

Abstract
Chelation therapy is frequently used to help reduce excess iron in the body, but current chelators such as Deferoxamine (DFO) are plagued by short blood circulation times, which necessitates infusions and can cause undesirable toxic side effects in patients. To address these issues, polyrotaxanes (PR) were synthesized by threading α-cyclodextrin (α-CD) onto poly(ethylene glycol) bis(amine) (PEG-BA, MW 3400 g/mol) capped with enzymatically cleavable bulky Z-L Phenylalanine (Z-L Phe) moieties. The resulting PR was conjugated to DFO and hydroxypropylated to generate the final polyrotaxane-DFO (hPR-DFO). The iron chelating capability of hPR-DFO was verified by UV-Vis absorption spectroscopy and the ability of materials to degrade into smaller CD-conjugated DFO fragments (hCD-DFO) in the presence of the protease was confirmed via Gel Permeation Chromatography (GPC). In vitro studies in iron-overloaded macrophages reveal that hPR-DFO can significantly reduce the cytotoxicity of the drug while maintaining its chelation efficacy, and that it is more rapidly endocytosed and trafficked to lysosomes of iron-overloaded cells in comparison to non-iron overloaded macrophages. In vivo studies indicate that iron-overloaded mice treated with hPR-DFO displayed lower serum ferritin levels (a measure of iron burden in the body) and could eliminate excess iron by both the renal and fecal routes. Moreover, there was no gross evidence of acute toxicological damage to the liver or spleen.
Keywords: Biodegradable, Polyrotaxane, Chelation Therapy, Iron Overload, Deferoxamine
Graphical abstract
Introduction
Iron is an essential biological element for the human body as it participates in many metabolic processes.1 Unfortunately, humans do not have an efficient means of excreting excess iron;2, 3 this can lead to eventual leakage of the metal ions from storage proteins (i.e. ferritin, hemosiderin) and iron saturation of transferrin, which ultimately results in the metal spilling into the bloodstream in the form of reactive non-transferrin bound iron (NTBI) that can then cause irreversible oxidative damage due to their preferential uptake by cardiomyocytes in the heart and hepatocytes in the liver.4-6 A large body of clinical evidence has demonstrated that iron overload conditions can further drive inflammation and increase the risk of cardiovascular disease in patients as well as encouraging infections. 7-9
Iron chelators such as DFO are often administered to patients suffering from iron overload conditions.10 Unfortunately, DFO needs to be administered under careful medical supervision to avoid undesirable toxic side effects.11 In addition, because of the extremely short residence time of DFO in the human body (ca. 20 min), the drug needs to be infused over prolonged periods of time and requires constant patient monitoring to avoid removing too much iron since it is a critical metal for downstream proteins involved in processes such as cellular respiration and metabolism.12 In an attempt to address these problems, we are exploring an environment-responsive carrier system capable of taking advantage of the inflammatory environment characteristic of iron-overload conditions. It is important for the carrier to maintain the iron chelation capability of DFO while minimizing its well-known cytotoxicity issues in cells13, 14 and to prolong drug residence time while incorporating suitable triggers for biodegradation of the carrier and elimination of iron- chelates from the body. In spite of the applicability of chelation therapy to iron overload conditions and other diseases characterized by metal imbalances, there have been few publications advancing this area of research over the years.15-18
Recently, cyclodextrin-based polyrotaxanes (PR) have attracted much attention as carriers for various drug delivery systems due to their biocompatibility and unique chemical composition.19-22 The presence of abundant hydroxy groups on the CD moiety makes it possible to easily attach various functional molecules and desirable drugs onto the carrier system depending on the strategy used.23-26 The degradable properties of PR can be designed and triggered either chemically or enzymatically for controlled release of drugs from the carrier backbone.27-29 Herein we report on a strategy for the preparation of iron-chelating PR capable of degrading via enzymatic cleavage of the endcaps (Scheme 1). Enzymatic cleavage was incorporated into the design due to the inflammatory nature of the iron overload condition and subsequent overexpression of various proteases; these factors should help speed up the biodegradation of PR in cells and allow for controlled elimination of iron from the body. In general, PR were formed by threading multiple α-CD rings onto PEG-BA chains and capping the ends with enzymatically cleavable bulky Z-L Phenylalanine (Z-L Phe) moieties.30 After forming PR, hydroxy groups on CD moieties were oxidized into aldehydes (PR-CHO) using Dess-Martin Periodinane (DMP) and the terminal amine of DFO was conjugated to oxidized PR via Schiff-base chemistry (PR-DFO). To further increase the water solubility of the carrier, remaining hydroxy groups on CDs were subjected to hydroxypropylation to afford the final hPR-DFO. The overall synthetic scheme for generating hPR-DFO is summarized in Figure 1.
Scheme 1.
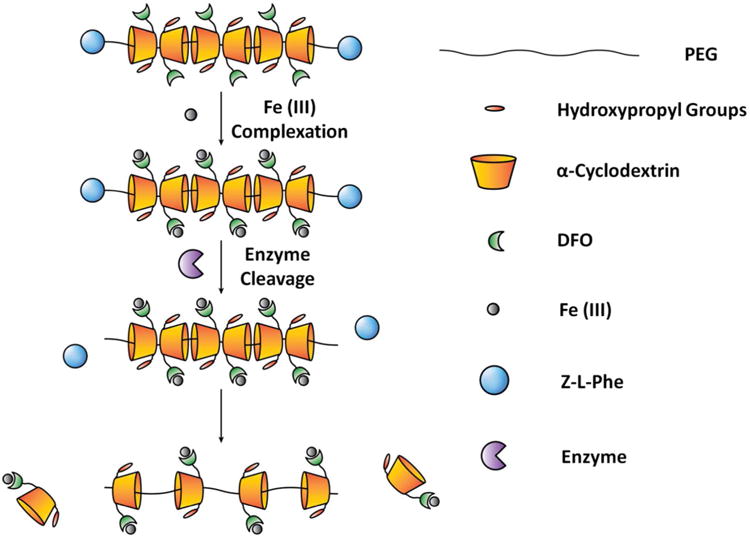
Schematic representation of hPR-DFO chelating iron, Fe(III), and controlled biodegradation of the nanomaterial via enzymatic degradation of the endcaps.
Figure 1.
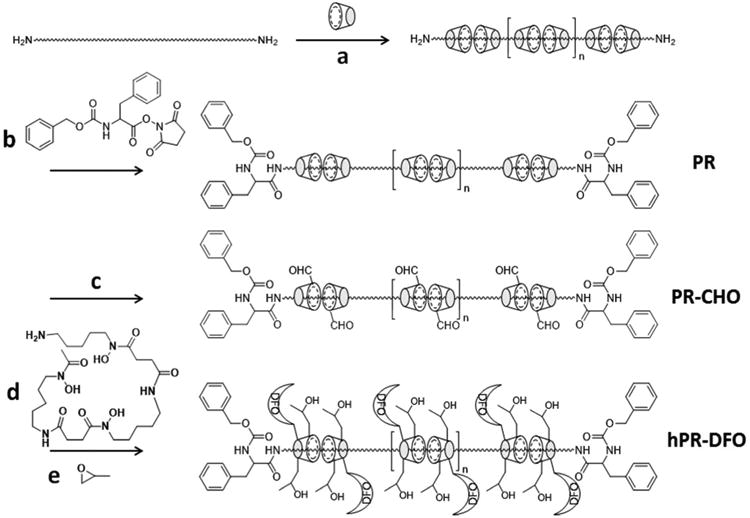
Synthesis of hPR-DFO: a) ddH2O, b) DMSO, c) DMP, DMSO, d) NaBH3CN, DMSO, e) NaOH, ddH2O.
