

Abstract
G protein-coupled receptors (GPCRs), which are modulated by a variety of endogenous and synthetic ligands, represent the largest family of druggable targets in the human genome. Recent structural and molecular studies have both transformed and expanded classical concepts of receptor pharmacology and begun to illuminate the distinct mechanisms by which structurally, chemically, and functionally diverse ligands modulate GPCR function. These molecular insights into ligand engagement and action have enabled new computational methods and accelerated the discovery of novel ligands and tool compounds —especially for understudied and orphan GPCRs. These advances promise to streamline the development of GPCR-targeted medications.
INTRODUCTION
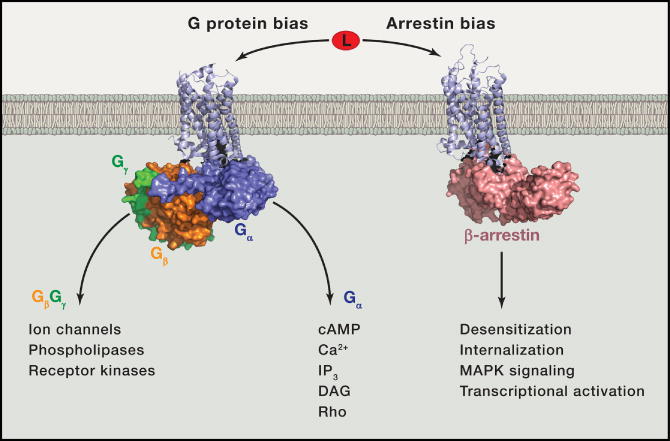
GPCRs are 7-transmembrane integral membrane proteins that typically translate extracellular stimulation into intracellular signals. GPCR activation is usually mediated by agonist binding which stabilizes receptor conformations that recruit and ultimately activate intracellular transducers. GPCR agonist ligands are physically and chemically diverse and can include: photons; ions (H+, Zn++, Ca++, etc.); odorants; tastants; vitamins (e.g. niacin, vitamin A1 aldehyde, etc.); peptidic and non-peptidergic hormones (estrogen, angiotensin, etc.); proteins (e.g. chemokines), neurotransmitters (dopamine, serotonin, etc.); natural products (morphine, salvinorin A, etc.); a large number of intermediary metabolites (ATP, ADP, fatty acids, bile acids, etc.); and products from human commensal bacteria [see (Allen et al., 2011a; Roth et al., 2015) for reviews]. Intracellularly, GPCR activation is translated into various signals mediated via hetereotrimeric G proteins, arrestins (Luttrell et al., 1999), kinases (Benovic et al., 1989) ion channels, and various scaffolding proteins (Brown et al., 2003) (Fig 1). In this context, arrestins function to abrogate G protein-mediated signaling (Lohse et al., 1990), as scaffolds for GPCR internalization via clathrin-coated vesicles (Goodman et al., 1996) and as scaffolds for signaling (Luttrell et al., 1999) (Fig 1).
Figure 1. Different ligand-stabilized GPCR conformations cause binding and activation of distinct signal transducers including G proteins and arrestins.
(Left) Crystal structure of β2AR (light blue cartoon) coupled to Gαs (blue) Gβ (orange) Gγ (green) heterotrimer (PDB ID: 3SN6 (Rasmussen et al., 2011b)) illustrates G protein-mediated signaling. Upon heterotrimer activation, subunits dissociate and Gα modulates second messenger production such as cAMP production through Gαs-mediated activation of adenylyl cyclase. Gβγ modulates separate Gα-independent downstream signaling networks such as ion channels, phospholipases, and receptor kinases.
(Right) Crystal structure of rhodopsin (light blue cartoon) coupled to β-arrestin (salmon) (PDB ID: 4ZWJ (Kang et al., 2015)) illustrates arrestin mediated effects such as receptor internalization or activation of kinase signaling networks.
GPCRs, of which there are more than 800 in humans (Fredriksson et al., 2003), comprise the largest family of membrane proteins in the human genome. Several classification schemes for GPCRs have been used, and in this review, we will adhere to the current International Union of Pharmacology (IUPHAR) classification that includes five main families: Rhodopsin family (Class A), Secretin family (Class B), Glutamate family (Class C), Frizzled/Taste family (Class F), and Adhesion Family [see (Alexander et al., 2015) for details]. In this review, we highlight findings that demonstrate how insights into ligand interactions at GPCRs are transforming molecular pharmacology and drug discovery.
Importance of GPCRs for physiology, disease and therapeutics
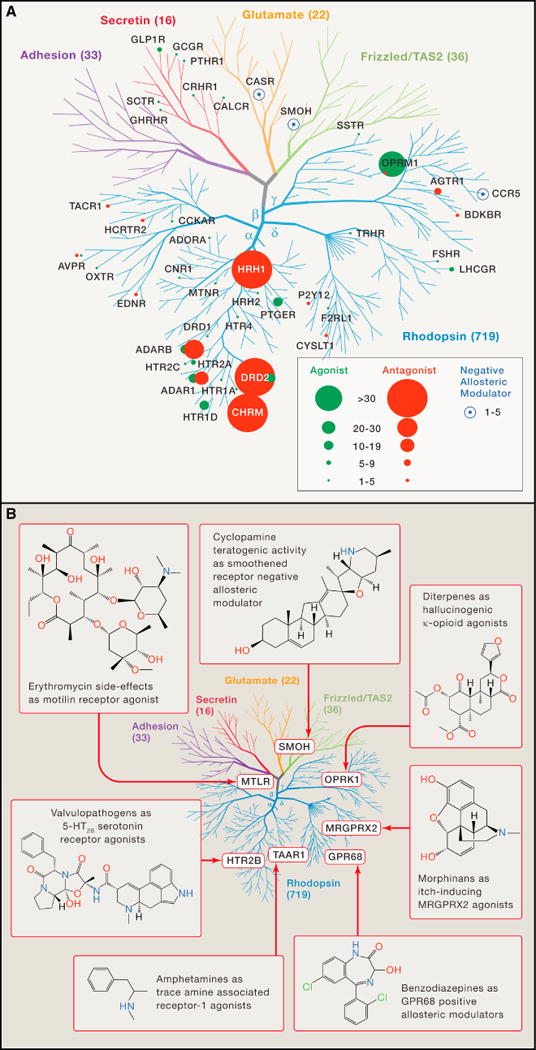
As of 2017, between 20 and 30% of FDA approved medications target GPCRs (Rask-Andersen et al., 2011). The popularity of GPCRs as drug targets is predominantly due to their physiological relevance, as GPCRs are expressed in most of the body’s tissues, are involved in cellular communication, and participate in virtually all aspects of human physiology via GPCR-mediated signal transduction; as well as their druggability, as GPCRs possess binding pockets with beneficial physiochemical properties that lend to the design of drug-like small molecules (Mason et al., 2012). Particularly prominent therapeutic applications involving GPCRs (see Fig 2A) include opioid analgesics (μ opioid receptor; OPRM1 receptor agonists), antihistamines (HRH1-histamine antagonists), anticholinergics (CHRM antagonists), typical and atypical antipsychotics (D2 dopamine receptor antagonists; DRD2), antimigraine drugs (5-HT1D serotonergic agonists; HTR1D), β2-agonists for asthma (ADBAR2) and anti-hypertensives (targeting α1 adrenergic and angiotensin II receptors; ADAR1, ATGR1).
Figure 2. GPCRome wide targets of approved and marketed medications and how ligands uncover unknown GPCR physiology towards potential therapeutic applications.
(A) Sphere size corresponds to number of approved drugs for highlighted therapeutic GPCR target with antagonists, agonists, and negative allosteric modulators shown in red, green, and blue, respectively. Phylogenetic tree of the GPCRome highlights the small fraction of GPCRs that are currently targeted by approved medications.
(B) Representative examples and their structures are shown for compounds used to identify previously unknown pharmacology at various receptors. Phylogenetic tree of the GPCRome highlights the diversity of GPCRs identified as off-targets.
In addition to being therapeutic targets for drug discovery, GPCR variants are occasionally implicated in disease processes. Indeed, dozens of monogenic diseases have been linked to constitutively activating mutations (CAMs), which augment GPCR constitutive activity. These include such conditions as congenital stationary night blindness caused by rhodopsin CAMs (McAlear et al., 2010), uveal melanoma by CAMs of the cysteinyl leukotriene receptor 2 (Moore et al., 2016), and many others (Smit et al., 2007). Additionally, loss-of-function GPCR mutations are occasionally linked to human disease. V2-vasopressin receptor mutations, for example, are associated with nephrogenic diabetes insipidus (Pan et al., 1992), and drugs which rescue the misfolded phenotype of V2-vasopressin receptor mutants via action as pharmacological chaperones have been proposed as therapeutics (Morello et al., 2000).
