

Summary
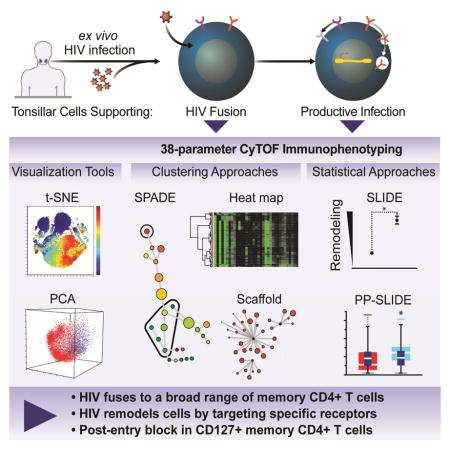
To characterize susceptibility to HIV infection, we phenotyped infected tonsillar T cells by single-cell mass cytometry and created comprehensive maps to identify which subsets of CD4+ T cells support HIV fusion and productive infection. By comparing HIV-fused and HIV-infected cells through dimensionality reduction, clustering, and statistical approaches to account for viral perturbations, we identified a subset of memory CD4+ T cells that support HIV entry but not viral gene expression. These cells express high levels of CD127, the IL-7 receptor, and are believed to be long-lived lymphocytes. In HIV-infected patients, CD127-expressing cells preferentially localize to extrafollicular lymphoid regions with limited viral replication. Thus, CyTOF-based phenotyping combined with analytical approaches to distinguish between selective infection and receptor modulation by viruses can be used as a discovery tool.
eTOC
Cavrois et al. conduct a global characterization of HIV entry and productive infection of tissue CD4+ T cells by CyTOF, and develop an analytical approach that takes advantage of the high-dimensional nature of CyTOF datasets to distinguish receptors modulated during infection from those differentially expressed on preferentially infected cells.
Introduction
CD4+ T cells are the primary targets of HIV-1, but not all are equally susceptible. Activated CD4+ T cells are more susceptible to productive infection than their naïve counterparts (Pan et al., 2013), and some subsets of memory CD4+ T cells, such as Th17 and follicular helper T cells (Tfh), are infected at higher rates than other subsets (El Hed et al., 2010; Kohler et al., 2016). However, a global assessment of which subsets of CD4+ T cells are susceptible to HIV has not been conducted. As different subsets have differences in trafficking, effector function, and longevity, a more detailed understanding of which subsets are most and least susceptible could lead to new insights into mechanisms of HIV pathogenesis and viral persistence.
Flow cytometry has been instrumental in establishing our current understanding of the types of cells that are susceptible to HIV infection but is limited by the spectral overlap of fluorophores. Recently, mass cytometry – or cytometry by time-of-flight (CyTOF) – was developed as a way to overcome these limitations. Phenotyping of immune cells by CyTOF has highlighted the diversity of immune cells, including primary CD4+ T cells. For example, Tfh-like cells, traditionally thought to be a relatively uniform subset of cells, can be sub-classified into 15 distinct subclusters when analyzed by CyTOF (Wong et al., 2015). While CyTOF was used recently to compare CD4+ T-cell subsets in uninfected and ART-suppressed HIV-infected subjects (Corneau et al., 2017), it has not been used to characterize cellular susceptibility to HIV infection.
When phenotyping HIV-infected cells, another layer of complexity arises from the fact that the expression pattern of antigens on an HIV-infected cell reflects not only the properties of the original CD4+ T cell targeted for viral entry, but also phenotypic changes that occur as a consequence of infection. Such changes include downregulation of a variety of cell-surface receptors by the viral accessory proteins Nef and Vpu (Matheson et al., 2015; Ross et al., 1999). Because markers used to classify subsets may be altered by the infection itself, the identification of cell subsets most susceptible to infection is not straightforward. Sen et al. recently demonstrated that remodeling of cells by viruses can be studied by combining CyTOF-based phenotyping of cells infected with varicella-zoster virus (VZV) with a statistical method termed Single Cell Linkage using Distance Estimation (SLIDE) (Sen et al., 2015). This analysis demonstrated that VZV reconfigures phenotypes and signaling pathways of human tonsil T cells to enhance skin homing, shedding light on VZV pathogenesis.
Here, we designed a 38-parameter CyTOF panel to phenotype tonsillar CD4+ T cells infected with HIV just after viral entry, and at a later phase after HIV genes are expressed. To distinguish between proteins downregulated by HIV from selective infection of particular subsets, we adapted SLIDE to predict the phenotype of each HIV-infected cell before it was infected. We compared expression levels of specific receptors on these cells, termed the “predicted precursor” cells, to those on the infected cells. By combining this analysis with other high-dimensional data analysis approaches, we identified a subset of long-lived memory CD4+ T cells that supports HIV entry but not replication, and further characterized the properties of these cells in vitro and in vivo.
Results
Generation of CyTOF-compatible HIV reporter virus
To characterize phenotypes of cells HIV-infected ex vivo, we re-engineered our HIV-1 reporter provirus (NL-LucR.6ATRi-Env.ecto), in which the LTR drove expression of Renilla luciferase (LucR) (Alberti et al., 2015). We inserted the gene encoding murine heat-stable antigen (HSA), a CyTOF-compatible cell-surface marker, between env and nef. At this location, HSA is followed by a modified IRES, called 6ATRi, that preserves wildtype levels of Nef expression and function, as measured by MHC class I modulation (Alberti et al., 2015). The provirus was then modified to encode the ectodomain of the R5-tropic transmitted/founder subtype C envelope, 109FPB4 (Derdeyn et al., 2004). This reporter virus (NL-HSA.6ATRi-C.109FPB4.ecto), termed HIV-F4.HSA, is fully described in Fig. S1A and the Supplemental Experimental Methods. HIV-F4.HSA expresses a functional Nef, as evidenced by down-modulation of MHC class I (Fig. S1B) and CD4 (Fig. S1C) in infected cells, and successfully infects human lymphocyte aggregate cultures (HLACs) in the absence of artificial mitogens (Fig. S1C). Ex vivo infection of HLACs was chosen as the system to further interrogate the properties of cells naturally permissive to infection by HIV-F4.HSA.
Strategy to characterize HIV-F4.HSA entry into tonsillar T cells
We began our study by focusing on the first step of infection: viral entry (Fig. 1A). The HIV fusion assay detects transfer of a BlaM-Vpr chimeric protein, co-packaged into virus particles, from the virion into the cytoplasm of target cells, and thereby allows measurement of HIV entry independent of viral gene expression (Cavrois et al., 2002). B cell-depleted HLACs were exposed to HIV-F4.HSA virions containing BlaM-Vpr for 2 h. After loading infected cultures with CCF2, the fluorescent substrate of BlaM, the cells supporting fusion, identified by a shift from green to blue fluorescence, were sorted following the gating strategy illustrated in Fig. S2. Mock-infected cells were sorted in parallel to control for possible changes in antigens during sorting. Sorted cells, confirmed to be >95% pure, were stained with a 38-parameter CD4+ T-cell panel (Table S1) that includes markers of T-cell differentiation and activation states, as well as adhesion and chemokine receptors. We validated our CyTOF panel by analyzing the expression profiles of select antigens on various T-cell subsets (Fig. S3A–D) and comparing expression levels of each antigen on T and B cells (Fig. S3E). To minimize cell loss during the staining and washing steps, we added carrier RAMOS (a B-cell line) to sorted cells. All data were acquired on a CyTOF2 instrument, and T cells were identified as events corresponding to live, intact cells expressing CD3 (T-cell marker) and not CD19 (B-cell marker) (Fig. S4A). After data acquisition, files corresponding to T-cell events were exported for analysis by multiple high-dimensional data analysis tools.
Figure 1. R5-tropic HIV enters a diverse array of memory CD4+ T cells.

