

Abstract
The mechanism for improved therapeutic efficacy of the combination therapy with albumin-bound paclitaxel (nAb-PTX) and gemcitabine (gem) for pancreatic ductal adenocarcinoma (PDAC) has been ascribed to enhanced gem transport by nAb-PTX. Here, we used an orthotopic mouse model of gem-resistant human PDAC in which increasing gem transport would not improve the efficacy, thus revealing the importance of nAb-PTX transport. We aimed to evaluate therapeutic outcomes and transport of nAb-PTX to PDAC as a result of: (1)encapsulating nAb-PTX in multistage nanovectors (MSV); (2)effect of gem on caveolin-1 expression. Treatment with MSV/nAb-PTX+gem was highly efficient in prolonging animal survival in comparison to other therapeutic regimens. MSV/nAb-PTX+gem also caused a substantial increase in tumor PTX accumulation and significantly reduced tumor growth and tumor cell proliferation, and increased apoptosis. Moreover, gem enhanced caveolin-1 expression in vitro and in vivo, thereby improving transport of nAb-PTX to PDAC. This data was confirmed by analysis of PDACs from patients who received gem-based neo-adjuvant chemotherapy. In conclusion, we found that nAb-PTX treatment of gem-resistant PDAC can be enhanced by: (1)gem through up-regulation of caveolin-1; (2)MSV through increasing accumulation of nAb-PTX in the tumor.
Keywords: Pancreatic cancer, gemcitabine, drug resistance, nab-paclitaxel, multistage nanovectors, transport
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is currently the fourth leading cause of cancer death in Europe and the US (1–3) with the five-year survival rate of 2% and the median survival less than six months (4). Multiple clinical trials have failed to improve the efficacy of the first line chemotherapeutic, gemcitabine (gem) by combining additional therapeutics (5–8). Extensive stroma and hypovascularization reduce drug transport in PDAC impeding treatment efficiency (9). Resistance to gem, frequently observed in patients with PDAC, is also responsible for limited survival (8). Recently, the combination therapy with gem and nanoparticle albumin-bound paclitaxel (nAb-PTX) has been shown to significantly improve therapeutic efficacy in patients with metastatic PDAC vs. gem monotherapy, thus, becoming a first-line treatment (10). Although nAb-PTX+gem improved overall survival (8.5 vs. 6.7 months), progression free survival (5.5 vs. 3.7 months), the one-year survival rate (35 vs. 22%), and the response rate (23 vs. 7%) as compared to gem monotherapy, better therapies are still necessary.
Studies investigating the transport-based synergistic mechanism of nAb-PTX and gem have revealed increased tumor accumulation of gem caused by nAb-PTX, which decreased the tumor stroma density (11–13). Interestingly, when comparing the physicochemical properties of the two drugs, nAb-PTX (≈ 70 kDa macromolecule-bound drug) should experience more difficulty being transported across the poorly vascularized tumor stroma versus gem (263 Da). This suggests that improved therapeutic efficacy of the combination may also account for nAb-PTX transport enhancement by gem. Previous studies indicate that in PDAC cells gem treatment increased the expression of a variety of transporters, including caveolin-1 (cav-1, albumin transporter) (14, 15). These clinical and pre-clinical findings prompted our hypothesis that increased efficacy of treatment with nAb-PTX+gem could be due to an enhancement of nAb-PTX transport, caused by gem. To prove it, we used a mouse model of orthotopic PDAC resistant to different dosages of gem monotherapy based on orthotopic injection of a low number of L3.6pL PDAC (16–18). The use of L3.6pL tumor model had a dual purpose. Since these tumors are resistant to gem therapy (16–18), in the L3.6pl PDAC model, increasing gem transport would not improve the therapeutic efficacy, thus isolating the effect of nAb-PTX transport component in the combination therapy. As opposed to L3.6pl PDAC model, other human PDAC mouse models are sensitive to gem treatment and, therefore, they could not be implemented in our experiments (19–21). Additionally, we have previously shown that this orthotopic model of slow developing PDAC that takes at least 30 days to develop, can produce a dense stroma comparable to the tumor microenvironment in the clinical PDAC specimens (16).
Interestingly, transport properties of nAb-PTX are important for its therapeutic efficacy (22), as tumor accumulation of nAb-PTX occurs due to albumin transport through cav-1 expressed on the tumor endothelium followed by caveolae-mediated transcytosis into the PDAC. Another possible mechanism involves nAb-PTX binding to a secreted protein acidic and rich in cysteine (SPARC), a glycoprotein secreted in the tumor extracellular matrix (23).
We have previously shown that loading various compounds into intravenously injected multistage nanovectors (MSV) enhances their accumulation in PDAC and other cancers (22, 24–32) without causing significant toxicity in major organs (29). For example, our recent study in hypoperfused breast and lung tumors’ liver metastases has shown that retention and release of MSV-bound nAb-PTX (MSV/nAb-PTX) from the tumor associated macrophages enabled pronounced therapeutic and survival benefits (31). Thus, to retain nAb-PTX in the PDAC microenvironment, we have used MSV.
The objectives of this study were: (1) to assess the retention of nAb-PTX in the tumor stroma by MSV and consequent therapeutic efficacy of MSV/nAb-PTX in combination with gem in gem-resistant PDAC in mice; (2) to evaluate the effect of gem on expression of albumin transporter protein caveolin-1 (cav-1) and transport of nAb-PTX to tumor.
2. Materials and Methods
2.1 Materials
Minimal essential medium (MEM), sodium pyruvate, nonessential amino acids (NEAA), L-glutamine, and MEM vitamin solution were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Fetal bovine serum (FBS) was purchased from Atlas Biologicals (Fort Collins, CO, USA) and DeadEnd Fluorometric TUNEL System from Promega Biosciences LLC (San Luis Obispo, CA, USA). Gemcitabine (Gemzar®, Eli Lilly Co. Indianapolis, IN, USA) and nAb-PTX (Abraxane®, Celgene, Summit, NJ, USA) were acquired from the pharmacy at Houston Methodist Hospital. Deuterated PTX (D5) was purchased from Santa Cruz Biotechnology (Dallas, Texas, USA). Anti-mouse Ki67 and cav-1 rabbit polyclonal antibody, and anti-mouse CD31 rat polyclonal antibody were purchased from Abcam (Cambridge, MA, USA). Alexa Fluor 488-conjugated AffiniPure Goat Anti-Rabbit IgG used for cav-1 detection, rhodamine (TRITC)-conjugated AffiniPure Donkey Anti-Rat IgG for CD31 detection, and secondary antibodies were purchased from Jackson ImmunoResearch Laboratories Inc. (West Grove, PA, USA).