Results and Discussion
Structural and Physical Characterizations of hPR-DFO
The synthesis of hPR-DFO was confirmed by 1H-NMR spectroscopy (Figure 2). In the 1H-NMR spectra of PR (Figure 2A), peaks belonging to PEG were found at 3.46 ppm (–CH2–) and peaks belonging to α-CD were observed at 3.10-3.94 ppm (C2–H, C3–H, C4–H, C5–H, C6–H), 4.39 ppm (C6–OH), 4.75 ppm (C1–H), 5.46 ppm (C3–OH), and 5.63 ppm (C2–OH); there was also a broad multiple peak observed at 7.13-7.30 ppm which belongs to the aromatic protons of Z-L-Phe. By integrating various corresponding CD peaks with respect to the PEG peak and assuming one CD threads over every two repeating units of ethylene glycol, it was estimated that there is an average of 28 CD units threaded onto the PEG backbone. This result is similar to what has previously been reported and confirms the formation of inclusion complexes between PEG and α-CD.31 Next, hydroxy groups of CD were converted to aldehydes via Dess-Martin oxidation and a peak corresponding to these aldehyde protons (-CHO) was observed at 9.70 ppm (Figure 2B). The formation of PR-DFO via Schiff-base chemistry was verified by noting the absence of the aldehyde groups at 9.70 ppm in the spectra, and the presence of new peaks at 1.15-1.50 ppm (– CH2-), 7.60-7.75 ppm (-NH-), and 9.51-9.64 ppm (-N-OH) corresponding to conjugated DFO (Figure 2C). Following hydroxypropylation, the -CH3 peak corresponding to hydroxypropyl groups were observed at 0.96 ppm in the final hPR-DFO product (Figure 2D); hydroxypropylated PR without DFO (hPR) served as a control for in vitro and in vivo studies. To further confirm peaks assigned to hPR-DFO, hydroxypropylated CD-DFO (hCD-DFO) was also synthesized as a reference and its 1H-NMR spectra is available in Figure S1. DMP is a mild oxidizer that allows for selective conversion of alcohols to aldehydes or ketones, with the length of the reaction as well as exposure time to the oxidizer playing an important role in controlling hydroxy to aldehyde conversion levels.32 This means that although we were able to detect the presence of the aldehyde proton in PR-CHO (Figure 2B), it is not possible to confirm that every threaded α-CD will have at least 1 aldehyde group available for conjugation to DFO. An indirect UV-Vis absorption methodology was therefore utilized to calculate DFO conjugation levels (see DFO Conjugation and Iron Chelation Properties).
Figure 2.
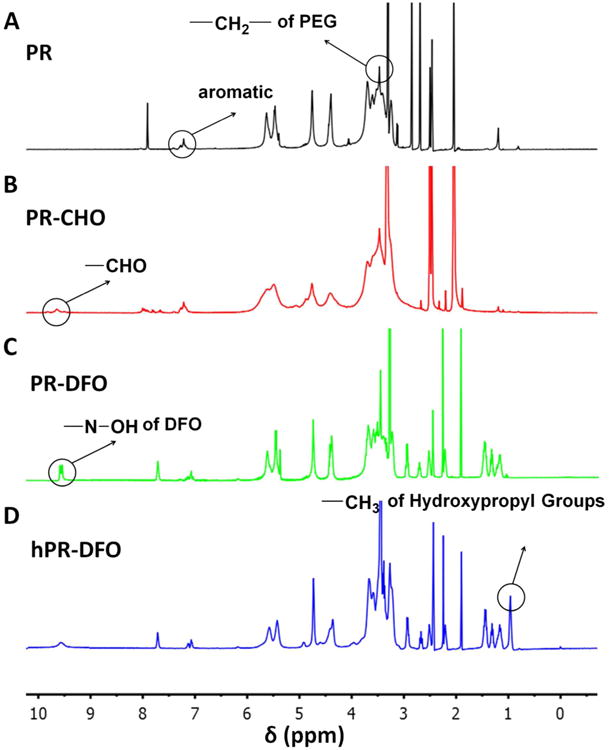
1H NMR spectra of PR (A), PR-CHO (B), PR-DFO (C) and hPR-DFO (D).
The morphological structure of hPR-DFO was examined by transmission electron microscopy (TEM) and reveals near-spherical shapes with diameters of ca. 4.0 nm (Figure 3A). Similarly, dynamic light scattering (DLS) displayed a single peak that was characterized by a z-average diameter of ca. 3.5 nm (Figure 3B). The polydispersity index (PDI) reported by DLS was 0.15 and indicates that resulting nanoparticles formed are reasonably monodisperse. These results are consistent with other reports which have demonstrated that PR behaves as random coils in good solvents, which explains their small sizes and spherical appearances.20
Figure 3.
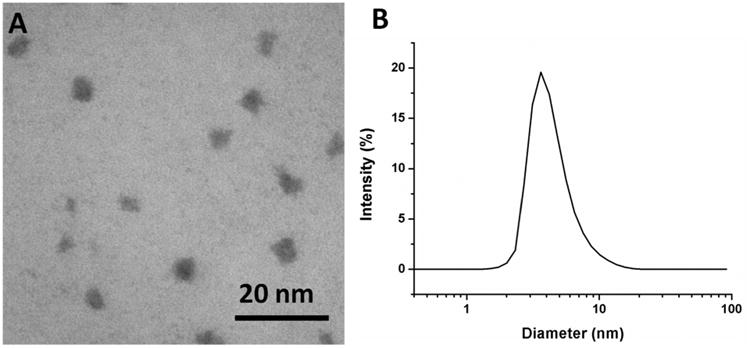
(A) Representative TEM image of hPR-DFO reveals ca. 4.0 nm spherical structures; (B) DLS size distribution of hPR-DFO dispersed in ddH2O displays a z-average diameter of ca. 3.5 nm with a PDI of 0.15.
DFO Conjugation and Iron Chelation Properties
To probe the iron chelating capability of this nanocarrier, UV-Vis was used to confirm the formation of a 1:1 complex between hPR-DFO and ferric iron, Fe(III), by monitoring its characteristic absorption peak at ca. 430 nm.33 After mixing the 1 mg/ml solution of hCD-DFO (as a control) or hPR-DFO with FeCl3 (1 mg/ml), a distinct clear yellow-brown color immediately formed, indicative of complex formation between hydroxylamine groups and iron (Figure 4). Although the characteristic complex formation peak at ca. 430 nm is broad, there is negligible absorption at that wavelength for hCD-DFO, hPR-DFO, and Fe(III) compared to mixtures of hCD-DFO/Fe(III) or hPR-DFO/Fe(III) (Figure 4A).
Figure 4.