(A) Experimental design for phenotyping HIV-fused cells by CyTOF. HLACs were ficolled to remove dead cells and mock-treated or incubated for 2 h with BlaM-Vpr-containing HIV-F4.HSA virions. After loading CCF2, cells supporting fusion (or uninfected cells) were sorted by FACS (Fig. S2) and stained with a 38-parameter panel for phenotyping by CyTOF. Live, CD3+CD19− events were exported for analysis by t-SNE or other dimension reduction methods.
(B) Representation of uninfected and HIV-fused cells by t-SNE based on expression of 35 surface markers. t-SNE plots corresponding to uninfected (red) or HIV-fused (blue) tonsillar T cells were overlaid to demonstrate that not all T cells support fusion to HIV-F4.HSA.
(C) Identification of the main T-cell subsets. Live, singlet CD3+CD19− cells were gated into subsets based on expression of CD4, CD8, the naïve marker CD45RA, and the memory marker CD45RO using standard dot plots. Populations corresponding to naïve CD4+ T cells (Tn), memory CD4+ T cells (Tm), CD4+ T cells expressing both isoforms of CD45 (Tdp), naïve CD8+ T cells, and memory CD8+ T cells were then overlaid onto the t-SNE plot of total T cells to map regions corresponding to these populations. Note regions lacking HIV-fused cells in panel B correspond to Tn and CD8+ T cells.
(D) Identification of Tm subsets that support fusion by HIV-F4.HSA. Live, singlet Tm that fused with HIV-F4.HSA were gated based on expression of CCR7, CD27, CCR4, CCR6, CD25, CD127, PD1, and CXCR5 to identify transitional memory cells (Ttm); central memory cells (Tcm); Th1-, Th2-, and Th17-like cells; regulatory T cells; and follicular helper T cells (Tfh). For each plot, two or three subpopulations of Tm were overlaid onto total HIV-fused and uninfected populations, thus displaying subpopulations of interest in color while remaining ungated HIV-fused cells (“Other HIV-fused”) are shown in dark grey.
(E) Preferential fusion of HIV-F4.HSA to Tm. Top: Gating strategy to identify Tn and Tm populations among live, singlet T cells. Bottom: Line graphs showing proportions of Tm and Tn in uninfected vs. HIV-fused cells in four independent donors. *p<0.05, **p<0.01 as assessed using a paired t-test.
(F) Preferential fusion of HIV-F4.HSA to the Th2-like, Th17-like, and Treg populations. Top: Gating strategy to identify the Tcm; Ttm; Th1, Th2, and Th17-like cells; Tfh, and Treg among Tm. Bottom: Line graphs showing proportions of the indicated Tm subset in uninfected vs. HIV-fused cells in four independent donors. *p<0.05, **p<0.01 as assessed using a paired t-test. See also Fig. S2, S4, and S5.
HIV-F4.HSA fuses to memory CD4+ T cells (Tm) and CD45RA+CD45RO+ double-positive CD4+ T cells (Tdp), but not naïve CD4+ T cells (Tn)
First, we compared the phenotypes of mock-infected cells to those that supported fusion using the t-Distributed Stochastic Neighbor Embedding (t-SNE) method of dimensionality reduction, recently implemented as Visualization of t-Distributed Stochastic Neighbor Embedding (vi-SNE) (Amir el et al., 2013; van der Maaten and Hinton, 2008) (Fig 1B). t-SNE converts high-dimensional CyTOF data into a two-dimensional (2D) plot in which cells (portrayed as dots) that are more similar to one another in high-dimensional space are placed closer together. Given that each parameter measured on cells by CyTOF creates one dimension, our CyTOF datasets have a total of 38 dimensions. We used 35 of these dimensions (Table S1) for t-SNE analysis: these included all cell surface proteins in the panel except for CD3 and CD19, used for identifying the T cells, and HSA, the marker for productive infection that is not expressed during the 2-h window of the fusion assay. For each HLAC donor, t-SNE analysis was performed simultaneously on uninfected (red) and HIV-fused (blue) T cells (Fig. 1B), to directly compare these populations. As evident in the t-SNE plot showing an overlay of the uninfected and HIV-fused populations of cells, not all T cells supported fusion to HIV-F4.HSA (Fig. 1B).
To identify T-cell subsets in these t-SNE plots, we assessed the locations within the t-SNE plot of five different subsets: naïve CD4+ T cells (Tn: CD4+CD45RA+CD45RO−), memory CD4+ T cells (Tm: CD4+CD45RA−CD45RO+), CD4+ T cells double-positive for both isoforms of CD45 (Tdp: CD4+CD45RA+CD45RO+), naïve CD8+ T cells (CD8+CD45RA+CD45RO−), and memory CD8+ T cells (CD8+CD45RA−CD45RO+) (Fig. 1C). These populations account for most T cells in HLACs, with very few T cells (labeled “Other”) not falling into one of these five categories (Fig. 1C). By comparing the plots in Fig. 1B and 1C, it was apparent that only Tm and Tdp supported efficient fusion by HIV-F4.HSA. The inability of Tn to support fusion is likely because Tn express insufficient levels of CCR5, the co-receptor for R5-tropic viruses like HIV-F4.HSA (Bleul et al., 1997).
Throughout this study, we used multiple high-dimensional analysis approaches to confirm our results and selected one of them to be presented in the main manuscript while compiling the complementary analyses in the accompanying Addendum. To accompany the t-SNE analysis of the fusion datasets, the Addendum (Section 1) presents analyses by principal component analysis (PCA) and agglomerative hierarchical clustering which verified the t-SNE-based conclusions drawn above, and additionally show high-dimensional analyses from 4 different donors to demonstrate reproducibility.
Characterization of HIV-F4.HSA-fused Tdp and Tm
To further characterize the two main populations of HIV-fused cells (Tdp and Tm), we first examined phenotypic features of Tdp. The Tdp population, previously described in tonsillar cultures (Picker et al., 1993; Sen et al., 2014), may be particularly susceptible to HIV infection (Yu et al., 2015). Standard 2D dot plot analysis of the expression levels of CD45RA and CD45RO on T cells in fact revealed two subsets of Tdp: one expressing intermediate levels of CD45RA and CD45RO (Tdpintermediate) and another expressing high levels of both antigens (Tdphigh) (Fig. S5A). The existence of these two distinct populations was previously demonstrated using standard flow cytometric techniques (Hamann et al., 1996; Picker et al., 1993), indicating that they were not an artifact of CyTOF analysis, which uses a different gating strategy to identify live, singlet cells. Because among HIV-fused cells Tdphigh were common (5.3%) but Tdpintermediate were rare (0.4%) (Fig. S5A), it became important to further compare these two subsets. Unexpectedly, image flow cytometry, which captures both fluorescence and morphological features of cells, revealed that the Tdphigh comprised mostly of two cells attached to each other, while the Tdpintermediate cells were singlets (Fig. S5B). Further analysis of our CyTOF dataset showed that the Tdphigh pairs did not appear randomly, but instead mainly between a CD45RA+ T cell and CD45RO+ T cell. Indeed, doublets between RAMOS carriers and T cells were never observed despite the over-representation of RAMOS in the samples (~100:1 ratio). The propensity for a subset of CD45RA+ T cells to associate with a subset of CD45RO+ T cells may have biological relevance, especially in light of their harboring a sizable proportion of HIV-fused cells. However, the challenges associated with characterizing pairs of cells by high-dimensional CyTOF led us to focus on the other major population of HIV-fused cells, the Tm.