Anti-smooth muscle actin antibody was acquired from Dako (Santa Clara, CA, USA), while anti-mouse HRP conjugated secondary antibody was purchased from Vector labs (Burlingame, CA, USA). Cav-1 ELISA kit was purchased from Cell Signaling Technology (Danvers, MA, USA). Optimal cutting temperature compound (OCT) was acquired from Sakura Finetek (Torrance, CA, USA).
2.2 MSV fabrication and characterization
Discoidal porous silicon particles 1000 × 400 nm (diameter × thickness) were fabricated as previously described (33). Briefly, a porous silicon film was created by electrochemical etching of p-type silicon wafers in hydrofluoric acid/ethanoic solution. Following deposition of low-temperature oxide (LTO) sacrificial layer on the wafer, cylindrical trenches were patterned. The wafers were sonicated in isopropyl alcohol to release the MSV. Next, MSV were dried, oxidized for 2 h in a piranha solution [1:2 H2O2: H2SO4 (v/v)] at 100–110°C, washed, and stored at 4°C in water. A Multisizer 4 Coulter Particle Counter (Beckman Coulter, Fuellerton, CA, USA) was used to determine MSV size, size distribution, and count. Zeta potential was measured in a phosphate buffer (PB, pH 7.4) using a Zetasizer Nano ZS (Malvern Instruments, Malvern, Worcestershire, UK). MSV were modified with 3-aminopropyl-triethoxysilane (APTES) as previously reported (24, 33, 34) and lyophilized utilizing Freezone Freeze Dry System (Labconco, Kansas City, MO). MSV/nAb-PTX were constructed as previously described (31).
2.3 Cell culture
L3.6pl human PDAC cells and mouse immortalized endothelial cells were kindly provided by Dr. I. J. Fidler (The University of Texas MD Anderson Cancer Center) and maintained in MEM supplemented with 10% FBS, sodium pyruvate, NEAA, L-glutamine, MEM vitamin, and a penicillin-streptomycin mixture (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) in humidified atmosphere at 37°C with 5% CO2. For in vivo injection, L3.6pl cell suspension was obtained from subconfluent cultures with >90% viability.
2.4 Orthotopic tumor implantation
Male nude mice (Nu/Nu, 43–56 days) were purchased from Charles River Laboratories (Wilmington, MA) and maintained in the animal facility at Houston Methodist Research Institute (HMRI) approved by the AAALAC and in accordance with the National Institutes of Health guidelines for the care and use of Laboratory animals (NIH Publications No. 8023, revised 1978). To establish orthotopic PDAC, 1 × 106 L3.6pl cells were injected in the mouse pancreas to create slowly growing tumors as described previously (36). All the procedures are approved by HMRI Institutional Animal Care and Use Committee (AUP-0514-0031, 6/3/2014).
2.5 Assessment of survival, therapeutic efficacy and transport of nAb-PTX
Slowly growing pancreatic tumors were created as described in 2.4. More specifically, palpable tumors (500–750 mm3) developed not earlier than 30 days after the inoculation of PDAC cells. We have previously shown that these dynamics of disease progression in L3.6pl PDAC model result in the formation of tumors characterized by a dense stroma comparable to the tumor microenvironment in the clinical PDAC specimens (36). Dense stroma formation in L3.6pl tumors was confirmed by pathological analysis of hematoxylin and eosin stained sections. Active fibrosis was also evaluated by immunohistochemical staining of alpha-smooth muscle actin (α-SMA), marker for activated myofibroblasts (Supplementary figure 1). The following studies were conducted: (1) survival study; (2) evaluation of therapeutic efficacy and (3) evaluation of gem effect on nAb-PTX transport. The following drugs were used solo or in combination as detailed in each study: (a) nAb-PTX (intravenously, 75 mg/kg equivalent to 7.5 mg/kg PTX); (b) MSV/nAb-PTX (nAb-PTX 75 mg/kg equivalent to 7.5 mg/kg PTX loaded into 1×109 particles/mouse intravenously); (c) gem (intraperitoneally 50 mg/kg); (d) phosphate buffered saline (PBS, intravenously); and (e) empty MSV (1×109 particles/mouse intravenously in PBS).
To evaluate the survival benefits of the therapies on gem-resistant tumors, the animals were randomized into 7 groups (n = 9–10) and treated weekly with: 1) PBS, 2) gem, 3) nAb-PTX, 4) MSV, 5) MSV/nAb-PTX, 6) nAb-PTX+gem, and 7) MSV/nAb-PTX+gem. The mice were sacrificed when moribund or at the maximum of four months from the initiation of the experiment (123 days) to confirm tumor incidence.
For evaluating the therapeutic efficacy by assessing PDAC tumor weight, the animals, randomized into three groups (n = 8) were treated weekly for three weeks with: 1) PBS, 2) nAb-PTX+gem, and 3) MSV/nAb-PTX+gem. The body weight was monitored. Twenty-four hours after the last therapy, the mice were sacrificed and their tumors were harvested, weighed, and processed for immunohistochemical analysis.
To evaluate the effect of gem or MSV on nAb-PTX accumulation in PDAC and liver, three groups (4 mice per group) were treated once with: 1) nAb-PTX, 2) nAb-PTX+gem, and 3) MSV/nAb-PTX+gem. The mice were sacrificed 6 hours later. Tumors and livers were collected, weighed, and levels of PTX were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
2.6 Immunohistochemical analysis of tumor samples
Tumor sections were either fixed in 10% buffered formalin or with acetone to perform the following immunohistochemical analysis. Proliferation and apoptosis of tumor cells were evaluated through Ki67 and TUNEL staining, respectively. For Ki67 staining, sections were incubated with anti-mouse Ki67 rabbit polyclonal antibody at 4°C overnight and consequently with corresponding secondary antibody. TUNEL assay was conducted according to the manufacturer’s protocol. Cav-1 staining was performed incubating the sections with rabbit polyclonal anti-mouse cav-1 antibody at 4°C overnight, followed by incubation with corresponding secondary antibody. Additionally, CD31 antibody was used for counterstaining of cav-1-stained tumor sections. Immunofluorescence was imaged using confocal microscope (Nikon, Tokyo, Japan) and the fluorescent images (n = 10 per sample) were analyzed for number of objects (TUNEL assay and Ki67) or mean intensity (cav-1) using NIS-Elements software (Nikon).
