

Abstract
Signaling by mTOR complex 1 (mTORC1) promotes anabolic cellular processes in response to growth factors, nutrients, and hormonal cues. Numerous clinical trials employing the mTORC1 inhibitor rapamycin (aka sirolimus) to immuno-suppress patients following organ transplantation have documented the development of hypertriglyceridemia and elevated serum free fatty acids (FFA). We therefore investigated the cellular role of mTORC1 in control of triacylglycerol (TAG) metabolism using cultured murine 3T3-L1 adipocytes. We found that treatment of adipocytes with rapamycin reduced insulin-stimulated TAG storage ~50%. To determine whether rapamycin reduces TAG storage by upregulating lipolytic rate, we treated adipocytes in the absence and presence of rapamycin and isoproterenol, a β2-adrenergic agonist that activates the cAMP/protein kinase A (PKA) pathway to promote lipolysis. We found that rapamycin augmented isoproterenol-induced lipolysis without altering cAMP levels. Rapamycin enhanced the isoproterenol-stimulated phosphorylation of hormone sensitive lipase (HSL) on Ser-563 (a PKA site), but had no effect on the phosphorylation of HSL S565 (an AMPK site). Additionally, rapamycin did not affect the isoproterenol-mediated phosphorylation of perilipin, a protein that coats the lipid droplet to initiate lipolysis upon phosphorylation by PKA. These data demonstrate that inhibition of mTORC1 signaling synergizes with the β-adrenergic-cAMP/PKA pathway to augment phosphorylation of HSL to promote hormone-induced lipolysis. Moreover, they reveal a novel metabolic function for mTORC1; mTORC1 signaling suppresses lipolysis, thus augmenting TAG storage.
Keywords: mTOR, mTORC1, Rapamycin, Lipid metabolism, Lipolysis, Adipocytes
Introduction
The mammalian target of rapamycin (mTOR), an evolutionarily conserved Ser/Thr protein kinase, coordinates a signal transduction network that controls a plethora of fundamental cellular functions [1–3]. mTOR forms the catalytic core of at least two functionally distinct signaling complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [3]. The better-understood complex, mTORC1, functions as an environmental sensor to promote anabolic cellular processes such as ribosomal gene transcription, protein synthesis, lipid synthesis, cell growth/size, and cell proliferation in response to sufficient levels of growth factors, nutrients, and hormones [2, 3]. Conversely, adverse cellular conditions rapidly downregulate mTORC1 signaling to promote catabolic functions such as autophagy [4]. mTORC1 and mTORC2 contain shared and unique partner proteins, possess unique cellular functions, and exhibit differing sensitivities to rapamycin, a clinically employed immunosuppressive drug [3]. Rapamycin (clinically known as sirolimus) associates with a cellular protein called FKBP12 [5]. Upon entering the cell, the rapamycin/FKBP12 complex directly binds to the mTOR FRB (FKBP12-rapamycin binding) domain, which lies immediately N-terminal to the kinase domain [5]. Rapamycin/FKBP12 binds assembled mTORC1 but not assembled mTORC2 [5]. Thus, rapamycin acutely inhibits mTORC1 but not mTORC2 signaling. While the mechanisms by which rapamycin allosterically inhibits mTORC1 signaling remain incompletely defined, the drug reduces the affinity between mTOR and raptor, a critical mTORC1 partner protein [5, 6]. Additionally, rapamycin reduces mTORC1 intrinsic kinase activity, as monitored by mTOR S2481 autophosphorylation [7]. The use of rapamycin in clinical medicine underscores the importance of mTORC1 for organismal physiology. Not only does rapamycin reduce organ transplant rejection [8], it reduces the intimal de-differentiation and hyperplasia of vascular smooth muscle cells that often follows coronary artery stent restenosis [9]. Strikingly, rapamycin extends rodent lifespan [10]. Rapamycin analogs and second-generation mTOR catalytic inhibitors also hold therapeutic promise as anti-cancer agents [5].
Rapamycin administration incurs adverse side effects, however. Various clinical trials have documented the development of hypertriglyceridemia or hypercholesterolemia in renal, hepatic, cardiac, and islet transplant patients [11, 12]. Additionally, rapamycin has also been shown to elevate serum free fatty acid (FFA) levels and induce glucose intolerance [13] in transplant patients as well as in mice lacking the mTORC1 downstream target S6K1 [14]. Clinical trials have also documented the occurrence of hyperlipidemia upon the administration of the rapamycin analog CCI-779 (also known as temsirolimus; Wyeth) in the treatment of metastatic melanoma and glioblastoma multiforme [15]. Consistent with rapamycin increasing levels of circulating FFAs and inducing hyperlipidemia, mice lacking S6K1 globally or lacking raptor in adipose are lean with reduced adipose mass and exhibit resistance to diet-induced obesity [14]. Thus, mTORC1 inhibition induced by pharmacological or genetic ablation disrupts lipid homeostasis in vivo.
In this study we set out to better understand how mTORC1 signaling impacts lipid metabolism in cultured adipocytes. Adipose tissue, once considered an inert energy storage depot, is now recognized as an important metabolic and endocrine organ that coordinates energy intake and expenditure critical for energy homeostasis [16]. Adipose tissue removes glucose from the circulation (via the action of the insulin-responsive glucose transporter GLUT4) and stores surplus energy in the form of triacylglycerol (TAG) within lipid droplets. The tissue releases energy as needed via the action of lipolysis, the process by which enzymes known as lipases break down TAG into FFA and glycerol, which are then released into the circulation. Diverse extracellular stimuli regulate lipolysis. Catecholamines (e.g., epinephrine) and glucocorticoids (e.g., Cortisol) promote lipolysis while insulin and FFA themselves suppress lipolysis [17]. Starvation or stress triggers the release of catecholamines, which promote lipolysis by activating G protein-coupled adrenergic receptors that activate adenylate cyclase (AC), which lead to increased production of cAMP and activation of protein kinase A (PKA). Several laboratories have established that PKA phosphorylates two lipolytic proteins, perilipin and hormone sensitive lipase (HSL) [18]. Perilipin functions as a master regulator of lipolysis [19, 20]. Under basal conditions, perilipin coats and protects lipid storage droplets. Upon agonist stimulation, PKA phosphorylates perilipin A, which induces a conformational change in the perilipin coating that enables the recruitment of HSL from the cytoplasm to the surface of the lipid droplet [21, 22]. Indeed, perilipin knockout mice have reduced adipose tissue mass and exhibit elevated basal lipolysis with resistance to β-adrenergic-stimulated lipolysis; moreover, the mice display resistance to diet-induced and genetic obesity [18, 22–24].