Among HIV-fused cells were several Tm subsets, including central memory (Tcm) cells; transitional memory (Ttm) cells; Th1-like, Th2-like, and Th17-like cells as defined by chemokine receptor expression levels; follicular helper T (Tfh) cells; and regulatory T cells as defined by memory CD127− cells expressing CD25 (Fig. 1D). To further study whether HIV-F4.HSA preferentially enters a particular Tm subset, we determined the proportion of each subset among the HIV-fused and uninfected populations in four HLAC donors. As expected, Tn were virtually absent among HIV-fused cells while Tm were well-represented (Fig. 1E). Among Tm, Th2-like, Th17-like, and Treg cells were present at significantly higher numbers within HIV-fused cells relative to uninfected cells (Fig. 1F), suggesting preferential entry of R5-tropic HIV into these Tm subsets. While HIV-F4.HSA also fused to the Tcm, Ttm, Th1-like, and Tfh subsets, their proportions were not significantly different from what was observed among uninfected cells, suggesting that these cells were not particularly more susceptible to HIV fusion. Thus, with the exception of Tn, HIV-F4.HSA fuses to a diverse array of CD4+ T cells, reflecting the ability of CCR5-tropic HIV-1 to use a coreceptor that is broadly expressed among Tm.
Global assessment of tonsillar T cells productively infected with HIV
Next, we surveyed the repertoire of cells that went on to become productively infected, as measured by HSA expression. HLACs were mock-treated or infected with HIV-F4.HSA for 4 d, and then stained with our CyTOF panel (Fig. 2A). The gating strategy to identify infected cells is illustrated in Fig. S4B. Compared to the fusion dataset, a different pattern emerged from the t-SNE analysis of productively-infected cells: most of the infected (HSA+) cells now resided in a unique region of the t-SNE plot, distinct from regions occupied by uninfected cells (Fig. 2B). This segregation of the infected cells indicates profound phenotypic differences between productively-infected cells and their non-infected counterparts. We confirmed this finding with both PCA and agglomerative hierarchical clustering (Addendum, Section 2).
Figure 2. HIV-infected tonsillar T cells are remodeled and include the Th17 and Tfh subsets.

(A) Experimental design for phenotyping cells productively infected with HIV-F4.HSA. HLACs were mock-treated or infected with HIV-F4.HSA and cultured for 4 d to allow for productive infection. Cells were then stained with a 38-parameter panel and processed for CyTOF. Productively-infected cells were identified as cells expressing high levels of the reporter gene HSA (Fig. S4B). Analyses were then performed on events corresponding to uninfected (CD3+CD19-HAS−) or infected (CD3+CD19−HSA+) cells.
(B) Representation of uninfected and productively-infected (HSA+) cells by t-SNE based on 35 surface markers. T-cell subsets identified among uninfected cells were used to locate the Tn, Tm, Tdp, CD8+ naïve cell, and CD8+ memory cell regions in the t-SNE map. Note the infected cells largely fall within the “Other” region not occupied by any of these five major subsets.
(C) Representation of uninfected and productively-infected (HSA+) cells by t-SNE based on expression of 31 surface markers (after excluding CD4, CCR5, CD28, and CD62L). T-cell subsets were identified among uninfected cells. Note that unlike in panel B, infected cells predominantly localize in a region of the t-SNE plot where Tm reside.
(D) HIV preferentially infects Tm. Live, singlet CD3+CD8− T cells were gated into Tn and Tm using the gating strategy in Fig. S6A and analyzed for percentage of infected (HSA+) cells. The line graph summarizes results from five independent donors. **p<0.01 as assessed using a paired t-test.
(E) t-SNE plot showing that Th17-like and Tfh subsets are among productively-infected cells. HIV-infected Tm, gated by standard dot plot analysis to identify Th17-like cells (CCR4+CCR6+) and Tfh (CXCR5+PD1+), were overlaid onto total infected (dark grey) and uninfected (light grey) cell populations.
(F) SPADE analysis of productively-infected cells expressing markers of Tfh and Th17. Infected (HSA+) cells were clustered by SPADE into 25 nodes using the 31 cell surface markers used for t-SNE analysis in panel C. Nodes were colored for expression levels of CXCR5, PD1, CCR6, or CCR4. Tfh and Th17-like clusters were defined as CXCR5highPD1high and CCR6highCCR4high clusters, respectively.
(G) HIV infects germinal center (GC) Tfh more efficiently than non-GC Tfh and non-Tfh. Live, singlet Tm were gated into GC Tfh, non-GC Tfh, and non-Tfh based on expression of CXCR5 and PD1 (Fig. S6B) and examined for percentage of HIV-infected cells. Note preferential infection of GC Tfh over non-GC Tfh in 4/5 donors, and preferential infection of GC Tfh over non-Tfh in 5/5 donors. *p<0.05 as assessed using a paired t-test.
(H) HIV infects Th17-like cells at higher efficiency than bulk Tm. Tm and Th17-like cells (Fig. S6C) were examined for percentages of HIV-infected cells. Note in 4/5 donors, HIV infected Th17-like cells at slightly higher rates than that in total Tm. *p<0.05 as assessed using a paired t-test.
(I) Infected Th17-like cells are highly activated while Tfh express a more limited number of activation markers. Top: Histograms showing expression of the indicated activation marker in infected Tfh and Th17-like cells compared to total infected Tm. Bottom: Analysis of productively-infected cells by SPADE, color-coded for the indicated activation marker. The single cluster of Th17 cells at the top expresses intermediate levels of CD38 and PD1, and high levels of CD25, CD69, HLADR, CD28, ICOS, and OX40. Tfh clusters express low levels of CD25 and OX40, intermediate levels of HLADR, CD28, and ICOS, and high levels of CD69, CD38, and PD1. See also Fig. S4 and S6.
We reasoned that the segregation of infected and uninfected cells may be a result of HIV’s ability to downregulate some cell surface antigens, creating what appears to be a unique and novel subset of cells. Four proteins in our panel—CD4, CCR5, CD28, and CD62L—were shown to be down-modulated by Nef (Garcia and Miller, 1991; Michel et al., 2005; Swigut et al., 2001; Vassena et al., 2015). Exclusion of these four markers during t-SNE analysis (i.e. by using 31 instead of 35 parameters) more broadly dispersed the HSA+ cells into a region that corresponded primarily to the Tm region of the t-SNE plot (Fig. 2C). To more precisely quantify the preferential infection of Tm over Tn, we analyzed the proportions of infected (HSA+) cells within each of these subsets (Fig. S6A). For the five HLAC donors tested, Tm harbored a significantly higher proportion of infected cells (Fig. 2D). These results confirm that HIV-F4.HSA preferentially infects Tm, and demonstrates that by excluding receptors modulated by HIV during t-SNE analysis, infected cells can more easily be assigned to cellular subsets found in the uninfected population.
Using this selective t-SNE analysis, we next investigated whether HIV-F4.HSA productively infected the Tfh and Th17-like subsets as reported (Alvarez et al., 2013; Kohler et al., 2016). Infected Tfh were identified by gating on HSA+ cells expressing high levels of CXCR5 and PD1 (Kohler et al., 2016), while infected Th17-like cells were defined as those expressing high levels of CCR4 and CCR6 (Acosta-Rodriguez et al., 2007). Overlaying infected Tfh and Th17-like cells onto the t-SNE plot highlighted their overall phenotypic differences (Fig. 2E). We confirmed that infected Tfh and Th17-like cells were among productively-infected cells via an independent analysis of our dataset with an algorithm called spanning-tree progression analysis of density-normalized event, or SPADE (Qiu et al., 2011) (Fig. 2F). SPADE uses agglomerative clustering and minimum spanning tree construction to build cluster trees in which closely related clusters (shown as colored nodes) are connected by lines. SPADE trees can then be colored to reflect expression of a chosen antigen, enabling identification of a cluster (or group of clusters) that corresponds to cellular subsets of interest. Unlike t-SNE, SPADE uses computational approaches to identify cell clusters and allows objective definition of cell populations. SPADE analysis of the HSA+ cells revealed multiple clusters of Tfh (CXCR5+PD1+) and a single cluster of Th17-like cells (CCR4+CCR6+) (Fig. 2F). As in the t-SNE analysis, the Tfh and Th17-like clusters did not overlap and were far apart on the SPADE tree, again highlighting their phenotypic dissimilarity.