In order to visualize tumor stroma, immunohistochemical analysis of paraffin-embedded tumors was performed with α-SMA antibody. The sections underwent heat-induced epitope retrieval with 10 mmol/L Tris buffer pH 9.0 and endogenous background elimination with 3% H2O2 for 10 minutes. Next, the sections were blocked, the primary antibody was added at a concentration of 1:100 for one hour at room temperature, and finally the anti-mouse HRP conjugated secondary antibody was added. In addition, some of the slides were stained with routine hematoxylin and eosin staining. 4× images were taken using a Nikon histological microscope.
2.7 Evaluation of the effect of gem on cav-1 expression in vitro
To determine if gem treatment increases nAb-PTX accumulation in the tumor by up-regulating cav-1 expression in endothelial cells, mouse immortalized endothelial cells were untreated or treated with gem 5, 10, and 20 nM for 48 hours. Next, untreated cells and cells treated with gem 20 nM were fixed with 4% paraformaldehyde in PBS, and stained for cav-1 as described above. 20× fluorescent images (n = 8–10 per group) were taken and analyzed for mean intensity (cav-1) using NIS-Elements software (Nikon). Also, cell lysates from untreated cells and cells treated with gem 5, 10, and 20 nM were used to perform cav-1 ELISA according to the manufacturer’s protocol in order to determine if the expression of cav-1 was dose-dependent.
2.8 Evaluation of cav-1 expression in resected patients’ PDAC specimens
The histologic sections of PDAC tissue (total 30 cases; 20 patients underwent upfront surgery and 10 patients received gem-based neoadjuvant chemotherapy before resection) were stained with a standard immunohistochemistry method with antibody to cav-1 (Abcam, USA). The study was approved by the MD Anderson Institutional Review Board (protocol PA14-0646). Patients’ characteristics are given in Supplementary Table 1. The stained slides were scanned using Aperio AT2 – High Volume, Digital Whole slide scanning (Leica Biosystems). Whole slide images were saved in the svs format and image analysis was performed with Definiens Tissue Studio 4.2 (Definiens, Munich, Germany). Marker Area detection algorithm was applied to detect cav1-positive stained component. We applied an image processing workflow by the following steps: preprocessing (Tissue background separation & annotation of pancreatic tumor), tissue segmentation (into cellular and stromal components) then marker area detection to quantify cav-1 expression levels (35). The marker area was calculated as the percent of positive IHC stained area to the whole tissue. We used SAS JMP Pro-12 for statistical analysis and graphs generation.
2.9 PTX quantification by LC-MS/MS
For LC-MS/MS analysis, the external standard consisted of PTX and the spiking internal standard consisted of deuterated PTX (D5). Plasma samples were analyzed whole, while PDAC and liver samples were homogenized with deionized water and the drug was extracted with acetonitrile containing the internal standard. Liquid chromatography was performed via Waters Acquity UPLC system (Waters Corporation, Milford, MA) using Acquity UPLC 2.1 mm × 50 mm BEH C-18 column (1.7μm) and matching guard column. Mass spectrometry was performed using Waters Xevo TQ tandem quadrupole mass spectrometer (Waters Corporation, Milford, MA) ran on SRM/MRM mode under positive ion electrospray conditions under capillary voltage of 3.5 kV, cone voltage of 16V, and transition dwell times of 0.105 sec. MassLynx v4.1 and TargetLynx v4.1 softwares (Waters Corporation, Milford, MA) were utilized to acquire data, and to quantify PTX, respectively. PTX was quantified based on the standard curve for the same molecular transition of the drug and normalized to percent recovery relative to the internal standard.
2.10 Statistical analysis
Statistical analysis was performed using GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA, USA). Data are presented as mean ± SD. Mann-Whitney U test was used for statistical analysis of the therapeutic efficacy study, gem effect on nAb-PTX transport study and gem effect on cav-1 expression in vitro, in vivo and in clinical samples from patients. The Kaplan-Meier method was used for survival analysis. P value <0.05 was considered to be statistically significant.
3. Results
3.1 Survival of animals as a function of therapy
To evaluate the effect of therapeutics on survival, tumor bearing mice were treated with 1) PBS, 2) gem, 3) nAb-PTX, 4) MSV, 5) MSV/nAb-PTX, 6) nAb-PTX+gem, or 7) MSV/nAb-PTX+gem (Figure 1). Animals from PBS, gem, and MSV groups showed a median survival between 37 and 45 days. As expected from the previous studies (16–18, 36), L3.6pl tumors are resistant to gem. Animals treated with nAb-PTX showed a significantly extended (P < 0.01) median survival of 53.5 days, demonstrating that this therapeutic affects gem resistant tumors. Because of MSV/nAb-PTX retention in the PDAC microenvironment, animal survival was significantly extended (P < 0.05) to 73 days vs. nAb-PTX alone. Similar survival was observed with a combination of nAb-PTX+gem (74 days), while the most extended survival was recorded for animals treated with MSV/nAb-PTX+gem (105 days, P < 0.005 vs. all other therapy groups). Half of the animals treated with MSV/nAb-PTX+gem did not become moribund due to the disease until the end of the experiment and were sacrificed at four months from the beginning of the experiment to confirm tumor incidence.
Figure 1.