Our previous work demonstrated that rapamycin administration induces hypertriglyceridemia in organ transplant patients [11], and increases TAG and plasma FFA in guinea pigs [25]. Here, we test the hypothesis that rapamycin promotes TAG lipolysis in cultured murine 3T3-L1 adipocytes. We found that inhibition of mTORC1 signaling with rapamycin augments lipolysis promoted by the β-adrenergic agonist isoproterenol without augmenting cellular cAMP levels. Additionally, we found that rapamycin enhances isoproterenol-induced phosphorylation of HSL. These data indicate that mTORC1 inhibition synergizes with the PKA pathway to promote lipolysis by modulating HSL function. Improved understanding of how mTORC1 inhibition promotes lipolysis to disrupt lipid metabolism may assist clinicians in the future to better handle the deleterious side effects incurred by therapeutic rapamycin treatment during immunosuppression, cancer chemotherapy, or following coronary artery angioplasty.
Experimental Procedures
Materials
Reagents were obtained from the following sources: CHAPS (3-[(3-cholamidopropyl)- dimethylammonio]-1-propanesulfonate) was from Pierce (Rockford, IL, USA); nitrocellulose membrane (0.45 microns) was from Schleicher and Schuell Bioscience Inc. (Keene, NH, USA); autoradiography film (HyBlot CL) was from Denville Scientific Inc. (Metuchen, NJ, USA); reagents for enhanced chemiluminescence (ECL) (Immobilon Western-Chemiluminescent HRP Substrate) were from Upstate/Millipore (Billerica, MA, USA); all standard chemicals were from either Fisher Chemicals (Pittsburgh, PA, USA), Calbiochem/EMD Chemicals (Gibbstown, NJ, USA), or Sigma-Aldrich (St. Louis, MI, USA).
Antibodies
The following antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA): P-S6K1-T389 (rabbit monoclonal 108D2); P-mTOR-S2481, HSL, ATGL, MGL. The perilipin antibody was from Chemicon/Millipore (Billerica, MA, USA) (#AB 10200), and the β-actin monoclonal antibody was from Sigma-Aldrich (St. Louis, MI, USA). Antibodies to detect the phosphorylation of PKA on its regulatory subunits RIIα (S96) and RIIβ (S114) were from Upstate/Millipore (Billerica, MA, USA). Custom, affinity-purified anti-peptide antibodies against mTOR and S6K1 were generated in rabbits, as described [7]. Sheep anti-mouse antibodies and donkey anti-rabbit HRP secondary antibodies were from GE-Healthcare (Piscataway, NJ, USA).
Differentiation of 3T3-L1 Fibroblasts into Adipocytes
3T3-L1 fibroblasts (kindly provided by Dr. Ormond Mac-Dougald; University of Michigan Medical School) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) that contained high glucose (4.5 g/L), glutamine (584 mg/L), and sodium pyruvate (110 mg/L) supplemented with 10% newborn calf serum (NCS) (Gibco/Invitrogen; Carlsbad, CA, USA) and maintained at 37 °C in 5% C02. 3T3-L1 fibroblasts were differentiated into adipocytes by a standard protocol. Briefly, differentiation was induced 2 days post confluency by the addition of DMEM containing 10% fetal bovine serum (FBS) (Hyclone) and dexamethasone (1 μM) (Sigma-Aldrich; St. Louis, MO, USA), 3-isobutyl-1-methylxanthine IBMX (0.5 mM) (Sigma-Aldrich; St. Louis, MI, USA) and insulin (100 nM) (Gibco/Invitrogen; Carlsbad, CA, USA) for 2 days. The cells were further incubated with DMEM/FBS supplemented with insulin for an additional 2 days. Fully differentiated adipocytes were maintained in DMEM/FBS at 37 °C in a humidified atmosphere containing 5% CO2 and used 9–14 days after the initiation of differentiation.
Cell Culture, Drug Treatment, Cell Lysis and Western Immunoblotting
3T3-L1 adipocytes were serum deprived for 20 h via incubation in DMEM supplemented with 20 mM Hepes (pH 7.2). Following serum deprivation, adipocytes were drug treated for 30 min by the addition of rapamycin (20 ng/mL) (22 nM) (Calbiochem/EMD Chemicals; Gibbstown, NJ, USA), or wortmannin (100 nM) (Upstate/Millipore; Billerica, MA, USA) prior to the addition of the β-adrenergic agonist isoproterenol (10 μM) (Chemicon/Millipore; Billerica, MA, USA) unless indicated otherwise, insulin (100 nM) (Invitrogen; Carlsbad, CA, USA), or both for 30 min. For cell lysis, adipocytes were washed 29 with ice cold PBS [pH 7.4] and collected in ice cold lysis Buffer A (10 mM KPO4 [pH 7.2]; 1 mM EDTA; 5 mM EGTA; 10 mM MgCl2; 50 mM β-glycerophosphate; 1 mM sodium orthovanadate (Na3V04); 5 μg/mL pepstatin A; 10 μg/mL leupeptin; 40 μg/mL PMSF) containing the detergent CHAPS (0.3%). Lysates were spun at 13,200 rpm for 5 min at 4 °C, and the post-nuclear supernatants were collected. Bradford assay was used to normalize protein levels for the measurement of triacylglycerol content, lipolysis, cAMP levels, and Western immunoblot analyses. For immunoblots, whole cell lysates were resuspended in 19 sample buffer (50 mM Tris–HCl pH 6.8, 10% glycerol, 2% SDS, 2% β-mercaptoethanol, 0.02% bromophenol blue). Samples were heated at 95 °C for 5 min, resolved on SDS–PAGE gels, and transferred to nitrocellulose membrane using Towbin transfer buffer (25 mM Tris; 192 mM glycine; 10% methanol; 0.02% SDS). Immunoblotting was performed by blocking membranes in TBST (40 mM Tris–HCl pH 7.5; 0.9% NaCl; 0.1% Tween-20) containing 3% nonfat milk and incubating the membranes in TBST with 2% BSA containing primary antibodies or secondary HRP-conjugated antibodies. Blots were developed by enhanced chemiluminescence (ECL).
Measurement of Cellular Triacylglycerol Content
Adipocyte triacylglycerol concentrations were measured by enzymatic methods following manufacturer instructions (Wako Chemicals, Inc.; Richmond, VA, USA) following cellular lysis and normalization of protein content. Briefly, samples and a set of triolein standards were incubated with triacylglycerol reagent A at 37 °C for 5 min followed by addition of triacylglycerol reagent B for 5 min at 37 °C. The colorimetric reaction was then measured at wavelength 590 nm and expressed as mg/mg protein.
Lipolysis Assays
Following serum deprivation and drug treatment, differentiated 3T3-L1 adipocytes were stimulated with isoproterenol for either 1 h or 4 h, as indicated in the figure legends. The media was collected and analyzed for free fatty acid release in the culture media using a NEFA C assay (Wako Chemicals; Richmond, VA, USA) and read by a spectrophotometer at wavelength 540 nm. Free fatty acid release was measured in millimole per microgram protein (mmol/ug) and expressed as relative free fatty acid percent. Glycerol release in the media was measured by colorimetric assay at wavelength 540 nm using a Chemicon lipolysis kit, following the manufacturer’s instructions (Chemicon/Millipore; Billerica, MA, USA).