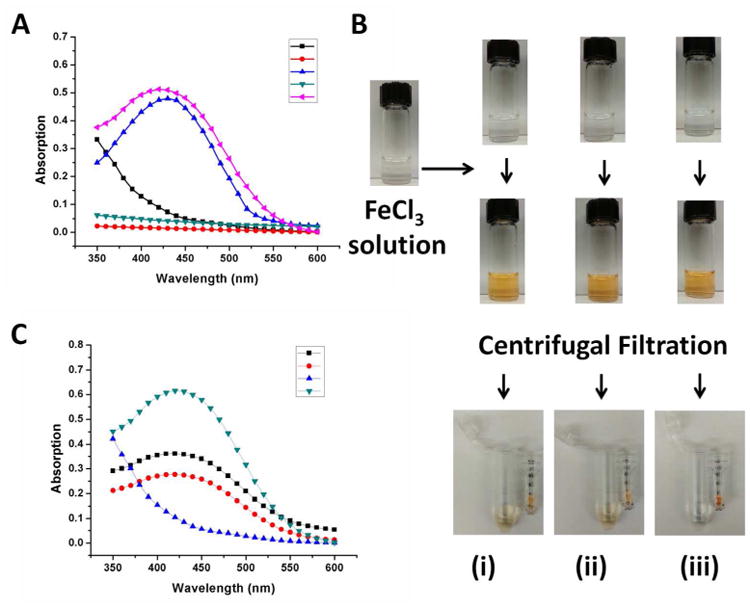
(A) UV-Vis absorption spectrum of Fe(III) in solution (black line), hCD-DFO in solution (red line), hCD-DFO in the presence of Fe(III) (blue line), hPR-DFO in solution (green line) and hPR-DFO in the presence of Fe(III) (pink line). (B) Optical images of DFO (i), hCD-DFO (ii) and hPR-DFO (iii) before addition of Fe(III) reveal clear solutions (first row of vials). After addition of iron, a distinct clear yellow-brown color forms immediately and is indicative of chelates (second row of vials). To further rule out the possibility that DFO was just loosely associated with PR, a microcentrifuge filter tube (MWCO 10,000 g/mol) was used to concentrate the material (third row of images); the yellow DFO/Fe(III) (i) or hCD-DFO/Fe(III) (ii) colored suspension passed through the filter into the bottom filtrate whereas the yellow colored suspension containing polymer chelates remained in the filter unit (iii). (C) UV-Vis absorption spectrum of the concentrate of hPR-DFO displays strong absorption at ca. 430 nm (green line) and no absorption for the filtrate (blue line) after extensive washing with the centrifugal filtration unit; the concentrates of DFO (black line) and hCD-DFO (red line) still absorb at ca. 430 nm but at lower magnitudes than before, reflecting their smaller sizes and elution through the membrane.
DFO is known to stoichiometrically bind iron with a stability constant on the order of 1031 M−1 at a 1:1 mole ratio,34 therefore peak intensity can be proportionally related to complex formation in order to indirectly quantify final conjugation levels of DFO to PR. First, a standard curve for free DFO and Fe(III) was generated by mixing solutions of 10 mM Fe(III) to increasing concentrations of DFO (up to 10 mM) and monitoring peak intensities (Figure S2). Next, hCD-DFO or hPR-DFO at the concentrations of 1 mg/ml was mixed with 10 mM Fe(III) at room temperature and the resulting absorbance at 430 nm was read. Final calculations revealed that ca. 35.7% (w/w) of the total weight of hPR-DFO contribution came from DFO, and assuming an average of 28 CD per PR, we calculated that there are approximately 28 DFO drug moieties per PR or an average of 1 DFO conjugated to every CD.
To further verify that DFO was indeed conjugated to PR and not just loosely associated, free DFO/Fe(III), hCD-DFO/Fe(III), and hPR-DFO/Fe(III) mixtures were washed extensively with a centrifugal filtration unit (MWCO 10,000). Small complexes such as those formed between DFO/Fe(III) and hCD-DFO/Fe(III) can easily pass through the membrane and this was visually confirmed by the presence of yellow-colored filtrates after the first wash; this was not the case for larger complexes formed between hPR-DFO/Fe(III) where the bottom filtrate remained a clear color (Figure 4B). To confirm this further, the absorption at 430 nm for hPR-DFO/Fe(III) was measured and repeatedly found to be very strong in the top concentrate solution but almost no signal was detected in the bottom filtrate after each successive wash (Figure 4C). In contrast, a signal was repeatedly detected (different intensities with each wash) in both the top concentrates and bottom filtrates of DFO/Fe(III) and hCD-DFO/Fe(III).
Enzyme-Triggered Degradation Studies
Most diseases characterized by inflammation are often associated with the overexpression of a variety of proteases such as cathepsin B, L, and H, and carboxypeptidases (which are present in Kupffer cells, macrophages, or other reticuloendothelial systems).30 Similarly, protracted iron overload conditions can cause chronic inflammation7 and the overexpression of many of these proteases. This can be particularly advantageous for more rapidly triggering the biodegradation of hPR-DFO. The use of a nanocarrier such as PR for DFO is important to help improve the overall residence time of the drug while minimizing toxic side effects. And the incorporation of enzymatically cleavable endcaps on PR provide a mechanism for triggering the release of iron-bound chelates from the carrier once they have been taken up by macrophages where a vast majority of ferritin and hemosiderin proteins are found. In this study, the kinetics of protease-induced degradation of hPR-DFO was monitored by GPC for up to 312 h (13 days). Papain of wide specificity was used in the degradation studies to simulate cathepsin, a commonly overexpressed protease found in lysosomes of cells.35 The intact hPR-DFO eluted at 11.8 min, in comparison to the elution time of PEG-BA, glutathione (GSH) (added to reduce papain), and hCD-DFO which occurred at 17.3 min, 20.6 min, and 23.2 min respectively (Figure 5A). As shown in Figure 5B, distinct peaks corresponding to PEG-BA, GSH, and hCD-DFO can clearly be observed at 48 h, but with increased incubation the area under the peak for hPR-DFO began to decrease over time until it completely disappeared after 312 h, indicative of hCD-DFOs threading off the PEG chains in response to enzymatic cleavage of the endcaps. The degradation study with papain was conducted in a closed stationary environment therefore it is difficult to draw predictive correlations between these studies and the rate at which the carrier would actually degrade in vivo. However it does show that the design for the carrier works and that endcaps of hPR-DFO were indeed eventually cleaved off to allow for hCD-DFO to release from the PEG chains.
Figure 5.
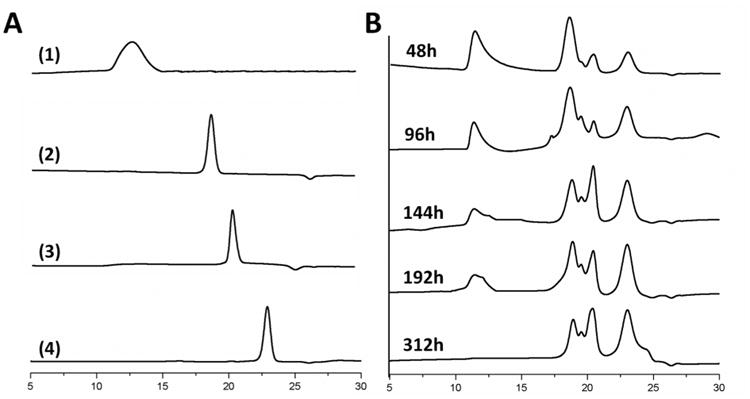
(A) Individual representative GPC curves for hPR-DFO (1), PEG-BA (2), GSH (3) and hCD-DFO (4). (B) hPR-DFO degradation in the presence of papain was evidenced by noting changes in the apparent molecular weight of the carrier as monitored by aqueous GPC at RT.