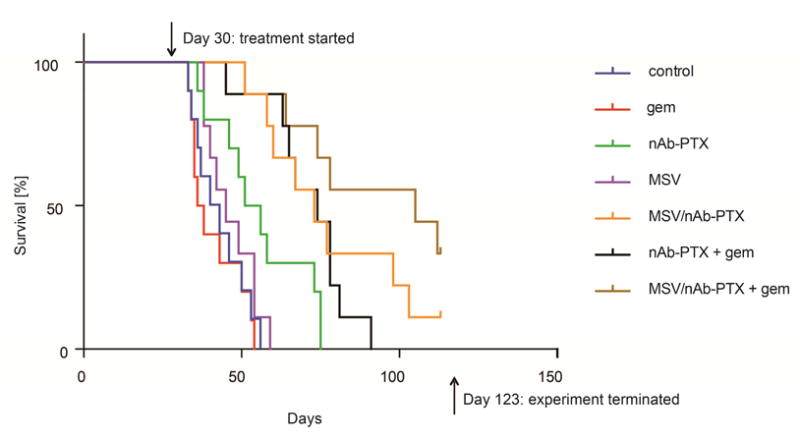
Survival of nude mice bearing L3.6pl orthotopic PDAC (n = 9–10) in response to therapy with control (PBS), gem, nAb-PTX, MSV, MSV/nAb-PTX, nAb-PTX + gem, and MSV/nAb-PTX + gem. Gem was given at a concentration of 50 mg/kg, nAb-PTX or MSV/nAb-PTX at a concentration of 75 mg/kg equivalent to 7.5 mg/kg PTX, and MSVs at a dose of 109 particles/mouse.
3.2 Evaluation of mechanisms involved in the therapeutic efficacy of the combination therapy
To evaluate the mechanisms that led to an increased survival of gem-resistant animals when treated with nAb-PTX+gem or MSV/nAb-PTX+gem combination, the drugs were administered three times and the animals were sacrificed for tumor analysis. PDAC tumor weights after treatment with MSV/nAb-PTX+gem were significantly lower in comparison to those after treatment with nAb-PTX+gem (P < 0.05, Figure 2A and Supplementary Figure 2B). Body weights were constant in all groups, showing no signs of toxicity (Supplementary Figure 2A). PDAC immunohistochemical analysis showed that cell proliferation (Ki67) in the MSV/nAb-PTX+gem group was reduced, while cell apoptosis (TUNEL) was increased, in comparison to two other groups (Supplementary Figure 3). The number of proliferating cancer cells was significantly lower, while apoptotic cancer cells number was significantly higher in the MSV/nAb-PTX+gem group in comparison to the other groups (Figure 2B–C, P < 0.01).
Figure 2.
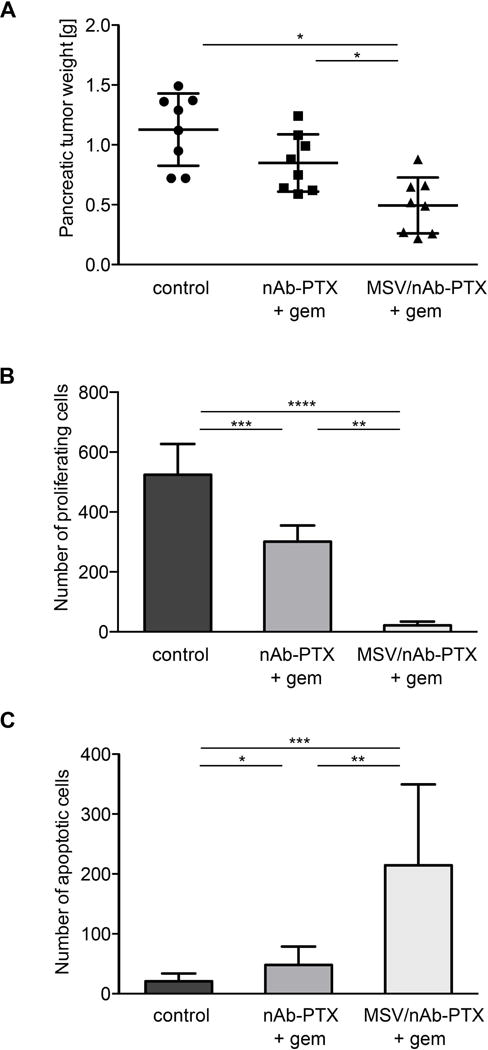
Suppression of tumor growth in response to nAb-PTX + gem or MSV/nAb-PTX + gem vs. no treatment control (PBS) in nude mice bearing L3.6pl orthotopic PDAC (n = 8): A) PDAC weight. B) Quantification of proliferating Ki67 positive cells. C) Quantification of apoptotic cells measured with the TUNEL assay. Field of view: 430 μm2. (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001).
3.3 Analysis of nAb-PTX accumulation in PDAC as a function of combined therapies
To assess the advantage of adding gem to nAb-PTX as well as loading nAb-PTX into MSV over nAb-PTX alone or nAb-PTX+gem, respectively, in terms of biodistribution of nAb-PTX, we quantified the amount of PTX in PDAC tumors and livers using LC-MS/MS. The concentration of PTX in PDAC was significantly higher (P < 0.05) in the animals treated with nAb-PTX+gem as compared to the mice treated with nAb-PTX alone. Furthermore, PTX concentration in tumor was higher in mice treated with MSV/nAb-PTX+gem vs. the two other groups (Figure 3A). In contrast, liver accumulation of PTX was similar in all three groups (Figure 3B). Thus, the advantage of adding gem to nAb-PTX on survival and therapeutic efficacy can be explained by increased accumulation of PTX in PDAC. Additionally, MSV showed preferential delivery of nAb-PTX into PDAC.
Figure 3.