Measurement of Cellular cAMP Levels
Following serum deprivation, drug treatment, and isoproterenol stimulation, cAMP levels were measured using a HitHunter cAMP EFC (enzyme fragment complementation) assay (Amersham/GE-Healthcare; Piscataway, NJ, USA) following the manufacturer’s instructions. All samples and standards were run in triplicates, and luminescence was detected using a multimode microplate reader (PerkinElmer, VICTOR 3; Waltham, MA, USA).
Statistics
ANOVA (Analysis of Variance) was used to determine overall statistical significance among various treatment groups. If significance was achieved at P < 0.05, then post-hoc secondary LSD (Least Significant Difference) tests were performed.
Results
Insulin-Mediated Activation of mTORC1 Promotes Triacylglycerol Accumulation in 3T3-L1 Adipocytes
To determine whether mTORC1 signaling controls triacylglycerol (TAG) content in adipocytes, we differentiated 3T3-L1 fibroblasts into adipocytes and treated them with insulin in the absence or presence of rapamycin for 18 h after a period of serum deprivation. As expected, chronic insulin treatment promoted triacylglycerol storage (Fig. 1a). We found that rapamycin treatment reduced insulin-stimulated TAG accumulation ~ 50% in a statistically significant manner (P< 0.05), thus demonstrating that mTORC1 signaling is required for insulin-stimulated TAG storage. To confirm that insulin mediated the activation of mTORC1 and downstream signaling in a rapamycin-sensitive manner in this experiment, we immunoblotted whole cell lysates with mTOR P-S2481 and S6K1 P-T389 antibodies (Fig. 1b). Upon activation, mTOR autophosphorylates on S2481; thus, mTOR P-S2481 immunoblotting monitors intrinsic mTOR catalytic activity [7]. Activated mTORC1 then directly phosphorylates S6K1 on T389 [26]; thus, S6K1 P-T389 immunoblotting monitors mTORC1 downstream signaling. As rapamycin inhibited insulin-stimulated autophosphorylation of mTOR on S2481 and the phosphorylation of S6K1 on T389, these data indicate that rapamycin indeed inhibited the insulin-stimulated activation of mTORC1.
Fig. 1.

Insulin-stimulated triacylglycerol (TAG) accumulation in 3T3-L1 adipocytes requires mTORC1 signaling. a 3T3-L1 adipocytes were serum-deprived for 20 h (Control) and then pre-treated without or with rapamycin (20 ng/mL) for 30 min. Cells were stimulated without (Control) or with insulin (INS) (100 nM) in the absence (R) or continued presence of rapamycin (R + INS) for 18 h. Whole cell lysates were analyzed for triacylglycerol content (mg/mg protein) via a colorimetric assay (see “Experimental Procedures”) after normalization for protein content. The graph represents the mean ± SD of triplicate samples from one experiment but is representative of three independent experiments; thus, each bar represents n = 3. Differences between treatment groups were analyzed using ANOVA, and differences were considered statistically different at a level of 0.05. Bars with different letters are statistically significant. b The whole cell lysates analyzed in (a) were resolved on SDS–PAGE and immunoblotted with the indicated antibodies to confirm the expected activation and/or inhibition of mTORC1 signaling
Rapamycin Augments β-Adrenergic-Stimulated Lipolysis in 3T3-L1 Adipocytes
As we found that rapamycin-mediated inhibition of mTORC1 signaling reduces cellular TAG content, we next asked whether rapamycin reduces TAG content by promoting lipolytic rate. As the insulin/PI3K pathway promotes mTORC1 signaling and also suppresses lipolysis (via Akt-mediated activation of phosphodiesterase [PDE], an enzyme that hydrolyzes cAMP to AMP) [27], we measured adipocyte lipolysis in the absence of serum growth factors in order to study lipolysis independently of PDE action. To stimulate lipolysis, we employed the catecholamine isoproterenol, a synthetic β2-adrenergic agonist. By treating serum deprived 3T3-L1 adipocytes with isoproterenol in the absence or presence of rapamycin, we found that inhibition of mTORC1 signaling augments isoproterenol stimulated lipolysis in a statistically significant manner, as measured by the release of FFA (Fig. 2a) into the cell culture media after 1 h of incubation. Based on our preliminary kinetic data (data not shown) as well as reported literature [28], a 1-h time point was optimal for measurements of glycerol release (Fig. 2b) while a 4-h time point was optimal for measurements of FFA release. We thus next measured FFA release utilizing a 4-h time point to determine whether mTORC1 inhibition with rapamycin augments basal lipolysis, whether insulin suppresses isoproterenol-stimulated lipolysis, and whether mTORC1 inhibition using the PI3K inhibitor wortmannin augments isoproterenol-stimulated lipolysis (similar to rapamycin). We found that rapamycin had no effect on basal lipolysis after 4 h of incubation, while, insulin indeed suppressed isoproterenol-stimulated lipolysis, as expected (Fig. 2c). Interestingly, mTORC1 inhibition with rapamycin rescued the suppression of lipolysis caused by insulin, suggesting a novel role for mTORC1 in suppression of lipolysis via the insulin/Akt/PDE pathway (Fig. 2c). Similar to rapamycin, wortmannin treatment augmented isoproterenol-stimulated lipolysis, suggesting a novel role for PI3K as well as mTORC1 in suppression of lipolysis (Fig. 2c). The finding that inhibition of PI3K with wortmannin phenocopies rapamycin treatment to augment isoproterenol-stimulated lipolysis is consistent with the well-known regulation of mTORC1 by PI3K [3]. These data suggest that the PI3K/mTORC1 pathway mediates lipolytic suppression.
Fig. 2.

Inhibition of mTORC1 with rapamycin augments β-adrenergic-activated lipolysis in 3T3-L1 adipocytes. a, b 3T3-L1 adipocytes were serum-deprived for 20 h and then pre-treated without or with rapamycin (R) (20 ng/mL) for 30 min. Cells were then incubated in the absence (Basal) or presence of isoproterenol (Iso) (10 μM) for 1 h in the absence or continued presence of rapamycin (Iso + R). Relative free fatty acid (FFA) release was measured in (a) and relative glycerol release was measured in (b). In graphs (a, b) each bar represents the mean value ± SD of 5 samples from three independent experiments; thus, each bar represents n = 5. The value from cells treated with Iso alone was set to 100%, and all other values were normalized to this value. c 3T3-L1 adipocytes were treated similarly as in (a, b) with minor modifications, and relative free fatty acid (FFA) release was measured, as in (a). Briefly, serum-deprived cells were pre-treated without or with rapamycin (20 ng/mL) or wortmannin (100 nM) for 30 min. Cells were then incubated in the absence (Basal lipolysis) or presence of isoproterenol (10 μM) for 4 h without (Iso) or with insulin (100 nM) (Iso + INS), rapamycin (Iso + R), wortmannin (Iso + W), or both insulin and rapamycin (Iso + INS + R), as indicated in the graph. Cells incubated in the absence of isoproterenol (Basal lipolysis) were also incubated with insulin (INS), rapamycin (R), wortmannin (W), and insulin and rapamycin (INS + R) for 4 h. In graph (c), each bar represents the mean value ± SD of nine samples from three independent experiments; thus, each bar represents n= 9. As in graphs (a, b), the value of Iso alone was set to 100%, and all other values were normalized to this value. Differences between treatment groups were analyzed using ANOVA, and differences were considered statistically different at a level of 0.05. Bars with different letters are statistically significant
Rapamycin Does Not Modulate Cellular cAMP Levels
To begin to understand the mechanism by which mTORC1 inhibition augments isoproterenol-stimulated lipolysis, we determined whether rapamycin augments isoproterenol-stimulated production of cAMP. Studies in yeast have shown that TOR may function upstream of PKA [29], thus lending support to such a hypothesis. We thus measured cellular levels of cAMP in adipocytes stimulated with isoproterenol in the absence or presence of rapamycin for 15 min (Fig. 3a) and 30 min (Fig. 3b). We found that rapamycin treatment had no effect on cAMP levels during basal or catecholamine-stimulated lipolysis. Thus, these data suggest that mTORC1 inhibition does not augment catecholamine-stimulated lipolysis by leading to increased levels of cAMP.