Cytotoxicity Studies in Macrophages
Although DFO is the oldest standard drug for treating iron overload conditions, it does possess undesirable cytotoxic effects and has been investigated as an anticancer agent against advanced hepatocellular carcinoma in clinical trials.36, 37 To investigate this issue, the cytotoxicity of free DFO and hPR-DFO were compared in J774A.1 non-iron overloaded mouse monocyte/macrophage cells as well as cells that had been iron-overloaded with 100 μM ferric ammonium citrate (FAC). The J774A.1 cells were selected for evaluation because excess iron due to transfusions tends to accumulate first in macrophages of the reticulendothelial system for storage in ferritin and hemosiderin.5
To evaluate the cytotoxicity of hPR-DFO, cells in complete DMEM medium were treated with equivalent amounts of free DFO, hCD-DFO, hPR, or hPR-DFO from 0.05-1000 μM and allowed to incubate for 48 h prior to evaluating cytotoxicity with a metabolism-based resazurin assay. As shown in Figure 6A, free DFO inhibited 50% viability of cells at concentration as low as ca. 8 μM, which is comparable to a previous cytotoxicity report.18 In contrast, hPR-DFO was 85-fold less toxic compared to free DFO with a 50% cell viability observed at ca. 700 μM. Similar data was obtained for the formulations in the iron-overloaded macrophages (Figure 6B), with DFO inhibiting 50% cell viability at ca. 9 μM in contrast to ca. 800 μM for hPR-DFO. The significant decrease in toxicity for hPR-DFO may be attributed to the absence of DFO's reactive primary amino group conjugated to PR. When equivalent concentrations of hCD-DFO and hPR were evaluated, the hPR backbone appeared non-toxic whereas hCD-DFO exhibited a toxicity profile very similar to hPR-DFO at the highest concentration evaluated; the toxicity may be attributed to chelating too much iron from critical intracellular proteins which can vastly disrupt metabolic and cellular respiration processes.
Figure 6.
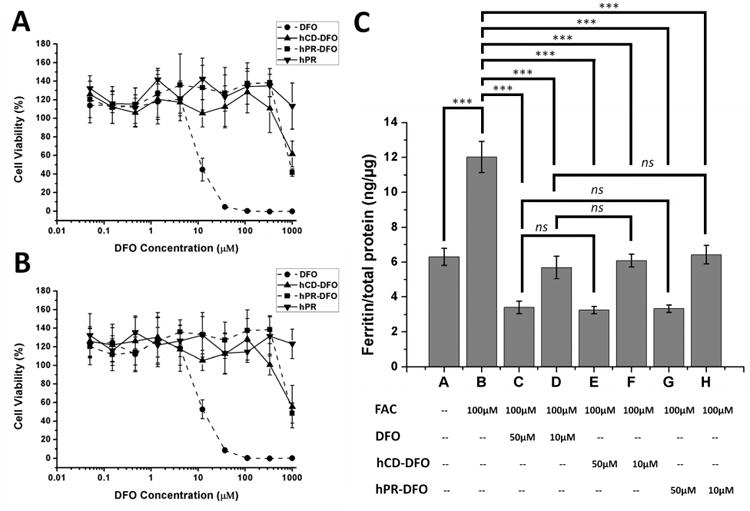
Cytotoxicity at 48 for DFO, hCD-DFO, hPR and hPR-DFO incubated in non-iron overloaded (A) and iron-overloaded macrophage cells (B); a representative set of data is shown where each data point is presented as the mean ± SD (n = 3). (C) Ferritin reduction assay to monitor iron chelation efficacy of DFO, hCD-DFO and hPR-DFO in iron-overloaded macrophage cells; cells were treated with DFO or hCD-DFO or hPR-DFO at 10 μM or 50 μM for 48 h. Cellular ferritin level was measured by a mouse ferritin ELISA assay. Results are normalized to total protein (ng/μg) and presented as mean ± SD (n = 3). “ns” means the difference was not significant. *** p < 0.001.
Iron Chelation Efficacy Studies in Macrophages
Macrophages were iron-overloaded with 100 μM FAC for 24 h, as done previously for the cytotoxicity studies, to increase their overall ferritin expression levels.2 We had previously found that treating macrophages with 100 μM FAC offered the best balance for inducing increased cellular ferritin expression without affecting cell viability (>80% cells were still viable).18 As shown in Figure 6C, 100 μM FAC treatment increased cellular ferritin expression in macrophages from 6.30 to 12.02 ng/μg total protein (p < 0.001). Next, iron-loaded cells were treated for 48 h with either 10 μM or 50 μM DFO, equivalent hCD-DFO, or hPR-DFO. DFO administered at 10 μM was able to reduce the cellular ferritin level from 12.02 to 5.69 ng/μg total protein (52.7% decrease, p < 0.001), and to 3.40 ng/μg total protein at 50 μM (71.7% decrease, p < 0.001). Treatment with hCD-DFO administered at 10 μM decreased the ferritin expression level from 12.02 to 6.08 ng/μg total protein (49.4% decrease, p < 0.001), and to 3.24 ng/μg total protein at 50 μM (73.0% decrease, p < 0.001). For hPR-DFO, the ferritin levels decreased from 12.02 to 6.42 ng/μg total protein at 10 μM (46.6% decrease, p < 0.001), and to 3.33 ng/μg total protein at 50 μM (72.3% decrease, p < 0.001).
Interestingly, free DFO, hCD-DFO and hPR-DFO generated similar treatment effects when comparing decreases in ferritin concentrations at both 10 and 50 μM equivalent DFO concentrations (ns); all the formulations were able to return iron-overloaded cell ferritin levels back to control baseline levels (non-iron overloaded) in spite of the conjugation chemistry performed. These results are consistent with the knowledge that the three hydroxylamines on the drug are solely responsible for the iron-chelating properties of DFO and thereby unaffected by conjugation chemistry to the primary amine.16 However, it is worthwhile to note that there was a drastic difference in cytotoxicity and safety between hPR-DFO (>100% cells were viable) and DFO (ca. 50% cells viable) (Figure 6A and 6B) at 10 μM concentration in spite of their similar iron chelation efficacies; at 50 μM concentration, the cytotoxicity of DFO was even more pronounced compared to hPR-DFO, with <50% cells viable compared to >100% cells treated with hPR-DFO. That means hPR-DFO appears to be not only as effective as DFO at chelating iron but also much less toxic to macrophages.
Cell Uptake Studies in Macrophages
Confocal laser scanning microscopy (CLSM) was used to track the cellular uptake behavior of hPR-DFO after labeling with fluorescein cadaverine (Fc). The primary amino group on the Fc fluorophore was conjugated to unreacted aldehyde groups present on PR-DFO and the final construct was hydroxypropylated to generate hPR-DFO-Fc. The fluorescently labeled hPR-DFO-Fc was incubated for 4 h at 37 °C with non-iron overloaded and iron-overloaded macrophages. Cells were stained for 30 min with the lysosomal probe LysoTracker Deep Red prior to imaging. Co-localization images confirm that hPR-DFO-Fc do appear to enter cells through endocytosis and are eventually trafficked to lysosomes of non-iron overloaded and iron-overloaded macrophages (Figure 7). Previous reports investigating the mechanisms of intracellular iron chelation between DFO and the new oral chelators Deferasirox (DFX) and Deferiprone (DFP) have indeed found differences in uptake behavior for these drugs. In contrast to DFX and DFP which exert their chelating effects in the cytosol of cells, DFO cannot pass through cell membranes and is taken into cells through endocytosis into lysosomes, where a large amount of iron-bound ferritin and hemosiderin storage proteins are found.38
Figure 7.
