

Abstract
Gyrase appears to be the primary cellular target for quinolone antibacterials in multiple pathogenic bacteria, including Bacillus anthracis, the causative agent of anthrax. Given the significance of this type II topoisomerase as a drug target, it is critical to understand how quinolones interact with gyrase and how specific mutations lead to resistance. However, these important issues have yet to be addressed for a canonical gyrase. Therefore, we utilized a mechanistic approach to characterize interactions of quinolones with wild-type B. anthracis gyrase and enzymes containing the most common quinolone resistance mutations. Results indicate that clinically relevant quinolones interact with the enzyme through a water–metal ion bridge in which a noncatalytic divalent metal ion is chelated by the C3/C4 keto acid of the drug. In contrast to other bacterial type II topoisomerases that have been examined, the bridge is anchored to gyrase primarily through a single residue (Ser85). Substitution of groups at the quinolone C7 and C8 positions generated drugs that were less dependent on the water–metal ion bridge and overcame resistance. Thus, by analyzing the interactions of drugs with type II topoisomerases from individual bacteria, it may be possible to identify specific quinolone derivatives that can overcome target-mediated resistance in important pathogenic species.
Quinolones are among the most commonly prescribed antibacterials worldwide and are used to treat infections caused by a broad spectrum of Gram-negative and Gram-positive bacteria in humans and animals.1−7 The cellular targets for quinolones are the bacterial type II topoisomerases, gyrase and topoisomerase IV.2,5,8,9
Gyrase functions ahead of replication forks and transcription complexes to alleviate torsional stress induced by DNA overwinding and also modulates the overall level of DNA supercoiling in the bacterial genome.10−12 Although topoisomerase IV may be involved in regulating DNA over- and underwinding, its primary function is to remove knots and tangles formed by recombination and replication.10,13−19 The two enzymes display sequence homology and function as heterotetramers (GyrA2GyrB2 and GrlA2GrlB2 for Gram-positive gyrase and topoisomerase IV, respectively).10,20 To perform their critical physiological functions, gyrase and topoisomerase IV induce transient double-stranded breaks in the DNA backbone. During the DNA cleavage event, the enzymes covalently attach to the newly generated 5′-termini via active-site tyrosine residues. These covalent enzyme-cleaved DNA complexes are termed “cleavage complexes”.6−8,12,21−23
Quinolones act by stabilizing cleavage complexes.7,8,24−27 As replication forks or other DNA-tracking systems encounter these protein-bound DNA roadblocks, transient cleavage complexes are converted to nonligatable strand breaks that must be repaired by DNA damage response pathways. At cytotoxic concentrations, quinolones overwhelm the bacteria with DNA strand breaks and trigger cell death.
Although gyrase and topoisomerase IV are both targets for quinolones, the relative contributions of each enzyme to cytotoxicity appear to be both species- and drug-dependent.7,28−33 As a generalization, gyrase is the primary cellular target for most quinolones in Gram-negative species, whereas topoisomerase IV plays a more important role in many Gram-positive bacteria. This trend notwithstanding, on the basis of laboratory studies in which quinolone-resistant cultures were selected, gyrase appears to be the primary target for clinically relevant quinolones in Bacillus anthracis, a Gram-positive organism that is the causative agent of anthrax.34−36
Unfortunately, the continued clinical efficacy of quinolones is threatened by the rising prevalence of resistance, which has been reported in nearly every infection treated with these drugs.4,5,7 Quinolone resistance is usually associated with mutations in gyrase and/or topoisomerase IV.2,5−8,33,35−38 For both enzymes, the most common resistance-conferring mutations occur at a highly conserved serine residue in the A subunit and, to a lesser extent, an acidic residue four amino acids downstream (originally described as Ser83 and Glu87 in Escherichia coli gyrase).5,7,37,39−42 Despite the prevalence of these mutations across a broad spectrum of quinolone-resistant isolates, the mechanistic basis by which they led to resistance remained an enigma for decades.
The first report proposing a potential mechanism by which the serine and acidic residues mediate quinolone action and resistance described the structure of a cleavage complex formed by Acinetobacter baumannii topoisomerase IV in the presence of the quinolone moxifloxacin.27 Although it had long been known that quinolones chelate divalent metal ions,43 the relevance of this chelation to drug–enzyme interactions was never addressed. The A. baumannii topoisomerase IV structure was the first to capture the drug-bound metal ion, a noncatalytic magnesium, which was chelated by the C3/C4 keto acid of moxifloxacin. In this structure, the bound metal ion was coordinated to four water molecules, and two of these water molecules appeared to form hydrogen bonds with the serine and acidic residues. However, because three other contemporary structures did not capture the metal ion,24−26 the basis of quinolone–topoisomerase IV interactions remained unclear.
To determine relationships between the reported structures and quinolone function, a biochemical approach was used to analyze interactions between quinolones and wild-type and quinolone-resistant B. anthracis and E. coli topoisomerase IV.44−46 This series of reports confirmed the existence of a “water–metal ion bridge” between quinolones and topoisomerase IV and demonstrated its function in mediating drug–enzyme interactions. These studies provided strong evidence that the serine and acidic residues anchored the quinolone to the enzyme through the water–metal ion bridge and that mutations at either residue caused quinolone resistance by disrupting the function of the bridge. Although B. anthracis and E. coli topoisomerase IV both utilize the bridge and require it for drug function, the bridge appears to play different roles in mediating the actions of quinolones in the two species. In B. anthracis topoisomerase IV, the bridge is required for the binding of quinolones to the enzyme, and loss of either bridge anchor dramatically decreases the affinity of the drug.44,45 Conversely, in E. coli topoisomerase IV, the loss of either bridge anchor has little effect on quinolone binding.46 This finding strongly suggests that the water–metal ion bridge is used for positioning the drug within its binding pocket in an orientation that stabilizes cleavage complexes.
Recent biochemical and structural studies extended work on the water–metal ion bridge to Mycobacterium tuberculosis gyrase.47,48 Once again, these studies demonstrated the existence of the bridge, its role in mediating quinolone–enzyme interactions, and how disruption of the bridge causes resistance. However, compared to most bacterial type II topoisomerases, M. tuberculosis gyrase is unusual in two respects. First, it lacks the highly conserved serine residue and depends solely on the acidic residue to anchor the bridge.47,48 Second, because M. tuberculosis does not encode topoisomerase IV, its gyrase is functionally distinct from “canonical” gyrases because it must carry out the cellular activities of both bacterial type II topoisomerases.49,50 Thus, it is not obvious whether quinolone interactions described for this enzyme reflect those of topoisomerase IV or are also inherent to canonical gyrase.
Given the importance of gyrase as the primary target for quinolones in many pathogenic species, it is critical to understand how these drugs interact with a canonical enzyme and how specific mutations lead to resistance. Because it is not yet known whether quinolones interact with a canonical gyrase through the water–metal ion bridge, we examined potential bridge function in B. anthracis gyrase. Results indicate that clinically relevant quinolones rely on the water–metal ion bridge to bind to the enzyme. However, in contrast to topoisomerase IV and M. tuberculosis gyrase, the bridge is anchored primarily through the serine residue. Finally, by introducing additional drug–enzyme interactions that are not mediated through the water–metal ion bridge, it is possible to overcome resistance.
Materials and Methods
Enzymes and Materials
Wild-type B. anthracis gyrase subunits (GyrA and GyrB) were expressed and purified using a modification45 of a previously published protocol.51 Genes were amplified via polymerase chain reaction (PCR) from chromosomal DNA of the Sterne strain of B. anthracis. PCR products were cloned into the pET21b vector, which added an N-terminal six-His tag to each protein subunit. The GyrAS85L, GyrAS85F, GyrAE89K, and GyrAE89A constructs were generated using a QuikChange kit (Stratagene). The identities of all constructs were confirmed by DNA sequencing. Each subunit construct was individually transformed into E. coli strain BL21(DE3), and subunits were purified as described previously.45 Subunits were stored in buffer containing 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 20% glycerol, and 5 mM dithiothreitol and kept at −80 °C.