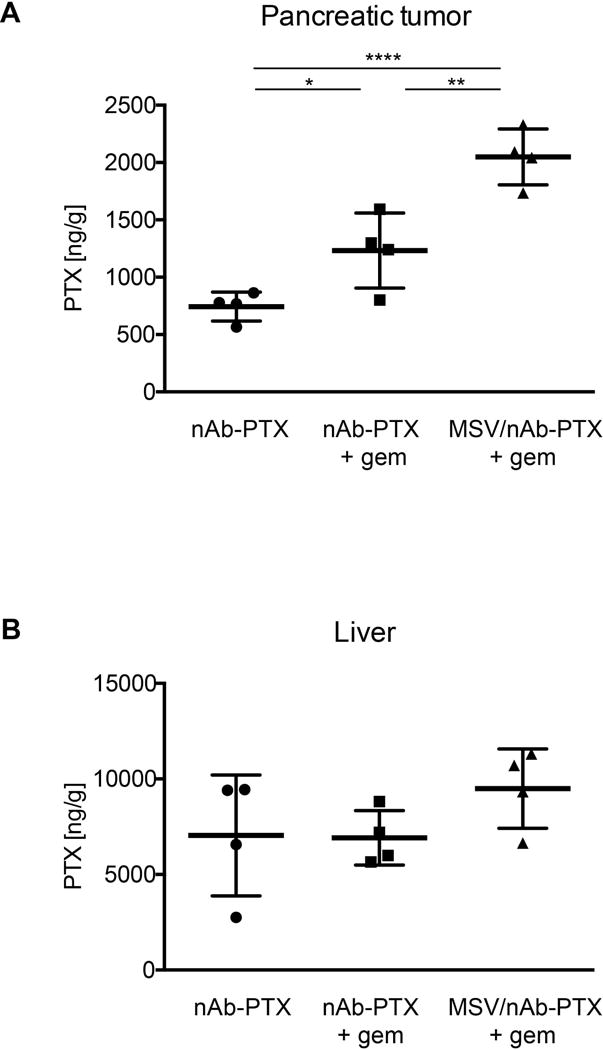
Accumulation of PTX in pancreatic tumor and liver of nude mice bearing L3.6pl orthotopic PDAC determined by LC-MS/MS (n = 5). (* P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001).
3.4 Expression of cav-1 as a function of gem administration in vitro, in vivo and in samples from patients
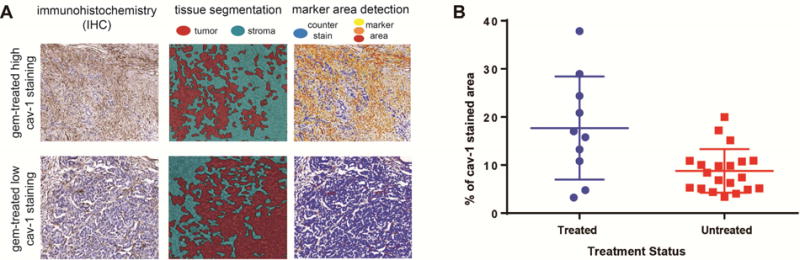
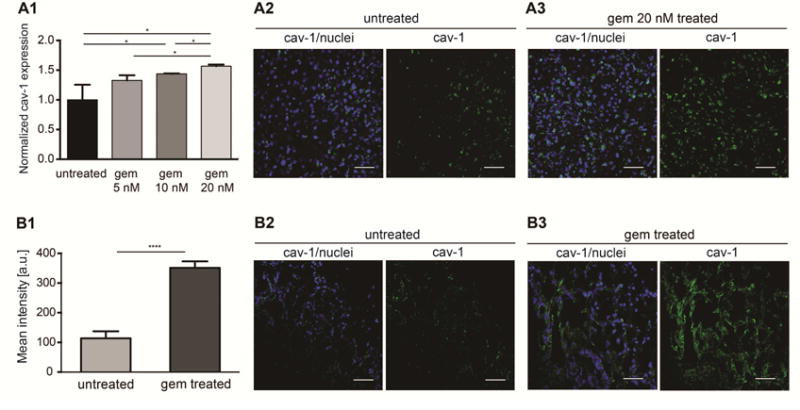
Cav-1 transports nAb-PTX, thus, immunohistochemical analysis was performed in vitro on mouse endothelial cells and in vivo on PDAC sections derived from animals treated with gem or PBS to determine the effect of gem on cav-1 expression. In both in vitro and in vivo studies, cav-1 expression (green) on endothelial cells and PDAC was significantly increased in response to treatment with gem in comparison to control treatment (Figure 4A2 and 4A3, and Figure 4B). In vitro results demonstrated that cav-1 expression in endothelial cells is dose-dependent, in fact we found that a higher dose of gem causes a higher expression of cav-1 (Figure 4A1). The number of endothelial CD31-positive cells in vivo was not affected (data not shown). Next, we validated the in vitro and in vivo data by evaluating 30 patient specimens (20 gem naive vs. 10 treated with gem). After staining with cav-1, the patient tumor slides were scanned and analyzed first for tissue segmentation in tumor and stroma, and then with a marker area detection algorithm, percentage of caveolin-positive tissue was detected (Figure 5A). We found that cav-1 expression is significantly increased in patients pre-treated with gem-based neoadjuvant chemotherapy as compared to non-treated patients, ANOVA (P = 0.0032) (Figure 5B).
Figure 4.
Increased expression of albumin transporter cav-1 in vitro (A), and in vivo (B) in response to gem treatment: (A1) Cav-1 expression obtained through ELISA of mouse endothelial cells lysates treated with gem 5, 10, and 20 nM in vitro (*P < 0.05); (A2) Fluorescent images of untreated mouse endothelial cells stained for cav-1 (green) and cell nuclei (DAPI, blue); (A3) Fluorescent images of mouse endothelial cells treated with gem 20 nM and stained for cav-1 (green) and cell nuclei (DAPI, blue). Scalebar: 100 μm; (B1) Quantification of mean fluorescent intensity of cav-1 immunofluorescence in tumor sections from nude mice bearing L3.6pl orthotopic PDAC in vivo. (**** P < 0.0001); (B2) Fluorescent images of PDAC from untreated mice (PBS group) stained for cav-1 (green) and cell nuclei (DAPI, blue); (B3) Fluorescent images of PDAC from mice treated with gem 50 mg/kg stained for cav-1 (green) and cell nuclei (DAPI, blue). Scalebar: 100 μm.
Figure 5.