GPCRs as deleterious off-targets in therapeutic drug discovery
GPCRs are also frequent medication ‘off-targets’ that display unpredicted interactions which can result in unanticipated therapeutic (Roth et al., 2004) or life-threatening side-effects (Rothman et al., 2000). The most infamous example is likely the anti-obesity drug fenfluramine, which was withdrawn because of association with valvular heart disease in many individuals (Allen et al., 2011a; Roth, 2007). Years after fenfluramine’s widely publicized withdrawal from the world-wide market and legal damages totaling over 10 billion dollars, it was discovered that the compound’s metabolite—norfenfluramine—activated cardiac 5-HT2B serotonin receptors, leading eventually to valvular heart disease (Rothman et al., 2000) (Roth, 2007). Since then, several more drugs have been withdrawn due to similar 5-HT2B —mediated valvular heart disease complications (Roth, 2007) (Allen et al., 2011a).
As GPCRs represent frequent off-targets for drugs that target kinases and other non-GPCR molecular targets, compounds are typically profiled against large numbers of cloned GPCRs prior to clinical trials in humans (Allen et al., 2011a). Importantly, the potent actions of sorafinib and many other approved and investigational kinase inhibitors on serotonergic, purinergic, and other GPCRs have been discovered via GPCR profiling (Elkins et al., 2016). Identification of potentially important off-target actions of drugs through GPCRs is also facilitated by large databases of drug-target information, including ChEMBL (https://www.ebi.ac.uk/chembl/), PubChem (https://pubchem.ncbi.nlm.nih.gov/), PHAROS (https://pharos.nih.gov/idg/index) and the Ki Database (http://kidbdev.med.unc.edu/databases/kidb.php). These cheminformatic datasets also have been useful for the in silico prediction and in vitro and in vivo confirmation of GPCRs as relevant and important drug off-targets (Keiser et al., 2009)
Understudied and orphan GPCRs and their therapeutic potential
Although there are more than 350 GPCR-targeted FDA approved drugs, they target only a small sector of the universe of potentially druggable GPCRs (Roth et al., 2015) (Rask-Andersen et al., 2011) (Fig 2A). Approximately 100 human GPCRs are currently active targets for late-stage preclinical development, and a total of nearly 400 small molecules are being actively investigated as therapeutics (Lafferty-Whyte et al., 2016). Current drug development, though, is geared mainly towards those GPCRs with extensive validation as potential therapeutic targets (Lafferty-Whyte et al., 2016). Conversely, only a few orphan or understudied GPCRs, “oGPCRs” – as defined by i) their comparatively low number of publications, ii) their low number of annotated small molecules interactors, or iii) absence of their known endogenous ligands (Roth et al., 2015) – are currently being investigated for therapeutic drug discovery. Significantly, the Adhesion, Tastant and Frizzled families of receptors are reported to have no annotated small drug-like molecules in clinical testing (Lafferty-Whyte et al., 2016).
The lack of drug development programs targeting oGPCRs stems mainly from risk-aversion, as little is known regarding oGPCR’s physiological roles and druggability. Although knock-out studies and replacement with chemogenetic mutant GPCRs, such as DREADDs (designer receptors exclusively activated by designer drugs) (Roth, 2016), or chimeric opsins for optogenetic studies, such as OptoXRs (Airan et al., 2009), represent strategies to identify the basic physiological roles of oGPCRs, the biggest bottleneck remains the lack of chemical tool compounds to reliably characterize receptor function in vitro and in vivo. Yet, oGPCRs have emerged as important receptors for natural products and synthetic drugs; drugs targeting oGPCRs have been used as tools to illuminate fundamental biological processes and therapeutic approaches mediated by oGPCRs (Fig 2B). For instance, the naturally occurring teratogen cyclopamine (Cooper et al., 1998) facilitated identification of the smoothened receptor (SMO) as a hedgehog signaling pathway modulator and target for cancer chemotherapy (Rudin et al., 2009). Similarly, the discovery that the hallucinogen salvinorin A from the sage Salvia divinorum is a selective κ-opioid receptor agonist, validated this receptor as a target for psychotomimetic compounds (Roth et al., 2002). Several benzodiazepines were found to also activate GPR68, suggesting that some of the side-effects of these anti-anxiety medications could be mediated by this receptor (Huang et al., 2015b). Additionally, the discovery of amphetamine actions at the TAAR1 trace amine receptor (Bunzow et al., 2001) identified TAAR1 as a potential target for neuropsychiatric diseases. The endogenous TAAR1 agonists known as thyronamines (Scanlan et al., 2004) revealed trace amine receptors as potential mediators of metabolic, thermogenic and neurologic processes. Clearly, expanding our understanding of GPCR on- and off-target pharmacology is important for both successful drug discovery as well as for illuminating basic biological and chemical processes in health and disease. As exemplified by oGPCRs, however, we are still far from a comprehensive molecular and physiological understanding of GPCR biology.
Next, we review how recent molecular insights from crystal structures have transformed classical receptor pharmacology and facilitated our understanding of the mechanisms by which ligands modulate GPCR function. We further highlight the utility of ligands in identifying and characterizing the physiological roles of poorly understood GPCRs, and provide an overview of current advances to computationally leverage molecular insight towards identifying novel GPCR ligands.
TOWARDS A STRUCTURE-BASED UNDERSTANDING OF GPCR-LIGAND PHARMACOLOGY
Key pharmacological concepts
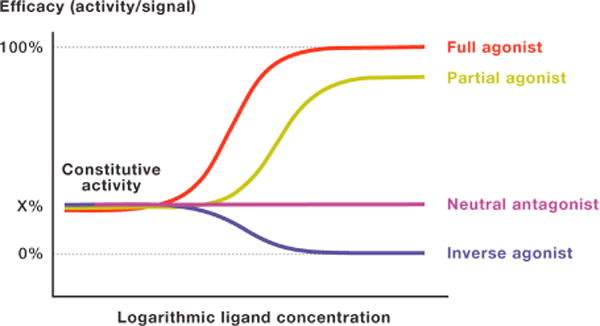
Historically, the concepts of agonism and antagonism arose from the observations of drug actions on isolated organs. One example is highlighted by the ordered and regular agonist activity of acetylcholine which is antagonized by atropine (Clark, 1926). Observations like these led to the initial concepts that agonists either induce or stabilize an ‘active’ state of a ‘receptor’, while antagonists have no effect on their own but block agonist access to this ‘receptor’. Once GPCRs were cloned and expressed in vitro it was observed that GPCRs also possessed variable degrees of basal or constitutive activity and could populate active signaling states in the absence of ligands. With a few notable exceptions (e.g. adhesion-, thrombin-, and some viral receptors like the KHSV related receptor), GPCRs typically require exogenous agonists to stabilize fully active states for maximal signaling. Endogenous agonists such as neurotransmitters or hormones usually, but not invariably, maximally activate their cognate GPCRs and are considered full agonists (see Box 1); agonists which do not induce 100% activation are defined as partial agonists. It is important to note that partial agonists can appear as full agonists when receptor reserve is present, and that a full response of the system can be elicited even when not all receptors are occupied. Antagonists on the other hand are compounds (either naturally occurring or synthetic) that block agonists activity; antagonists are classified as inverse agonists (antagonists that decrease constitutive activity) or neutral antagonists [antagonists that inhibit agonist effects but do not interfere with constitutive activity; Box 1, for further details and see (Roth, 2016)].
Box 1.
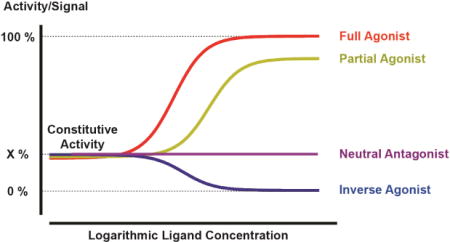
Constitutive Activity
Receptor mediated signaling in the absence of ligand due to spontaneous population of active receptor states
Full Agonists
Ligands that elicit maximum signal at the interrogated pathway (endogenous ligands are per definition full agonists)
Partial Agonists
Ligands that elicit activity below maximum level
Inverse Agonists
Ligands that inhibit constitutive receptor activity
Neutral Antagonists
Ligands that bind the receptor but do not affect constitutive receptor activity
Receptor Reserve
Receptors not coupled to the system resulting in maximum signal by activation of only a fraction of total receptors
Orthosteric Site
Binding pocket accommodating endogenous receptor ligand
Allosteric Site
Pocket distinct from the orthosteric site that can modulate ligand binding and receptor activity
Positive and negative allosteric modulators (PAMs and NAMs)
PAMs increase and NAMs decrease a receptor’s activity in response to an orthosteric ligand, while binding at a site distinct from the orthosteric site
Bitopic Ligands
Ligands that possess both orthosteric and allosteric moieties
Functional Selectivity/Biased Signaling
Ability of ligands to impart different degrees of activation in distinct pathways downstream of the receptor
Operational model
Mathematical description of receptor activation to quantify and predict drug action
Agonists, partial agonists, and antagonists interact with the so-called orthosteric site, which represents the binding site through which endogenous agonists activate the GPCR. Some GPCRs, most notably the adhesion (Hamann et al. 2015) and protease activated receptors (Coughlin, 200) lack classical endogenous agonists, although following proteolysis an N-terminal fragment occupies the orthosteric site.