Negatively supercoiled pBR322 DNA was prepared from E. coli using a Plasmid Mega Kit (Qiagen) as described by the manufacturer. Relaxed pBR322 plasmid DNA was generated by treating negatively supercoiled pBR322 with calf thymus topoisomerase I (Invitrogen) and purified as described previously.46
Ciprofloxacin was obtained from LKT Laboratories, stored at 4 °C as a 40 mM stock solution in 0.1 N NaOH, and diluted 5-fold with 10 mM Tris-HCl (pH 7.9) immediately prior to use. Moxifloxacin was obtained from LKT Laboratories and stored at 4 °C as a 20 mM stock solution in 100% dimethyl sulfoxide (DMSO). 8-Methyl-cipro [1-cyclopropyl-6-fluoro-1,4-dihydro-8-methyl-7-(1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid], 8-methoxy-cipro [1-cyclopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid], cipro-dione [3-amino-7-(1-piperazinyl)-1-cyclopropyl-6-fluoro-2,4(1H,3H)-quinazolinedione], 8-H-moxi {1-cyclopropyl-6-fluoro-1,4-dihydro-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid}, 8-methyl-moxi {1-cyclopropyl-6-fluoro-1,4-dihydro-8-methyl-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid}, moxi-dione {3-amino-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-1-cyclopropyl-6-fluoro-8-methoxy-2,4(1H,3H)-quinazolinedione}, 3′-(AM)P-quinolone {1-cyclopropyl-6-fluoro-1,4-dihydro-8-methyl-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-4-oxo-3-quinolinecarboxylic acid}, and 3′-(AM)P-dione {3-amino-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-8-methyl-2,4(1H,3H)-quinazolinedione} were synthesized as reported previously.44,52 8-Methyl-cipro and 8-methoxy-cipro were stored at −20 °C as 40 mM stock solutions in 0.1 N NaOH and diluted 5-fold with 10 mM Tris-HCl (pH 7.9) immediately prior to use. All other compounds were stored at 4 °C as 20 mM stock solutions in 100% DMSO. All other chemicals were analytical reagent grade.
DNA Supercoiling
DNA supercoiling assays were based on previously published protocols.44,47 GyrA and GyrB (400 nM enzyme at a 1:2 GyrA:GyrB ratio) were incubated for 5 min at 37 °C in 100 mM Tris-HCl (pH 7.5), 350 mM KGlu, and 100 μg/mL bovine serum albumin (BSA, Sigma) and then diluted 2-fold with a mixture containing DNA, Mg2+, and ATP to reach a final reaction volume of 20 μL. The final concentrations of reactants were 200 nM gyrase, 5 nM relaxed DNA, and 1.5 mM ATP in 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 175 mM KGlu, and 50 μg/mL BSA. Reaction mixtures were incubated at 37 °C for various times and reactions were stopped by the addition of 3 μL of a mixture of 0.77% SDS and 77.5 mM Na2EDTA. Samples were mixed with 2 μL of agarose loading dye [60% sucrose in 10 mM Tris-HCl (pH 7.9), 0.5% bromophenol blue, and 0.5% xylene cyanol FF] and subjected to electrophoresis in 1% agarose gels in 100 mM Tris-borate (pH 8.3) and 2 mM EDTA. Gels were stained with 1 μg/mL ethidium bromide for 30 min. DNA bands were visualized with medium-range ultraviolet light on an Alpha Innotech digital imaging system.
DNA Cleavage
DNA cleavage reactions were based on the procedure of Aldred et al.44 Reaction mixtures contained 500 nM wild-type GyrA, GyrAS85L, GyrAS85F, GyrAE89K, or GyrAE89A gyrase at a 1:2 GyrA:GyrB ratio and 10 nM negatively supercoiled pBR322 in a total volume of 20 μL of 50 mM Tris-HCl (pH 7.5), 100 mM KGlu, 5 mM MgCl2, and 50 μg/mL BSA. In some cases, a range of MgCl2 concentrations (0–3 mM) was tested or MgCl2 was replaced with BaCl2 (2 mM), NiCl2 (1 mM), or MnCl2 (1 mM). Alternatively, MgCl2 was replaced with CaCl2 (20 mM), and a range of enzyme concentrations was tested. Reaction mixtures were incubated at 37 °C for 30 min. Enzyme–DNA cleavage complexes were trapped by adding 2 μL of 5% SDS followed by 2 μL of 250 mM Na2EDTA and 2 μL of 0.8 mg/mL Proteinase K. Reaction mixtures were incubated at 45 °C for 30 min to digest the enzyme. Samples were mixed with 2 μL of agarose loading dye and incubated at 45 °C for 2 min before being loaded on the gels. Reaction products were subjected to electrophoresis in 1% agarose gels in 40 mM Tris-acetate (pH 8.3), 2 mM Na2EDTA, and 0.5 μg/mL ethidium bromide and visualized as described above. DNA cleavage was monitored by the conversion of supercoiled plasmid to linear molecules and quantified by comparison to a control reaction in which an equal mass of DNA was digested by EcoRI (New England BioLabs).
In assays that monitored competition between ciprofloxacin (0–50 μM), moxifloxacin (0–50 μM), cipro-dione (0–250 μM), or moxi-dione (0–250 μM) and 3′-(AM)P-dione (5 μM), drugs were added simultaneously to reaction mixtures. The percent DNA cleavage induced by the competing drug only was subtracted from the percent DNA cleavage induced by 3′-(AM)P-dione. DNA cleavage levels were then normalized to levels seen with 3′-(AM)P-dione alone (set to 1.0). The competition studies were used to assess the relative abilities of different drugs to interact with a series of wild-type gyrase and mutant enzymes. Quinolones have been shown to bind to individual subunits of gyrase, to DNA, to an enzyme–DNA complex in which the enzyme is not capable of cleaving the DNA, and to the enzyme–DNA cleavage complex.53−56 At any given time, the DNA cleavage complex (which is the complex of interest) represents a relatively small percentage of the total gyrase in reaction mixtures. Thus, we used a competition assay that specifically provides information about how drugs interact with cleavage complexes. Utilizing 3′-(AM)P-dione (which induced nearly identical high levels of DNA cleavage with each of the mutant enzymes) as the drug being competed out provided an “internal” standard for each competition experiment that normalized results from the wild-type and mutant enzymes.
Results and Discussion
Characterization of Wild-Type B. anthracis Gyrase and GyrA Mutants
As reported for other bacterial species, the most common gyrase mutations in quinolone-resistant strains of B. anthracis are found at the conserved serine and glutamic acid residues (GyrAS85 and GyrAE89). In laboratory strains selected for resistance against ciprofloxacin and/or moxifloxacin (two widely prescribed quinolone antibacterials), approximately 80% of the isolates carried a GyrAS85L mutation (either alone or in combination with other gyrase/topoisomerase IV amino acid changes).35,38 The only other mutation reported to cause resistance without any other gyrase/topoisomerase IV changes was a GyrAE89K substitution.35,38 Therefore, we focused our studies on gyrase containing these two mutations. In addition, we characterized mutant enzymes carrying GyrAS85F or GyrAE89A. The S85F mutation recapitulates the most common quinolone resistance mutation seen in B. anthracis topoisomerase IV, which has been examined previously.44 This mutation is not observed in gyrase because of differences in the codon used for the serine residue. The E89A mutation represents a control for the charge change in GyrAE89K to determine whether resistance is due to the loss of glutamic acid or the conversion of a negative to a positive charge at that position.
As a first step toward comparing the wild-type and mutant enzymes, we assessed their ability to supercoil relaxed DNA. As seen in Figure 1, the serine mutant enzymes and the E89A mutant gyrase maintain a wild-type ability to supercoil DNA. Consistent with previous reports, the overall catalytic activity of the E89K mutant enzymes was decreased compared to that of wild-type gyrase. This may be the reason why mutations at this position are observed less frequently than those at the serine residue.
Figure 1.

DNA supercoiling activities of wild-type B. anthracis gyrase and mutant enzymes. A time course is shown for the introduction of negative supercoils into relaxed DNA by wild-type gyrase (top), enzymes with S85L and S85F mutations (middle), and enzymes with E89K and E89A mutations (bottom). Positions of nicked, relaxed (Rel), and negatively supercoiled [(−)SC] DNA are marked on the top image. Gel images are representative of at least three independent experiments.
Because quinolones kill cells primarily by increasing levels of gyrase- and/or topoisomerase IV-mediated DNA cleavage, we examined the ability of the enzymes discussed above to cleave DNA in the absence of drugs (Figure 2). CaCl2 was used instead of MgCl2 in these reactions because this metal ion increases baseline levels of cleavage mediated by type II topoisomerases without affecting other properties of the DNA scission reaction.44,57−60 All of the mutant enzymes displayed an activity similar to or higher than that of wild-type gyrase. These DNA cleavage patterns are similar to those reported for the corresponding mutations in topoisomerase IV44,45 and strongly suggest that quinolone resistance in B. anthracis gyrase is not due to a general loss of enzymatic activity.
Figure 2.