To assess whether Tfh and Th17-like cells were infected at higher rates than other subsets, we compared infection rates in non-Tfh and Tfh. We further divided Tfh into germinal center (GC) and non-GC Tfh, which express high and intermediate levels of PD1 and CXCR5, respectively (Fig. S6B). Averaging data from five donors, infection rates were highest among GC Tfh and lowest among non-Tfh (Fig. 2G, S6B), and Th17-like cells were infected in higher proportions than bulk Tm (Fig. 2H, S6C).
Because HIV preferentially replicates in activated T cells, we next examined the activation status of the infected Tfh and Th17-like cells by assessing their expression of the eight antigens in our panel associated with T-cell activation. As shown by standard histogram analysis, Th17-like cells exhibited intermediate or high levels of all 8 activation markers, while Tfh expressed high levels of some activation markers (e.g., CD69, PD1, and CD38) but low levels of others (e.g., CD25 and OX40) (Fig. 2I). SPADE analysis of the Tfh and Th17 clusters (Fig. 2F) confirmed that Th17 cells expressed high levels of all eight activation markers, while the Tfh exhibited a more diverse pattern of activation marker expression (Fig. 2I). Therefore, the high rates of productive infection in Tfh cannot be entirely explained by the activation state of the cells. For a deeper analysis of the findings reported in this section, we present an independent t-SNE analysis showing the raw expression values of all the antigens described here (Addendum, Section 2).
SLIDE analysis reveals both Nef-dependent and Nef-independent remodeling in HIV-infected cells
Analysis by t-SNE revealed that HIV-infected and uninfected cells are phenotypically distinct, largely due to variations in CD4, CCR5, CD28, and CD62L expression (Fig. 2B, C). To quantitate the remodeling of cells by HIV, we used SLIDE, a two-step, nearest-neighbor algorithm that quantifies the similarity between each virally-infected cell to its most closely-related cell found in the uninfected sample (Sen et al., 2014). To implement SLIDE on our datasets, for every HIV-infected cell, the most phenotypically similar nearest-neighbor cell in the mock-infected sample was identified and the dissimilarity between the cells was defined by a distance (d1) (Fig. 3A). The identified uninfected cell was then subjected to its own nearest-neighbor analysis to identify a second distance (d2) defining the dissimilarity between the two uninfected cells in the mock-infected sample. The SLIDE score is represented as the ratio d1/d2 and, in the complete absence of remodeling, equals to 1. By contrast, remodeling raises the SLIDE score to >1. We conducted SLIDE on ~2 × 103 infected (HSA+) cells against each of the ~105 uninfected T cells. To study the role of Nef in remodeling, we generated new fusion and replication datasets using the isogenic reporter virus lacking Nef (HIV-F4.HSA.dNef), engineered and tested as described in Fig. S1. The impact of both viruses on remodeling was assessed within both the fusion and infection datasets (cumulative SLIDE analysis shown in Fig. 3B). While the SLIDE score for HIV-F4.HSA was close to 1 (1.136) in the fusion dataset, the score for productively-infected cells was significantly greater (1.433; p<0.0001). Infection with HIV-F4.HSA.dNef decreased the SLIDE score to 1.343 (p<0.0001 relative to score of 1.433 for HIV-F4.HSA), highlighting Nef’s partial role in HIV remodeling. Interestingly, when CD4, CCR5, CD28, and CD62L were removed from the SLIDE analysis, the scores for both HIV-F4.HSA and HIV-F4.HSA.dNef decreased significantly [from 1.433 to 1.204 (p<0.0001) for HIV-F4.HSA, and 1.343 to 1.193 (p<0.0001) for HIV-F4.HSA.dNef], but remained higher than the corresponding fusion datasets [1.204 vs. 1.136 (p<0.0001) for HIV-F4.HSA, and 1.193 vs. 1.135 (p<0.0001) for HIV-F4.HSA.dNef]. This finding suggests that antigens other than these four proteins are modulated by HIV independently of Nef. Taken together, the SLIDE data provide statistical evidence for both Nef-dependent and -independent remodeling in HIV-infected cells.
Figure 3. SLIDE analysis reveals both Nef-dependent and Nef-independent remodeling.

(A) Principle of SLIDE analysis. The final SLIDE score corresponds to the average of the d1/d2 ratios determined for each fused or infected cell.
(B) SLIDE scores for the fusion and infection datasets. SLIDE analysis for fused cells was conducted using all 35 parameters used for t-SNE in Fig. 1. SLIDE analysis for infected cells was conducted with either 35 or 31 parameters (excluding CD4, CCR5, CD28, and CD62L). Data for each condition are representative of data averaged from at least 3 tonsillar donors. **** p<0.0001, n.s. = non-significant, as assessed by a two-sample Wilcoxon test (see Statistics section in Supplemental Experimental Procedures). Error bars correspond to standard deviations.
PP-SLIDE analysis identifies the specific receptors modulated during HIV infection
We next aimed to determine which surface proteins were actively altered by infection. Assessing HIV modulation of a particular receptor is confounded by the fact that HIV does not replicate equivalently in different cell types. Therefore, it is difficult to distinguish between active downregulation of an antigen in infected cells from preferential infection of cells expressing this antigen at low levels, as both processes can lead to similar overlay plots at the population level (Fig. S7A). However, by comparing infected cells to their precursors (i.e., the cells that HIV initially targeted) instead of to the bulk population of uninfected T cells, we would be able to distinguish receptor down-modulation by HIV from preferential infection (Fig. S7B). The reason for this is that when infection triggers downregulation of an antigen, the infected cells will express lower levels of this antigen relative to their precursors. Conversely, when HIV preferentially infects cells with lower levels of an antigen, both the infected cells and their precursors will express this antigen at equivalent levels (Fig. S7B).
The identification of the precursor of an infected cell can essentially be achieved by finding the nearest neighbor of an infected cell in the uninfected population (Step 1 of SLIDE, Fig. 3A). This is because in our datasets, each cell is defined by more than 30 parameters, most of which we expect to remain unaffected upon HIV infection. It is therefore likely that an uninfected cell phenotypically similar to a given infected cell can be found in the uninfected population and will closely resemble the cell that the virus initially targeted. To highlight this resemblance, we now change the term “nearest neighbor” to “predicted precursor” identified by SLIDE (PP-SLIDE) (Fig. S7B). After identifying the population of predicted precursor cells with PP-SLIDE, we displayed, in box plot format, expression levels of each antigen of interest in infected vs. predicted precursor cells, and used statistical t-tests (see Supplemental Experimental Procedures) to determine whether these populations exhibit significant differences in expression levels of the antigen.
We first determined whether PP-SLIDE analysis could successfully identify CD4 as a receptor modulated by HIV infection. Indeed, infected cells expressed significantly lower levels of CD4 than their predicted precursors (Fig. 4A). Likewise and as expected (Swigut et al., 2001), PP-SLIDE analysis identified CD28 as a receptor that is down-modulated upon infection with HIV-F4.HSA expressing Nef but not with the corresponding Nef-deficient virus (Fig. 4B). Having validated PP-SLIDE analysis on CD4 and CD28, we then assessed whether other receptors in our panel were also downregulated. We found that HIV-F4.HSA infection modified expression of OX40, a member of the TNFR-superfamily of receptors. In HSA+ cells, OX40 was expressed at higher levels relative to the bulk of uninfected cells (Fig. S8A, left), but at significantly lower levels than in their predicted precursors (Fig. S8A, right), suggesting that HIV-F4.HSA preferentially infects cells with higher levels of OX40, but downregulates OX40 upon infection. Similar analysis with the fusion datasets showed that HIV-F4.HSA fused to cells expressing high levels of OX40 (Fig. S8B, left), but could not down-modulate OX40 within the 2-h infection window as evidenced by similar expression of OX40 in HIV-fused cells and their predicted precursors (Fig. S8B, right). Flow cytometric analysis of infected HLACs revealed that while uninfected tonsillar Tm expressed either high or low levels of OX40, productively-infected cells expressed OX40 at slightly lower levels than the population of OX40high cells (Fig. S8C), also implicating HIV-induced OX40 downregulation. Interestingly, downregulation of OX40 on infected cells occurs independently of Nef, as HSA+ cells infected with HIV-F4.HSA.dNef still show significantly lower levels of OX40 compared to their predicted precursors (Fig. S8D, right).