Additionally, GPCRs may also be modulated allosterically by molecules that bind at a site distinct from the orthosteric site. Generally, allosteric modulators are classified as negative allosteric modulators or positive allosteric modulators (NAMs and PAMs, respectively) (Christopoulos et al., 2014) (Box 1). Allosteric modulators do not directly interact with the orthosteric site, but modulate the function of orthosteric ligands in a negative (NAMs) or positive (PAM) way. Molecules that interact with both the orthosteric and allosteric sites are defined as bitopic ligands and may be either agonists or antagonists. Allosteric modulators can be endogenous, as in the case of the nearly universal allosteric modulators sodium [which is a NAM (Fenalti et al., 2014)] and cholesterol [which could function as a NAM or PAM(Katritch et al., 2013)] or exogenous natural products or synthetic compounds (Kenakin et al., 1989).
GPCR ligands also often display functional selectivity or biased signaling [Box 1; (Urban et al., 2007)], a process by which ligands will direct or bias the signaling towards one pathway or another (Fig 1). Related to this, although GPCRs were initially classified based on the presumed main G protein with which they interact [e.g. Gs-, Gi-,Gq- and G12/13-coupled (Simon et al., 1991)], the schema was abandoned due to observations that GPCRs can couple to multiple G proteins (Asano et al., 1984). Many theoretical models have arisen to explain the agonist, allosteric and antagonist actions and biased signaling including the highly useful, albeit phenomenological, operational model (Box 1) of Black and Leff (Black et al., 1983) These simplified models continue to be useful for quantifying and predicting drug actions at GPCRs (Kenakin et al., 2012). With the discovery that GPCRs require G proteins for activation, more detailed models have arisen including the so-called ternary (De Lean et al., 1980) and extended ternary complex models ((Samama et al., 1993), as well as other models incorporating arrestin signaling (Roth, 2016) (Fig 3). The simple ternary and extended ternary complex models are called ‘ternary’ because they have three members: receptor (R), ligand (L) and hetereotrimec G protein (G). Extended versions incorporating other effectors like arrestins (Fig 3) are becoming validated by more mechanistic approaches which incorporate structure-based insights into ligand pharmacology.
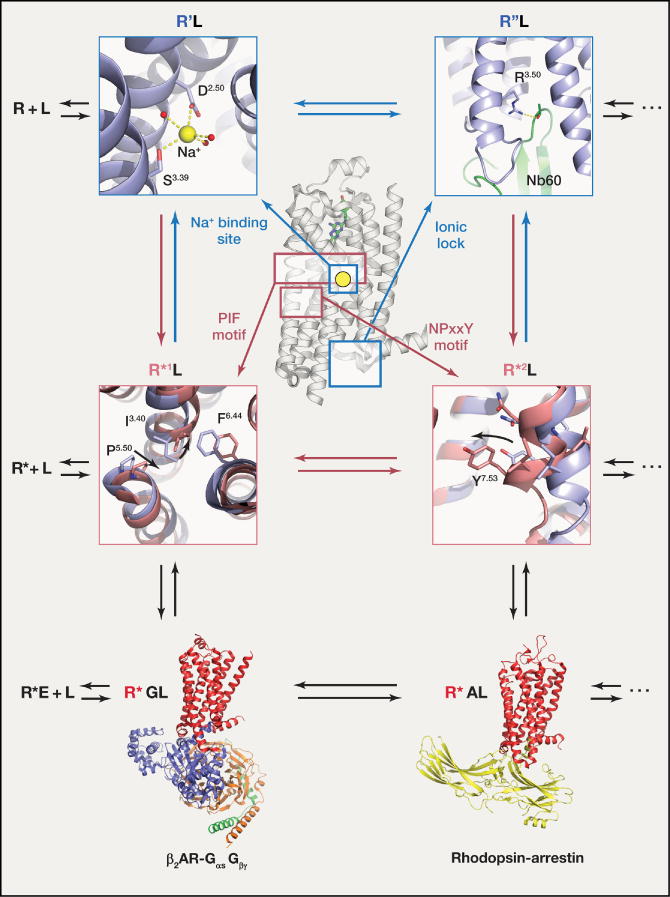
Figure 3. Molecular and structural pharmacology extend the ternary complex model for quantitative description of drug action at GPCRs.
Several ligand bound inactive receptor states (R’L, R”L …) and ligand bound active receptor states (R*1L, R*2L …), as well as ternary complex structures of ligand and effector bound active receptor states (R*GL, R*AL) (PDB ID: 3SN6 (Rasmussen et al., 2011b), PDB ID: 4ZWJ (Kang et al., 2015)) have been structurally characterized by x-ray crystallography. Distinct conformational characteristics such as the sodium binding site of the A2AAR (PDB ID: 4EIY (Liu et al., 2012b)), the ionic lock of a nanobody stabilized β2AR (PDB ID: 5JQH (Staus et al., 2016)), the PIF motif of the 5-HT2B receptor (PDB ID: 4IB4 (Wacker et al., 2013), PDB ID: 3NY8 (Wacker et al., 2010)), and the NPxxY motif of a nanobody bound β2AR (PDB ID: 3NY8 (Wacker et al., 2010), PDB ID: 3P0G (Rasmussen et al., 2011a)), highlight diverse GPCR activation states.
From a conceptual perspective, the ternary (De Lean et al., 1980) and extended (Samama et al., 1993) complex models were important because they predicted that GPCRs will spontaneously adapt multiple active (R*1, R*2…) and inactive (R’, R”…) conformations. As also predicted, these transient states can spontaneously interact with G protein or arrestin transducers to yield signaling complexes in the absence of agonist (R*E with E=G protein, Arrestin, or other transducers) (Fig 3). Indeed, this constitutive activity, has been amply documented in both recombinant (Burns et al., 1997) and endogenous (Arvanitakis et al., 1997) expression systems. These models also predict that antagonists do not simply ‘antagonize’ GPCRs, but that they stabilize inactive states (R’L) and thereby inhibit constitutive activity by acting as inverse agonists. The models further predict that so-called neutral antagonists are likely to be rare, as any ligand will bias the ensemble of spontaneously arising conformations at least to some extent. The predictions that antagonists are actually inverse agonists have been extensively validated in recombinant systems in vitro (Chidiac et al., 1993) and in vivo (Dillon et al., 2011). These models also predict that the active states stabilized by agonist (R*L) might differ conformationally from ternary signaling complexes (R*GL; R*AL). These predictions have been validated by biochemical studies demonstrating that G protein binding allosterically enhances agonist binding affinity (Cerione et al., 1984)
The impact of molecular insights into GPCR-ligand interactions illuminate both empirically based and classical concepts of receptor pharmacology
Historically, most of the initial functional concepts describing binding interactions were based on phenomenological observations from ligand binding and signaling studies done in recombinant or endogenous systems. It wasn’t until technological advances enabled the study of ligand-receptor interactions by x-ray crystallography, NMR, and other biophysical assays, that high resolution insights enabled the molecular characterization of these distinct states (Fig 3). Initial biochemical studies suggested that GPCR activation involves helical movements (Farrens, 1996); since then crystallographic and now cryo-EM structures are greatly enhancing our understanding of the molecular characteristics that define distinct GPCR conformations and signaling states. Most GPCRs have been crystallized in apparently inactive conformations (R’L), bound to antagonists or agonists (Tesmer, 2016). These inactive-state structures highlight distinct features of inactive GPCRs, such as binding of the endogenous NAM sodium (Fenalti et al., 2014; Liu et al., 2012b), and the closed ‘ionic lock’ (Staus et al., 2016). Intermediate active states in the absence of bound effector (R*L) have been described for the 5-HT1B serotonin (Wang et al., 2013), the A2A adenosine (A2AAR) (Allen et al., 2011b), and neurotensin (White et al., 2012) receptors. Structures of the 5-HT2B serotonin receptor bound to the agonists ergotamine (Wacker et al., 2013) and lysergic acid diethylamide (LSD) (Wacker et al., 2017) have identified features of an arrestin-biased intermediate state (R*L). They particularly include ‘active-like’ conformations of selected “trigger” motifs, including the ‘PIF’ and ‘NPxxY’ motifs [Fig 3; see (Venkatakrishnan et al., 2013) (Wacker et al., 2017) for details], which are structural elements found in many GPCRs that are critical for receptor activation. Active state receptor structures stabilized by nanobodies have been particularly helpful in characterizing structural hallmarks of GPCR activation (Huang et al., 2015a; Kruse et al., 2013; Rasmussen et al., 2011a). Crystal structures have also been determined for ternary signaling complexes, such as that of β2AR with agonist and hetereotrimeric G protein (R*GL) (Rasmussen et al., 2011b) and rhodopsin with arrestin (R*AL) (Kang et al., 2015) (Fig 3). Recent cryo-EM structures of R*GL states (Liang et al., 2017; Zhang et al., 2017) have confirmed some of the key features of the β2AR—G protein complex. Finally, although not associated with a high resolution structure, a recent report indicates that under some circumstances a ‘megaplex’ of GPCR, heterotrimeric G protein and arrestin may exist as a functioning signaling entity (Thomsen et al., 2016) mediating endosomally-based GPCR signaling. Thus, several conformational ensembles predicted by these models have finally been revealed structurally, and greatly improve our molecular understanding of GPCR activation and regulation.