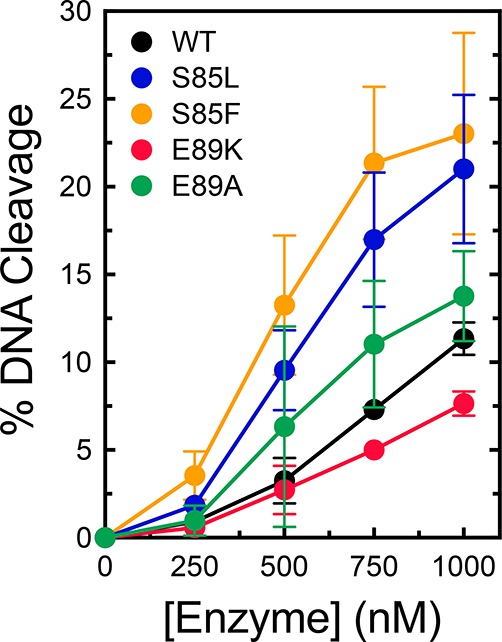
DNA cleavage activities of wild-type B. anthracis gyrase and mutant enzymes. The abilities of wild-type (black), S85L (blue), S85F (orange), E89K (red), and E89A (green) gyrase to cleave DNA in the absence of drugs are shown. Error bars represent the standard deviation of at least three independent experiments.
Effects of Quinolones and Related Compounds on DNA Cleavage Mediated by Wild-Type Gyrase and Mutant Enzymes
Although GyrA mutations at S85 and E89 have been observed in quinolone-resistant B. anthracis cultures, a direct causal link between these mutations and quinolone resistance in gyrase has yet to be established. Therefore, we determined the ability of ciprofloxacin and moxifloxacin to enhance DNA cleavage mediated by wild-type, S85L, S85F, E89K, and E89A gyrase. The S85L, S85F, and E89K mutant enzymes all displayed high resistance to both quinolones (Figures 3 and 4). These mutations decreased the relative potencies of the drugs >10-fold. The mutations had a weaker effect on the potency of moxifloxacin as compared to ciprofloxacin. At 250 μM, moxifloxacin could induce levels of cleavage with the mutant enzymes close to those generated with wild-type gyrase and 20 μM drug (the highest drug concentration that could be used without producing multiple cleavage events per plasmid). In contrast, ciprofloxacin could not induce wild-type levels of cleavage with the mutant enzymes, even at concentrations as high as 500 μM.
Figure 3.
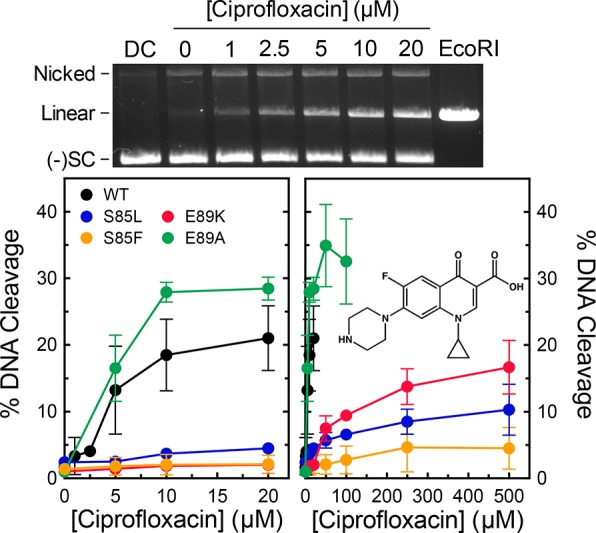
Effects of ciprofloxacin on DNA cleavage mediated by wild-type gyrase and mutant enzymes. A representative gel of DNA cleavage induced by wild-type gyrase in the presence of ciprofloxacin is shown (top). The gel includes a DNA control in the absence of enzyme (DC) and DNA digested by EcoRI (EcoRI). The positions of nicked, linear, and negatively supercoiled [(−)SC] DNA are indicated. Graphs show the abilities of wild-type (black), S85L (blue), S85F (orange), E89K (red), and E89A (green) gyrase to cleave DNA in the presence of ciprofloxacin over either low (left) or high (right) concentration ranges. The structure of ciprofloxacin is shown in the right panel. Error bars represent the standard deviation of at least three independent experiments.
Figure 4.
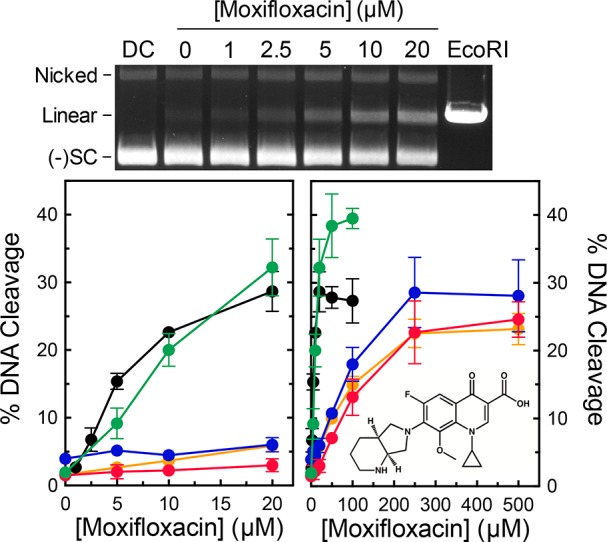
Effects of moxifloxacin on DNA cleavage mediated by wild-type gyrase and mutant enzymes. A representative gel of DNA cleavage induced by wild-type gyrase in the presence of moxifloxacin is shown (top). The gel includes a DNA control in the absence of enzyme (DC) and DNA digested by EcoRI (EcoRI). The positions of nicked, linear, and negatively supercoiled [(−)SC] DNA are indicated. Graphs show the abilities of wild-type (black), S85L (blue), S85F (orange), E89K (red), and E89A (green) gyrase to cleave DNA in the presence of moxifloxacin over either low (left) or high (right) concentration ranges. The structure of moxifloxacin is shown in the right panel. Error bars represent the standard deviation of at least three independent experiments.
The E89K mutation introduces a negative to positive change in the vicinity of quinolone binding. To determine whether resistance associated with this mutation is due to the loss of the glutamic acid or to the introduction of an opposite charge at this position, we examined the effects of quinolones on DNA cleavage mediated by GyrAE89A gyrase (Figures 3 and 4). Unlike the other mutants examined, this enzyme displayed no resistance to either ciprofloxacin or moxifloxacin. This implies that the basis for quinolone resistance caused by the E89K mutation is the disruptive change in charge. Alternatively, the increased length of the lysine side chain may contribute steric effects. This is in marked contrast to results with B. anthracis topoisomerase IV, in which the equivalent E → K and E → A mutations displayed similar levels of resistance.45 These findings suggest potentially important differences in the way that quinolones interact with gyrase and topoisomerase IV in B. anthracis.
In addition to ciprofloxacin and moxifloxacin, we also examined the activity of 3′-(AM)P-quinolone against the wild-type and mutant enzymes. Unlike the C7 groups of clinically relevant quinolones, on the basis of crystallography and docking studies, the 3′-(AM)P substituent can interact directly with a glutamic acid residue in GyrB, which would correspond to Glu472 in B. anthracis GyrB.61,62 This interaction potentially obviates the requirement for the proposed water–metal ion bridge in mediating drug–enzyme interactions.44−46,52 Consistent with this hypothesis, 3′-(AM)P-quinolone displayed high activity against all of the mutant enzymes with little change in potency compared to that of the wild type (Figure 5, top left). Even when the 3′-(AM)P group was moved to a quinazolinedione skeleton (which lacks the C3/C4 keto acid required to form a water–metal ion bridge), the compound retained high activity against wild-type gyrase as well as quinolone-resistant mutant enzymes (Figure 5, top right). Conversely, when the C7/C8 substituents of ciprofloxacin and moxifloxacin (piperazinyl/H and diazabicyclononyl/methoxyl groups, respectively) were transferred to a quinazolinedione core, the resulting diones lost the majority of their activity against all of the enzymes, including the wild-type gyrase (Figure 5, bottom). These findings with “cipro-dione” and “moxi-dione” emphasize the importance of the C3/C4 keto acid in mediating the activity of clinically relevant quinolones against B. anthracis gyrase.
Figure 5.