Fig. 3.

Rapamycin does not augment cAMP levels over β-adrenergic agonist alone. cAMP levels were measured using the HitHunter cAMP EFC chemiluminescent detection assay (Amersham, Inc.), according to manufacturer’s directions. Briefly, 3T3-L1 adipocytes were serum deprived 20 h and pre-treated without or with rapamycin (R) (20 ng/mL) for 30 min. Cells were then incubated in the absence (DMEM) or presence of isoproterenol (Iso) (1 μM or 10 μM) for 15 min (a) or (10 μM) 30 min (b) in the absence or presence of rapamycin (Iso + R). “No ED” (without enzyme donor) and “PBS” represent negative controls for this assay. Bars with different letters are statistically significant at a level of 0.05
Rapamycin Does Not Affect PKA Phosphorylation
To begin to study the regulation of PKA activity in intact cells, we investigated the phosphorylation of PKA on its regulatory subunits, RIIα (S96) and RIIβ (S114), utilizing phospho-specific antibodies. Studies have shown that autophosphorylation of S96 on the regulatory subunit RIIα promotes local PKA activity in cardiac muscle [30]. Additionally, point mutation of the S114 autophosphorylation site on RIIβ has been shown to block the nuclear localization of PKA and to inhibit PKA function in T cells both in vitro [31] and in vivo [32]. We found that isoproterenol-stimulated PKA phosphorylation on regulatory subunit RIIα (S96) or regulatory subunit RIIβ (S114) was not altered by rapamycin treatment at various isoproterenol concentrations (Fig. 4a). While by no means conclusive, these data suggest that mTORC1 inhibition may promote isoproterenol-stimulated lipolysis downstream of PKA. Alternatively, it remains possible that mTORC1 inhibition inhibits PKA activity via a mechanism independent of regulatory subunit phosphorylation.
Fig. 4.

Rapamycin augments isoproterenol-stimulated phosphorylation of HSL on S563 in the absence of effects on PKA regulatory subunit or perilipin phosphorylation, a Rapamycin does not modulate the phosphorylation of PKA on the regulatory subunits RIIα (S196) or RIIβ (S114): 3T3-L1 adipocytes were serum-deprived for 20 h and then pre-treated without (−) or with (+) rapamycin (20 ng/mL) for 30 min. Cells were incubated in the absence (−) or presence of various concentrations of isoproterenol (Iso) ranging from 10 μM to 1 nM for 15 min, as indicated. After lysis, whole cell lysate was resolved on SDS–PAGE and immunoblotted with the indicated antibodies. b Rapamycin does not augment isoproterenol-stimulated phosphorylation of perilipin: 3T3-L1 adipocytes were serum-deprived for 20 h and pre-treated without (−) or with (+) rapamycin (Rapa) (20 ng/mL) for 30 min. Cells were then incubated in the absence (−) or presence (+) of isoproterenol (Iso) (10 μM) in the absence or presence of rapamycin for 15 min. After lysis, whole cell lysate was resolved on SDS–PAGE and immunoblotted with perilipin antibodies. Note: To observe perilipin electrophoretic mobility shift, a measure of perilipin phosphorylation, a 21 cm long 8% gel was run for 3:30 h. To visualize total perilipin, a 13-cm long 12% gel was run for 1:45 h. c Rapamycin augments isoproterenol-stimulated phosphorylation of HSL on S563 (a PKA phosphorylation site) but not on S565 (an AMPK phosphorylation site): Upper panel: Schematic of the HSL sites phosphorylated by PKA and AMPK Lower panel: 3T3-L1 adipocytes were serum-deprived and pre-treated without (−) or with (+) rapamycin, as in (a, b) above. Cells were then incubated in the absence (−) or presence of various concentrations of isoproterenol (Iso) ranging from 10 μM to 1 nM for 15 min, as indicated. After lysis, whole cell lysates were resolved on SDS–PAGE and immunoblotted with the indicated antibodies. Cells were also lysed immediately after serum deprivation (Basal). Two P-HSL-S563 film exposures are shown (SE-short exposure, LE-long exposure)
Rapamycin Modestly Augments the Phosphorylation of HSL
To determine whether PKA signaling and mTORC1 inhibition synergistically modulate the activation state of lipolytic targets, we assayed the phosphorylation of two well known PKA dependent targets, hormone sensitive lipase (HSL) and perilipin. In response to starvation and stress, catecholamine induced activation of the cAMP/PKA pathway leads to PKA-mediated phosphorylation of both perilipin and HSL [23]. PKA-induced phosphorylation of perilipin on at least 6 sites triggers a conformational change that disassembles the perilipin protective coat and induces the translocation of HSL from the cytoplasm to the surface of lipid droplet, whereby HSL docks to phosphorylated perilipin [23, 33]. To determine whether rapamycin augments isoproterenol-stimulated phosphorylation of perilipin, we employed a commonly used SDS–PAGE mobility shift assay to determine the phosphorylation state of perilipin. Serum deprived 3T3-L1 adipocytes were pretreated with rapamycin and then stimulated in the absence of isoproterenol or with a series of isoproterenol concentrations ranging from maximal to sub-maximal (10 μM down to 1 nM). As expected, isoproterenol strongly promoted the phosphorylation of perilipin in a dose-dependent manner, as shown by decreased electrophoretic mobility on SDS–PAGE (Fig. 4b). Rapamycin treatment had no effect on isoproterenol-induced perilipin phosphorylation, even at sub-maximal doses (0.1 μM, 10 nM). At the limited resolution of this assay, these data suggest that mTORC1 inhibition with rapamycin does not synergize with catecholamine induced PKA signaling to promote significant phosphorylation of perilipin. Although mobility shift is a commonly used method in detection of perilipin phosphorylation state, we acknowledge the possibility that rapamycin could promote the phosphorylation of individual sites on perilipin whose phosphorylation state may not be measurable by mobility shift. We next investigated the phosphorylation of the lipase HSL, which functions as the rate-limiting enzyme in the lipolytic cascade. To date, three PKA-mediated phosphorylation sites (Ser563; Ser659; Ser660) and one AMPK-mediated phosphorylation site (Ser565) have been identified on rat HSL, as illustrated in Fig. 4c [34, 35]. Phosphorylation of HSL on Ser563 by PKA is thought to promote the intrinsic catalytic activity of HSL [36], while phosphorylation of HSL on Ser659 and Ser660 by PKA are thought to promote the translocation of cytosolic HSL to the surface of the lipid droplet [23]. Conversely, phosphorylation of HSL on Ser565 by AMPK is thought to inhibit HSL activity [37]. By treating 3T3-L1 adipocytes with various concentrations of isoproterenol in the absence and presence of rapamycin, we found that rapamycin augmented HSL Ser563 phosphorylation (Fig. 4c). On the contrary, there was no additional effect of rapamycin on isoproterenol-stimulated phosphorylation of HSL Ser660 (Fig. 4c). Neither isoproterenol nor rapamycin modulated AMPK Ser565 phosphorylation, which appears to be constitutively activated under basal conditions [37]. Taken together, these data indicate that mTORC1 inhibition with rapamycin synergizes with PKA signaling to augment the phosphorylation of HSL on Serine 563, which may accelerate lipolysis by augmenting HSL catalytic activity.