Increased expression of albumin transporter cav-1 in PDAC samples from patients (see Supplementary Table 1) in response to gem treatment: (A) image processing workflow applied to analyze 30 patient specimens stained with cav-1. Tissue segmentation was employed in order visualize tumoral and stromal components, while a marker area detection algorithm was used to identify the cav-1 positive tissue. (B) Immunohistological quantification of cav-1 in the PDAC tumors of the patients pre-treated with gem-based neoadjuvant chemotherapy compared to the patients without neoadjuvant chemotherapy, ANOVA (P = 0.0032)
4. Discussion
This study focuses on the effects of gem treatment on the transport of nAb-PTX. Additionally, we investigate the effect of loading nAb-PTX into MSV for combination therapy with gem in nude mice bearing orthotopic L3.6pl pancreatic cancer. This PDAC model was chosen due to previously shown resistance to gem therapy (16–18) and could represent patients with gemcitabine-resistant PDAC, thus providing a better understanding of treatment options for patients who do not respond to the first line monotherapy (8). Accordingly, gem had no effect on median survival (37, 41.5 and 45 days for gem, PBS and empty MSV treated mice, respectively) (Figure 1). Treatment with nAb-PTX alone resulted in enhanced median survival (53.5 days), further increased by combining nAb-PTX with gem (74 days). An additional effect was achieved by loading nAb-PTX into the MSV, resulting in a median survival of 73 and 105 days when they were given alone or with gem, respectively. The data show an increased PTX accumulation in tumor in animals treated with nAb-PTX+gem in comparison to nAb-PTX alone (Figure 3A). Similarly to the survival study, loading nAb-PTX into the MSV and combining it with gem resulted in a further increase of PTX tumor accumulation. As a result of enhanced PTX accumulation in pancreatic tumors in vivo, we also observed increased median survival (Figure 1), increased tumor cell apoptosis (Figure 2C), and reduced tumor cell proliferation (Figure 2B) when animals bearing PDAC were administered with nAb-PTX+gem. These effects were further enhanced when mice were treated with MSV/nAb-PTX with gem. These data provide direct evidence for: (1) role of gem in increasing transport of nAb-PTX to the PDAC tumor and (2) advantage of the MSV in nAb-PTX transport to tumors.
To understand the mechanism of enhanced accumulation of PTX in PDAC when combined with gem treatment, analysis of cav-1 expression as a function of gem therapy was performed in in vitro, in vivo, and in clinical samples. Considering that treatment with gem has been previously shown to increase the expression of cav-1 in PDAC (14), we hypothesized that nAb-PTX accumulation could be mediated by an increased cav-1 expression induced by gem. Accordingly, cav-1 stainings demonstrated that gem treatment enhanced the expression of cav-1 both in mouse immortalized endothelial cells in vitro and orthotopic L3.6pl PDAC in vivo, thus increasing the ability for nAb-PTX transport into tumor tissue (Figure 4). These results were strongly supported by the findings in patient resected tumors, showing that cav-1 expression was also significantly increased in the PDAC tumors of the patients pre-treated with gem-based chemotherapy compared to the patients without neo-adjuvant chemotherapy (Figure 5).
Cav-1 expression has been shown to be affected by several molecular pathways. As an example, HRAS or ABL1-induced oncogenic transformation of mouse fibroblasts caused the transcriptional downregulation of cav-1 (37, 38). In another work, mutations in TP53 gene, encoding p53, resulted in a reduced expression of cav-1 (39). Studies in prostate cancer cells have shown that cav-1 expression can be induced by protein kinase C ε (PKC ε) through the sequential activation of mitogen-activated protein kinases (MAPK), c-Myc, and androgen receptor (40). Molecular mechanisms of cav-1 regulation in pancreatic cancer are yet to be studied. There are several reports suggesting that cav-1 expression in breast, prostate, bladder, and other cancers is affected by epigenetic factors, in particular DNA methylation (41–44). In human breast cancer cells, 5′ promoter of cav-1 is methylated as opposed to normal human mammary epithelial cells (41, 45). In studies on clinical samples from breast tumors, hypermethylation of Cav1 promoter was accompanied by the downregulation of cav-1 expression in comparison to the adjacent unaffected breast tissue (42, 46). Gem has been shown to function as DNA methyltransferase inhibitor reactivating epigenetically silenced genes (47). Additionally, a recent work demonstrated the ability of gem to affect the expression of multiple genes, including p73, IP6K3, MAP2K6, and others (48). These effects of gem can be related to the overexpression of cav-1 in animals and patients treated with the drug.
Our findings suggest that, following enhanced delivery to the tumor vasculature by MSV, PTX is transported to PDAC microenvironment by albumin transporters, whose expression is increased by gem (Figure 6). MSV has been shown to allow preferential accumulation of drugs into tumor tissue by preventing payload degradation and localizing in the tumor microenvironment (49–52). Following intravenous administration, a drug encounters multiple biological barriers (i.e. enzymatic degradation, endothelial barrier) preventing the transport to the site of action (22, 53, 54). MSV was developed to sequentially circumnavigate such biological barriers and achieve preferential accumulation of second- and third-stage compounds in tumor tissue by concurrently taking advantage of hemodynamics of tumor vasculature, interfacial interactions with the cells in the tumor microenvironment (e.g. endothelial cells and macrophages (24, 55, 56), and the enhanced permeability and retention (EPR) effect. MSVs are micrometer-size nanoporous silicon particles characterized by a non-spherical shape, which allows particle margination and adhesion to the tumor endothelium (25). Subsequently, as a result of porous silicon degradation into harmless orthosilicic acid, the nano-sized compound loaded into the particles is released to the tumor interstitium (57, 58). Accordingly, several in vivo studies have shown no subacute toxicity, acute toxicity and immunotoxicity of MSV (29, 50, 59–61). In the study by Tanaka et al. in an immunocompetent mouse model (FBV mice), MSV did not alter renal and liver functions (evaluated by plasma BUN/creatinine and LDH levels, respectively) as well as 23 plasma cytokines and chemokines (61). Additionally, MSV had no effect on infiltration of leukocytes into the liver, spleen, kidney, lung, brain, heart, and thyroid. In the study by Godin et al MSV were shown to degrade in serum within 24h, didn’t stimulate cytokine release from human macrophages or affect human endothelial cell viability in vitro (62). Tanei et al. have shown that in the immunocompetent models of Balb/C and C57BL/6 mice there were no changes in the number of macrophages in the unaffected liver and body weight of the animals following the treatment with MSV or MSV-nAb-PTX (31).