As predicted by extended versions of the ternary complex model (Roth, 2016; Samama et al., 1993) and verified biophysically (Manglik et al., 2015) multiple additional conformational intermediates exist. These include an R’L ‘ground-state’, which resembles inactive rhodopsin by maintaining an intact ionic lock (Palczewski et al., 2000) between conserved residues in TM III (typically basic like Asn or Gln) and TM VI (typically acidic like Asp or Glu). Additionally, there are likely to be many intermediate states between the active (R*L) and coupled (R*GL; R*AL) (Manglik et al., 2015) receptor. As well, these extended models predict that holo-GPCR complexes in the inactive (R’) and active (R*) states exist; again, there is emerging biophysical evidence for these conformational intermediates (Manglik et al., 2015.) Finally, these models predict that GPCRs and G proteins could exist in a precoupled active-like state (R*G); although such states have not yet been observed by direct structural studies, there is evidence for GPCR-G protein precoupling [RG; (Nobles et al., 2005)]. From the previously discussed observations, it is clear that multiple intermediate conformational states exist for GPCRs (alone and in complex with their effectors); elucidating the full complement of these effectors will be important for future efforts to design drugs to stabilize unique conformations and signaling intermediates.
Multiple modes of ligand recognition: orthosteric, bitopic and allosteric GPCR-ligand interactions
Although a plethora of previous structure-function studies identified residues critical for binding and activation of many GPCRs, the molecular details of ligand-GPCR interactions remained speculative until the first crystal structure of a GPCR, rhodopsin (Palczewski et al., 2000), provided the initial insight. Only a few GPCRs have been crystalized when bound to their endogenous ligands: rhodopsin and other opsins in complex with retinal (Palczewski et al., 2000) (Fig 4), glutamate in complex with the extracellular domains of the mGluRs (Kunishima et al., 2000), the adenosine-bound A2AAR (Lebon et al., 2011), the neurotensin-bound NT-1 neurotensin receptor (White et al., 2012), the endothelin-bound endothelin ETB receptor (Shihoya et al., 2016), and adrenaline-bound β2AR (Ring et al., 2013). Since rhodopsin is an exception, as it is covalently bound to its endogenous ligand, the first structural evidence for a more general orthosteric binding site shared amongst many Class A GPCRs came from the structures of A2AAR and β2AR bound to their diffusible endogenous ligands (Lebon et al., 2011; Ring et al., 2013). For peptidergic GPCRs—based on structures of NT-1 neurotensin receptor in complex with neurotensin and the δ-opioid receptor in complex with a modified peptide agonist, the orthosteric site likely overlaps with the shared Class A orthosteric site and extends to include extensive contacts with the extracellular loops. Recent cryo-EM studies showed that endogenous peptides of several Class B receptors occupy a similar orthosteric site as in Class A receptors, but appear to prefer an even larger and more extended binding pocket (Liang et al., 2017; Zhang et al., 2017).
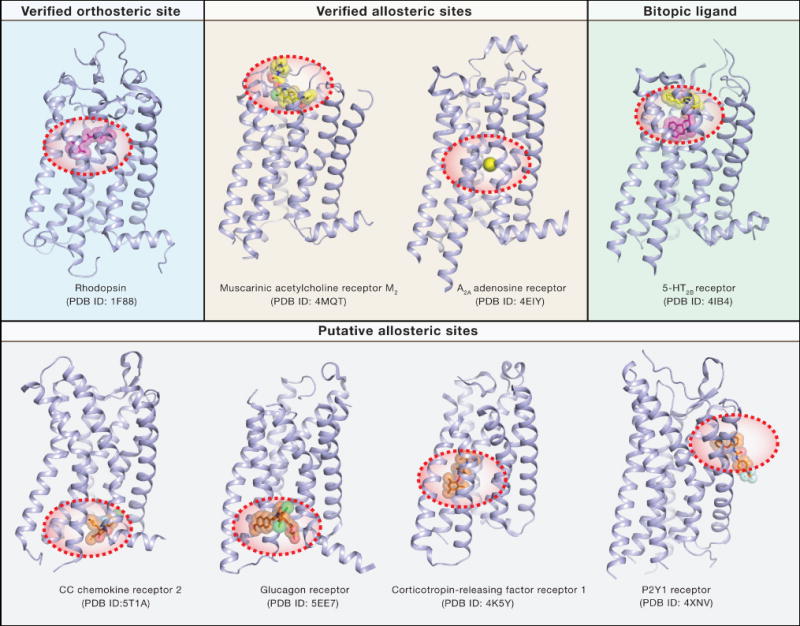
Figure 4. Crystal structures of different GPCR-ligand complexes highlight the diverse locations of ligand binding sites.
Ligands are shown as stick models with transparent surfaces, receptors are shown in cartoon representation in light blue. Complexes show retinal-bound RHO, LY2119620-bound CHRM2, sodium-bound ADORA2A, ergotamine-bound HTR2B, CCR2-RA-[R]-bound CCR2, MK-0893-bound GCGR, CP-376395-bound CRHR1, and BPTU-bound P2RY1.
Frequently, the orthosteric binding site of Class A GPCRs resides in the middle of the seven-transmembrane helical bundle, located between the extracellular loops and the middle plane of the membrane. Orthosteric sites exhibit different shapes and chemostatic make up depending on the nature of ligand. For instance, more lipophilic ligands, such as cannabinoids, likely enter the receptor from the membrane and the hydrophobic binding site is thus covered with residues from the extracellular loops to provide a barrier to the hydrophilic extracellular space (Hua et al., 2016). Conversely, peptide ligands —particularly for Class B receptors—are often large and possess considerable flexibility (O’Connor et al., 2015), and require an orthosteric site open to the extracellular space (Liang et al., 2017; Zhang et al., 2017). Interestingly, while the nature of orthosteric sites is critical to accommodate the ligands’ chemostatic and steric properties, entry to the binding site through a network of receptor specific residues can present the largest energetic barrier to compound binding (Dror et al., 2011). This finding is particularly important for the design of receptor selective ligands and the control of receptor binding and dissociation rates.
Several studies have also provided critical structural insights into ligand binding sites that are structurally distinct from the orthosteric pocket as observed in Class A GPCRs (Fig 4). These include the M2 muscarinic receptor bound to a PAM located in a vestibule above the orthosteric agonist binding site (Kruse et al., 2013), and the corticotropin-releasing factor 1 receptor bound to a presumed allosteric antagonist situated deep within the cytoplasmic portion of the helical bundle (Hollenstein et al., 2013). Allosteric ligands have also been found bound to the putative intracellular binding site of the C-terminal tail of the Gα subunit in the chemokine receptors CCR9 (Oswald et al., 2016), and extra-helical binding sites in the glucagon (Jazayeri et al., 2016) and purinergic P2Y1 receptor (Zhang et al., 2015) (Fig 4). Lastly, general allosteric modulators of GPCR function include cholesterol, which binds to different extra helical binding sites in different receptors (Gimpl, 2016), and sodium, which is bound in a highly conserved pocket in the center of the helical bundle below the orthosteric site (Katritch et al., 2014) (Fig 4).
Surprisingly, and despite decades of work exploring GPCR allosteric modulation (Kenakin et al., 1989; Lanzafame et al., 1997), currently approved drugs mainly target orthosteric sites (Fig 2A). Only a handful of FDA-approved GPCR allosteric modulators exist: cinacalcet, a NAM for the Calcium-sensing receptor (CASR), maraviroc, a CCR5 chemokine receptor NAM (CCR5), and the Smoothened receptor (SMO) NAMs sonedigib and vismodegib. Despite these limited numbers, targeting GPCRs allosterically remains a promising approach for therapeutic drug development (Changeux et al., 2016), as allosteric modulators are often more selective for their targets, and allosterically modulating the actions of endogenous ligands provides substantial therapeutic benefits (Aitken et al., 2009).
One important reason for the lack of allosteric therapeutics could be that it is challenging to design compounds with sufficient efficacy, as for most GPCRs only the orthosteric pocket has evolved to govern receptor modulation. It is important to note that while endogenous ions, lipids, adapter proteins and effectors modulate GPCR function (van der Westhuizen et al., 2015), their interacting surfaces rarely possess the physiochemical properties necessary for the structure-informed design of synthetic modulators. Other reasons for the lack of available allosteric modulators may include the lack of tool compounds, as well as difficulties associated with developing suitable assays to test for allosteric modulation. The increasing abundance of crystal structures in combination with computational methods should greatly facilitate the identification and characterization of potential allosteric sites and patches, which in turn could greatly accelerate the targeted design of allosteric modulators.