Rapamycin Has No Effect on ATGL, HSL, or MGL Protein Expression
To determine whether mTORC1 inhibition with rapamycin additionally promotes lipolysis by modulating the expression of the lipases critical for the breakdown of TAG into FFAs and glycerol, we examined the protein expression of adipose triacylglycerol lipase (ATGL), hormone sensitive lipase (HSL), and monoacyl lipase (MGL). It is currently believed that ATGL initiates basal as well as hormone sensitive lipolysis of triacylglycerol [38, 39] to FFA and diacylglycerol. Subsequently, HSL acts on diacylglycerol particles to release FFA and monoacylglycerol. The final step in lipolysis is catalyzed by the hormone-insensitive MGL, which acts on monoacylglycerol to release FFA and glycerol [39]. Some investigators [40] also suggest a role for triacylglycerol hydrolase (TGH) in lipolysis; the relative contribution of TGH in the lipolytic cascade within the adipocyte remains unclear, however. We treated 3T3-L1 adipocytes with isoproterenol in the absence or presence of rapamycin for 18 h and measured the protein expression of ATGL, HSL, and MGL by immunoblot (Fig. 5). We found no differences in the abundance of these 3 important lipases under any treatment condition. Thus, these data indicate that mTORC1 inhibition with rapamycin does not augment lipolytic rate by increasing the expression of a critical lipase.
Fig. 5.
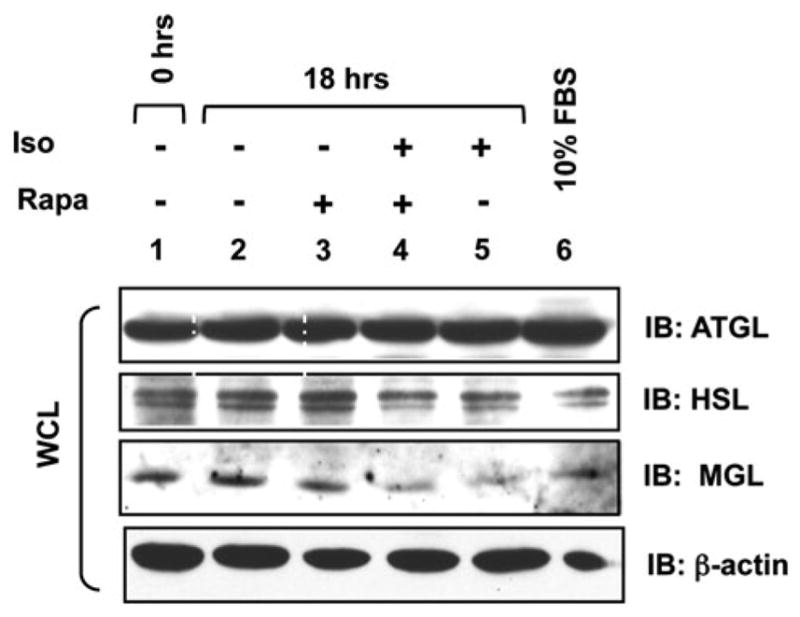
Rapamycin does not modulate the protein expression of ATGL, HSL, or MGL. 3T3-L1 adipocytes were serum-deprived for 20 h (lanes 1–5) and pre-treated without (lanes 1–2, 5) or with (lanes 3, 4) rapamycin (R) (20 ng/mL) for 30 min. Cells were then incubated in the absence (−) (lanes 2–3) or presence (+) (lanes 4–5) of isoproterenol (10 μM) for 18 h in the absence or continued presence of rapamycin, as indicated. In lane 1, cells were lysed after the 20 h serum deprivation period (t = 0). In lane 6, cells were cultured in DMEM/10% FBS (steady-state, non-starved conditions). After lysis, whole cell lysate was resolved on SDS-PAGE and immunoblotted with the indicated antibodies
Discussion
Here, we investigated the biochemical mechanism by which mTORC1 inhibition via the immunosuppressive drug rapamycin induces hypertriglyceridemia and increases circulating free FFAs in organ transplant patients [11] as well as in guinea pigs [25]. We report that treatment of 3T3-L1 adipocytes with the mTORC1 inhibitor rapamycin decreases TAG storage by augmenting lipolysis induced by isoproterenol, a β-adrenergic agonist, without altering basal lipolysis. While, the measured effect of rapamycin on hormone-induced lipolysis was modest, the combined effect of increased lipolysis and decreased TAG synthesis incurred by mTOR inhibition likely has a more significant impact on TAG metabolism. Our data further elucidate the mechanism by which mTORC1 inhibition cooperates with PKA signaling to promote lipolysis: We found that rapamycin augments isoproterenol-induced phosphorylation of HSL (on S563) via a mechanism that does not involve increased production of cAMP. These data indicate that mTORC1 inhibition and PKA signaling synergize either downstream of PKA or at the level of PKA to promote lipolysis (Fig. 6). Whether, mTORC1 inhibition and PKA converge on common lipolytic targets (e.g. HSL, ATGL) via parallel pathways or, whether mTORC1 inhibition converges on PKA itself to enhance its function (via increased enzymatic activity, altered subcellular localization, etc.) remains currently unknown (see model in Fig. 6). In support of the former model, TOR and PKA signal via parallel pathways in budding yeast to regulate the transcriptional expression of ribosomal protein genes and stress-responsive genes [41]. In support of the latter model, TORC1 signaling in budding yeast was reported recently to regulate PKA activity [42]. Lastly, we found that rapamycin treatment (18 h) had no effect on the expression of various lipases, including ATGL, HSL, or MGL under basal or isoproterenol-stimulated conditions.
Fig. 6.