Figure 4. PP-SLIDE analysis of effects of HIV infection on expression levels of cell surface receptors.

(A) PP-SLIDE analysis of CD4 on cells productively infected with HIV-F4.HSA. The left plot shows CD4 expression on infected (dark blue) vs. bulk uninfected (red) cells. The right plot shows CD4 expression on infected (dark blue) vs. predicted precursor (light blue) cells. Levels of CD4 are significantly lower on infected cells relative to their predicted precursors. ****p<0.0001.
(B) PP-SLIDE analysis of CD28 on cells productively infected with HIV-F4.HSA (left) or Nef-deficient HIV-F4.HSA.dNef (right). Box plots show CD28 expression on infected cells (dark blue) relative to predicted precursors (light blue). Significant downregulation of CD28 by HIV-F4.HSA but not HIV-F4.HSA.dNef was observed. **p<0.01, ****p<0.0001, n.s.: non-significant.
(C) PP-SLIDE analysis of CD69, CD57, and CD127 on cells productively infected with HIV-F4.HSA. Left plots show infected (dark blue) vs. bulk uninfected (red) cells, while right plots show infected (dark blue) vs. predicted precursor (light blue) cells. n.s. non-significant as assessed by rejecting the null hypothesis that median antigen levels in infected and uninfected nearest-neighbor cells are statistically equivalent (see Statistics section in Supplemental Experimental Procedures). See also Fig. S7 and S8.
In contrast to CD4, CD28, and OX40, many markers in our panel were differentially expressed between infected and uninfected cells, but not between infected cells and their predicted precursors. For example, CD69 and CD57 were higher on infected cells than uninfected cells, but similar between infected cells and their predicted precursors (Fig. 4C), suggesting that HIV-F4.HSA selectively infects cells with high levels of CD69 and CD57 without changing their expression levels. Conversely, CD127, a component of the IL7 receptor, was expressed at lower levels on infected cells than uninfected cells, but at the same levels between infected cells and their predicted precursors (Fig. 4C). This suggests that HIV-F4.HSA preferentially replicates in cells expressing low levels of CD127 and does not down-regulate this receptor.
HIV-F4.HSA enters both CD57+ and CD127+ Tm but HIV gene expression only occurs in CD57+ Tm
To further characterize the cells supporting HIV replication, we assessed whether certain subsets were under- or over-represented among productively-infected cells. With t-SNE, it was apparent that although CD57+ Tm and CD127+ Tm were well-represented among uninfected T cells (Fig. 5A, left), a high proportion of the infected cells expressed CD57 while very few expressed CD127 (Fig. 5A, right). These results suggest that HIV replication occurs in CD57+ but not CD127+ Tm, which we confirmed in multiple donors (Addendum, Section 2). Surprisingly, analysis of the fusion dataset revealed a good representation of CD127+ Tm among HIV-fused cells (Fig. 5B). Standard 2D plot analysis confirmed that while CD127+ cells were rare among productively-infected cells (Fig. 5C), they were as abundant as CD57+ cells among HIV-fused cells (Fig. 5D). Analysis of infected CD57+ and CD127+ Tm from five different donors revealed infected CD127+ Tm at significantly lower proportions than infected CD57+ Tm, which was not observed among HIV-fused cells (Fig. 5E). Because neither CD57 nor CD127 are remodeled by HIV after infection (Fig. 4C), our results suggest that HIV efficiently enters both CD57+ and CD127+ Tm, but only replicates efficiently in the former subset. Independent analysis by Scaffold, a recently developed clustering and data visualization approach (Spitzer et al., 2015), confirmed that infected cells do not bear any phenotypic features of CD127+ Tm, further supporting that CD127+ Tm are not susceptible to productive infection by HIV-F4.HSA (Addendum, Section 3). Interestingly, cellular activation state did not account for the preferential HIV replication in CD57+ Tm over CD127+ Tm, since clustering of CD57+ and CD127+ Tm by SPADE revealed that clusters within both subsets included unactivated and activated cells, as assessed by expression of CD38 and HLA-DR (Fig. 5F).
Figure 5. CD57+ and CD127+ Tm exhibit differential susceptibility to productive HIV infection.

(A) CD57+ but not CD127+ Tm are well-represented among cells productively infected with HIV-F4.HSA. t-SNE overlays of uninfected (left) or infected (right) Tm expressing CD57 or CD127. Note the paucity of CD127+ Tm (magenta) among infected cells.
(B) Both CD57+ and CD127+ Tm are well-represented among cells that support fusion to HIV-F4.HSA. t-SNE overlay of uninfected (left) or HIV-fused (right) Tm expressing CD57 or CD127. Note the prevalence of CD127+ Tm (magenta) among HIV-fused cells.
(C) Standard FACS plots of CD57 and CD127 expression in uninfected (left) and infected (HSA+) (right) cells. Events shown were pre-gated on live, singlet Tm. Note the paucity of CD127+ cells (bottom right-hand quadrant) among infected but not uninfected cells.
(D) Standard FACS plots of CD57 and CD127 expression in uninfected (left) and HIV-fused (right) cells. The plots shown were pre-gated on live, singlet Tm. Note that unlike in panel C, CD127+ cells are abundant in both samples.
(E) Proportion of CD57+ and CD127+ Tm among infected (HSA+) and HIV-fused cells. Results correspond to data from 4–5 donors. Error bars correspond to standard deviations. ****p<0.0001. Note that while CD57+ Tm and CD127+ Tm were equally represented among HIV-fused cells (right), the proportion of CD127+ Tm was significantly lower among productively-infected cells (left).
(F) CD38 and HLADR are expressed by both CD57+ and CD127+ Tm. HIV-fused Tm were distributed by SPADE into 25 nodes based on 35 cell-surface markers and subsequently colored by levels of the indicated markers. Note expression of CD38 and HLADR in clusters of both CD57+ and CD127+ Tm.
Confirmation of lack of productive infection in CD127+ Tm in vitro and in vivo
To ensure that the lack of productively-infected CD127+ Tm did not result from differences in kinetics of HIV replication, we conducted a time-course experiment (Fig. 6A). HLACs were infected with BlaM-Vpr-containing HIV-F4.HSA and samples were taken at different time points and analyzed either for fusion (2 h) or productive infection (2–5 days). Gating strategies to measure the proportion of HIV-fused and HIV-infected cells in Tm, CD57+ Tm, and CD127+ Tm are described in Fig. S9. At 2 h post-infection, HIV-F4.HSA had entered CD127+ Tm with slightly higher efficiency than CD57+ Tm (Fig. 6A: 3.5% versus 2.4%). However, HIV-F4.HSA failed to express the HSA reporter in CD127+ Tm throughout the entire 5-day time course. Consistent with the inability of CD127+ Tm to support efficient viral replication, Tm enriched for CD127+ Tm replicated HIV less efficiently than bulk Tm, even though both cultures supported similar levels of fusion (Fig. S10).
Figure 6. Confirmation of differential susceptibility of CD57+ and CD127+ Tm to productive HIV infection.