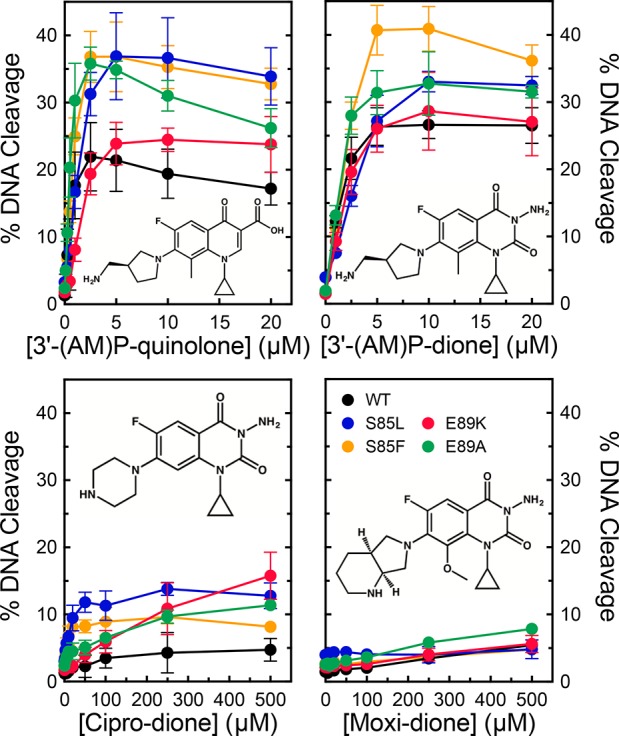
Effects of 3′-(AM)P-quinolone and quinazolinediones on DNA cleavage mediated by wild-type gyrase and mutant enzymes. The abilities of wild-type (black), S85L (blue), S85F (orange), E89K (red), and E89A (green) gyrase to cleave DNA in the presence of 3′-(AM)P-quinolone (top left), 3′-(AM)P-dione (top right), cipro-dione (bottom left), and moxi-dione (bottom right) are shown. Drug structures are shown in each panel. Error bars represent the standard deviation of at least three independent experiments.
On the basis of results with B. anthracis(44,45) and E. coli(46) topoisomerase IV, the critical role of the C3/C4 keto acid of quinolones may be to facilitate binding to the enzyme or to position the drug appropriately for stabilizing cleavage complexes. To distinguish between these possibilities, we assessed the ability of cipro-dione and moxi-dione to compete with 5 μM 3′-(AM)P-dione in a DNA cleavage assay. If the C3/C4 keto acid is important for binding, the removal of this group should prevent effective competition.44,45 Conversely, if the group is used to position the drug in the active site, the drugs should compete efficiently.46 As seen in Figure 6, neither drug could substantially decrease levels of DNA cleavage induced by 3′-(AM)P-dione. Even at concentrations as high as 250 μM (corresponding to a 50-fold molar excess), cipro-dione and moxi-dione decreased the level of cleavage by <50%. These results provide evidence that the C3/C4 keto acid is crucial for the binding of clinically used quinolones to B. anthracis gyrase.
Figure 6.
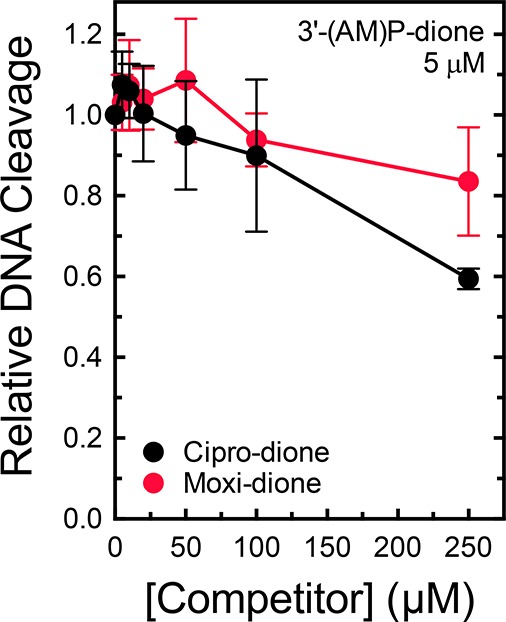
Ability of cipro-dione and moxi-dione to compete with 3′-(AM)P-dione in DNA cleavage reactions mediated by wild-type gyrase. The abilities of increasing concentrations of cipro-dione (black) and moxi-dione (red) to compete with and prevent cleavage enhancement by 5 μM 3′-(AM)P-dione were determined. Drugs were added simultaneously to reaction mixtures. The low levels of cleavage seen in the presence of cipro-dione or moxi-dione alone were subtracted from the levels of cleavage seen with both drugs, and the resulting corrected cleavage level is plotted relative to the level observed with 3′-(AM)P-dione alone (set to 1.0). Error bars represent the standard deviation of at least three independent experiments.
Evidence of a Water–Metal Ion Bridge in Mediating Interactions between Clinically Relevant Quinolones and B. anthracis Gyrase
The importance of the C3/C4 keto acid for drug binding coupled with the effects of the resistance mutations strongly suggest that B. anthracis gyrase utilizes the water–metal ion bridge to mediate interactions with quinolones. Therefore, three approaches were used to determine whether this was the case. First, we examined the requirement for metal ions to support quinolone-stabilized DNA cleavage mediated by wild-type gyrase. Type II topoisomerases use a two-metal ion mechanism to mediate DNA cleavage and religation.59,60,63−65 Although B. anthracis gyrase could utilize a variety of metal ions other than Mg2+ to support DNA scission (data not shown), not all ions could support DNA cleavage induced by quinolones. For example, as seen in Figure 7, ciprofloxacin and moxifloxacin could not induce cleavage in the presence of Ba2+ or Ni2+. In contrast, substantial levels of cleavage were observed with 3′-(AM)P-dione, which does not require a metal ion to enhance DNA cleavage. These findings indicate that quinolones require a noncatalytic divalent metal ion in addition to those at the active site of the enzyme to function against B. anthracis gyrase.
Figure 7.
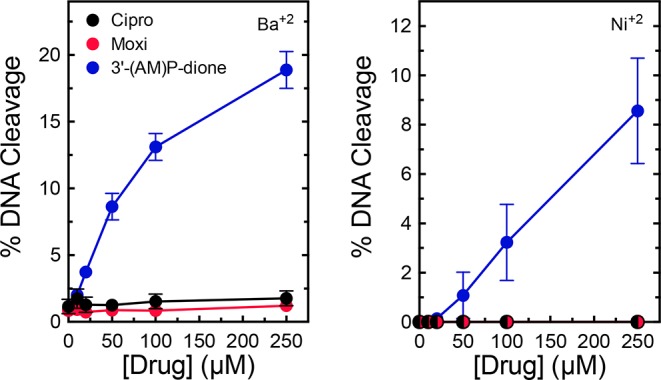
Effects of alternative metal ions on drug-induced DNA cleavage mediated by wild-type gyrase. The abilities of ciprofloxacin (black), moxifloxacin (red), and 3′-(AM)P-dione (blue) to induce cleavage by wild-type gyrase in the presence of 2 mM Ba2+ (left) or 1 mM Ni2+ (right) are shown. Error bars represent the standard deviation of at least three independent experiments.
Second, we determined whether mutations at S85 or E89 of GyrA altered the metal ion requirements for quinolone-induced DNA cleavage. In the presence of Mg2+, high quinolone concentrations could (at least partially) overcome drug resistance with the S85L and E89K mutants (see Figures 3 and 4). However, in the presence of Mn2+, ciprofloxacin and moxifloxacin could not induce cleavage with these mutant enzymes (Figure 8). This finding is despite the fact that Mn2+ supported the activity of both quinolones against the wild-type enzyme and the nonresistant E89A mutant and also supported the activity of 3′-(AM)P-dione against all of the enzymes examined. The finding that the quinolone-resistant S85L and E89K mutations restrict the variety of metal ions that clinically relevant quinolones can use to enhance DNA cleavage further implicates a role for a noncatalytic divalent metal ion in mediating interactions between the drug and B. anthracis gyrase.
Figure 8.
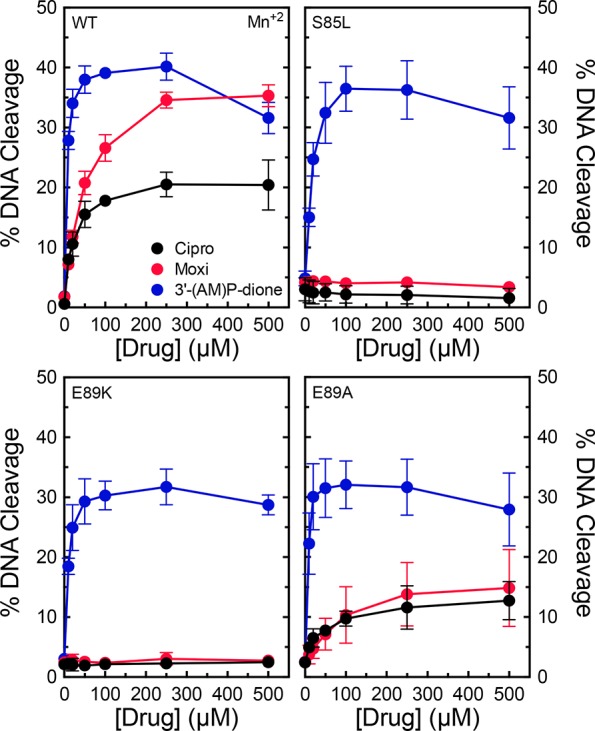
Effects of Mn2+ on drug-induced DNA cleavage mediated by wild-type gyrase and mutant enzymes. The abilities of ciprofloxacin (black), moxifloxacin (red), and 3′-(AM)P-dione (blue) to induce cleavage by wild-type (top left), S85L (top right), E89K (bottom left), or E89A (bottom right) gyrase in the presence of 1 mM Mn2+ are shown. Error bars represent the standard deviation of at least three independent experiments.