Biased signaling and kinetics as drivers of ligand-encoded activities
GPCR signaling has both contextual and kinetic aspects and these can be exploited – ultimately from structural approaches – to fine-tune signaling for basic science and therapeutic applications. As classically illustrated for β2AR, G protein signaling generally occurs rapidly within a second or so of agonist administration, while it typically lasts only a few minutes. Signal termination occurs via desensitization through receptor phosphorylation, arrestin binding and internalization (Lefkowitz et al., 2005). By contrast, arrestin binding and G protein-independent arrestin-ergic signaling events, such as MAP kinase activation, typically occur on the minute to hour time scale (Lefkowitz et al., 2005). The delineation of these two signaling pathways has led to a major reconceptualization of how GPCRs mediate their actions in both normal physiology and disease and has ushered in a new era of GPCR drug discovery to identify agonists biased for either G protein (White et al., 2015) or arrestin (Allen et al., 2011b) signaling with the promise of improved therapeutic properties and reduced side-effects (DeWire et al., 2011). It should be noted, however, that identification and characterization of GPCR effectors remains an active area of investigation (Paek et al., 2017), and that there are likely many more unidentified intracellular proteins that interact with signaling complexes and modulate GPCR signaling (Paek et al., 2017).
From a mechanistic perspective, it remains largely unclear how receptors mediate biased signaling, although recent structures and biophysical studies have begun to clarify this issue. For instance, the structure of the arrestin-biased drug ergotamine bound 5-HT2B serotonin receptor (Wacker et al., 2013) revealed how ergotamine stabilizes a distinct receptor conformation in which motifs essential for arrestin-biased signaling (e.g. NPxxY) are activated while others associated with G protein signaling (e.g. DRY or PIF) remain in the inactive state. NMR (Liu et al., 2012a) and fluorescent spectroscopy studies (Rahmeh et al., 2012) highlight how ligands selectively engage distinct motifs to stabilize GPCR conformations that are more conducive to accommodating one effector over the other.
GPCR conformations associated with signal transduction along specific pathways have further been shown to depend on ligand binding rates. In recent studies of LSD’s interactions with 5-HT2B and 5-HT2A serotonin receptors, slow ligand kinetics were found to be pivotal for arrestinergic signaling (Wacker et al., 2017). Studies with D2 dopamine receptor further revealed how kinetics modulate patterns of biased signaling (Klein Herenbrink et al., 2016). Taken together, these findings highlight a crucial kinetic component to functional selectivity at the level of compound association, stabilization of distinct receptor conformations, and intracellular signal progression, which, together, appear to dramatically influence cellular responses.
The development and identification of drugs with differential patterns of biased signaling represents a major area of investigation, as these chemical tools help to delineate the downstream signaling network of GPCRs and are particularly useful to explore the physiological role of oGPCRs. Thus for instance, the hallucinogen LSD (Wacker et al., 2017), appears to display bias towards β-arrestin signaling (Box 1, Fig 1), while the synthetic opioids TRV-130 (DeWire et al., 2013) and PZM-21 (Manglik et al., 2016) are biased towards G protein signaling (Box 1, Fig 1). Allosteric modulators have also manifested biased potentiation in instances in which a single GPCR may interact with multiple G proteins, as in the case of the proton-sensing GPCR GPR68 (Huang et al., 2015b). Due to the differential expression of key signal transducers, signal transduction, and thus signaling bias, will vary between cell-types (Urs et al., 2016). Exploiting signaling bias for drug discovery promises to yield much improved compounds that specifically target therapeutic pathways while avoiding pathologic events downstream of the same receptor (Allen et al., 2011b; Violin et al., 2007).
EXPLOITING GPCR-LIGAND STRUCTURES FOR DRUG DISCOVERY
With the increasing number of GPCR structures, we can anticipate the generation of new chemical tools for the study of GPCRs by providing platforms for the structure-guided design of improved therapeutics. Since GPCRs are routinely crystallized in complex with ligands, structure-guided drug design is a promising alternative to classical methods of ligand discovery, which employ cycles of medicinal chemistry-based modification of existing scaffolds. However, although true structure-guided drug discovery and optimization has been routinely done for protein kinase inhibitors for more than a decade (Noble et al., 2004), the successful examples for GPCRs are rare. For example, structure-based drug design for A2AAR yielded new 1,2,4-triazine derivatives that were shown cystallographically to adopt novel binding modes (Congreve et al., 2012). Similarly, new mGluR5 metabotropic glutamate receptor NAMs were initially discovered via a combination of fragment-based screening and medicinal chemistry, and their binding modes subsequently identified crystallographically (Christopher et al., 2015). It is important to note that while this kind of structure-guided drug discovery and optimization is relatively routine for other drug targets, where several leading compounds are serially crystallized with the molecular target and derivatives synthesized based on insights gained from the structures, routine crystallization of GPCRs remains highly challenging.
Structure-based virtual discovery of novel chemotypes for GPCRs
The availability of GPCR structures has inspired a large number of successful computational campaigns aimed at discovering novel chemical probes with unique chemical structures (e.g. chemotypes) for GPCRs (e.g. virtual screening). One of the earliest successes was automated molecular docking to β2AR that yielded nanomolar potency inverse agonists (Kolb et al., 2009). One of the resulting compounds when crystallized revealed a novel β2AR ligand binding conformation (Wacker et al., 2010).
Given this and other successes in structure-guided discovery of new GPCR ligands, one might ask whether it might be possible to use molecular models in lieu of experimentally-determined structures as templates for docking in other receptors. An early example of this approach with the D3 dopamine receptor (Carlsson et al., 2011) featured 3.3 million compounds docked initially against a homology model of the D3 dopamine receptor that used β2AR as a template. Several nanomolar potency antagonists were discovered. Docking was also performed using the D3 receptor x-ray structure as template (Chien et al., 2010) and, surprisingly, a distinct set of nanomolar potency antagonists was identified. What emerged from this exercise was the observation that the slightly different conformations sampled by the crystal structure and the models of this receptor yielded chemically distinct sets of active molecules.
An exciting extension of this overall approach—and one which may prove to provide a template going forward—is the serial structure-guided and docking-based optimization of active molecules into potential therapeutic entities. In one instance, 3 million commercially available compounds from the ZINC database (Irwin et al., 2005) were docked against an inactive conformation of the μ opioid receptor (Manglik et al., 2016), the molecular receptor for morphine. Initial lead compounds from the docking campaign were experimentally tested and commercially available analogs were purchased to obtain ligands of higher affinity. Active compounds were then optimized through modest medicinal chemistry, guided by their docking pose, structural considerations, and experimental pharmacology. This process ultimately yielded PZM-21, a selective, high affinity μ opioid agonist with modest G protein bias that was ultimately found to be analgesic with fewer side-effects compared to morphine (Manglik et al., 2016).
Structure-inspired discovery of chemical probes for oGPCRs
That homology models can be used to identify novel chemical matter using virtual ligand screening has proven to be particularly useful for oGPCRs, for which conventional ligand screening campaigns have not yielded useful probes. For instance, selective allosteric modulators (both NAMs and PAMs) were identified for the oGPCRs GPR68 and GPR65, and further optimized by a combination of physical screening and in silico docking (Huang et al., 2015b). One GPR68 PAM, ogerin, was demonstrated to have on-target activity in vivo by modulating conditioned fear responses in mice. A similar approach has recently been used to identify small molecule ligands for the oGPCRs GPR171 (Wardman et al., 2016) and MRGPRX2 (Lansu et al., 2017), and these compounds were subsequently used to further characterize the receptor’s role in feeding behavior and itch, respectively.
The successful use of homology models, however, largely depends on the accuracy of the models. The analysis of docking results is a process that considers previous biochemical and pharmacological information regarding ligand-receptor interactions as validated from crystal structures. Accordingly, the description of key residues accessible for ligand binding in class A GPCRs (Gloriam et al., 2009) or the definition of so-called protein-ligand interaction fingerprints [reviewed in (Vass et al., 2016)], not only greatly enhances the identification of relevant molecules, but also highlights the importance of using tool compounds to study GPCRs interactions and function.
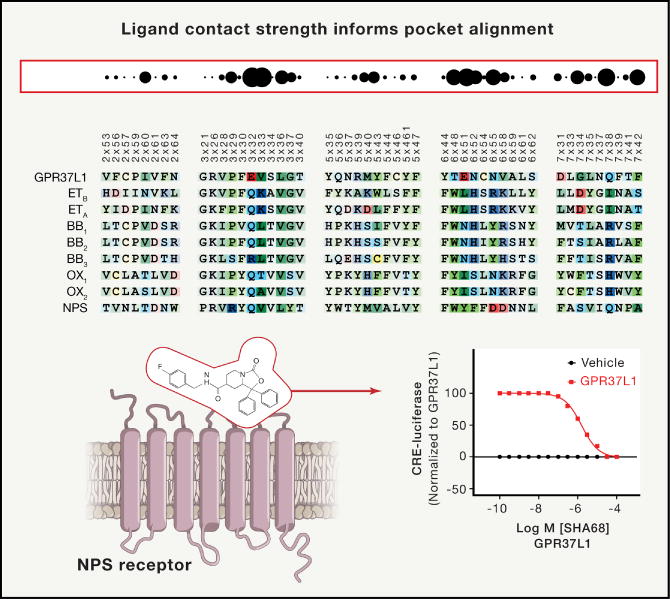
An alternative approach dubbed ‘pickpocketing’, analyzes ligand-residue contact strength to identify potential ligand-binding similarity between non-homologous receptors. This method allowed the authors of a recent paper to identify new chemotypes for the oGPCR GPR37L, by “stealing” ligands from orexin and neuropeptide receptors that possess sufficient chemical matter and exhibit substantial contact-strength similarity with GPR37L (Ngo et al., 2016) (Fig 5). Taken together these recent successes indicate that homology model-based approaches can be powerful and useful approaches for discovering agonists, antagonists and allosteric modulators of oGPCRs.