(A) Kinetic assessment of HIV fusion and productive infection. HLACs were infected with BlaM-Vpr-containing HIV-F4.HSA. After 2 h, the cells were washed and split into 2 sets. A set corresponding to 20% of the culture was used to quantify fusion, while the remainder was cultured for 2 – 5 d to quantify productive infection. The first row of plots shows the proportion of HIV-fused cells within total Tm, CD57+ Tm, and CD127+ Tm, demonstrating similar levels of fusion in the 3 populations. The remaining plots show the proportion of productively-infected cells within total Tm, CD57+ Tm, and CD127+ Tm, demonstrating consistent low proportions of HSA+ cells within CD127+ Tm after 2, 3, 4, and 5 d. Gating strategies for defining HIV-fused and infected cells are presented in Fig. S9. Shown is one of two representative experiments (using cells from different donors) that gave similar results. See also Fig. S9 and S10.
(B) In situ hybridization detection of HIV RNA and immunofluorescence detection of CD57 and CD127 in lymph node tissues of an uninfected and one of two representative HIV-infected subjects. The infected lymph node comes from a viremic 48-year-old HIV+ male subject with lymphadenopathy. Scale bar = 100 μm.
In vivo, CD57+ Tm localize preferentially in the GC while CD127+ Tm are found in the extrafollicular region (Lim and Kim, 2007), suggesting that these subsets serve different functions. Indeed, besides differentially expressing CD57 and CD127, these subsets express completely different patterns of other cell surface proteins (Addendum, Section 4), suggesting that they are indeed phenotypically distinct. To evaluate whether productive infection in infected patients is found preferentially in the CD57+ zone, we analyzed regions of active HIV replication in a lymph node obtained from a viremic HIV-infected patient. Immunostaining identified the CD57+ GC and CD127+ extrafollicular regions, while RNAscope identified areas of viral replication. While RNAscope revealed only background signal in control samples from an uninfected individual, distinct regions of HIV replication were observed in the lymph nodes of HIV+ individuals. These regions containing a high density of virions localized where CD57+ cells were enriched and CD127+ cells were under-represented (Fig. 6B). These data suggest that active HIV replication largely occurs in lymph node regions where CD57+ cells are abundant and CD127+ cells are scarce, consistent with our in vitro findings that Tm expressing CD57 but not CD127 are targets of active HIV replication.
Discussion
We used high-dimensional CyTOF analysis to characterize the phenotypes of tissue CD4+ T cells after exposure to HIV. By applying a 38-parameter panel that spans activation, differentiation, and polarization markers, we created an atlas of the types of tonsillar CD4+ T cells that support HIV entry, and the smaller subset of cells that support productive infection. These datasets were mined using a combination of dimensionality reduction visualization (t-SNE, PCA), clustering (agglomerative hierarchical clustering, SPADE, Scaffold), and quantitative statistical approaches (SLIDE and the subanalysis, PP-SLIDE) to assess HIV-induced remodeling and identify cellular subsets with differential susceptibility to HIV infection.
Characterizing the phenotypes of virally-infected cells is challenging because viruses can selectively enter and/or replicate in particular cell subsets while, at the same time, alter the phenotype of infected cells. To distinguish between these processes, we analyzed both early and late phases of HIV infection and characterized viral-induced remodeling. The fusion dataset, measuring the early phase of HIV infection, was relatively straightforward to analyze given the limited remodeling observed during a 2-h incubation period. We found all Tm – including Tcm, Ttm, Tfh, Treg, and the Th1-, Th2-, and Th17-like subsets – supported fusion, with the Th2-like, Th17-like, and Treg subsets being preferentially targeted (Fig. 1). Analysis of productive infection required more extensive approaches to discern the effects of selective replication from viral-induced changes. To limit the contribution of viral-induced changes in the analysis, we removed from t-SNE analysis markers reported to be altered by HIV infection (e.g., CD4, CCR5, CD62L, and CD28). This approach, which allowed productively-infected cells to be matched to their uninfected counterparts in t-SNE plots (Fig. 2C), demonstrated that productively-infected Tm are heterogeneous with regards to activation state, and include previously described HIV-permissive cells such as the Tfh and Th17-like subsets (Alvarez et al., 2013; Kohler et al., 2016), which were preferentially targeted for productive infection.
A second set of approaches using SLIDE allowed a more thorough analysis of overall virus-induced remodeling. SLIDE was developed to assess VZV-mediated remodeling of cells (Sen et al., 2014), and allows quantification of remodeling by first identifying an uninfected nearest neighbor cell most phenotypically similar to each infected cell (and therefore referred to as the predicted precursor cell) and then measuring the relative dissimilarity between these two cells. The SLIDE score calculated for HIV-fused cells was marginally but significantly greater than 1, the theoretical SLIDE score of cells in the complete absence of remodeling. Because fusion was measured after just 2 h of infection, a timeframe too short to see CD4 downregulation and likely other remodeling effects, we speculate that the slight increase in SLIDE score may be due to free virions bound to the cell surface, affecting detection of some markers by CyTOF antibodies. By contrast, the SLIDE score for productively-infected cells was significantly higher than that for HIV-fused cells, highlighting significant levels of remodeling induced by HIV-F4.HSA after 4 d of infection. This remodeling was partially mediated by Nef and affected receptors other than the previously described CD4, CCR5, CD62L, and CD28 (Fig. 3).
To identify other markers modulated by HIV, we applied SLIDE to directly compare any given marker on an infected cell to its predicted precursor (PP-SLIDE). Using this method, we found that most markers expressed at different levels on infected relative to uninfected cells (e.g., CD69) were in fact expressed at similar levels between infected and predicted precursor cells, indicating preferential infection of these cells as opposed to HIV-induced changes in receptor expression. Consistent with previous reports (Garcia and Miller, 1991; Swigut et al., 2001), CD4 and CD28 were actively downregulated by HIV during infection. OX40, a TNF receptor family member, was identified as a receptor remodeled by HIV, although the molecular pathways involved in this remodeling are unknown. We envision that SLIDE will be a broadly useful tool to assess how HIV impacts expression of any receptor quantifiable by CyTOF or flow cytometry, due to its dual application in quantifying remodeling and predicting precursor cells (via PP-SLIDE), as shown in this study. This bioinformatics-based approach will complement experimental approaches of infecting cells sorted based on expression levels of a receptor of interest to assess infection-induced receptor modulation.
In future work, the nearest-neighbor approaches described here could be used in conjunction with Wanderlust, a method recently developed to establish the trajectory of B-cell development from analysis of a mixed population of cells (Bendall et al., 2014). While SLIDE identifies a single nearest neighbor cell and compares cells between two different datasets (i.e., infected vs. uninfected), Wanderlust receives as input a single dataset and uses k-nearest neighbor approaches to score each cell. These scores place similar cells more closely to each other along a trajectory. By running Wanderlust on an asynchronous population of HIV-infected cells, we could additionally gain information on the path a permissive cell takes as it progresses from the pre-infection state to the early, intermediate, and final stages of infection.