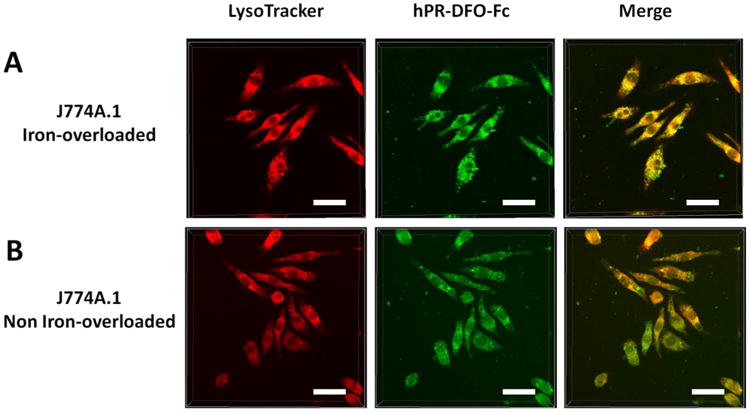
Iron-overloaded and non-overloaded macrophage cells were incubated with hPR-DFO-Fc for 4 h prior to adding LysoTracker and imaging. From left to right, LysoTracker red fluorescence, hPR-DFO-Fc green fluorescence, and the overlay of images (scale bar: 20 μm).
There was more evidence of co-localization between hPR-DFO-Fc with LysoTracker in lysosomes of iron-overloaded cells in comparison to non-iron overloaded macrophages at the 4 h time point. As shown in Figure 7A, the labeled carrier was rapidly internalized by iron overloaded macrophages after 4 h incubation at 37 °C, with cell morphology still appearing normal. Interestingly, the labeled carrier did not seem to internalize as rapidly or to the same extent in the non-iron overloaded cells (Figure 7B). After washing the cells, replacing the culture media, and incubating cells for another 12 h, the intracellular co-localization of hPR-DFO and Lysotracker in lysosomes was even more pronounced in the iron-overloaded cells compared to non-iron overloaded cells, as shown in Figure S3. Since adding excess iron to macrophages does result in a doubling of ferritin expression levels (Figure 6C), our studies seem to point to a correlation between rapid trafficking of hPR-DFO-Fc to lysosomes when there is more ferritin expressed. It has indeed previously been observed that DFO alone appears to induce ferritin to accumulate in lysosomes, in contrast to DFX and DFP, and that this mechanism may perhaps be unique to how the drug exerts its iron chelating effects.38
Efficacy and Elimination Studies in Iron-Overloaded Mice
Efficiency of iron binding and elimination was investigated in vivo in iron-overloaded mice. Normal female Balb/C mice (6 weeks old) were intravenously iron overloaded by a single tail vein injection of 150 mg/kg dextran/Fe (Day 1) as previously reported.15 After a week of monitoring animals to ensure adequate iron levels, mice were treated with either saline, free DFO, hPR-DFO at an equivalent dose of 150 mg/kg DFO, or control hPR at the same w/v ratio of hPR-DFO. Treatment began on Day 8, with an additional two doses of treatments administered on Day 10 and Day 12, for a total of three treatments. On Day 19, mice were sacrificed, organs collected, and blood drawn for the serum ferritin assay.
Ferritin, which is found to varying degrees in almost all cells, is one of the main storage proteins for iron and a good indicator of overall iron levels in the body.4-6 The elevated level of ferritin in the serum is therefore an important parameter to monitor as it often correlates with the degree and severity of systemic iron overload.39, 40 By Day 19, the serum ferritin level decreased significantly only in mice that had been treated with hPR-DFO compared to control treatments (p < 0.001). The serum ferritin concentrations of animals that had been treated with DFO was 8160.26 +/- 439.02 ng/ml ferritin, a value not statistically significant from control saline (7502.92 +/- 266.05), but was 6331.15 +/- 291.52 ng/ml ferritin for hPR-DFO, which was statistically significant compared to both control saline and DFO (p < 0.001) (Figure 8A). To assess the safety of the carrier following the three treatments, organs (lungs, heart, spleen, kidneys, brain, and liver) were harvested and weighed after sacrificing animals on Day 19 (Figure 8B). Especially important are the spleen and liver of mice receiving DFO, hPR, and hPR-DFO, which were all found to weigh within the normal range (ns). This is encouraging and suggests that hPR and hPR-DFO may not accumulate at levels sufficient to cause acute toxicological damage to those organs.
Figure 8.
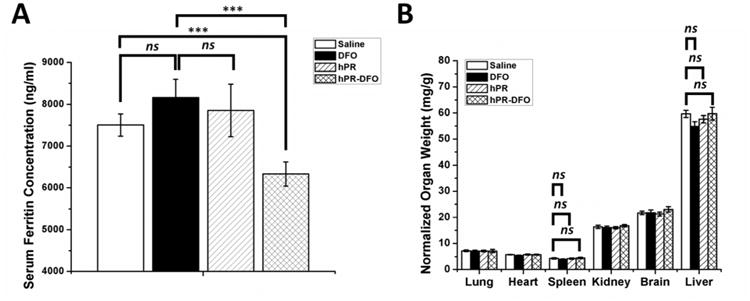
(A) Ferritin level in serum of animals was measured via a colorimetric mouse Ferritin ELISA assay. (B) Acute toxicological effect of DFO, hPR and hPR-DFO on various organs based on weight; note the lack of inflammation observed in the liver and spleen of animals. Results are presented as mean ± SD (n = 3). “ns” means the difference was not significant. *** p < 0.001.
Elimination of iron-bound chelates from the body is a critical parameter to investigate in order to minimize iron redistribution in the body. The urine and feces of mice were collected each day starting on Day 9 up until Day 19. Due to very low iron concentrations, urine for each formulation was pooled to generate a total iron urine exclusion value by the use of inductively coupled plasma-mass spectrometry (ICP–MS). As expected, due to its smaller size, DFO-treated mice were able to excrete iron chelates through the urine in significantly greater amounts than hPR-DFO treated animals (p < 0.01) (Figure 9A). Since the feces collected had higher concentrations of iron, it could therefore be measured daily by atomic absorption spectroscopy (AAS). A significant increase in total iron exclusion was observed for both DFO and hPR-DFO treatment groups compared to saline and hPR-treated mice (p < 0.001) (Figure 9B). The daily concentration of iron (Figure 9C) and the daily amount of Fe (Figure 9D) excreted from feces are included to demonstrate the overall trends of iron excretion over time. The amount of food eaten by animals on any particular day can affect the overall concentration of iron excreted (even though mice were fed a low-iron diet), however it is possible to observe a general trend where we see significant increased iron elimination from feces on the day of treatment or the day after each treatment with DFO and hPR-DFO; these trends were not readily observed with saline or hPR-treated groups. From these elimination studies, it is possible to conclude that hPR-DFO can increase excretion of some iron via the renal route into urine; however a larger majority of carrier metabolism and degradation does appear to occur in the liver since there is evidence of more iron being excreted into the intestinal tract and into the bile for elimination via the fecal route. Although we did not analyze the metabolites or the form of iron chelates eliminated, the significantly reduced serum ferritin concentration of animals treated with hPR-DFO compared to DFO (Figure 8A), coupled with a lack of acute toxicological damage in the liver and spleen (Figure 8B), and encouraging renal and fecal elimination (Figure 9), do suggest that hPR-DFO may be a good candidate for further evaluations.
Figure 9.
