan A, et al. Geometric Parameters in Nucleic Acids: Nitrogenous Bases. J Am Chem Soc. 1996;118:509–518. [Google Scholar]
- 48.Gelbin A, Schneider B, Clowney L, et al. Geometric Parameters in Nucleic Acids: Sugar and Phosphate Constituents. J Am Chem Soc. 1996;118:519–529. [Google Scholar]
- 49.Kleywegt GJ, Harris MR, Zou JY, et al. The Uppsala Electron-Density Server. Acta Crystallogr. 2004;D60:2240–2249. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
- 50.Schumacher MA, Tonthat NK, Lee J, et al. Structures of archaeal DNA segregation machinery reveal bacterial and eukaryotic linkages. Science. 2015;349:1120–1124. doi: 10.1126/science.aaa9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brünger AT. The free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 52.Wlodawer A, Minor W, Dauter Z, et al. Protein crystallography for non-crystallographers or how to get the best (but not more) from the published macromolecular structures. FEBS J. 2008;275:1–21. doi: 10.1111/j.1742-4658.2007.06178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng H, Chordia MD, Cooper DR, et al. Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat Protoc. 2014;9:156–170. doi: 10.1038/nprot.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lubkowski J, Dauter M, Aghaiypour K, et al. Atomic resolution structure of Erwinia chrysanthemi L-asparaginase. Acta Crystallogr. 2003;D59:84–92. doi: 10.1107/s0907444902019443. [DOI] [PubMed] [Google Scholar]