Third, we determined whether mutations at S85 and E89 affected the affinity of the metal ion required for quinolone actions against gyrase. These experiments monitored DNA cleavage levels over a range of 0–3 mM Mg2+. To facilitate direct comparisons between the wild-type and mutant enzymes, DNA cleavage levels were normalized to those observed in reaction mixtures containing 3 mM MgCl2. Compared to wild-type gyrase, the S85L and E89K mutants decreased the affinity of Mg2+ in reaction mixtures that contained ciprofloxacin (Figure 9, left). Concentrations of the metal ion required to induce half-maximal and maximal levels of cleavage increased ∼2.5-fold. In contrast, the E89A mutation, which does not cause quinolone resistance, had no effect on metal ion usage. As a control, parallel titrations were performed with 3′-(AM)P-dione (Figure 9, right). No decrease in metal ion affinity was observed with any of the mutant enzymes. In fact, the quinazolinedione required lower levels of Mg2+ with the E89 mutants.
Figure 9.
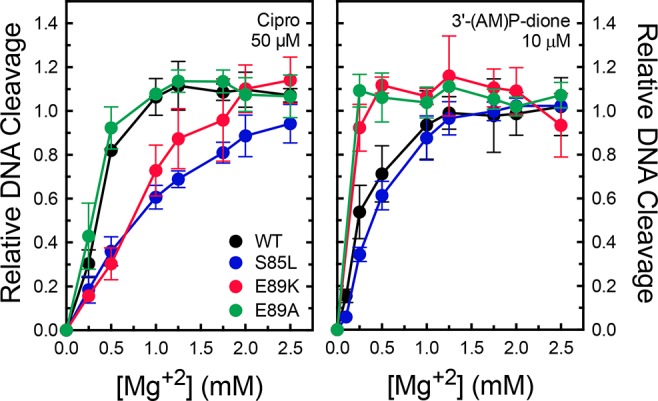
Effects of GyrA mutants on Mg2+ concentrations required to support maximal levels of drug-induced DNA cleavage. Results are shown for ciprofloxacin (left) and 3′-(AM)P-dione (right) with wild-type (black), S85L (blue), E89K (red), or E89A (green) gyrase. Cleavage levels with each drug–enzyme pair were normalized to levels observed with 3 mM MgCl2 (set to 1.0). Error bars represent the standard deviation of at least three independent experiments.
The studies described above indicate that clinically relevant quinolones such as ciprofloxacin and moxifloxacin require a divalent metal ion for their actions and that resistance mutations restrict the usage and decrease the affinity of the coordinating metal ion. Taken together, these findings provide strong evidence that interactions of these quinolones with B. anthracis gyrase are mediated by a water–metal ion bridge. However, these results point to an important distinction between the amino acids used to anchor the bridges in B. anthracis gyrase and topoisomerase IV. Whereas the serine residue is a critical bridge anchor in both enzymes, it appears that the glutamic acid residue is used only in topoisomerase IV. This is based on the fact that the conversion of this residue to an alanine causes resistance and altered utilization of metal ions in topoisomerase IV45 but does neither in gyrase.
Role of the Water–Metal Ion Bridge in Mediating Quinolone–Gyrase Interactions
Two lines of evidence imply that the water–metal ion bridge is used primarily to mediate binding interactions between clinically relevant quinolones and B. anthracis gyrase: high concentrations of quinolones can overcome the effects of resistance mutations (Figures 3 and 4), and removal of the C3/C4 keto acid greatly diminishes the affinity of the drug for the enzyme (Figure 6). To determine whether this is the case, a competition assay that utilized S85L or E89K gyrase was employed. These experiments monitored the ability of 0–50 μM ciprofloxacin and moxifloxacin to compete with DNA cleavage induced by 5 μM 3′-(AM)P-dione. These three drugs display similar potencies against wild-type B. anthracis gyrase (see Figures 3–5). Therefore, if the resistance mutations do not reduce drug affinity, cleavage levels induced by 3′-(AM)P-dione should decrease by 50% at ∼5 μM quinolone. As seen in Figure 10, this was not the case. Even at a 10-fold molar excess of ciprofloxacin or moxifloxacin, the level of DNA scission decreased <50%. These results provide strong evidence that the water–metal ion bridge provides the primary binding contact between clinically relevant quinolones and B. anthracis gyrase.
Figure 10.
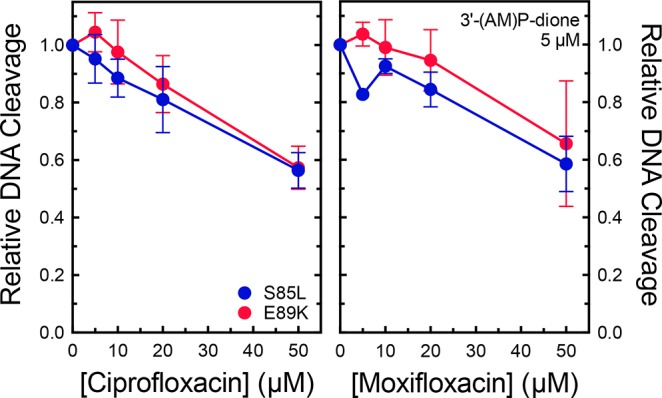
Abilities of ciprofloxacin and moxifloxacin to compete with 3′-(AM)P-dione in DNA cleavage reactions mediated by quinolone-resistant enzymes. The abilities of increasing concentrations of ciprofloxacin (left) and moxifloxacin (right) to compete with and prevent cleavage enhancement by 5 μM 3′-(AM)P-dione with S85L (blue) and E89K (red) gyrase were determined. Drugs were added simultaneously to reaction mixtures. The levels of cleavage seen in the presence of ciprofloxacin or moxifloxacin alone were subtracted from the levels of cleavage seen with both drugs, and the resulting corrected cleavage level is plotted relative to the level observed with 3′-(AM)P-dione alone (set to 1.0). Error bars represent the standard deviation of at least three independent experiments.
Effects of the C8 Substituent on Quinolone Activity and Resistance
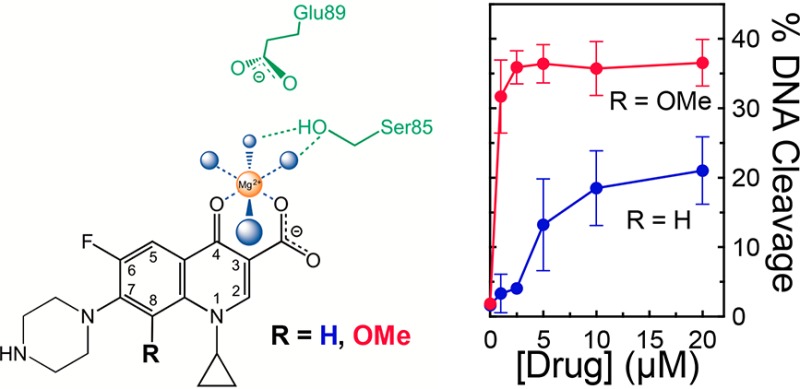
Previous studies with B. anthracis topoisomerase IV52 and M. tuberculosis gyrase47 demonstrated that the substituent at C8 can make important contributions to quinolone activity and have the potential to overcome resistance caused by the most common mutations. Therefore, we examined a series of ciprofloxacin- and moxifloxacin-based compounds containing a -H, -methyl, or -methoxyl substituent at C8 for their activity against wild-type, S85L, and E89K gyrase. Compared to ciprofloxacin (which contains a C8–H group), 8-methyl-cipro and 8-methoxy-cipro displayed considerably higher potencies and efficacies against the wild-type and mutant enzymes (Figure 11). In fact, 8-methoxy-cipro could overcome resistance at ∼20 μM drug. Although less striking, similar results were seen with the moxifloxacin series (Figure 12). Compared to the parent compound (which contains a C8-methoxyl group), 8-H-moxi displayed a much lower efficacy against all of the enzymes and could not overcome resistance even at high concentrations. In contrast, 8-methyl-moxi displayed high potency and efficacy that were similar to (or slightly higher than) those of moxifloxacin. These results demonstrate that the substituent at C8 affects the actions of quinolones against B. anthracis gyrase.