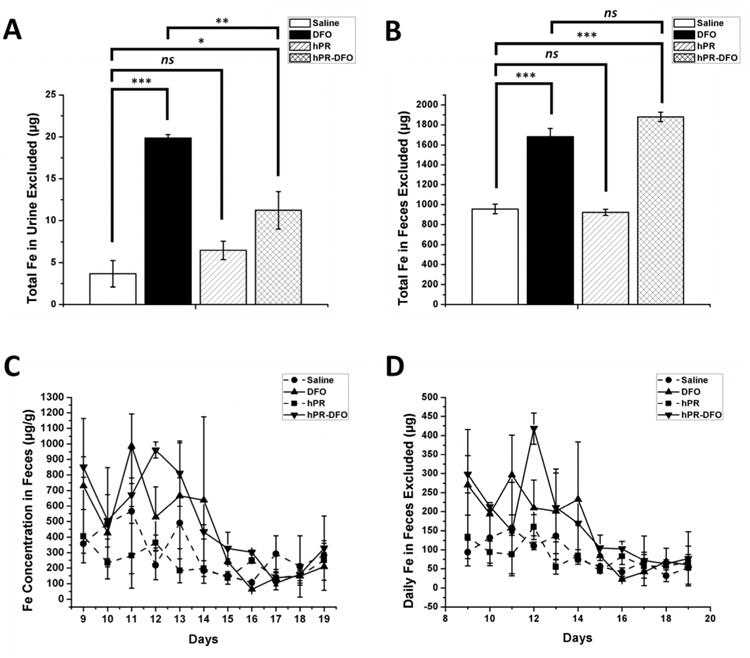
Iron elimination trends in urine and feces of mice: (A) Total iron (Fe) excluded through urine; (B) Total Fe excluded through feces; (C) Daily Fe concentration excluded from feces; (D) Daily Fe amount (weight) excluded through feces. Results are presented as mean ± SD (n = 3). “ns” means the difference was not significant. * p < 0.05, ** p < 0.01, *** p < 0.001.
Conclusions
In summary, we have synthesized biodegradable nanomaterials capable of chelating iron by incorporating enzyme-sensitive caps onto the ends of PR and conjugating the iron chelator DFO onto the PR scaffold. This nanomaterial was shown to degrade into smaller hCD-DFO fragments upon cleavage of the endcaps by papain. hPR-DFO exhibited negligible cytotoxicity compared to the free drug, retained the iron-chelating efficacy of DFO, and was demonstrated to more rapidly co-localize to lysosomes of iron-overloaded macrophages. Iron-overloaded mice treated with hPR-DFO exhibited significantly lower serum ferritin concentrations and were able to utilize both the renal and fecal elimination routes to excrete excess iron. hPR-DFO warrants further investigations and optimization of the structural features of this system is currently in progress.
Methods
Synthesis of Components and Material Precursors
See Supporting Information for details.
Structural and Physical Characterizations of hPR-DFO
To investigate the structural composition of PR formed, 1H-NMR experiments were carried out on a Varian Unity-Inova 400 MHz NMR spectrometer. The iron chelation capability of the final hPR-DFO was investigated via UV-Vis absorption spectroscopy in the presence of a surplus concentration of Fe(III) by scanning between 350–600 nm (SpectraMax Plus, Molecular Devices). The magnitude of the absorbance peak at 430 nm is characteristic of degree of complexes formed between DFO and Fe (III) and was used to determine the amount of DFO monomers incorporated into the PR scaffold. More specifically, a series of solutions containing fixed concentrations of Fe(III) (10 mM) and increasing concentrations of free DFO up to 10 mM were prepared and the absorption at 430 nm measured. Since free DFO chelates Fe(III) at a 1:1 mole ratio, a standard curve relating absorbance to DFO concentration was generated:
where x is the concentration of free DFO (mg/mL) and y is the absorption measured. To determine DFO conjugation levels on hPR-DFO, a 1.0 mg/mL solution of the carrier was mixed with Fe(III) (10 mM) at room temperature and the absorption at 430 nm was measured. The final concentration of DFO in hPR-DFO was then back-calculated based on known parameters obtained via NMR and known final polymer weight. Note that DFO and hPR-DFO samples in the absence of Fe(III) do not absorb at 430 nm.
To investigate the morphology of resulting hPR-DFO, transmission electron microscopy (TEM) images were taken on a Philips CM120 instrument with an acceleration voltage of 120 kV. Sample was prepared by air-drying a drop of 0.01 mg/mL hPR-DFO suspension on copper grid. Thereafter, the copper grid was inverted onto a drop of aqueous solution of sodium phosphotungstate (2%, w/v, pH = 7) and rinsed gently with deionized water.
To investigate the hydrodynamic size and PDI of hPR-DFO, DLS measurements were collected on a Zetasizer Nano ZS (Malvern Instruments, UK) and analyzed with Zetasizer software v7.10. Briefly, hPR-DFO was suspended in ddH2O at about 1 mg/mL and the cumulant analysis method was used to calculate the z-average diameter and PDI. Measurements were conducted on three batches of samples and results are reported as mean ± standard deviation (SD). Prior to measurements, hPR-DFO solutions were clarified by filtering through Millipore membranes with a 0.45 μm pore size.
Enzyme-Triggered Degradation Studies
Aqueous phase GPC was used to measure the apparent molecular weight changes of hPR-DFO incubated with papain. More specifically, 20 mg of hPR-DFO was dissolved in 0.5 ml McIlvaine's Buffer (100 mM of citric acid, 50 mM of Na2HPO4, 2 mM EDTA, 10 mM GSH, and pH 7.0), followed by the addition of 1 mg papain in 0.5 mL McIlvaine's Buffer and stirred at 37°C. A fresh dose of 1 mg papain was added to the buffer solution every 72 h. The reaction mixture (0.2 ml) was sampled at various time points and immediately heated to precipitate the enzyme before storing at −20°C until ready for analysis by aqueous GPC. Samples were clarified by filtering through Millipore membranes with a 0.45 μm pore size and GPC data acquisition was conducted on a Shimadzu UFLC system equipped with Shodex OHpak SB-806M HQ column (8.0 × 300 mm) and a refractive index detector (RID), and eluted with corresponding McIlvaine's Buffer at a flow rate of 0.5 mL/min. GPC data was analyzed with Shimadzu LCsolution GPC post-run software.
Cytotoxicity Studies in Macrophages
J774A.1 macrophage cells were seeded in 96-well plates at a density of 3,000 cells/well, cultured at 37°C, and 5% CO2 in complete DMEM (supplemented with 10% (v/v) heat-inactivated FBS, 100 I.U./mL penicillin and 100 μg/mL streptomycin), and allowed to settle for 24 h. Cells were then treated with DFO or hCD-DFO or hPR-DFO at equivalent DFO concentrations up to 1 mM for 48h. Equivalent hPR based on w/v to hPR-DFO concentrations was also investigated as a control.
Cell viability was measured with the metabolism-based resazurin assay. Briefly, the substrate resazurin was dissolved in cell culture medium at a concentration of 44 μM, added to each well (100 μl) and incubated at 37°C for 4 h. The fluorescence was measured with excitation at 560 nm and emission at 590 nm, on a SpectraMax Gemini EM microplate reader. Readings from the wells without cells were used as Eblank, and the readings from control cells without treatment (Econtrol) were used to represent 100% cell viability. The viability of treated cells at different concentrations can be calculated by the following equation:
Cytotoxicity was also similarly evaluated in iron-overloaded cells. Briefly, macrophage cells were iron overloaded for 24 h by incubating with culture medium containing 100 μM FAC (cells >80% viable, data not shown) prior to adding formulations.