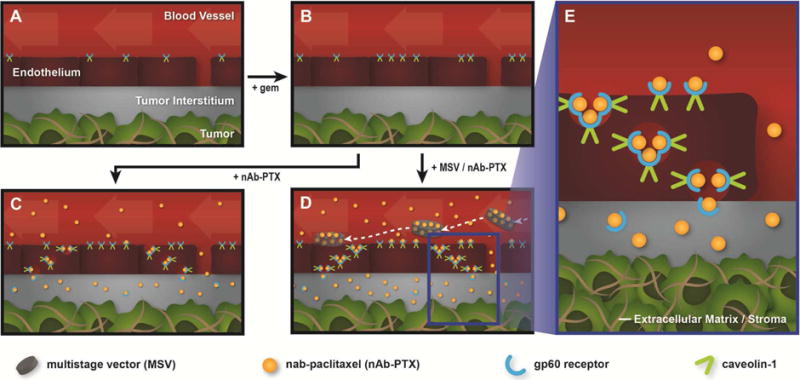
Figure 6.
Proposed mechanism for gem and MSV-induced preferential accumulation of nAb-PTX in tumor tissue. (A) normal tumor vasculature, (B) upon intraperitoneal injection of gem the expression of gp60 and caveolin-1 increases, (C) after intravenous injection, nAb-PTX accumulates in tumor, (D) MSV delivery further improves preferential localization of nAb-PTX in the tumor due to the formation of vascular depots in tumor blood vessels, (E) Mechanism for nAb-PTX transport across the endothelial barrier through transcytosis mediated by gp60 receptor and caveolin-1.
In a powder form, nAb-PTX consists of 130 nm in diameter nanoparticles, which decreases to 12 nm when the compound is suspended in water. Resuspended nAb-PTX can be successfully loaded into 20–50 nm pores of MSV (31). After systemic injection of MSV/nAb-PTX, nAb-PTX is released in proximity of tumor vasculature due to MSV degradation. Next, the albumin-bound PTX crosses the endothelial barrier by taking advantage of both an endogenous albumin transport pathway and the EPR effect. The latter is the phenomenon by which tumor vasculature displays increased permeability and nanoparticle retention due to enhanced number of fenestrations, atypical blood flow, and lack of proper lymphatic drainage (63–65), while the former involves albumin binding proteins such as 60-kDa glycoprotein (gp60), cav-1, and SPARC (Figure 6e) (23). It was recently reported that SPARC expression levels were not associated with efficacy of nAb-PTX plus gem or gem alone in patients with PDAC (66). However, while the same trend regarding SPARC expression and therapeutic efficacy was found in patients with advanced lung cancer treated with nAb-PTX plus carboplatin, an increase in the stromal cav-1 expression was significantly associated with therapeutic response and patients’ survival (67).
In conclusion, in our work, we have seen that gem treatment significantly increased cav-1 levels on endothelial cells in vitro and in the tumor microenvironment in vivo. This increase was in agreement with the data obtained from the clinical samples from patients with PDAC. The therapeutic regimen based on the combination of MSV/nAb-PTX and gem shows the best therapeutic efficacy, increased median survival, and enhanced PTX accumulation in tumors as a result of: (1) increased expression of albumin transporter cav-1; and (2) preferential delivery by MSVs. These results suggest that MSV/nAb-PTX+gem has a potential to be translated to the clinic for the treatment of gem-resistant PDAC.
Supplementary Material
Supplementary table I. Characteristics of patients with PDAC.
Supplementary figure 1. Presence of tumor stroma in mouse PDAC samples. (A) Alpha-smooth muscle actin staining (B) Hematoxylin and eosin staining. Scalebar: 200 μm.
Supplementary figure 2. Effect of treatment with nAb-PTX + gem with or without MSV delivery in nude mice bearing L3.6pl pancreatic cancer: A) Animal body weight. B) Image of pancreatic tumors.
Supplementary figure 3. Reduction of tumor proliferation and increase of tumor apoptosis in response to nAb-PTX + gem with or without MSV delivery in nude mice bearing L3.6pl pancreatic cancer. Confocal images of pancreatic tumors with Ki67 staining and TUNEL assay to determine tumor proliferation and tumor apoptosis, respectively. Scalebar: 50 μm.
{kind=link}
{kind=link}
{kind=link}
Highlights.
Multistage nanovectors target PDAC stroma
A combination therapy exploiting nanovectors for PDAC is proposed
Gemcitabine increases the expression of albumin transporter protein caveolin-1
Gemcitabine increases the transport of albumin-bound paclitaxel to PDAC
Acknowledgments
We thank Matt Landry for preparation of Figure 5. The authors gratefully acknowledge funding from the NCI 1-U54-CA143837 and NIH 1-U54-CA151668-01. M. Ferrari would like to acknowledge support via the Ernest Cockrell Jr. Presidential Distinguished Chair. E. Koay would like to acknowledge support from MD Anderson Cancer Center including the Cancer Moonshots program and the Center for Advanced Biomedical Imaging with equipment support from GE Healthcare.