Figure 11.
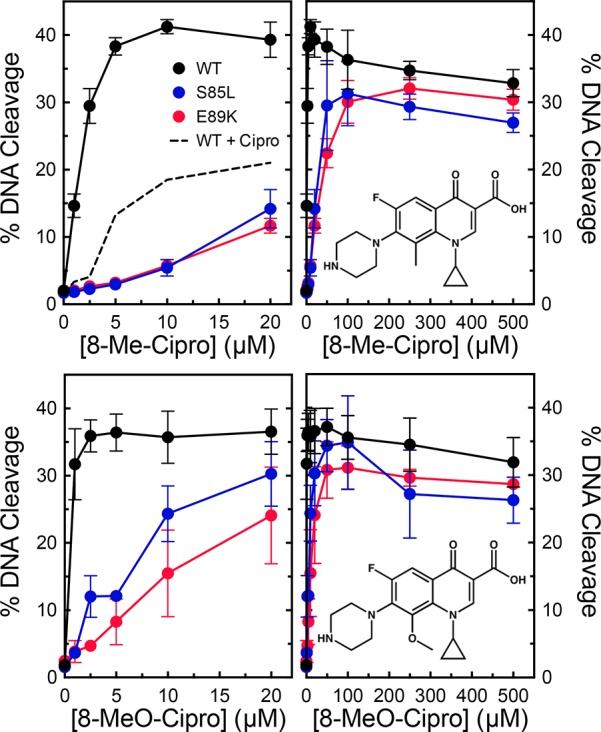
Effects of C8 substituents on ciprofloxacin-induced DNA cleavage mediated by wild-type gyrase and quinolone-resistant mutants. The abilities of low (left) or high (right) concentrations ranges of 8-methyl-ciprofloxacin (top) and 8-methoxy-ciprofloxacin (bottom) to enhance cleavage mediated by wild-type (black), S85L (blue), and E89K (red) gyrase were determined. Cleavage induced by ciprofloxacin with wild-type gyrase is shown by the dotted black line in the top left panel. Drug structures are shown in the right panels. Error bars represent the standard deviation of at least three independent experiments.
Figure 12.
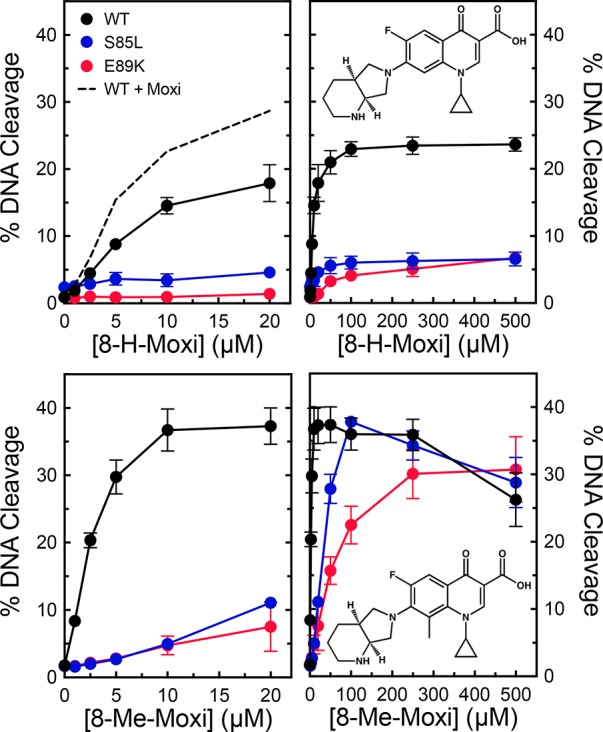
Effects of C8 substituents on moxifloxacin-induced DNA cleavage mediated by wild-type gyrase and quinolone-resistant mutants. The abilities of low (left) or high (right) concentrations ranges of 8-H-moxifloxacin (top) and 8-methyl-moxifloxacin (bottom) to enhance cleavage mediated by wild-type (black), S85L (blue), and E89K (red) gyrase were determined. Cleavage induced by moxifloxacin with wild-type gyrase is shown by the dotted black line in the top left panel. Drug structures are shown in the right panels. Error bars represent the standard deviation of at least three independent experiments.
Conclusions
The findings presented above provide strong evidence that the primary interaction between clinically relevant quinolones and B. anthracis gyrase occurs through the water–metal ion bridge (Figure 13). Furthermore, the function of the bridge in gyrase is to facilitate binding of the drug to the enzyme. This is as opposed to E. coli topoisomerase IV, which uses the bridge to position quinolones but does not require it for drug binding.46
Figure 13.
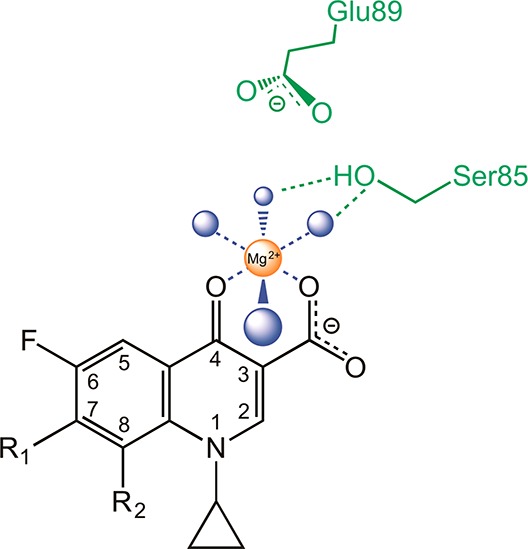
Simplified schematic of the water–metal ion bridge used to mediate interactions between quinolones and B. anthracis gyrase. Only interactions with the protein (and not DNA) are shown. A representative quinolone core is depicted in black, the noncatalytic Mg2+ in orange, water molecules in blue, and the serine and acidic residues in green. Blue lines indicate the octahedral coordination sphere of the divalent metal ion interacting with four water molecules and the C3/C4 keto acid of the quinolone. The green dashed lines represent hydrogen bonds between the serine side chain hydroxyl group and the water molecules. In contrast to B. anthracis and E. coli topoisomerase IV, the glutamic acid residue does not appear to act as a bridge anchor. Adapted from ref (44).
The bridge “anchors” in B. anthracis gyrase are distinct from those in topoisomerase IV and in M. tuberculosis gyrase. In B. anthracis and E. coli topoisomerase IV, both the serine and the glutamic acid residues are used to anchor the bridge, and loss of either anchor causes quinolone resistance.44,45 Because M. tuberculosis gyrase lacks the serine residue, the only available anchor is the glutamic acid, so this residue provides the principal contact for quinolone binding.47 Conversely, in B. anthracis gyrase, the water–metal ion bridge seems to be primarily coordinated through the serine residue. In this case, quinolone resistance in gyrase harboring an E89K mutation is likely due to the disruptive change in charge rather than the loss of an anchor. These findings point to subtle, but critical, differences in the drug binding pockets of gyrase and topoisomerase IV from different species that give rise to distinct quinolone interactions.
In accord with the findings described above, it is possible to introduce interactions that overcome drug resistance differentially. For example, changing the C8 substituent of ciprofloxacin (which is the primary prophylactic and first-line drug used to treat anthrax)66,67 from a hydrogen to a methoxyl group greatly improves the efficacy of the drug against wild-type and quinolone-resistant B. anthracis gyrase. However, in M. tuberculosis gyrase, the drug derivative that can best overcome resistance is the C8-methyl form of moxifloxacin.47
Taken together, these results present an opportunity to shift the paradigm for the development and use of new quinolones. Historically, these drugs were developed as broad-spectrum antibacterials. However, by analyzing the interactions of drugs with gyrase and topoisomerase IV from individual bacteria, it should be possible to identify specific quinolone derivatives that can overcome target-mediated resistance in important pathogenic species.
Acknowledgments
We are grateful to Lorena Infante Lara and Elizabeth G. Gibson for critical reading of the manuscript. R.H.L. initiated this project and performed several preliminary experiments that characterized B. anthracis gyrase. Unfortunately, he lost his valiant battle against cancer before this project was fully underway.
This work was supported by the U.S. Veterans Administration (Merit Review Award I01 Bx002198 to N.O.), the National Institutes of Health (Grants AI81775 to C.L.T., AI87671 to R.J.K., and GM033944 to N.O.), and the National Science Foundation (Grant DGE-0909667 to R.E.A.).
The authors declare no competing financial interest.