Figure 5. Computational approaches generate novel tool compounds for GPCRs.
As shown here, the “Pickpocketing” approach identifies GPCRs with potentially related ligand binding properties according to ligand contact strength informed pocket alignment. Similar ligand binding properties between the Neuropeptide S receptor (NPS) and the orphan GPCR GPR37L1 identified the NPS receptor ligand SHA68 as a novel ligand to interrogate GPR37L1 function. The figure is extensively modified from Ngo et al (2017) with permission.
CONCLUSIONS AND FUTURE DIRECTIONS
As is clear, the past 10 years have witnessed a renaissance in GPCR research catalyzed by structural insights into the most basic aspects of GPCR ligand binding and signaling. Concepts of ligand recognition previously considered to be theoretical—such as allosteric modulation (Kenakin et al., 1989)—are now firmly validated from structural and functional perspectives. Additionally, we are beginning to get hints of how different ligands might stabilize distinct conformational ensembles leading to a variety of active, inactive, and biased states as was predicted many years ago (Urban et al., 2007). Finally, the plethora of structures has provided computational biologists tremendous opportunities for both structure-guided and structure-inspired drug discovery.
Although these advances are breathtaking when considered from a historical perspective, huge gaps remain in our understanding of GPCR structure, function, signaling and pharmacology. Clearly, our understanding of GPCR functional selectivity from a mechanistic, molecular perspective is inadequate and remains phenomenological. Thus, for instance, there are no structures of GPCRs with G proteins in complex with G protein biased agonists, nor are there structures of GPCRs with arrestin-biased ligands in complex with arrestin.
Additionally, how biased ligands might stabilize distinct states is unknown and approaches to discover and develop such ligands is discovery- rather than mechanistically-based. Consider, for instance, that although the structures of more than 40 GPCRs have been solved by x-ray crystallography, in most cases this has been achieved with only one ligand and in only an inactive state. For some receptors including β2AR and A2AAR multiple ligand complexes are available, but only minimal plasticity within the binding pocket is evident. In our recent studies on 5-HT2B receptors, however, we observed extensive plasticity within the binding pocket of a GPCR when examining structures stabilized by similar compounds (Wacker et al., 2017). From the foregoing, we predict that depending upon the particular GPCR and ligands that are examined a range conformational rearrangements within the binding site will be observed. Further, it is clear that regions of GPCRs normally considered to be ‘undruggable’ (e.g. intracellular loops, interfaces and so on) (Oswald et al., 2016) are sites of action for current and potential therapeutics. Thus, understanding and predicting how GPCR binding sites change in complex with specific ligands, and how these alterations ultimately lead to differential signaling and physiological outcomes remains a major grand challenge for structural biologists and molecular pharmacologists.
Finally, although GPCRs continue to be the most popular family of molecular targets for therapeutic drug discovery, current medications target a vanishingly small number of GPCRs. Although drugs are in development targeting approximately 100 GPCRs, huge gaps remain regarding our understanding of oGPCRs and how they might be useful as therapeutic targets. As was recently emphasized, for more than half of the druggable GPCRs in the human genome, little useful information is available regarding their roles in normal physiology much less their utility as therapeutic targets (Roth et al., 2015). Additionally, many GPCRs remain “orphan” by having no bona fide endogenous agonists identified (Roth et al., 2015). While classical knock-out studies and/or chemo- and optogenetic approaches may provide some insight into oGPCR function, it is tool compounds that are desperately required to delineate their physiological roles, characterize their function on a molecular level, and perhaps thereby identify novel therapeutic targets. Application of technologies such as virtual ligand screening and a variety of computational approaches to build ligands de novo, both enabled by molecular GPCR studies, are already beginning to yield valuable chemical tools. With the implementation of machine learning technology to better predict compound activity in a complex biological system, the speed and success rate of computational screening is more than likely to drastically increase over the coming years. Based on an ever-increasing number of GPCR structures, computational approaches are thus poised to augment or even replace manual high throughput drug screening, while accelerating the generation of new tool compounds to study GPCR function or develop drug design platforms. Ligands, synthetic or natural, are the single most powerful tool for elucidating GPCR mechanisms and physiology, data that will not only help to better understand the single largest class of membrane proteins, but likely also translate into better therapies.
Figure 6.
Acknowledgments
This review was supported by grants from the National Institute of Health (RO1MH112205; UO1MH104974; U19MH82441; and PO1DA035764) and the Michael Hooker Distinguished Professorship to BLR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Airan RD, Thompson KR, Fenno LE, Bernstein H, Deisseroth K. Temporally precise in vivo control of intracellular signalling. Nature. 2009;458:1025–1029. doi: 10.1038/nature07926. [DOI] [PubMed] [Google Scholar]
- Aitken M, Berndt ER, Cutler DM. Prescription drug spending trends in the United States: looking beyond the turning point. Health Aff (Millwood) 2009;28:w151–160. doi: 10.1377/hlthaff.28.1.w151. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Southan C, Davies JA, Collaborators, C. The Concise Guide to PHARMACOLOGY 2015/16: G protein-coupled receptors. Br J Pharmacol. 2015;172:5744–5869. doi: 10.1111/bph.13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JA, Roth BL. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2011a;51:117–144. doi: 10.1146/annurev-pharmtox-010510-100553. [DOI] [PubMed] [Google Scholar]
- Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, Jensen NH, Che X, Bai X, Frye SV, Wetsel WC, Caron MG, Javitch JA, Roth BL, Jin J. Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci U S A. 2011b;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature. 1997;385:347–350. doi: 10.1038/385347a0. [DOI] [PubMed] [Google Scholar]
- Asano T, Katada T, Gilman AG, Ross EM. Activation of the inhibitory GTP-binding protein of adenylate cyclase, Gi, by beta-adrenergic receptors in reconstituted phospholipid vesicles. J Biol Chem. 1984;259:9351–9354. [PubMed] [Google Scholar]
- Benovic JL, DeBlasi A, Stone WC, Caron MG, Lefkowitz RJ. Beta-adrenergic receptor kinase: primary structure delineates a multigene family. Science. 1989;246:235–240. doi: 10.1126/science.2552582. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, Pike NB, Strum JC, Steplewski KM, Murdock PR, Holder JC, Marshall FH, Szekeres PG, Wilson S, Ignar DM, Foord SM, Wise A, Dowell SJ. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278:11312–11319. doi: 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- Bunzow JR, Sonders MS, Arttamangkul S, Harrison LM, Zhang G, Quigley DI, Darland T, Suchland KL, Pasumamula S, Kennedy JL, Olson SB, Magenis RE, Amara SG, Grandy DK. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol Pharmacol. 2001;60:1181–1188. doi: 10.1124/mol.60.6.1181. [DOI] [PubMed] [Google Scholar]
- Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat Chem Biol. 2011;7:769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerione RA, Codina J, Benovic JL, Lefkowitz RJ, Birnbaumer L, Caron MG. The mammalian beta 2-adrenergic receptor: reconstitution of functional interactions between pure receptor and pure stimulatory nucleotide binding protein of the adenylate cyclase system. Biochemistry. 1984;23:4519–4525. doi: 10.1021/bi00315a003. [DOI] [PubMed] [Google Scholar]
- Changeux JP, Christopoulos A. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell. 2016;166:1084–1102. doi: 10.1016/j.cell.2016.08.015. [DOI] [PubMed] [Google Scholar]
- Chidiac P, Hebert T, Valiquette M, Dennis M, Bouvier M. Inverse agonist activity of Beta-adrenergic antagonists. Molecular Pharmacology. 1993;45:490–499. [PubMed] [Google Scholar]