Iron Chelation Efficacy Studies in Iron-Overloaded Macrophages
J774A.1 macrophage cells were seeded in 6-well plates at a density of 30,000 cell/well and allowed to settle for 24 h at 37°C, 5% CO2 in DMEM complete medium before treatment. First the cells were treated with 100 μM FAC (added to DMEM complete medium) for 24 h to induce iron overload. Subsequently, cells were washed with PBS and treated with DFO or hCD-DFO or hPR-DFO at both 10 μM and 50 μM for 48 h. Control group A cells were not iron-overloaded with FAC; cells in control group B were iron-overloaded with FAC but not treated with DFO or hCD-DFO or hPR-DFO. After 48 h incubation with DFO or hCD-DFO or hPR-DFO, cells were lysed with cell lysis buffer (150 mM NaCl, 10 mM Tris, 1% Triton X-100 and protease inhibitor cocktail, pH 7.4) and total protein concentration was measured with the BCA protein assay kit. Cellular ferritin concentration was measured with a mouse ferritin ELISA kit. The results are plotted as the ratio of ng of ferritin per μg total protein concentration.
Cell Uptake Studies in Macrophages
Macrophage cells were seeded on cover slips at a density of 1×105 cells per well and incubated at 37°C, 5% CO2 in DMEM culture medium (with or without 100 μM FAC to iron-overload the cells) for 24 h. Next, cells were washed three times with PBS at room temperature before adding 2 mg/mL hPR-DFO-Fc in culture medium. Cells under non-iron and iron-overloaded conditions were incubated with the labeled carrier at 37 °C for 4 h or for 16 h (4 h + 12 h). In general, prior to imaging, cells were washed three times with PBS and then LysoTracker® Red DND-99 in culture medium at a final concentration of 1 μg/mL was added for 30 min incubation at 37 °C. Afterwards, cells were washed three times with PBS and fixed with 1 mL 4% paraformaldehyde (PFA) for 10 min at room temperature. Fixed slides were stored in PBS at 4 °C before imaging with a confocal microscope.
Efficacy and Elimination Studies in Iron-Overloaded Mice
All efficacy experiments were conducted in accordance with University of Wisconsin Animal Care and Use Committee guidelines and the NIH Guide for the Care and Use of Laboratory Animals. Female Balb/C mice, 6 weeks old, were housed in Innovive static microisolator cages in a room maintained at 20 ± 1°C and with 12 h light and dark cycles. Feed (Harlan Teklad 8604 Rodent Diet) and water were available ad libitum. Iron overload was achieved by single tail vein injection of dextran/Fe (Anem-X 100, Aspen Veterinary Resources, Ltd; 150 mg/kg of Fe, 10 μl/g body weight, BW, in normal saline) on Day 1. It was previously shown that this amount of dextran/Fe is sufficient to produce iron overload in mice based on previous studies.15 Four groups of mice (n = 3 mice per metabolic chamber) were started on iron-deficient powder diet (Teklad TD.80396.PWD) ad libitum on Day 6. On Day 8, 10, and 12, the following 4 formulations were administered via single tail vein injections to mice; group 1 received normal saline injections, group 2 was administered DFO, group 3 was administered hPR, and group 4 was administered hPR-DFO at an equivalent dose of 150 mg/kg DFO. The final administration volumes were all adjusted to 10 μl/g BW in normal saline. Mice were necropsied 7 days after the last dose (Day 19).
Daily feces and urine from metabolic cages were collected and weighed from Day 6 onward till necropsy day, and iron content measured on each day. Iron content in urine was measured by magnetic-sector ICP-MS (Thermo-Finnigan Element 2) using the m/z 56 isotope in medium resolution. Specifically, 0.1 mL aliquot of samples were placed into cleaned 15 mL polypropylene tubes and diluted with 5 mL of 2% Optima nitric acid. Iron concentration in feces was measured by standard AAS (GBC Scientific Equipment model 932AA). Briefly, fecal material was homogenized in distilled water and iron was extracted by adding 5% trichloroacetic acid (TCA)/1.5 N HCl. The fecal extract was heated to 70°C for 90 min and clarified before AAS measurements.
Mice were euthanized by CO2 overdose. Blood was collected directly from cardiac puncture and put into anticoagulating tubes. Blood samples were added to microcentrifuge tubes for serum collection and ferritin measurement by ELISA assay according to the manufacturer's instruction (Immunology Consultants Laboratory, Inc.). The lungs, heart, spleen, kidneys, brain, and liver of each animal were collected, rinsed with fresh PBS, blotted dry with kimwipes, and then weighed.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism 5.0 software. Statistical significance between groups was assessed with one way ANOVA; p < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work is supported by NIH grant R01DK099596.
Footnotes
Supporting Information. Synthesis details, compound characterization data, CLSM images. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes: The authors declare no competing financial interest.
References
- 1.Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell. 2010;142:24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 2.De Domenico I, Ward DM, Kaplan J. Regulation of iron acquisition and storage: consequences for iron-linked disorders. Nat Rev Mol Cell Bio. 2008;9:72–81. doi: 10.1038/nrm2295. [DOI] [PubMed] [Google Scholar]
- 3.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: Molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 4.Kremastinos DT, Farmakis D. Iron Overload Cardiomyopathy in Clinical Practice. Circulation. 2011;124:2253–2263. doi: 10.1161/CIRCULATIONAHA.111.050773. [DOI] [PubMed] [Google Scholar]
- 5.Fleming RE, Ponka P. Iron Overload in Human Disease. New Engl J Med. 2012;366:348–359. doi: 10.1056/NEJMra1004967. [DOI] [PubMed] [Google Scholar]
- 6.Brissot P, Ropert M, Le Lan C, Loreal O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim Biophys Acta. 2012;1820:403–410. doi: 10.1016/j.bbagen.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 7.Wessling-Resnick M. Iron Homeostasis and the Inflammatory Response. Annu Rev Nutr. 2010;30:105–122. doi: 10.1146/annurev.nutr.012809.104804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kletzmayr J, Horl WH. Iron overload and cardiovascular complications in dialysis patients. Nephrol Dial Transpl. 2002;17:25–29. doi: 10.1093/ndt/17.suppl_2.25. [DOI] [PubMed] [Google Scholar]
- 9.Gujja P, Rosing DR, Tripodi DJ, Shizukuda Y. Iron Overload Cardiomyopathy Better Understanding of an Increasing Disorder. J Am Coll Cardiol. 2010;56:1001–1012. doi: 10.1016/j.jacc.2010.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poggiali E, Cassinerio E, Zanaboni L, Cappellini MD. An update on iron chelation therapy. Blood Transfus. 2012;10:411–422. doi: 10.2450/2012.0008-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porter JB. Practical management of iron overload. Brit J Haematol. 2001;115:239–252. doi: 10.1046/j.1365-2141.2001.03195.x. [DOI] [PubMed] [Google Scholar]