References
- Hooper D. C. (1998) Clinical applications of quinolones. Biochim. Biophys. Acta, Gene Struct. Expression 1400, 45–61. 10.1016/S0167-4781(98)00127-4. [DOI] [PubMed] [Google Scholar]
- Hooper D. C. (2001) Mechanisms of action of antimicrobials: focus on fluoroquinolones. Clin. Infect. Dis. 32 (Suppl. 1), S9–S15. 10.1086/319370. [DOI] [PubMed] [Google Scholar]
- Linder J. A.; Huang E. S.; Steinman M. A.; Gonzales R.; Stafford R. S. (2005) Fluoroquinolone prescribing in the United States: 1995 to 2002. Am. J. Med. 118, 259–268. 10.1016/j.amjmed.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Andriole V. T. (2005) The quinolones: past, present, and future. Clin. Infect. Dis. 41 (Suppl. 2), S113–119. 10.1086/428051. [DOI] [PubMed] [Google Scholar]
- Drlica K.; Hiasa H.; Kerns R.; Malik M.; Mustaev A.; Zhao X. (2009) Quinolones: action and resistance updated. Curr. Top. Med. Chem. 9, 981–998. 10.2174/156802609789630947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; Leo E.; Zhang H.; Marchand C. (2010) DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 17, 421–433. 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Kerns R. J.; Osheroff N. (2014) Mechanism of quinolone action and resistance. Biochemistry 53, 1565–1574. 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson V. E.; Osheroff N. (2001) Type II topoisomerases as targets for quinolone antibacterials: turning Dr. Jekyll into Mr. Hyde. Curr. Pharm. Des. 7, 337–353. 10.2174/1381612013398013. [DOI] [PubMed] [Google Scholar]
- Drlica K.; Malik M.; Kerns R. J.; Zhao X. (2008) Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 52, 385–392. 10.1128/AAC.01617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine C.; Hiasa H.; Marians K. J. (1998) DNA gyrase and topoisomerase IV: biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys. Acta, Gene Struct. Expression 1400, 29–43. 10.1016/S0167-4781(98)00126-2. [DOI] [PubMed] [Google Scholar]
- Koster D. A.; Crut A.; Shuman S.; Bjornsti M. A.; Dekker N. H. (2010) Cellular strategies for regulating DNA supercoiling: a single-molecule perspective. Cell 142, 519–530. 10.1016/j.cell.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos S. M.; Tretter E. M.; Schmidt B. H.; Berger J. M. (2011) All tangled up: how cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 12, 827–841. 10.1038/nrm3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiasa H.; Marians K. J. (1994) Topoisomerase IV can support oriC DNA replication in vitro. J. Biol. Chem. 269, 16371–16375. [PubMed] [Google Scholar]
- Zechiedrich E. L.; Khodursky A. B.; Bachellier S.; Schneider R.; Chen D.; Lilley D. M.; Cozzarelli N. R. (2000) Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J. Biol. Chem. 275, 8103–8113. 10.1074/jbc.275.11.8103. [DOI] [PubMed] [Google Scholar]
- Crisona N. J.; Strick T. R.; Bensimon D.; Croquette V.; Cozzarelli N. R. (2000) Preferential relaxation of positively supercoiled DNA by E. coli topoisomerase IV in single-molecule and ensemble measurements. Genes Dev. 14, 2881–2892. 10.1101/gad.838900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Reyes-Lamothe R.; Sherratt D. J. (2008) Modulation of Escherichia coli sister chromosome cohesion by topoisomerase IV. Genes Dev. 22, 2426–2433. 10.1101/gad.487508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Deibler R. W.; Chan H. S.; Zechiedrich L. (2009) The why and how of DNA unlinking. Nucleic Acids Res. 37, 661–671. 10.1093/nar/gkp041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi M. C.; Magnan D.; Montminy T. P.; Lies M.; Stepankiw N.; Bates D. (2013) Regulation of sister chromosome cohesion by the replication fork tracking protein SeqA. PLoS Genet. 9, e1003673. 10.1371/journal.pgen.1003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawadzki P.; Stracy M.; Ginda K.; Zawadzka K.; Lesterlin C.; Kapanidis A. N.; Sherratt D. J. (2015) The localization and action of topoisomerase IV in Escherichia coli chromosome segregation is coordinated by the SMC complex, MukBEF. Cell Rep. 13, 2587–2596. 10.1016/j.celrep.2015.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J.; Nishimura Y.; Imamura R.; Niki H.; Hiraga S.; Suzuki H. (1990) New topoisomerase essential for chromosome segregation in E. coli. Cell 63, 393–404. 10.1016/0092-8674(90)90172-B. [DOI] [PubMed] [Google Scholar]
- Champoux J. J. (2001) DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70, 369–413. 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- Deweese J. E.; Osheroff N. (2009) The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res. 37, 738–749. 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitiss J. L. (2009) DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 9, 327–337. 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laponogov I.; Sohi M. K.; Veselkov D. A.; Pan X. S.; Sawhney R.; Thompson A. W.; McAuley K. E.; Fisher L. M.; Sanderson M. R. (2009) Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 16, 667–669. 10.1038/nsmb.1604. [DOI] [PubMed] [Google Scholar]
- Laponogov I.; Pan X. S.; Veselkov D. A.; McAuley K. E.; Fisher L. M.; Sanderson M. R. (2010) Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS One 5, e11338. 10.1371/journal.pone.0011338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax B. D.; Chan P. F.; Eggleston D. S.; Fosberry A.; Gentry D. R.; Gorrec F.; Giordano I.; Hann M. M.; Hennessy A.; Hibbs M.; Huang J.; Jones E.; Jones J.; Brown K. K.; Lewis C. J.; May E. W.; Saunders M. R.; Singh O.; Spitzfaden C. E.; Shen C.; Shillings A.; Theobald A. J.; Wohlkonig A.; Pearson N. D.; Gwynn M. N. (2010) Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466, 935–940. 10.1038/nature09197. [DOI] [PubMed] [Google Scholar]
- Wohlkonig A.; Chan P. F.; Fosberry A. P.; Homes P.; Huang J.; Kranz M.; Leydon V. R.; Miles T. J.; Pearson N. D.; Perera R. L.; Shillings A. J.; Gwynn M. N.; Bax B. D. (2010) Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 17, 1152–1153. 10.1038/nsmb.1892. [DOI] [PubMed] [Google Scholar]
- Khodursky A. B.; Zechiedrich E. L.; Cozzarelli N. R. (1995) Topoisomerase IV is a target of quinolones in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 92, 11801–11805. 10.1073/pnas.92.25.11801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aedo S.; Tse-Dinh Y.-C. (2012) Isolation and quantitation of topoisomerase complexes accumulated on Escherichia coli chromosomal DNA. Antimicrob. Agents Chemother. 56, 5458–5464. 10.1128/AAC.01182-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.-S.; Fisher L. M. (1997) Targeting of DNA gyrase in Streptococcus pneumoniae by sparfloxacin: selective targeting of gyrase or topoisomerase IV by quinolones. Antimicrob. Agents Chemother. 41, 471–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.-S.; Fisher L. M. (1999) Streptococcus pneumoniae DNA gyrase and topoisomerase IV: overexpression, purification, and differential inhibition by fluoroquinolones. Antimicrob. Agents Chemother. 43, 1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.-S.; Ambler J.; Mehtar S.; Fisher L. M. (1996) Involvement of topoisomerase IV and DNA gyrase as ciprofloxacin targets in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 40, 2321–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier B.; Zhao X.; Lu T.; Drlica K.; Hooper D. C. (2000) Selective targeting of topoisomerase IV and DNA gyrase in Staphylococcus aureus: different patterns of quinolone-induced inhibition of DNA synthesis. Antimicrob. Agents Chemother. 44, 2160–2165. 10.1128/AAC.44.8.2160-2165.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook I.; Elliott T. B.; Pryor H. I. 2nd; Sautter T. E.; Gnade B. T.; Thakar J. H.; Knudson G. B. (2001) In vitro resistance of Bacillus anthracis Sterne to doxycycline, macrolides and quinolones. Int. J. Antimicrob. Agents 18, 559–562. 10.1016/S0924-8579(01)00464-2. [DOI] [PubMed] [Google Scholar]