Model for the role of mTORC1 signaling in suppression of lipolysis in adipocytes. The β-adrenergic agonist isoproterenol, a synthetic catecholamine, promotes triacylglycerol (TAG) breakdown (lipolysis) by activating a large heterotrimeric Gαs-protein that activates adenylate cyclase (AC), which converts ATP into cAMP. cAMP binds the regulatory subunits of protein kinase (PKA), thus dissociating the repressive regulatory subunits from the catalytic subunits. Catalytically active PKA phosphorylates downstream targets, including perilipin and hormone sensitive lipase (HSL), which contribute to initiation of lipolysis together with several other lipases including ATGL (adipose triacylglycerol lipase) and MGL (monoacyl lipase), ultimately leading to the breakdown of TAG into free fatty acids (FFA) and glycerol. mTORC1 signaling suppresses lipolysis by either acting on common lipolytic targets (e.g., HSL, others?) via a pathway parallel to PKA or by converging on PKA to inhibiting its function. By inhibiting mTORC1 signaling, rapamycin synergizes with the PKA pathway to promote lipolysis, thus reducing TAG storage; therefore, mTORC1 signaling suppresses lipolysis, thus augmenting TAG storage
Our data are consistent with the recent work of Chak-rabarti et al., who showed that inhibition of mTORC1 signaling via rapamycin or knockdown of the critical mTORC1 partner raptor decreases TAG storage by increasing lipolysis in 3T3-L1 adipocytes [43]. Consistently, this work demonstrated that overexpression of the mTORC1 activator Rheb augments TAG storage and suppresses lipolysis [43]. Our data differ in that Chakrabarti et al. found that rapamycin also modestly augments basal lipolysis and modestly reduces the mRNA and protein expression of the lipase ATGL [43]. We found no evidence for alterations in basal lipolytic rate or ATGL expression upon rapamycin treatment. Perhaps differences in experimental conditions account for these apparent discrepancies.
Emerging data identify mTORC1 an important novel controller of both anabolic and catabolic lipid metabolism by regulating lipogenesis and lipolysis, respectively [44, 45]. mTORC1 signaling induces adipogenic differentiation and maintains the adipogenic program by promoting the expression and activation state of the transcription factor PPARγ (peroxisome proliferator-activated receptor-γ), a nuclear hormone receptor that induces the expression of genes that promote fatty acid uptake, synthesis, esterification, and storage [46]. Moreover, mTORC1 signaling promotes lipid biosynthesis by cleaving the transcription factor SREBP-1 (sterol regulatory element binding protein-1) into a mature form that translocates to the nucleus to induce the expression of lipogenic genes that promote fatty acid and TAG synthesis [45, 47].
As increased levels of circulating FFA are well documented as an etiological factor in the development of insulin resistance, the cumulative data suggest that rapamycin administration to patients may exacerbate insulin resistance by promoting TAG lipolysis [48]. Indeed, studies in a diabetic rodent model showed that injection of rapamycin led to worsened hyperglycemia and diabetes [48]. Impaired TOR signaling also appears to promote lipolysis in model organisms. For example, in Drosophila, loss-of-function mutations in melted leads to decreased dTOR signaling and a 10% decrease in animal size, consistent with a role of mTOR in cell growth control; strikingly, melted flies possess 40% less fat relative to control animals in the fat body, the primary energy storage organ [49]. This phenotype is fat-body autonomous, as re-expression of wild type melted in the fat body rescues the leanness of melted mutants [49]. Additionally, global knockout of the mTORC1 substrate S6K1 in mice produces animals with increased levels of plasma FFA that exhibit mild glucose intolerance and resistance to diet-induced obesity [14, 50]. Consistently, increased mTORC1 signaling to eukaryotic initiation factor 4E (eIF4E) via global knockout of the eIF4E inhibitors 4EBP-1 and 4EBP-2 leads to increased adipogenesis, decreased lipolysis, and increased FFA re-esterification, which cooperatively induce obesity [51].
In conclusion, these data demonstrate that cellular inhibition of mTORC1 signaling with rapamycin, as well as inhibition of PI3K with wortmannin, synergizes with the PKA pathway to augment the phosphorylation of HSL leading to enhanced hormone-induced lipolysis. Thus, mTORC1 signaling antagonizes the PKA pathway to suppress hormone-induced lipolysis. Many questions remain regarding the molecular mechanism by which mTORC1 signaling impacts lipolytic targets. Does mTORC1 directly or indirectly (via downstream substrates) antagonize PKA signaling? Does mTORC1 signaling suppress PKA activity or does mTORC1 signal along a parallel pathway that converges on a common lipolytic target that lies downstream of PKA? Our data and that of others reveal a novel role for PI3K/mTORC1 signaling in control of cellular TAG and FFA metabolism: mTORC1 signaling reduces lipolytic rate, thus promoting TAG storage. Elucidating the molecular mechanisms by which mTORC1 signaling modulates lipid metabolism may enable clinicians in the future to better manage the deleterious side effects incurred by therapeutic rapamycin or analog treatment employed for immunosuppression, stent restenosis, and cancer. Additionally, as altered lipid homeostasis contributes to diverse human diseases including type II diabetes, obesity, cancer, and cardiovascular disease, elucidation of the biochemical mechanisms by which mTORC1 signaling modulates lipid metabolism is important both biologically and clinically.
Acknowledgments
The authors would like to express their gratitude to Drs. Nancy Weigel (Baylor College of Medicine) and Victoria Knutson (University of Texas) for sharing reagents, encouragement, support, and advice. Funding: This work was funded by grants from the National Institutes of Health (KOI DK60654) and the American Heart Association (0750060Z) to GS and NIH-R01 (DK-078135) to DCF.
Abbreviations
- ATGL
Adipocyte triacylglycerol lipase
- AMPK
AMP activated protein kinase
- ATP
Adenosine triphosphate
- cAMP
Cyclic adenosine monophosphate
- eIF4E
Eukaryotic initiation factor-4E
- 4EBP1/PHAS-I
eIF-4E-binding protein or protein-1/heat and acid stable-activated by insulin
- HEAT
Huntington elongation factor 3, the A subunit of protein phosphatase 2A, and TOR1
- HSL
Hormone sensitive lipase
- FBS
Fetal bovine serum
- FRAP
FKBP-12 rapamycin associated protein
- FRB
FKBP12-rapamycin binding domain
- FFA
Free fatty acids
- FKBP12
FK506 binding protein 12
- GAP
GTPase activating protein
- GβL
G protein β subunit-like protein also known as mLST8
- GLUT 4
Glucose transporter 4
- IRS
Insulin receptor substrate
- LKB
Tumor suppressor protein
- LPAAT
Lysophosphatidic acid acyl transferase
- MEFs
Mouse embryonic fibroblasts
- MGL
Monoacylglycerol lipase
- mTOR
Mammalian target of rapamycin (TOR)
- mTOR P-S2481
mTOR phosphorylated on serine 2481
- mTORC1
Mammalian target of rapamycin complex 1
- mTORC2
Mammalian target of rapamycin complex 2
- NCS
Newborn calf serum
- NEFA
Nonesterified fatty acids
- RAPA
Rapamycin
- Raptor
Regulatory associated protein of mammalian target of rapamycin
- Rheb
Ras homolog enriched in brain
- Rictor
Rapamycin-insensitive companion of mTOR
- RII
Regulatory subunit II of PKA
- TOS
TOR signaling motif
- PA
Phosphatidic acid
- PDE
Phosphodiesterase
- PLD
Phospholipase D
- PI3K
Phosphatidylinositol 3-OH kinase
- Pol I
Polymerase I
- PKA
Protein kinase A
- PKB/AKT
Protein kinase B
- PPAR γ
Peroxisome proliferator-activated receptor-γ
- PTEN
Phosphatase and tensin homologue deleted on chromosome 10
- S6K1
p70 ribosomal protein S6 kinase 1
- S6K1 P-T389
S6K1 phosphorylated on threonine 389
- TAG
Triacylglycerol
- TSC
Tuberous sclerosis complex
- VLDL
Very low density lipoprotein
Footnotes
Conflict of interest Nothing to disclose; there are no commercial or other associations that may pose a conflict of interest.