After globally analyzing the cell types susceptible to HIV entry, infection, and remodeling, we focused on two cell subsets that emerged from the datasets: Tm expressing CD57 and Tm expressing CD127. Although both supported HIV-F4.HSA fusion, only CD57+ Tm were found among productively-infected cells (Fig. 5). By PP-SLIDE, we found that lack of CD127 expression among productively-infected cells was not due to downregulation of CD127 upon infection (Fig. 4). Furthermore, CD127+ Tm were not delayed in HIV gene expression, and Tm enriched for CD127+ cells showed low levels of viral replication, despite allowing viral entry (Fig. 6A and S10). Our multidimensional analysis highlighted profound dissimilarities between CD57+ and CD127+ Tm, since these subsets always segregated apart (Fig. 5), whether or not CD57 and CD127 were used in the analysis (Addendum, Section 4) thereby highlighting phenotypic differences independent of CD57 and CD127 expression. Interestingly, in vivo, CD57+ and CD127+ Tm are also spatially segregated, with CD57+ and CD127+ cells residing primarily in the GC and extrafollicular region, respectively (Lim and Kim, 2007). The microenvironment and/or distinct functions of these two types of Tm may provide an explanation as to why these cells are so phenotypically distinct. Consistent with our in vitro findings that HIV efficiently replicates in CD57+ but not CD127+ Tm, viral replication localized primarily to regions where CD57+ Tm reside (Fig. 6B). However, most detected viruses were part of the extracellular network and we did not detect HIV-infected CD57+ T cells in the HIV+ lymph nodes, which may reflect a sampling bias or issues with assay sensitivity. Future studies from larger, diverse cohorts may resolve our findings in vivo.
The molecular basis for the block in HIV replication in CD127+ Tm is unknown. CD127+ Tm could naturally resist HIV by harboring high levels of active SAMHD1, an enzyme that depletes intracellular nucleotides and blocks reverse transcription. Indeed, SAMHD1-expressing cells primarily localize to the extrafollicular region (Baldauf et al., 2012) where CD127+ cells tend to reside (Lim and Kim, 2007) (Fig. 6B). However, blocks other than SAMHD1-mediated restriction are likely at play, given that the SAMHD1 block occurs primarily in resting cells (Baldauf et al., 2012) and CD127+ Tm harbor a subset of activated cells (Fig. 5F). CD127+ and CD57+ Tm likely harbor major differences in transcription factors, which could profoundly affect LTR activity. It would be intriguing to study whether CD127+ Tm could harbor integrated, transcriptionally silent HIV, especially because CD127 is expressed by long-lived cells and could therefore potentially constitute a long-term stable reservoir. Consistent with expression of CD127 on latently-infected cells are reports that IL-7, a cytokine that stimulates homeostatic proliferation of cells through CD127, drives persistence of latently-infected cells (Chomont et al., 2009).
Experimental Procedures
Processing, infection, and analysis of samples by CyTOF
Human tonsils obtained from CHTN (IRB#12-08429) were processed into HLACs as previously described (Jekle et al., 2003). For viral fusion, HLACs were depleted of B cells using CD19 microbeads (Miltenyi) and incubated for 2 h with BlaM-Vpr virions of HIV-F4.HSA or HIV-F4.HSA.dNef whose construction are described in the Supplemental Experimental Procedures. Viral fusion was assessed as described (Cavrois et al., 2002; Cavrois et al., 2014). Fused cells from virus-exposed cultures, and control cells from unexposed cultures, were sorted and processed for CyTOF. For viral infection experiments, HLACs were incubated with HIV-F4.HSA or HIV-F4.HSA.dNef for 2 h, washed, and cultured for 4 d unless otherwise noted, before assessment of infection.
To process cells for CyTOF, 6 × 106 cells from infected or non-infected cultures were stained with the cocktail of metal isotope-conjugated antibodies (Table S1), similar to methods described (Bendall et al., 2014). Given that only ~150,000 fused cells were sorted from each donor, “carrier cells” of the B-cell line RAMOS were added to minimize cell loss during processing. Antibody staining was performed in a total volume of 100 μl for 45 min, followed by treatment with 139In-DOTA maleimide (Macrocyclics) as a live/dead marker, and fixation overnight in PBS supplemented with 2% PFA. Cells were permeabilized for 45 min at 4°C with Permeabilization Buffer (eBioscience) and resuspended in 1:4,000 191/193Ir DNA intercalator (Fluidigm). Cells were washed, resuspended in distilled water, and analyzed on a CyTOF2 (Fluidigm) at the Stanford Shared FACS Facility. Between 300K and 900K events were acquired per sample. EQ™ calibration beads (Fluidigm) were included and used for normalization across runs. Data were analyzed with FlowJo (Treestar), Cytobank, Partek Genomics Suite, SLIDE and the PP-SLIDE subanalysis, and Scaffold as detailed below and in the Supplemental Experimental Procedures and Addendum.
Flow cytometric analysis of samples
To characterize HIV fusion and infection among Tm, CD57+ Tm, and CD127+ Tm, HLACs were infected with HIV-F4.HSA containing BlaM-Vpr as described above, and analyzed for fusion after 2 h, while infection was monitored from 2 – 5 d post-infection by staining cells with flow cytometric antibodies against CD3, CD19, CD4, CD45RA, CD45RO, CD57, CD127, HSA, and a LIVE/DEAD fixable Green Dead Cell Stain kit (Molecular Probes) (Table S2).
Immunofluorescence Analysis
FFPE lymph node blocks from an uninfected and a viremic HIV-infected individual (IRB#14-13840) were detected by ISH using the RNASCOPE® 2.5 HD brown kit (Advanced Cell Diagnostics). For IF and ISH multiplexing, sections were treated with 10% FBS followed by overnight incubation with mouse anti-CD57 (eBioscience) and rabbit anti-CD127 (LifeSpan Biosciences). Following incubation with Alexa Fluor 650-conjugated anti-rabbit or Alexa Fluor 488-conjugated anti-mouse secondary antibodies (Thermo Fisher Scientific), sections were washed, incubated with DAPI, and scanned using a Zeiss Axio Scan.Z1.
Statistical Analysis
Pairwise statistical comparisons between different cellular subsets in multiple donors were compared using a paired one-sided t-test. To test the statistical hypothesis of no remodeling versus remodeling, SLIDE (Sen et al., 2015) was implemented. Specifically, uninfected cells phenotypically most similar to HIV-fused and -infected cells were identified using a nearest neighbor approach wherein empirical distances based on the agglomerative L1 norm across proteins was used to determine remodeling scores. To compare the extent of remodeling between different populations, we tested whether the average remodeling score of one population was significantly different from that of the other using a one-sided two-sample Wilcoxon test (Fay and Proschan, 2010). Statistical tests for expression differences in specific receptors between the fused/infected cells and their predicted precursors (PP-SLIDE sub-analysis) were conducted based on an intersection test involving a two-sided binomial test of equality of proportions, and a two-sample Wilcoxon test.
Supplementary Material
Highlights.
CyTOF-generated atlas of CD4+ T cells susceptible to HIV entry and infection
High-dimensional analysis to distinguish remodeling from preferential infection
Development of statistical method to identify receptors modulated on infected cells
Identification of CD4+ T memory subset with post-entry block
Acknowledgments
This work was supported by NIH (R00AI104262, R01AI127219 to NRR, DP1DA036502 to WCG, U19AI96109 to JMM) and the amfAR Institute for HIV Cure Research (109301). CO and JJJ were supported by CAVIMC (1032144). TB and GM were supported by the Zumberge award from USC’s Zumberge faculty research and innovation fund. We acknowledge NIH for the sorter (S10-RR028962); DOD for the Imagestream (W81XWH-11-1-0562); the James B. Pendelton Charitable Trust; and CFAR (P30-AI-027763, P30-AI-27767). We thank B. Carter, M. Leipold, and H. Maecker for advice on CyTOF, D. Soler-Ferran for the 1B5 clone, J.Y. Lee for microscopy assistance, T. Roberts for graphics, C. Herron and G. Howard for editorial assistance, and S. Cammack and R. Givens for administrative assistance. We thank the Stanford Shared FACS Facility, and the Molecular Imaging Center facility at UC Berkeley.
Footnotes
Author Contributions
MC and NRR conceived the study. MC, NR, RH, BR, JV, JJJ, and NRR conducted experiments. MC, TB, GM, RH, MHS, NHL, CO, JMM, ECB, AMA, NS, WCG, and NRR analyzed data. MC and NRR wrote the manuscript. NRR supervised the study.
Accession Codes
Raw FCS CyTOF data files can be accessed through Cytobank Community (Experiment “Cavrois-et-al_CellReports_2017”).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nature immunology. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- Alberti MO, Jones JJ, Miglietta R, Ding H, Bakshi RK, Edmonds TG, Kappes JC, Ochsenbauer C. Optimized Replicating Renilla Luciferase Reporter HIV-1 Utilizing Novel Internal Ribosome Entry Site Elements for Native Nef Expression and Function. AIDS Res Hum Retroviruses. 2015 doi: 10.1089/aid.2015.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez Y, Tuen M, Shen G, Nawaz F, Arthos J, Wolff MJ, Poles MA, Hioe CE. Preferential HIV infection of CCR6+ Th17 cells is associated with higher levels of virus receptor expression and lack of CCR5 ligands. Journal of virology. 2013;87:10843–10854. doi: 10.1128/JVI.01838-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe’er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nature medicine. 2012;18:1682–1687. doi: 10.1038/nm.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Davis KL, Amir el AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, Pe’er D. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157:714–725. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:1925–1930. doi: 10.1073/pnas.94.5.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol. 2002;20:1151–1154. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- Cavrois M, Neidleman J, Greene WC. HIV-1 Fusion Assay. Bio Protoc. 2014:4. doi: 10.21769/bioprotoc.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nature medicine. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corneau A, Cosma A, Even S, Katlama C, Le Grand R, Frachet V, Blanc C, Autran B. Comprehensive Mass Cytometry Analysis of Cell Cycle, Activation, and Coinhibitory Receptors Expression in CD4 T Cells from Healthy and HIV-Infected Individuals. Cytometry B Clin Cytom. 2017;92:21–32. doi: 10.1002/cyto.b.21502. [DOI] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, Denham SA, Heil ML, Kasolo F, Musonda R, Hahn BH, et al. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science (New York, NY. 2004;303:2019–2022. doi: 10.1126/science.1093137. [DOI] [PubMed] [Google Scholar]
- El Hed A, Khaitan A, Kozhaya L, Manel N, Daskalakis D, Borkowsky W, Valentine F, Littman DR, Unutmaz D. Susceptibility of human Th17 cells to human immunodeficiency virus and their perturbation during infection. The Journal of infectious diseases. 2010;201:843–854. doi: 10.1086/651021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay MP, Proschan MA. Wilcoxon-Mann-Whitney or t-test? On assumptions for hypothesis tests and multiple interpretations of decision rules. Stat Surv. 2010;4:1–39. doi: 10.1214/09-SS051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JV, Miller AD. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature. 1991;350:508–511. doi: 10.1038/350508a0. [DOI] [PubMed] [Google Scholar]
- Hamann D, Baars PA, Hooibrink B, van Lier RW. Heterogeneity of the human CD4+ T-cell population: two distinct CD4+ T-cell subsets characterized by coexpression of CD45RA and CD45RO isoforms. Blood. 1996;88:3513–3521. [PubMed] [Google Scholar]
- Jekle A, Keppler OT, De Clercq E, Schols D, Weinstein M, Goldsmith MA. In vivo evolution of human immunodeficiency virus type 1 toward increased pathogenicity through CXCR4-mediated killing of uninfected CD4 T cells. J Virol. 2003;77:5846–5854. doi: 10.1128/JVI.77.10.5846-5854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler SL, Pham MN, Folkvord JM, Arends T, Miller SM, Miles B, Meditz AL, McCarter M, Levy DN, Connick E. Germinal Center T Follicular Helper Cells Are Highly Permissive to HIV-1 and Alter Their Phenotype during Virus Replication. J Immunol. 2016;196:2711–2722. doi: 10.4049/jimmunol.1502174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HW, Kim CH. Loss of IL-7 receptor alpha on CD4+ T cells defines terminally differentiated B cell-helping effector T cells in a B cell-rich lymphoid tissue. J Immunol. 2007;179:7448–7456. doi: 10.4049/jimmunol.179.11.7448. [DOI] [PubMed] [Google Scholar]
- Matheson NJ, Sumner J, Wals K, Rapiteanu R, Weekes MP, Vigan R, Weinelt J, Schindler M, Antrobus R, Costa AS, et al. Cell Surface Proteomic Map of HIV Infection Reveals Antagonism of Amino Acid Metabolism by Vpu and Nef. Cell host & microbe. 2015;18:409–423. doi: 10.1016/j.chom.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel N, Allespach I, Venzke S, Fackler OT, Keppler OT. The Nef protein of human immunodeficiency virus establishes superinfection immunity by a dual strategy to downregulate cell-surface CCR5 and CD4. Curr Biol. 2005;15:714–723. doi: 10.1016/j.cub.2005.02.058. [DOI] [PubMed] [Google Scholar]
- Pan X, Baldauf HM, Keppler OT, Fackler OT. Restrictions to HIV-1 replication in resting CD4+ T lymphocytes. Cell research. 2013;23:876–885. doi: 10.1038/cr.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picker LJ, Treer JR, Ferguson-Darnell B, Collins PA, Buck D, Terstappen LW. Control of lymphocyte recirculation in man. I. Differential regulation of the peripheral lymph node homing receptor L-selectin on T cells during the virgin to memory cell transition. J Immunol. 1993;150:1105–1121. [PubMed] [Google Scholar]
- Qiu P, Simonds EF, Bendall SC, Gibbs KD, Jr, Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross TM, Oran AE, Cullen BR. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr Biol. 1999;9:613–621. doi: 10.1016/s0960-9822(99)80283-8. [DOI] [PubMed] [Google Scholar]
- Sen N, Mukherjee G, Arvin AM. Single cell mass cytometry reveals remodeling of human T cell phenotypes by varicella zoster virus. Methods. 2015;90:85–94. doi: 10.1016/j.ymeth.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Mukherjee G, Sen A, Bendall SC, Sung P, Nolan GP, Arvin AM. Single-cell mass cytometry analysis of human tonsil T cell remodeling by varicella zoster virus. Cell reports. 2014;8:633–645. doi: 10.1016/j.celrep.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer MH, Gherardini PF, Fragiadakis GK, Bhattacharya N, Yuan RT, Hotson AN, Finck R, Carmi Y, Zunder ER, Fantl WJ, et al. IMMUNOLOGY. An interactive reference framework for modeling a dynamic immune system. Science (New York, NY. 2015;349:1259425. doi: 10.1126/science.1259425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swigut T, Shohdy N, Skowronski J. Mechanism for down-regulation of CD28 by Nef. The EMBO journal. 2001;20:1593–1604. doi: 10.1093/emboj/20.7.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L, Hinton G. Visualizing Data using t-SNE. J Mach Learn Res. 2008;9:2579–2605. [Google Scholar]
- Vassena L, Giuliani E, Koppensteiner H, Bolduan S, Schindler M, Doria M. HIV-1 Nef and Vpu Interfere with L-Selectin (CD62L) Cell Surface Expression To Inhibit Adhesion and Signaling in Infected CD4+ T Lymphocytes. J Virol. 2015;89:5687–5700. doi: 10.1128/JVI.00611-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MT, Chen J, Narayanan S, Lin W, Anicete R, Kiaang HT, De Lafaille MA, Poidinger M, Newell EW. Mapping the Diversity of Follicular Helper T Cells in Human Blood and Tonsils Using High-Dimensional Mass Cytometry Analysis. Cell reports. 2015;11:1822–1833. doi: 10.1016/j.celrep.2015.05.022. [DOI] [PubMed] [Google Scholar]
- Yu H, Khalid M, Heigele A, Schmokel J, Usmani SM, van der Merwe J, Munch J, Silvestri G, Kirchhoff F. Lentiviral Nef proteins manipulate T cells in a subset-specific manner. J Virol. 2015;89:1986–2001. doi: 10.1128/JVI.03104-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.