- Price L. B.; Vogler A.; Pearson T.; Busch J. D.; Schupp J. M.; Keim P. (2003) In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob. Agents Chemother. 47, 2362–2365. 10.1128/AAC.47.7.2362-2365.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grohs P.; Podglajen I.; Gutmann L. (2004) Activities of different fluoroquinolones against Bacillus anthracis mutants selected in vitro and harboring topoisomerase mutations. Antimicrob. Agents Chemother. 48, 3024–3027. 10.1128/AAC.48.8.3024-3027.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan-Linnell S. K.; Becnel Boyd L.; Steffen D.; Zechiedrich L. (2009) Mechanisms accounting for fluoroquinolone resistance in Escherichia coli clinical isolates. Antimicrob. Agents Chemother. 53, 235–241. 10.1128/AAC.00665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bast D. J.; Athamna A.; Duncan C. L.; de Azavedo J. C.; Low D. E.; Rahav G.; Farrell D.; Rubinstein E. (2004) Type II topoisomerase mutations in Bacillus anthracis associated with high-level fluoroquinolone resistance. J. Antimicrob. Chemother. 54, 90–94. 10.1093/jac/dkh294. [DOI] [PubMed] [Google Scholar]
- Drlica K.; Zhao X. (1997) DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61, 377–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H.; Kojima T.; Yamagishi J.; Nakamura S. (1988) Quinolone-resistant mutations of the gyrA gene of Escherichia coli. Mol. Gen. Genet. 211, 1–7. 10.1007/BF00338386. [DOI] [PubMed] [Google Scholar]
- Cullen M. E.; Wyke A. W.; Kuroda R.; Fisher L. M. (1989) Cloning and characterization of a DNA gyrase A gene from Escherichia coli that confers clinical resistance to 4-quinolones. Antimicrob. Agents Chemother. 33, 886–894. 10.1128/AAC.33.6.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Deguchi T.; Yasuda M.; Kawamura T.; Kanematsu E.; Nishino Y.; Ishihara S.; Kawada Y. (1998) Alteration in the GyrA subunit of DNA gyrase and the ParC subunit of DNA topoisomerase IV in quinolone-resistant clinical isolates of Staphylococcus epidermidis. Antimicrob. Agents Chemother. 42, 3293–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uivarosi V. (2013) Metal complexes of quinolone antibiotics and their applications: an update. Molecules 18, 11153–11197. 10.3390/molecules180911153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; McPherson S. A.; Wang P.; Kerns R. J.; Graves D. E.; Turnbough C. L.; Osheroff N. (2012) Drug interactions with Bacillus anthracis topoisomerase IV: biochemical basis for quinolone action and resistance. Biochemistry 51, 370–381. 10.1021/bi2013905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; McPherson S. A.; Turnbough C. L.; Kerns R. J.; Osheroff N. (2013) Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: mechanistic basis of quinolone resistance. Nucleic Acids Res. 41, 4628–4639. 10.1093/nar/gkt124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Breland E. J.; Vlčková V.; Strub M. P.; Neuman K. C.; Kerns R. J.; Osheroff N. (2014) Role of the water-metal ion bridge in mediating interactions between quinolones and Escherichia coli topoisomerase IV. Biochemistry 53, 5558–5567. 10.1021/bi500682e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Blower T. R.; Kerns R. J.; Berger J. M.; Osheroff N. (2016) Fluoroquinolone interactions with Mycobacterium tuberculosis gyrase: enhancing drug activity against wild-type and resistant gyrase. Proc. Natl. Acad. Sci. U. S. A. 113, E839–E846. 10.1073/pnas.1525055113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower T. R.; Williamson B. H.; Kerns R. J.; Berger J. M. (2016) Crystal structure and stability of gyrase-fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 113, 1706–1713. 10.1073/pnas.1525047113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S. T.; Brosch R.; Parkhill J.; Garnier T.; Churcher C.; Harris D.; Gordon S. V.; Eiglmeier K.; Gas S.; Barry C. E. 3rd; Tekaia F.; Badcock K.; Basham D.; Brown D.; Chillingworth T.; Connor R.; Davies R.; Devlin K.; Feltwell T.; Gentles S.; Hamlin N.; Holroyd S.; Hornsby T.; Jagels K.; Krogh A.; McLean J.; Moule S.; Murphy L.; Oliver K.; Osborne J.; Quail M. A.; Rajandream M. A.; Rogers J.; Rutter S.; Seeger K.; Skelton J.; Squares R.; Squares S.; Sulston J. E.; Taylor K.; Whitehead S.; Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Bouige A.; Darmon A.; Piton J.; Roue M.; Petrella S.; Capton E.; Forterre P.; Aubry A.; Mayer C. (2013) Mycobacterium tuberculosis DNA gyrase possesses two functional GyrA-boxes. Biochem. J. 455, 285–294. 10.1042/BJ20130430. [DOI] [PubMed] [Google Scholar]
- Dong S.; McPherson S. A.; Wang Y.; Li M.; Wang P.; Turnbough C. L. Jr.; Pritchard D. G. (2010) Characterization of the enzymes encoded by the anthrose biosynthetic operon of Bacillus anthracis. J. Bacteriol. 192, 5053–5062. 10.1128/JB.00568-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Schwanz H. A.; Li G.; McPherson S. A.; Turnbough C. L.; Kerns R. J.; Osheroff N. (2013) Overcoming target-mediated quinolone resistance in topoisomerase IV by introducing metal-ion-independent drug–enzyme interactions. ACS Chem. Biol. 8, 2660–2668. 10.1021/cb400592n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palu G.; Valisena S.; Ciarrocchi G.; Gatto B.; Palumbo M. (1992) Quinolone binding to DNA is mediated by magnesium ions. Proc. Natl. Acad. Sci. U. S. A. 89, 9671–9675. 10.1073/pnas.89.20.9671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmott C. J.; Maxwell A. (1993) A single point mutation in the DNA gyrase A protein greatly reduces binding of fluoroquinolones to the gyrase-DNA complex. Antimicrob. Agents Chemother. 37, 126–127. 10.1128/AAC.37.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchlow S. E.; Maxwell A. (1996) DNA cleavage is not required for the binding of quinolone drugs to the DNA gyrase-DNA complex. Biochemistry 35, 7387–7393. 10.1021/bi9603175. [DOI] [PubMed] [Google Scholar]
- Kumar R.; Madhumathi B. S.; Nagaraja V. (2014) Molecular basis for the differential quinolone susceptibility of mycobacterial DNA gyrase. Antimicrob. Agents Chemother. 58, 2013–2020. 10.1128/AAC.01958-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osheroff N.; Zechiedrich E. L. (1987) Calcium-promoted DNA cleavage by eukaryotic topoisomerase II: trapping the covalent enzyme-DNA complex in an active form. Biochemistry 26, 4303–4309. 10.1021/bi00388a018. [DOI] [PubMed] [Google Scholar]
- Barnard F. M.; Maxwell A. (2001) Interaction between DNA gyrase and quinolones: effects of alanine mutations at GyrA subunit residues Ser(83) and Asp(87). Antimicrob. Agents Chemother. 45, 1994–2000. 10.1128/AAC.45.7.1994-2000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese J. E.; Burgin A. B.; Osheroff N. (2008) Human topoisomerase IIα uses a two-metal-ion mechanism for DNA cleavage. Nucleic Acids Res. 36, 4883–4893. 10.1093/nar/gkn466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts S. L.; Liou G. F.; Mitchenall L. A.; Burgin A. B.; Maxwell A.; Neuman K. C.; Osheroff N. (2011) Use of divalent metal ions in the DNA cleavage reaction of topoisomerase IV. Nucleic Acids Res. 39, 4808–4817. 10.1093/nar/gkr018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X. S.; Gould K. A.; Fisher L. M. (2009) Probing the differential interactions of quinazolinedione PD 0305970 and quinolones with gyrase and topoisomerase IV. Antimicrob. Agents Chemother. 53, 3822–3831. 10.1128/AAC.00113-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drlica K.; Mustaev A.; Towle T. R.; Luan G.; Kerns R. J.; Berger J. M. (2014) Bypassing fluoroquinolone resistance with quinazolinediones: studies of drug-gyrase-DNA complexes having implications for drug design. ACS Chem. Biol. 9, 2895–2904. 10.1021/cb500629k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble C. G.; Maxwell A. (2002) The role of GyrB in the DNA cleavage-religation reaction of DNA gyrase: a proposed two metal-ion mechanism. J. Mol. Biol. 318, 361–371. 10.1016/S0022-2836(02)00049-9. [DOI] [PubMed] [Google Scholar]
- Schmidt B. H.; Burgin A. B.; Deweese J. E.; Osheroff N.; Berger J. M. (2010) A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature 465, 641–644. 10.1038/nature08974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese J. E.; Osheroff N. (2010) The use of divalent metal ions by type II topoisomerases. Metallomics 2, 450–459. 10.1039/c003759a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi P.; Tegnell A.; Baka A.; Van Loock F.; Hendriks J.; Werner A.; Maidhof H.; Gouvras G. (2004) Bichat guidelines for the clinical management of anthrax and bioterrorism-related anthrax. Eurosurveillance 9, 1–7. [DOI] [PubMed] [Google Scholar]
- Waterer G. W.; Robertson H. (2009) Bioterrorism for the respiratory physician. Respirology 14, 5–11. 10.1111/j.1440-1843.2008.01446.x. [DOI] [PubMed] [Google Scholar]