Contributor Information
Ghada A. Soliman, Division of Metabolism, Endocrinology, and Diabetes, Department of Medicine, University of Michigan Medical School, 109 Zina Pitcher Place, Ann Arbor, MI 48109-2200, USA. Department of Family and Consumer Sciences, Western Michigan University, 1903 W. Michigan Avenue, Kalamazoo, MI 49008-5322, USA
Hugo A. Acosta-Jaquez, Department of Cell and Developmental Biology, University of Michigan Medical School, 109 Zina Pitcher Place, Ann Arbor, MI 48109-2200, USA
Diane C. Fingar, Division of Metabolism, Endocrinology, and Diabetes, Department of Medicine, University of Michigan Medical School, 109 Zina Pitcher Place, Ann Arbor, MI 48109-2200, USA. Department of Cell and Developmental Biology, University of Michigan Medical School, 109 Zina Pitcher Place, Ann Arbor, MI 48109-2200, USA
References
- 1.Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23:3151–3171. doi: 10.1038/sj.onc.1207542. [DOI] [PubMed] [Google Scholar]
- 2.Soliman GA. The mammalian target of rapamycin signaling network and gene regulation. Curr Opin Lipidol. 2005;16:317–323. doi: 10.1097/01.mol.0000169352.35642.06. [DOI] [PubMed] [Google Scholar]
- 3.Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–14077. doi: 10.1074/jbc.R109.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- 6.Oshiro N, Yoshino K, Hidayat S, Tokunaga C, Hara K, Eguchi S, Avruch J, Yonezawa K. Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells. 2004;9:359–366. doi: 10.1111/j.1356-9597.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- 7.Soliman GA, Acosta-Jaquez HA, Dunlop EA, Ekim B, Maj NE, Tee AR, Fingar DC. mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J Biol Chem. 2010;285:7866–7879. doi: 10.1074/jbc.M109.096222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kahan BD. Sirolimus: a ten-year perspective. Transplant Proc. 2004;36:71–75. doi: 10.1016/j.transproceed.2003.11.039. [DOI] [PubMed] [Google Scholar]
- 9.Morice MC, Serruys PW, Sousa JE, Fajadet J, Ban Hayashi E, Perin M, Colombo A, Schuler G, Barragan P, Guagliumi G, Molnar F, Falotico R. A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization. N Engl J Med. 2002;346:1773–1780. doi: 10.1056/NEJMoa012843. [DOI] [PubMed] [Google Scholar]
- 10.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrisett JD, Abdel-Fattah G, Hoogeveen R, Mitchell E, Ballantyne CM, Pownall HJ, Opekun AR, Jaffe JS, Oppermann S, Kahan BD. Effects of sirolimus on plasma lipids, lipoprotein levels, and fatty acid metabolism in renal transplant patients. J Lipid Res. 2002;43:1170–1180. [PubMed] [Google Scholar]
- 12.Mathe D, Adam R, Malmendier C, Gigou M, Lontie JF, Dubois D, Martin C, Bismuth H, Jacotot B. Prevalence of dyslipidemia in liver transplant recipients. Transplantation. 1992;54:167–170. [PubMed] [Google Scholar]
- 13.Teutonico A, Schena PF, Di Paolo S. Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol. 2005;16:3128–3135. doi: 10.1681/ASN.2005050487. [DOI] [PubMed] [Google Scholar]
- 14.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 15.Margolin K, Longmate J, Baratta T, Synold T, Christensen S, Weber J, Gajewski T, Quirt I, Doroshow JH. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer. 2005;104:1045–1048. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- 16.Ahima RS. Adipose tissue as an endocrine organ. Obesity (Silver Spring) 2006;14(Suppl 5):242S–249S. doi: 10.1038/oby.2006.317. [DOI] [PubMed] [Google Scholar]
- 17.Cawthorn WP, Sethi JK. TNF-alpha and adipocyte biology. FEBS Lett. 2008;582:117–131. doi: 10.1016/j.febslet.2007.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egan JJ, Greenberg AS, Chang MK, Wek SA, Moos MC, Jr, Londos C. Mechanism of hormone-stimulated lipolysis in adipocytes: translocation of hormone-sensitive lipase to the lipid storage droplet. Proc Natl Acad Sci USA. 1992;89:8537–8541. doi: 10.1073/pnas.89.18.8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Soni KG, Semache M, Casavant S, Fortier M, Pan L, Mitchell GA. Lipolysis and the integrated physiology of lipid energy metabolism. Mol Genet Metab. 2008;95:117–126. doi: 10.1016/j.ymgme.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 20.Cohen AW, Razani B, Schubert W, Williams TM, Wang XB, Iyengar P, Brasaemle DL, Scherer PE, Lisanti MP. Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes. 2004;53:1261–1270. doi: 10.2337/diabetes.53.5.1261. [DOI] [PubMed] [Google Scholar]
- 21.Su CL, Sztalryd C, Contreras JA, Holm C, Kimmel AR, Londos C. Mutational analysis of the hormone-sensitive lipase translocation reaction in adipocytes. J Biol Chem. 2003;278:43615–43619. doi: 10.1074/jbc.M301809200. [DOI] [PubMed] [Google Scholar]
- 22.Saha PK, Kojima H, Martinez-Botas J, Sunehag AL, Chan L. Metabolic adaptations in the absence of perilipin: increased beta-oxidation and decreased hepatic glucose production associated with peripheral insulin resistance but normal glucose tolerance in perilipin-null mice. J Biol Chem. 2004;279:35150–35158. doi: 10.1074/jbc.M405499200. [DOI] [PubMed] [Google Scholar]
- 23.Brasaemle DL, Rubin B, Harten IA, Gruia-Gray J, Kimmel AR, Londos C. Perilipin A increases triacylglycerol storage by decreasing the rate of triacylglycerol hydrolysis. J Biol Chem. 2000;275:38486–38493. doi: 10.1074/jbc.M007322200. [DOI] [PubMed] [Google Scholar]
- 24.Martinez-Botas J, Anderson JB, Tessier D, Lapillonne A, Chang BH, Quast MJ, Gorenstein D, Chen KH, Chan L. Absence of perilipin results in leanness and reverses obesity in Lepr(db/db) mice. Nat Genet. 2000;26:474–479. doi: 10.1038/82630. [DOI] [PubMed] [Google Scholar]
- 25.Aggarwal D, Fernandez ML, Soliman GA. Rapamycin, an mTOR inhibitor, disrupts triglyceride metabolism in guinea pigs. Metabolism. 2006;55:794–802. doi: 10.1016/j.metabol.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 26.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFTl phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA. 1998;95:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holm C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem Soc Trans. 2003;31:1120–1124. doi: 10.1042/bst0311120. [DOI] [PubMed] [Google Scholar]