- 12.Porter JB, Faherty A, Stallibrass L, Brookman L, Hassan I, Howes C. A trial to investigate the relationship between DFO pharmacokinetics and metabolism and DFO-related toxicity. Ann NY Acad Sci. 1998;850:483–487. doi: 10.1111/j.1749-6632.1998.tb10528.x. [DOI] [PubMed] [Google Scholar]
- 13.Olivieri NF, Buncic JR, Chew E, Gallant T, Harrison RV, Keenan N, Logan W, Mitchell D, Ricci G, Skarf B, Taylor M, Freedman MH. Visual and auditory neurotoxicity in patients receiving subcutaneous deferoxamine infusions. N Engl J Med. 1986;314:869–873. doi: 10.1056/NEJM198604033141402. [DOI] [PubMed] [Google Scholar]
- 14.Bentur Y, McGuigan M, Koren G. Deferoxamine (desferrioxamine). New toxicities for an old drug. Drug Saf. 1991;6:37–46. doi: 10.2165/00002018-199106010-00004. [DOI] [PubMed] [Google Scholar]
- 15.ul-haq MI, Hamilton JL, Lai BFL, Shenoi RA, Horte S, Constantinescu I, Leitch HA, Kizhakkedathu JN. Design of Long Circulating Nontoxic Dendritic Polymers for the Removal of Iron in vivo. Acs Nano. 2013;7:10704–10716. doi: 10.1021/nn4035074. [DOI] [PubMed] [Google Scholar]
- 16.Hallaway PE, Eaton JW, Panter SS, Hedlund BE. Modulation of Deferoxamine Toxicity and Clearance by Covalent Attachment to Biocompatible Polymers. Proc Natl Acad Sci USA. 1989;86:10108–10112. doi: 10.1073/pnas.86.24.10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossi NAA, Mustafa I, Jackson JK, Burt HM, Horte SA, Scott MD, Kizhakkedathu JN. In vitro chelating, cytotoxicity, and blood compatibility of degradable poly(ethylene glycol)-based macromolecular iron chelators. Biomaterials. 2009;30:638–648. doi: 10.1016/j.biomaterials.2008.09.057. [DOI] [PubMed] [Google Scholar]
- 18.Liu Z, Wang Y, Purro M, Xiong MP. Oxidation-Induced Degradable Nanogels for Iron Chelation. Sci Rep. 2016;6:20923. doi: 10.1038/srep20923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenz G, Han BH, Muller A. Cyclodextrin rotaxanes and polyrotaxanes. Chem Rev. 2006;106:782–817. doi: 10.1021/cr970027+. [DOI] [PubMed] [Google Scholar]
- 20.Yu SL, Zhang YJ, Wang X, Zhen X, Zhang ZH, Wu W, Jiang XQ. Synthesis of Paclitaxel-Conjugated β-Cyclodextrin Polyrotaxane and Its Antitumor Activity. Angew Chem Int Ed. 2013;52:7272–7277. doi: 10.1002/anie.201301397. [DOI] [PubMed] [Google Scholar]
- 21.Li J, Yang C, Li HZ, Wang X, Goh SH, Ding JL, Wang DY, Leong KW. Cationic supramolecules composed of multiple oligoethylenimine-grafted β-cyclodextrins threaded on a polymer chain for efficient gene delivery. Adv Mater. 2006;18:2969–2974. [Google Scholar]
- 22.Li J, Loh XJ. Cyclodextrin-based supramolecular architectures: Syntheses, structures, and applications for drug and gene delivery. Adv Drug Deliver Rev. 2008;60:1000–1017. doi: 10.1016/j.addr.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 23.Yan Q, Yuan JY, Cai ZN, Xin Y, Kang Y, Yin YW. Voltage-Responsive Vesicles Based on Orthogonal Assembly of Two Homopolymers. J Am Chem Soc. 2010;132:9268–9270. doi: 10.1021/ja1027502. [DOI] [PubMed] [Google Scholar]
- 24.Harada A, Kobayashi R, Takashima Y, Hashidzume A, Yamaguchi H. Macroscopic self-assembly through molecular recognition. Nat Chem. 2011;3:34–37. doi: 10.1038/nchem.893. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y, Yu CY, Jin HB, Jiang BB, Zhu XY, Zhou YF, Lu ZY, Yan DY. A Supramolecular Janus Hyperbranched Polymer and Its Photoresponsive Self-Assembly of Vesicles with Narrow Size Distribution. J Am Chem Soc. 2013;135:4765–4770. doi: 10.1021/ja3122608. [DOI] [PubMed] [Google Scholar]
- 26.Liu Z, Qiao J, Tian Y, Wu M, Niu ZW, Huang Y. Polymeric Supra-amphiphiles Based on Terminal Group Electrostatic Interactions: Fabrication of Micelles with Modifiable Surfaces. Langmuir. 2014;30:8938–8944. doi: 10.1021/la501936a. [DOI] [PubMed] [Google Scholar]
- 27.Ooya T, Choi HS, Yamashita A, Yui N, Sugaya Y, Kano A, Maruyama A, Akita H, Ito R, Kogure K, Harashima H. Biocleavable polyrotaxane - Plasmid DNA polyplex for enhanced gene delivery. J Am Chem Soc. 2006;128:3852–3853. doi: 10.1021/ja055868+. [DOI] [PubMed] [Google Scholar]
- 28.Ohya Y, Takamido S, Nagahama K, Ouchi T, Katoono R, Yui N. Polyrotaxane Composed of Poly-L-lactide and α-Cyclodextrin Exhibiting Protease-Triggered Hydrolysis. Biomacromolecules. 2009;10:2261–2267. doi: 10.1021/bm900440v. [DOI] [PubMed] [Google Scholar]
- 29.Moon C, Kwon YM, Lee WK, Park YJ, Yang VC. In vitro assessment of a novel polyrotaxane-based drug delivery system integrated with a cell-penetrating peptide. J Control Release. 2007;124:43–50. doi: 10.1016/j.jconrel.2007.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ooya T, Yui N. Synthesis and characterization of biodegradable polyrotaxane as a novel supramolecular-structured drug carrier. J Biomat Sci Polym Ed. 1997;8:437–455. doi: 10.1163/156856297x00371. [DOI] [PubMed] [Google Scholar]
- 31.Ceccato M, LoNostro P, Baglioni P. α-Cyclodextrin/Polyethylene Glycol Polyrotaxane: A Study of the Threading Process. Langmuir. 1997;13:2436–2439. [Google Scholar]
- 32.Cornwell MJ, Huff JB, Bieniarz C. A one-step synthesis of cyclodextrin monoaldehydes. Tetrahedron Lett. 1995;36:8371–8374. [Google Scholar]
- 33.Goodwin JF, Whitten CF. Chelation of Ferrous Sulphate Solutions by Desferrioxamine B. Nature. 1965;205:281–283. doi: 10.1038/205281b0. [DOI] [PubMed] [Google Scholar]
- 34.Keberle H. The Biochemistry of Desferrioxamine and Its Relation to Iron Metabolism. Ann NY Acad Sci. 1964;119:758–68. doi: 10.1111/j.1749-6632.1965.tb54077.x. [DOI] [PubMed] [Google Scholar]
- 35.Turk V, Turk B, Turk D. Lysosomal cysteine proteases: facts and opportunities. Embo J. 2001;20:4629–4633. doi: 10.1093/emboj/20.17.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamasaki T, Terai S, Sakaida I. Deferoxamine for Advanced Hepatocellular Carcinoma. New Engl J Med. 2011;365:576–578. doi: 10.1056/NEJMc1105726. [DOI] [PubMed] [Google Scholar]
- 37.Buss JL, Torti FM, Torti SV. The role of iron chelation in cancer therapy. Curr Med Chem. 2003;10:1021–1034. doi: 10.2174/0929867033457638. [DOI] [PubMed] [Google Scholar]
- 38.Theil EC. Mining ferritin iron: 2 pathways. Blood. 2009;114:4325–4326. doi: 10.1182/blood-2009-08-239913. [DOI] [PubMed] [Google Scholar]
- 39.Lipschit Da, Cook JD, Finch CA. Clinical Evaluation of Serum Ferritin as an Index of Iron Stores. New Engl J Med. 1974;290:1213–1216. doi: 10.1056/NEJM197405302902201. [DOI] [PubMed] [Google Scholar]
- 40.Cook JD, Lipschit Da, Miles LEM, Finch CA. Serum Ferritin as a Measure of Iron Stores in Normal Subjects. Am J Clin Nutr. 1974;27:681–687. doi: 10.1093/ajcn/27.7.681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.