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher JA, Aves SJ, Bennett KA, Dore AS, Errey JC, Jazayeri A, Marshall FH, Okrasa K, Serrano-Vega MJ, Tehan BG, Wiggin GR, Congreve M. Fragment and Structure-Based Drug Discovery for a Class C GPCR: Discovery of the mGlu5 Negative Allosteric Modulator HTL14242 (3-Chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile) J Med Chem. 2015;58:6653–6664. doi: 10.1021/acs.jmedchem.5b00892. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Changeux JP, Catterall WA, Fabbro D, Burris TP, Cidlowski JA, Olsen RW, Peters JA, Neubig RR, Pin JP, Sexton PM, Kenakin TP, Ehlert FJ, Spedding M, Langmead CJ. International Union of Basic and Clinical Pharmacology. XC. multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev. 2014;66:918–947. doi: 10.1124/pr.114.008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AJ. The antagonism of acetyl choline by atropine. J Physiol. 1926;61:547–556. doi: 10.1113/jphysiol.1926.sp002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M, Andrews SP, Dore AS, Hollenstein K, Hurrell E, Langmead CJ, Mason JS, Ng IW, Tehan B, Zhukov A, Weir M, Marshall FH. Discovery of 1,2,4-triazine derivatives as adenosine A(2A) antagonists using structure based drug design. J Med Chem. 2012;55:1898–1903. doi: 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem. 1980;255:7108–7117. [PubMed] [Google Scholar]
- DeWire SM, Violin JD. Biased ligands for better cardiovascular drugs: dissecting G-protein-coupled receptor pharmacology. Circ Res. 2011;109:205–216. doi: 10.1161/CIRCRESAHA.110.231308. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344:708–717. doi: 10.1124/jpet.112.201616. [DOI] [PubMed] [Google Scholar]
- Dillon GM, Lubbers LS, Ferguson MT, Lao JZ, Huang RR, Xiao JC, Fong TM, Hale JJ, Rupprecht K, Miao S, Rowe BA, Kornecook TJ, Dodart JC. MK-7128, a novel CB1 receptor inverse agonist, improves scopolamine-induced learning and memory deficits in mice. Behav Pharmacol. 2011;22:91–100. doi: 10.1097/FBP.0b013e3283423d7e. [DOI] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci U S A. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JM, Fedele V, Szklarz M, Abdul Azeez KR, Salah E, Mikolajczyk J, Romanov S, Sepetov N, Huang XP, Roth BL, Al Haj Zen A, Fourches D, Muratov E, Tropsha A, Morris J, Teicher BA, Kunkel M, Polley E, Lackey KE, Atkinson FL, Overington JP, Bamborough P, Muller S, Price DJ, Willson TM, Drewry DH, Knapp S, Zuercher WJ. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat Biotechnol. 2016;34:95–103. doi: 10.1038/nbt.3374. [DOI] [PubMed] [Google Scholar]
- Farrens DAC, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. Molecular control of delta-opioid receptor signalling. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Gimpl G. Interaction of G protein coupled receptors and cholesterol. Chem Phys Lipids. 2016;199:61–73. doi: 10.1016/j.chemphyslip.2016.04.006. [DOI] [PubMed] [Google Scholar]
- Gloriam DE, Foord SM, Blaney FE, Garland SL. Definition of the G protein-coupled receptor transmembrane bundle binding pocket and calculation of receptor similarities for drug design. J Med Chem. 2009;52:4429–4442. doi: 10.1021/jm900319e. [DOI] [PubMed] [Google Scholar]
- Goodman O, Krupnick J, Santini F, Gurevich V, Penn R, Gagnon A, Keen J, Benovic J. b-arrestin acts as a clathrin adaptor in endocytosis of the b2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- Hamann J, Aust G, Arac D, Engel FB, Formstone C, Fredriksson R, Hall RA, Harty BL, Kirchhoff C, Knapp B, Krishnan A, Liebscher I, Lin HH, Martinelli DC, Monk KR, Peeters MC, Piao X, Promel S, Schoneberg T, Schwartz TW, Singer K, Stacey M, Ushkaryov YA, Vallon M, Wolfrum U, Wright MW, Xu L, Langenhan T, Schioth HB. International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol Rev. 2015;67:338–367. doi: 10.1124/pr.114.009647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenstein K, Kean J, Bortolato A, Cheng RK, Dore AS, Jazayeri A, Cooke RM, Weir M, Marshall FH. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, Zhao S, Shui W, Li S, Korde A, Laprairie RB, Stahl EL, Ho JH, Zvonok N, Zhou H, Kufareva I, Wu B, Zhao Q, Hanson MA, Bohn LM, Makriyannis A, Stevens RC, Liu ZJ. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell. 2016;167:750–762e714. doi: 10.1016/j.cell.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Manglik A, Venkatakrishnan AJ, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, Kobilka BK. Structural insights into micro-opioid receptor activation. Nature. 2015a;524:315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XP, Karpiak J, Kroeze WK, Zhu H, Chen X, Moy SS, Saddoris KA, Nikolova VD, Farrell MS, Wang S, Mangano TJ, Deshpande DA, Jiang A, Penn RB, Jin J, Koller BH, Kenakin T, Shoichet BK, Roth BL. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature. 2015b;527:477–483. doi: 10.1038/nature15699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Shoichet BK. ZINC—a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A, Dore AS, Lamb D, Krishnamurthy H, Southall SM, Baig AH, Bortolato A, Koglin M, Robertson NJ, Errey JC, Andrews SP, Teobald I, Brown AJ, Cooke RM, Weir M, Marshall FH. Extra-helical binding site of a glucagon receptor antagonist. Nature. 2016;533:274–277. doi: 10.1038/nature17414. [DOI] [PubMed] [Google Scholar]
- Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, Xu Q, de Waal PW, Ke J, Tan MH, Zhang C, Moeller A, West GM, Pascal BD, Van Eps N, Caro LN, Vishnivetskiy SA, Lee RJ, Suino-Powell KM, Gu X, Pal K, Ma J, Zhi X, Boutet S, Williams GJ, Messerschmidt M, Gati C, Zatsepin NA, Wang D, James D, Basu S, Roy-Chowdhury S, Conrad CE, Coe J, Liu H, Lisova S, Kupitz C, Grotjohann I, Fromme R, Jiang Y, Tan M, Yang H, Li J, Wang M, Zheng Z, Li D, Howe N, Zhao Y, Standfuss J, Diederichs K, Dong Y, Potter CS, Carragher B, Caffrey M, Jiang H, Chapman HN, Spence JC, Fromme P, Weierstall U, Ernst OP, Katritch V, Gurevich VV, Griffin PR, Hubbell WL, Stevens RC, Cherezov V, Melcher K, Xu HE. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC. Allosteric sodium in class A GPCR signaling. Trends Biochem Sci. 2014;39:233–244. doi: 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, Whaley R, Glennon RA, Hert J, Thomas KL, Edwards DD, Shoichet BK, Roth BL. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Boselli C. Pharmacologic discrimination between receptor heterogeneity and allosteric interaction: resultant analysis of gallamine and pirenzepine antagonism of muscarinic responses in rat trachea. J Pharmacol Exp Ther. 1989;250:944–952. [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci. 2012;3:193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Herenbrink C, Sykes DA, Donthamsetti P, Canals M, Coudrat T, Shonberg J, Scammells PJ, Capuano B, Sexton PM, Charlton SJ, Javitch JA, Christopoulos A, Lane JR. The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun. 2016;7:10842. doi: 10.1038/ncomms10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK. Structure-based discovery of beta2-adrenergic receptor ligands. Proc Natl Acad Sci U S A. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra AJ, Roumen L, Leurs R, de Esch IJ, de Graaf C. From heptahelical bundle to hits from the Haystack: structure-based virtual screening for GPCR ligands. Methods Enzymol. 2013;522:279–336. doi: 10.1016/B978-0-12-407865-9.00015-7. [DOI] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- Lafferty-Whyte K, Mormeneo D, Del Fresno Marimon M. Trial watch: Opportunities and challenges of the 2016 target landscape. Nat Rev Drug Discov. 2016 doi: 10.1038/nrd.2016.263. [DOI] [PubMed] [Google Scholar]
- Lansu K, Karpiak J, Liu J, Huang XP, McCorvy JD, Kroeze WK, Che T, Nagase H, Carroll FI, Jin J, Shoichet BK, Roth BL. In silico design of novel probes for the atypical opioid receptor MRGPRX2. Nat Chem Biol. 2017;13:529–536. doi: 10.1038/nchembio.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzafame A, Christopoulos A, Mitchelson F. Three allosteric modulators act at a common site, distinct from that of competitive antagonists, at muscarinic acetylcholine M2 receptors. J Pharmacol Exp Ther. 1997;282:278–285. [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Liang YL, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, Tarrasch J, Thal DM, Furness SGB, Christopoulos G, Coudrat T, Danev R, Baumeister W, Miller LJ, Christopoulos A, Kobilka BK, Wootten D, Skiniotis G, Sexton PM. Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature. 2017;546:118–123. doi: 10.1038/nature22327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science. 2012a;335:1106–1110. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, AP IJ, Cherezov V, Stevens RC. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012b;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M, Benovic J, Codina J, Caron M, Lefkowitz R. Beta-Arrestin: a protein that regulates Beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, Prosser RS, Kobilka BK. Structural Insights into the Dynamic Process of beta2-Adrenergic Receptor Signaling. Cell. 2015;161:1101–1111. doi: 10.1016/j.cell.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hubner H, Huang XP, Sassano MF, Giguere PM, Lober S, Da D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK. Structure-based discovery of opioid analgesics with reduced side effects. Nature. 2016;537:185–190. doi: 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JS, Bortolato A, Congreve M, Marshall FH. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol Sci. 2012;33:249–260. doi: 10.1016/j.tips.2012.02.005. [DOI] [PubMed] [Google Scholar]