- 28.Kishida K, Kuriyama H, Funahashi T, Shimomura I, Kihara S, Ouchi N, Nishida M, Nishizawa H, Matsuda M, Takahashi M, Hotta K, Nakamura T, Yamashita S, Tochino Y, Matsuzawa Y. Aquaporin adipose, a putative glycerol channel in adipocytes. J Biol Chem. 2000;275:20896–20902. doi: 10.1074/jbc.M001119200. [DOI] [PubMed] [Google Scholar]
- 29.Chen JC, Powers T. Coordinate regulation of multiple and distinct biosynthetic pathways by TOR and PKA kinases in S. cerevisiae. Curr Genet. 2006;49:281–293. doi: 10.1007/s00294-005-0055-9. [DOI] [PubMed] [Google Scholar]
- 30.Manni S, Mauban JH, Ward CW, Bond M. Phosphorylation of the cAMP-dependent protein kinase (PKA) regulatory subunit modulates PKA-AKAP interaction, substrate phosphorylation, and calcium signaling in cardiac cells. J Biol Chem. 2008;283:24145–24154. doi: 10.1074/jbc.M802278200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Budillon A, Cereseto A, Kondrashin A, Nesterova M, Merlo G, Clair T, Cho-Chung YS. Point mutation of the autophosphorylation site or in the nuclear location signal causes protein kinase A RII beta regulatory subunit to lose its ability to revert transformed fibroblasts. Proc Natl Acad Sci USA. 1995;92:10634–10638. doi: 10.1073/pnas.92.23.10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elliott MR, Shanks RA, Khan IU, Brooks JW, Burkett PJ, Nelson BJ, Kyttaris V, Juang YT, Tsokos GC, Kammer GM. Down-regulation of IL-2 production in T lymphocytes by phosphorylated protein kinase A-RIIbeta. J Immunol. 2004;172:7804–7812. doi: 10.4049/jimmunol.172.12.7804. [DOI] [PubMed] [Google Scholar]
- 33.Sztalryd C, Xu G, Dorward H, Tansey JT, Contreras JA, Kimmel AR, Londos C. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J Cell Biol. 2003;161:1093–1103. doi: 10.1083/jcb.200210169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garton AJ, Campbell DG, Cohen P, Yeaman SJ. Primary structure of the site on bovine hormone-sensitive lipase phosphorylated by cyclic AMP-dependent protein kinase. FEBS Lett. 1988;229:68–72. doi: 10.1016/0014-5793(88)80799-3. [DOI] [PubMed] [Google Scholar]
- 35.Anthonsen MW, Ronnstrand L, Wernstedt C, Degerman E, Holm C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J Biol Chem. 1998;273:215–221. doi: 10.1074/jbc.273.1.215. [DOI] [PubMed] [Google Scholar]
- 36.Shen WJ, Patel S, Natu V, Kraemer FB. Mutational analysis of structural features of rat hormone-sensitive lipase. Biochemistry. 1998;37:8973–8979. doi: 10.1021/bi980545u. [DOI] [PubMed] [Google Scholar]
- 37.Donsmark M, Langfort J, Holm C, Ploug T, Galbo H. Contractions induce phosphorylation of the AMPK site Ser565 in hormone-sensitive lipase in muscle. Biochem Biophys Res Commun. 2004;316:867–871. doi: 10.1016/j.bbrc.2004.02.140. [DOI] [PubMed] [Google Scholar]
- 38.Donsmark M, Langfort J, Holm C, Ploug T, Galbo H. Regulation and role of hormone-sensitive lipase in rat skeletal muscle. Proc Nutr Soc. 2004;63:309–314. doi: 10.1079/PNS2004359. [DOI] [PubMed] [Google Scholar]
- 39.Zechner R, Kienesberger PC, Haemmerle G, Zimmermann R, Lass A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J Lipid Res. 2009;50:3–21. doi: 10.1194/jlr.R800031-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Wei E, Gao W, Lehner R. Attenuation of adipocyte triacylglycerol hydrolase activity decreases basal fatty acid efflux. J Biol Chem. 2007;282:8027–8035. doi: 10.1074/jbc.M605789200. [DOI] [PubMed] [Google Scholar]
- 41.Zurita-Martinez SA, Cardenas ME. Tor and cyclic AMP-protein kinase A: two parallel pathways regulating expression of genes required for cell growth. Eukaryot Cell. 2005;4:63–71. doi: 10.1128/EC.4.1.63-71.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soulard A, Cremonesi A, Moes S, Schutz F, Jeno P, Hall MN. The rapamycin-sensitive phosphoproteome reveals that TOR controls protein kinase A toward some but not all substrates. Mol Biol Cell. 2010;21:3475–3486. doi: 10.1091/mbc.E10-03-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chakrabarti P, English T, Shi J, Smas CM, Kandror KV. Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes. 2010;59:775–781. doi: 10.2337/db09-1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122:3589–3594. doi: 10.1242/jcs.051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008;8:224–236. doi: 10.1016/j.cmet.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim JE, Chen J. regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes. 2004;53:2748–2756. doi: 10.2337/diabetes.53.11.2748. [DOI] [PubMed] [Google Scholar]
- 47.Laplante M, Sabatini DM. mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc Natl Acad Sci USA. 2010;107:3281–3282. doi: 10.1073/pnas.1000323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leibowitz G, Cerasi E, Ketzinel-Gilad M. The role of mTOR in the adaptation and failure of beta-cells in type 2 diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):157–169. doi: 10.1111/j.1463-1326.2008.00952.x. [DOI] [PubMed] [Google Scholar]
- 49.Teleman AA, Chen YW, Cohen SM. Drosophila Melted modulates FOXO and TOR activity. Dev Cell. 2005;9:271–281. doi: 10.1016/j.devcel.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 50.Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y, Klumperman J, Thorens B, Thomas G. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature. 2000;408:994–997. doi: 10.1038/35050135. [DOI] [PubMed] [Google Scholar]
- 51.Le Bacquer O, Petroulakis E, Paglialunga S, Poulin F, Richard D, Cianflone K, Sonenberg N. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J Clin Invest. 2007;117:387–396. doi: 10.1172/JCI29528. [DOI] [PMC free article] [PubMed] [Google Scholar]