Financial support
This work was supported by the National Cancer Institute (NCI 1-U54-CA143837); the National Institutes of Health (NIH 1-U54-CA151668-01). M. Ferrari would like to acknowledge support via the Ernest Cockrell Jr. Presidential Distinguished Chair. E. Koay would like to acknowledge support from MD Anderson Cancer Center including the Cancer Moonshots program and the Center for Advanced Biomedical Imaging with equipment support from GE Healthcare. The funding sources had no involvement in the conduct of the research and/or the preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors’ contributions
Conception and design: C. Borsoi, M. Ferrari, B. Godin, K. Yokoi
Development of methodology: C. Borsoi, X. Liu, B. Godin, K. Yokoi
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): C. Borsoi, F. Leonard, Y.T. Liu, X. Liu, J.F. Alexander, M Kai, Y Lee, M. Zaid, D. Elganainy, Y. Kang, E. Koay, B. Godin, K. Yokoi
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): C. Borsoi, Y. Lee, M. Zaid, D. Elganainy, E. Koay, M. Ferrari, B. Godin, K. Yokoi
Writing, review, and/or revision of the manuscript: C. Borsoi, Y. Lee, M. Zaid, D. Elganainy, E. Koay, M. Ferrari, B. Godin, K. Yokoi
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): C. Borsoi, B. Godin, K. Yokoi
Study supervision: B. Godin, K. Yokoi
Conflicts of interest
M. Ferrari is the founding scientist and a member of the Board of Directors of NanoMedical Systems, and a member of Board of Directors of Arrowhead Research Corporation, and hereby discloses potential financial interests in the companies. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Malvezzi M, Bertuccio P, Rosso T, Rota M, Levi F, La Vecchia C, et al. European cancer mortality predictions for the year 2015: does lung cancer have the highest death rate in EU women? Ann Oncol. 2015;26(4):779–86. doi: 10.1093/annonc/mdv001. [DOI] [PubMed] [Google Scholar]
- 2.American Cancer Society. Cancer Facts & Figures 2015. Atlanta: American Cancer Society; 2015. [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 4.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 5.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25(15):1960–6. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 6.Louvet C, Labianca R, Hammel P, Lledo G, Zampino MG, Andre T, et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J Clin Oncol. 2005;23(15):3509–16. doi: 10.1200/JCO.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 7.Heinemann V, Quietzsch D, Gieseler F, Gonnermann M, Schonekas H, Rost A, et al. Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J Clin Oncol. 2006;24(24):3946–52. doi: 10.1200/JCO.2005.05.1490. [DOI] [PubMed] [Google Scholar]
- 8.Bernhard J, Dietrich D, Scheithauer W, Gerber D, Bodoky G, Ruhstaller T, et al. Clinical benefit and quality of life in patients with advanced pancreatic cancer receiving gemcitabine plus capecitabine versus gemcitabine alone: a randomized multicenter phase III clinical trial—SAKK 44/00-CECOG/PAN.1.3.001. J Clin Oncol. 2008;26(22):3695–701. doi: 10.1200/JCO.2007.15.6240. [DOI] [PubMed] [Google Scholar]
- 9.Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol. 2012;9(8):454–67. doi: 10.1038/nrgastro.2012.115. [DOI] [PubMed] [Google Scholar]
- 10.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29(34):4548–54. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Awasthi N, Zhang C, Schwarz AM, Hinz S, Wang C, Williams NS, et al. Comparative benefits of Nab-paclitaxel over gemcitabine or polysorbate-based docetaxel in experimental pancreatic cancer. Carcinogenesis. 2013;34(10):2361–9. doi: 10.1093/carcin/bgt227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frese KK, Neesse A, Cook N, Bapiro TE, Lolkema MP, Jodrell DI, et al. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012;2(3):260–9. doi: 10.1158/2159-8290.CD-11-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chatterjee M, Ben-Josef E, Thomas DG, Morgan MA, Zalupski MM, Khan G, et al. Caveolin-1 is Associated with Tumor Progression and Confers a Multi-Modality Resistance Phenotype in Pancreatic Cancer. Sci Rep. 2015;5:10867. doi: 10.1038/srep10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kondratska K, Kondratskyi A, Yassine M, Lemonnier L, Lepage G, Morabito A, et al. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim Biophys Acta. 2014;1843(10):2263–9. doi: 10.1016/j.bbamcr.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 16.Yokoi K, Sasaki T, Bucana CD, Fan D, Baker CH, Kitadai Y, et al. Simultaneous inhibition of EGFR, VEGFR, and platelet-derived growth factor receptor signaling combined with gemcitabine produces therapy of human pancreatic carcinoma and prolongs survival in an orthotopic nude mouse model. Cancer Res. 2005;65(22):10371–80. doi: 10.1158/0008-5472.CAN-05-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker CH, Trevino JG, Summy JM, Zhang F, Caron A, Nesbit M, et al. Inhibition of PDGFR phosphorylation and Src and Akt activity by GN963 leads to therapy of human pancreatic cancer growing orthotopically in nude mice. Int J Oncol. 2006;29(1):125–38. [PubMed] [Google Scholar]
- 18.Hwang RF, Yokoi K, Bucana CD, Tsan R, Killion JJ, Evans DB, et al. Inhibition of platelet-derived growth factor receptor phosphorylation by STI571 (Gleevec) reduces growth and metastasis of human pancreatic carcinoma in an orthotopic nude mouse model. Clin Cancer Res. 2003;9(17):6534–44. [PubMed] [Google Scholar]
- 19.Chapiro J, Sur S, Savic LJ, Ganapathy-Kanniappan S, Reyes J, Duran R, et al. Systemic delivery of microencapsulated 3-bromopyruvate for the therapy of pancreatic cancer. Clin Cancer Res. 2014;20(24):6406–17. doi: 10.1158/1078-0432.CCR-14-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao M, Tominaga Y, Ohuchida K, Mizumoto K, Cui L, Kozono S, et al. Significance of combination therapy of zoledronic acid and gemcitabine on pancreatic cancer. Cancer Sci. 2012;103(1):58–66. doi: 10.1111/j.1349-7006.2011.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al Shemaili J, Mensah-Brown E, Parekh K, Thomas SA, Attoub S, Hellman B, et al. Frondoside A enhances the antiproliferative effects of gemcitabine in pancreatic cancer. Eur J Cancer. 2014;50(7):1391–8. doi: 10.1016/j.ejca.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Ferrari M. Frontiers in cancer nanomedicine: directing mass transport through biological barriers. Trends Biotechnol. 2010;28(4):181–8. doi: 10.1016/j.tibtech.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hawkins MJ, Soon-Shiong P, Desai N. Protein nanoparticles as drug carriers in clinical medicine. Adv Drug Deliv Rev. 2008;60(8):876–85. doi: 10.1016/j.addr.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 24.Yokoi K, Godin B, Oborn CJ, Alexander JF, Liu X, Fidler IJ, et al. Porous silicon nanocarriers for dual targeting tumor associated endothelial cells and macrophages in stroma of orthotopic human pancreatic cancers. Cancer Lett. 2013;334(2):319–27. doi: 10.1016/j.canlet.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Godin B, Tasciotti E, Liu X, Serda RE, Ferrari M. Multistage nanovectors: from concept to novel imaging contrast agents and therapeutics. Acc Chem Res. 2011;44(10):979–89. doi: 10.1021/ar200077p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolfram J, Shen H, