

Abstract
Selective tuning of arylethynyl urea scaffolds for anionic guests requires an understanding of preferred binding motifs of the host–guest interaction. To investigate the binding preference of receptors without a pre-organized binding pocket, two electron-deficient phenylacetylene receptors with a single urea moiety have been prepared and were found to bind halides as 2:1 host–guest complexes that feature key CH–anion or anion–π interactions. These supporting interactions also appear to influence the mechanism of the 2:1 binding event.
Keywords: anions, host–guest systems, hydrogen bonds, supramolecular chemistry
For the past few decades, the field of anion sensing has been dominated by supramolecular receptors.[1] Supramolecular hosts have been shown to bind anionic guests through a variety of host–guest interactions, including anion–π interactions, hydrogen bonds, and weak σ interactions.[1–3] Disregarding the larger molecular structure or type of guest, supramolecular hosts are currently designed to include some degree of preorganization and an attractive binding pocket.[1,2,4] Ideally, such probes can be easily tuned for analyte specificity and optoelectronic responses.[1–5]
Arylethynyl urea scaffolds make up the foundation of the supramolecular anion-sensing scaffolds in our studies. A preorganized binding cavity is formed by a rigid alkyne linkage between arene rings and urea-based hydrogen-bond (HB) donors.[6,7] The easily functionalized core and pendant arenes are strategic designs, because they can be modified with electron-donating or electron-withdrawing substituents to modulate the acidity of the HB donors.[7b] Our previously reported arylethynyl bis-urea (e.g., 1) and tris-urea receptors (2) have exhibited a variety of binding motifs for anions.[6–9] The majority of the bipodal hosts bind anions by aryl CH or pyridinium HB donors at the core of the host, with the urea groups forming a U-shaped pocket that dominates the binding event, as shown in Figure 1.[7,8] This binding pattern is altered in trifluorophenyl tripodal receptor 2, however, in which anion–π interactions influence selectivity in favor of binding nitrate over chloride.[9] Furthermore, the crystal structure of the tris-urea host indicated that only two of the three available urea “arms” were interacting with an anionic guest.[9] This suggested that the number of urea donors may influence anion binding as much as the type of binding motif utilized in the host–guest interaction. Anion–π interactions have been observed in a myriad of receptors, and the ability to tune arenes to increase their selectivity for anion–π binding has been shown in other arene-based hosts.[1,9,10]
Figure 1.

Previously reported bipodal bis-urea (1) and tripodal tris-urea (2) receptors along with the new monopodal arylethynyl mono-urea scaffolds (3, 4).
To elucidate both the degree of tunability of the anion-binding motifs and the number of arylethynyl urea recognition elements necessary to bind an anion, we designed mono-urea host scaffolds 3 and 4 (Figure 1). The single “arm” permits more aggressive tuning of the arene core than what is synthetically accessible on the bis-arylethynyl scaffold, and the increased rotational freedom around the single ethynyl unit permits the core arene to rotate to facilitate the preferred binding motif (i.e., anion–π, aryl CH H bond, or weak σ interactions). Berryman et al. utilized dinitro-substituted arenes in a tris-arene scaffold to host anions.[11] It was calculated that the 3,5-dinitro groups sterically block the aryl CH, preventing an H-bond interaction between the phenyl core and an anionic guest.[11] With the additional rotational freedom of scaffold 3, we hypothesized that the 3,5-dinitrobenzene substitution pattern would promote anion–π or weak σ interactions between the host scaffold and an anionic guest. Similarly, the pentafluoroarene scaffold 4 was inspired by the trifluorophenyl tripodal receptor 2.[9] We hypothesized that the combination of an electron-deficient aromatic ring and the removal of aryl H-bond donors would result in a scaffold that hosts anionic guests exclusively through an anion–π interaction in combination with the urea HB donors.
Monopodal hosts 3 and 4 were synthesized as shown in Scheme 1. Desilylation of known ethynylaniline 5[8b,f] and subsequent Sonogashira cross-coupling with 1-iodo-3,5-dinitrobenzene or iodopentafluorobenzene gave cores 6 and 7 in 87 and 73 % yield, respectively. Reaction of 6 or 7 with p-nitrophenyl isocyanate gave receptors 3 and 4 in 73 and 71 % yield, respectively. The final compounds were fully characterized by 1H, 13C, and 19F NMR spectroscopy, and 2D 1H/13C heteronuclear single quantum coherence (HSQC) NMR spectroscopy was used to assign the aryl and urea proton resonances of 3.
Scheme 1.

Synthesis of arylethynyl mono-urea receptors 3 and 4.
The anion-binding characteristics of 3 and 4 were investigated with tetrabutylammonium (TBA) halide salts in 10 % DMSO/CHCl3 or the perdeutero equivalent. Titration experiments were performed at 1.0 mM concentration of chosen host (Figure 2).[12] Association constants (Ka) for 3 and 4 with halides Cl−, Br−, and I− were calculated by using non-linear regression, non-cooperative fitting models in MatLab by simultaneously fitting the downfield shifting of the urea protons (Hb, Hc for 3; Ha, Hb for 4).[13] The internal aryl proton (Ha) resonance shifts were also included in the fitting of 3.
Figure 2.

a) 1H NMR titration of 3 with TBA+Cl− at 298 K; [3] =0.4 mM in 10% water-saturated [D6]DMSO/CDCl3. b) 1H NMR titration of 4 with TBA+Br− at 298 K; [4] =1.0 mM in 10 % water-saturated [D6]DMSO/CDCl3. Peak assignments refer to labelled hydrogen atoms presented in Figure 1.
Titrations were initially fit to a 1:1 host–guest model, but residual errors were large, indicating a poor fit. In addition, the serpentine-like shift of urea proton Hc in the titrations of 3 hinted at the possibility of higher-order binding stoichiometry (Figure 2a).[14–16] Job’s plot analysis revealed a 2:1 host–guest model might be more appropriate for the binding stoichiometry (see the Supporting Information). Indeed, titrations fit to a 2:1 host–guest model provided minimalized residual errors.[15] The previous arylethynyl urea probes studied by our lab included at least two urea-recognition motifs to host a guest anion, and the fit of the mono-arylethynyl urea probes 3 and 4 to a 2:1 host–guest system further signifies the necessity of including multiple urea recognition motifs in a scaffold’s design.
The stepwise Ka1 and Ka2 values for both 3 and 4 with the various halides were determined across three titrations with less than a 15 % error (Table 1). The Ka1 values for 3 are within error of each other, but the Ka2 values increase by an order of magnitude with increasing guest size. The trend could be related to the ability of the recognition scaffolds to donate increasingly linear hydrogen bonds in the assembled binding pocket. Interestingly, there is a clear statistically significant difference in the Ka1 values for 4 with the halides, and the overall trend of association constants appears to be opposite in 4 versus 3; that is, in 3 there is a slight reverse Hofmeister trend in anion binding of I−>Br−>Cl−, and in 4 the opposite is true: Cl−> Br−>I−. The change in anion preference could be due to the formation of an anion–π interaction in 4·X−, and the smaller anions are capable of a closer interaction with the π systems.[2] The preference for larger halides in 3 could be the result of both aryl CH hydrogen bonds becoming more linear, increasing the strength of the interactions.
Table 1.
Anion-association constants (Ka) for receptors 3 and 4 obtained by fitting titration data to a stepwise 2:1 host–guest model in MatLab.[a]
| Host | Cl−/[M−1][a] | Br−/[M−1][a] | I−/[M−1][a] | |||
|---|---|---|---|---|---|---|
| Ka1 | Ka2 | Ka1 | Ka2 | Ka1 | Ka2 | |
| 3 | 300 | 740 | 320 | 1040 | 360 | 6570 |
| 4 | 11 8000[b] | 10200 | 930 | 2500 | 130 | 750 |
Anions were added as tetrabutylammonium salts in 10 % water-saturated [D6]DMSO/CDCl3. Values represent an average of three 1H NMR titrations. Error is approximately ± 15 %.
See Reference [17].
The order, in which the anion binds the two hosts, could shed additional light on the nature of the interactions of these hosts with anions. There are two likely mechanisms, in which a 2:1 host–guest complex can form: two hosts associate, then an anion binds in the dimer pocket (Figure 3a), or one host binds the anion, followed by a second host binding the 1:1 complex (Figure 3b).[13,14] If a complex initially dimerizes/aggregates, the Ka1 value would likely be independent of the nature of anion present; this rings true for scaffold 3. Additionally, these Ka1 values are on the same order of magnitude as the dimerization constant for 3 in the absence of an anion/salt, suggesting that Ka13 might resemble a receptor dimerization event.[18] It is also possible the supporting “weak” interaction in 3 (e.g., CH anion from the dinitrophenyl ring) creates a competing trend in anion binding that prefers the softer iodide over chloride/bromide, and thus mechanism (b) is still at play, but this competing selectivity cancels the anion-binding dependence in Ka1.
Figure 3.
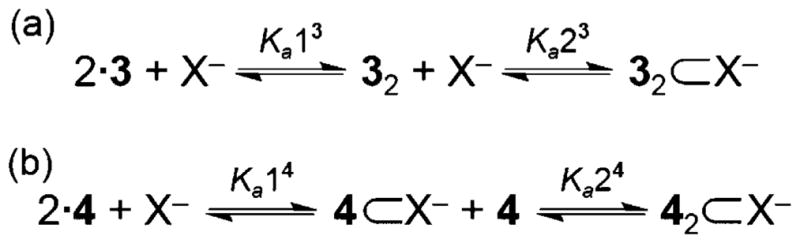
Simplified equilibrium equations illustrating the two possible modes for formation of a 2:1 host–guest complex: a) initially a dimer forms, followed by the anion addition to form the 2:1 complex, or b) a 1:1 host–guest complex forms, and a second host binds to form a 2:1 complex.
The 2:1 assembly situation is much more clear for the anion complexes of 4. Both Ka1 and Ka2 values of 4 change across the anion series, as was predicted by relative anion basicity, supporting the 2:1 complex forming via a step-wise mechanism dominated by traditional hydrogen-bonding interactions with the ureas and possible supporting anion–π interactions with the pendant pentafluorphenyl rings (Figure 3b).
The 2:1 host–guest stoichiometry was further confirmed in the solid-state by X-ray crystallography. Single crystals of 3 grown in the presence of TBA+Br− were obtained by slow evaporation from CHCl3/DMSO.[19] Two receptors asymmetrically encapsulate the Br− atom through a total of six weak hydrogen bond contacts (Figure 4). Each receptor donates two hydrogen bonds through the urea moiety, and another weak CH hydrogen bond through the dinitrophenyl core with distances Nd–Br 3.27(1) Å, Nc–Br 3.63(2) Å, Cb–Br 3.62(2) Å, Nd′–Br 3.29(2) Å, Nc′–Br 3.64(2) Å, and Cb′–Br 3.69(2) Å; and angles Nd-Hd···Br 141.3(4)°, Nc-Hc···Br 146.3(2)°, Cb-Hb···Br 161.8(6)°, Nd′-Hd′···Br 172.6(4)°, Nc′-Hc′···Br 151.2(9)°, and Cb′-Hb′ ···Br 133.3(1)°.
Figure 4.

X-ray crystal structure of 32·Br−. Hydrogen-bond interactions shown as dotted line. TBA+ countercation and solvent molecules have been omitted for clarity.
Although previous calculations predicted the aryl CH HBs would be inaccessible due to the steric hindrance of the nitro substituents,[11] the importance of aryl CH HBs is not lost in the crystal structure of scaffold 3. The ability for two equivalents of 3 to encapsulate an anionic guest by six weak hydrogen bonds also contributes to the large association constants seen in the solution-state studies. Though solid-state data has yet to be obtained, it is reasonable to hypothesize that 4 shows a similar binding interaction as 3, with the CH HBs replaced by anion–π interactions, because 4 lacks CH HB donors. A color change was not seen upon the addition of anion, indicating that a weak σ interaction/charge-transfer complex is not involved. This lends further credence to our speculation that anion–π is the most probable supporting interaction in the host–guest complex of 4, whereas CH–anion interactions are present in 3. An anionic guest can interact with 4 through two anion–π interactions, along with the four urea HB donors.
In summary, the solid-state data in combination with the solution-phase Ka values provided a convincing argument for the necessity of at least two urea recognition motifs in a strong arylethynyl receptor scaffold. The inclusion of a phenyl core with the ability to host an anionic guest by an aryl CH HB or an anion–π interaction also appears to influence the order of the halide binding within the self-assembled binding pocket. Further research on the effect of cooperativity of these monopodal arylethynyl urea scaffolds is currently in progress.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01-GM087398, which also funded early-stage intellectual property that was licensed by SupraSensor Technologies, a company co-founded by the principal investigators. We thank the NSF for an NMR spectrometer grant (CHE-1427987). The authors acknowledge the Biomolecular Mass Spectrometry Core of the Environmental Health Sciences Core Center at Oregon State University (NIH P30ES000210).
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: http://dx.doi.org/10.1002/chem.201605452.
Conflict of interest
This work was supported by NIH grant R01-GM087398, which also funded early stage intellectual property that was licensed by SupraSensor Technologies, a company co-founded by the principal investigators.
References
- 1.a) Sessler JL, Gale PA, Cho W-S. Anion Receptor Chemistry. Royal Society of Chemistry; Cambridge: 2006. [Google Scholar]; b) Gale PA, Dehaen W. Anion Recognition in Supramolecular Chemistry. Springer; Berlin: 2010. [Google Scholar]
- 2.a) Giese M, Albrecht M, Rissanen K. Chem Rev. 2015;115:8867. doi: 10.1021/acs.chemrev.5b00156. [DOI] [PubMed] [Google Scholar]; b) Busschaert N, Caltagirone C, Van Rossom W, Gale PA. Chem Rev. 2015;115:8038. doi: 10.1021/acs.chemrev.5b00099. [DOI] [PubMed] [Google Scholar]
- 3.Berryman OB, Johnson DW. Chem Commun. 2009:3143. doi: 10.1039/b823236a. [DOI] [PubMed] [Google Scholar]
- 4.Bryantsev VS, Hay BP. Org Lett. 2005;7:5031. doi: 10.1021/ol0520119. [DOI] [PubMed] [Google Scholar]
- 5.Carroll CN, Naleway JJ, Haley MM, Johnson DW. Chem Soc Rev. 2010;39:3875. doi: 10.1039/b926231h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vonnegut CL, Tresca BW, Johnson DW, Haley MM. Chem Asian J. 2015;10:522. doi: 10.1002/asia.201403212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Tresca BW, Zakharov LN, Carroll CN, Johnson DW, Haley MM. Chem Commun. 2013;49:7240. doi: 10.1039/c3cc44574g. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tresca BW, Hansen RJ, Chau CV, Hay BP, Zakharov LN, Haley MM, Johnson DW. J Am Chem Soc. 2015;137:14959. doi: 10.1021/jacs.5b08767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Gavette JV, Evoniuk CJ, Zakharov LN, Carnes ME, Haley MM, Johnson DW. Chem Sci. 2014;5:2899. doi: 10.1039/c4sc00950a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Carroll CN, Coombs BA, McClintock SP, Johnson CA, II, Berryman OB, Johnson DW, Haley MM. Chem Commun. 2011;47:5539. doi: 10.1039/c1cc10947b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gavette JV, Mills NS, Zakharov LN, Johnson CA, II, Johnson DW, Haley MM. Angew Chem Int Ed. 2013;52:10270. doi: 10.1002/anie.201302929. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2013;125:10460. [Google Scholar]; d) Engle JM, Lakshminarayanan PS, Carroll CN, Zakharov LN, Haley MM, Johnson DW. Cryst Growth Des. 2011;11:5144. doi: 10.1021/cg201074v. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Engle JM, Carroll CN, Johnson DW, Haley MM. Chem Sci. 2012;3:1105. doi: 10.1039/C2SC00975G. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Watt MM, Engle JM, Fairley KC, Robitshek TE, Haley MM, Johnson DW. Org Biomol Chem. 2015;13:4266. doi: 10.1039/c4ob02409e. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Hartle MD, Hansen RJ, Tresca BW, Prakel SS, Zakharov LN, Haley MM, Pluth MD, Johnson DW. Angew Chem Int Ed. 2016;55:11480. doi: 10.1002/anie.201605757. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2016;128:11652. [Google Scholar]
- 9.Watt MM, Zakharov LN, Haley MM, Johnson DW. Angew Chem Int Ed. 2013;52:10275. doi: 10.1002/anie.201303881. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2013;125:10465. [Google Scholar]
- 10.a) de Hoog P, Gamez P, Mutikainen H, Turpeinen U, Reedijk J. Angew Chem Int Ed. 2004;43:5815. doi: 10.1002/anie.200460486. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2004;116:5939. [Google Scholar]; b) Demeshko S, Dechert S, Meyer F. J Am Chem Soc. 2004;126:4508. doi: 10.1021/ja049458h. [DOI] [PubMed] [Google Scholar]; c) Rosokha YS, Lindeman SV, Rosokha SV, Kochi JK. Angew Chem Int Ed. 2004;43:4650. doi: 10.1002/anie.200460337. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2004;116:4750. [Google Scholar]; d) Berryman OB, Hof F, Hynes MJ, Johnson DW. Chem Commun. 2006:506. doi: 10.1039/b511570a. [DOI] [PubMed] [Google Scholar]; e) Gamez P. Inorg Chem Front. 2014;1:35. [Google Scholar]; f) Giese M, Albrecht M, Rissanen K. Chem Commun. 2016;52:1778. doi: 10.1039/c5cc09072e. [DOI] [PubMed] [Google Scholar]; g) Ballester P. Acc Chem Res. 2013;46:874. doi: 10.1021/ar300080f. [DOI] [PubMed] [Google Scholar]
- 11.Berryman OB, Sather AC, Hay BP, Meisner JS, Johnson DW. J Am Chem Soc. 2008;130:10895. doi: 10.1021/ja8035652. [DOI] [PubMed] [Google Scholar]
- 12.Host concentration for titrations of 3 with TBA+Cl− was held constant at 0.4 mM. All other titrations were performed with host concentration of 1.0 mM.
- 13.Thordarson P. Chem Soc Rev. 2011;40:1305. doi: 10.1039/c0cs00062k. [DOI] [PubMed] [Google Scholar]
- 14.a) Li Y, Pink M, Karty JA, Flood AH. J Am Chem Soc. 2008;130:17293. doi: 10.1021/ja8077329. [DOI] [PubMed] [Google Scholar]; b) Chen H, Yang H, Xu W, Ta Y. RSC Adv. 2013;3:13311. [Google Scholar]; c) Giese M, Albrecht M, Valkonen A, Rissanen K. Eur J Org Chem. 2013:3247. [Google Scholar]; d) Zhou H, Zhao Y, Gao G, Li S, Lan J, You J. J Am Chem Soc. 2013;135:14908. doi: 10.1021/ja406638b. [DOI] [PubMed] [Google Scholar]
- 15.Ulatowski F, Da browa K, Balakier T, Jurczak J. J Org Chem. 2016;81:1746. doi: 10.1021/acs.joc.5b02909. [DOI] [PubMed] [Google Scholar]
- 16.During the titrations of 3, the saturation point was determined by loss of signals of urea protons Hc and Hb. Further analysis with a 2:1 host–guest system indicates NMR time-scale averaging across two asymmetric probes binding to a single guest. This is consistent with the conformation and stoichiometry found in the crystal structure shown in Figure 4.
- 17.Value is at the limit of detection provided by 1H NMR titrations and would be more appropriately determined by UV/Vis spectroscopy.[10,11] The μM concentrations needed to obtain UV/Vis spectroscopy titration data dilute out the expected 2:1 host–guest model, however, leading to titrations only appropriately fit to a 1:1 host–guest model. Association constants determined by these UV/Vis titrations gave a Ka of 32800 M−1 (see the Supporting Information).
- 18.Value of Kdimer 3 is 500 M−1. Although the experiments were done in the same solvent system, it is important to note that the dimerization constants were determined in a solution with different ionic strengths compared to the titrations that produced the association constants. Additionally, the concentration at which the titrations were performed is low, leading to approximately 10 % of “free” (non-anion-bound) dimer formed in solution at any given time. Thermodynamic equilibria could drive more dimer to form to promote the sandwich-like interaction with the anion.
- 19.CCDC 1507418 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.