- McAlear SD, Kraft TW, Gross AK. 1 rhodopsin mutations in congenital night blindness. Adv Exp Med Biol. 2010;664:263–272. doi: 10.1007/978-1-4419-1399-9_30. [DOI] [PubMed] [Google Scholar]
- Moore AR, Ceraudo E, Sher JJ, Guan Y, Shoushtari AN, Chang MT, Zhang JQ, Walczak EG, Kazmi MA, Taylor BS, Huber T, Chi P, Sakmar TP, Chen Y. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet. 2016;48:675–680. doi: 10.1038/ng.3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello JP, Salahpour A, Laperriere A, Bernier V, Arthus MF, Lonergan M, Petaja-Repo U, Angers S, Morin D, Bichet DG, Bouvier M. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000;105:887–895. doi: 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo T, Ilatovskiy AV, Stewart AG, Coleman JL, McRobb FM, Riek RP, Graham RM, Abagyan R, Kufareva I, Smith NJ. Orphan receptor ligand discovery by pickpocketing pharmacological neighbors. Nat Chem Biol. 2016 doi: 10.1038/nchembio.2266. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- Nobles M, Benians A, Tinker A. Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proc Natl Acad Sci U S A. 2005;102:18706–18711. doi: 10.1073/pnas.0504778102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor C, White KL, Doncescu N, Didenko T, Roth BL, Czaplicki G, Stevens RC, Wuthrich K, Milon A. NMR structure and dynamics of the agonist dynorphin peptide bound to the human kappa opioid receptor. Proc Natl Acad Sci U S A. 2015;112:11852–11857. doi: 10.1073/pnas.1510117112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald C, Rappas M, Kean J, Dore AS, Errey JC, Bennett K, Deflorian F, Christopher JA, Jazayeri A, Mason JS, Congreve M, Cooke RM, Marshall FH. Intracellular allosteric antagonism of the CCR9 receptor. Nature. 2016;540:462–465. doi: 10.1038/nature20606. [DOI] [PubMed] [Google Scholar]
- Paek J, Kalocsay M, Staus DP, Wingler L, Pascolutti R, Paulo JA, Gygi SP, Kruse AC. Multidimensional Tracking of GPCR Signaling via Peroxidase-Catalyzed Proximity Labeling. Cell. 2017;169:338–349 e311. doi: 10.1016/j.cell.2017.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Pan Y, Metzenberg A, Das S, Jing B, Gitschier J. Mutations in the V2 vasopressin receptor gene are associated with X-linked nephrogenic diabetes insipidus. Nat Genet. 1992;2:103–106. doi: 10.1038/ng1092-103. [DOI] [PubMed] [Google Scholar]
- Pert CB, Pasternak G, Snyder SH. Opiate agonists and antagonists discriminated by receptor binding in brain. Science. 1973;182:1359–1361. doi: 10.1126/science.182.4119.1359. [DOI] [PubMed] [Google Scholar]
- Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, Sharma KS, Durand G, Pucci B, Trinquet E, Zwier JM, Deupi X, Bron P, Baneres JL, Mouillac B, Granier S. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci U S A. 2012;109:6733–6738. doi: 10.1073/pnas.1201093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rask-Andersen M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nat Rev Drug Discov. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011a;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011b;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature. 2013;502:575–579. doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL. Drugs and valvular heart disease. N Engl J Med. 2007;356:6–9. doi: 10.1056/NEJMp068265. [DOI] [PubMed] [Google Scholar]
- Roth BL. DREADDs for Neuroscientists. Neuron. 2016;89:683–694. doi: 10.1016/j.neuron.2016.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc Natl Acad Sci U S A. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Kroeze WK. Integrated Approaches for Genome-wide Interrogation of the Druggable Non-olfactory G Protein-coupled Receptor Superfamily. J Biol Chem. 2015;290:19471–19477. doi: 10.1074/jbc.R115.654764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, Hufeisen SJ, Roth BL. Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation. 2000;102:2836–2841. doi: 10.1161/01.cir.102.23.2836. [DOI] [PubMed] [Google Scholar]
- Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, Low JA. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- Scanlan TS, Suchland KL, Hart ME, Chiellini G, Huang Y, Kruzich PJ, Frascarelli S, Crossley DA, Bunzow JR, Ronca-Testoni S, Lin ET, Hatton D, Zucchi R, Grandy DK. 3-Iodothyronamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat Med. 2004;10:638–642. doi: 10.1038/nm1051. [DOI] [PubMed] [Google Scholar]
- Shihoya W, Nishizawa T, Okuta A, Tani K, Dohmae N, Fujiyoshi Y, Nureki O, Doi T. Activation mechanism of endothelin ETB receptor by endothelin-1. Nature. 2016;537:363–368. doi: 10.1038/nature19319. [DOI] [PubMed] [Google Scholar]
- Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science. 1991;252:802–808. doi: 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- Smit MJ, Vischer HF, Bakker RA, Jongejan A, Timmerman H, Pardo L, Leurs R. Pharmacogenomic and structural analysis of constitutive g protein-coupled receptor activity. Annu Rev Pharmacol Toxicol. 2007;47:53–87. doi: 10.1146/annurev.pharmtox.47.120505.105126. [DOI] [PubMed] [Google Scholar]
- Staus DP, Strachan RT, Manglik A, Pani B, Kahsai AW, Kim TH, Wingler LM, Ahn S, Chatterjee A, Masoudi A, Kruse AC, Pardon E, Steyaert J, Weis WI, Prosser RS, Kobilka BK, Costa T, Lefkowitz RJ. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature. 2016;535:448–452. doi: 10.1038/nature18636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahtaoui C, Parrot I, Klotz P, Guillier F, Galzi JL, Hibert M, Ilien B. Fluorescent pirenzepine derivatives as potential bitopic ligands of the human M1 muscarinic receptor. J Med Chem. 2004;47:4300–4315. doi: 10.1021/jm040800a. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ. Hitchhiking on the heptahelical highway: structure and function of 7TM receptor complexes. Nat Rev Mol Cell Biol. 2016;17:439–450. doi: 10.1038/nrm.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen AR, Plouffe B, Cahill TJ, 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, Huang L, Breton B, Heydenreich FM, Sunahara RK, Skiniotis G, Bouvier M, Lefkowitz RJ. GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell. 2016;166:907–919. doi: 10.1016/j.cell.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Urs NM, Gee SM, Pack TF, McCorvy JD, Evron T, Snyder JC, Yang X, Rodriguiz RM, Borrelli E, Wetsel WC, Jin J, Roth BL, O’Donnell P, Caron MG. Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc Natl Acad Sci U S A. 2016;113:E8178–E8186. doi: 10.1073/pnas.1614347113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Westhuizen ET, Valant C, Sexton PM, Christopoulos A. Endogenous allosteric modulators of G protein-coupled receptors. J Pharmacol Exp Ther. 2015;353:246–260. doi: 10.1124/jpet.114.221606. [DOI] [PubMed] [Google Scholar]
- Vass M, Kooistra AJ, Ritschel T, Leurs R, de Esch IJ, de Graaf C. Molecular interaction fingerprint approaches for GPCR drug discovery. Curr Opin Pharmacol. 2016;30:59–68. doi: 10.1016/j.coph.2016.07.007. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, Stevens RC. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc. 2010;132:11443–11445. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Seng W, McCorvy JD, Betz RM, Venkatakrishnan AJ, Levit A, Lansu K, Schools ZL, Che T, Nichols DE, Shoichet BK, Dror RO, Roth BL. Crystal structure of an LSD-bound human serotonin receptor. Cell. 2017 doi: 10.1016/j.cell.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, Liu W, Xu HE, Cherezov V, Roth BL, Stevens RC. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, McCorvy JD, Gao X, Zhou XE, Melcher K, Zhang C, Bai F, Yang H, Yang L, Jiang H, Roth BL, Cherezov V, Stevens RC, Xu HE. Structural basis for molecular recognition at serotonin receptors. Science. 2013;340:610–614. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardman JH, Gomes I, Bobeck EN, Stockert JA, Kapoor A, Bisignano P, Gupta A, Mezei M, Kumar S, Filizola M, Devi LA. Identification of a small-molecule ligand that activates the neuropeptide receptor GPR171 and increases food intake. Sci Signal. 2016;9:ra55. doi: 10.1126/scisignal.aac8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, Gvozdenovic-Jeremic J, Shah P, Shiloach J, Tate CG, Grisshammer R. Structure of the agonist-bound neurotensin receptor. Nature. 2012;490:508–513. doi: 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, Roth BL. The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther. 2015;352:98–109. doi: 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Gao ZG, Zhang K, Kiselev E, Crane S, Wang J, Paoletta S, Yi C, Ma L, Zhang W, Han GW, Liu H, Cherezov V, Katritch V, Jiang H, Stevens RC, Jacobson KA, Zhao Q, Wu B. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature. 2015;520:317–321. doi: 10.1038/nature14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sun B, Feng D, Hu H, Chu M, Qu Q, Tarrasch JT, Li S, Sun Kobilka T, Kobilka BK, Skiniotis G. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature. 2017 doi: 10.1038/nature22394. [DOI] [PMC free article] [PubMed] [Google Scholar]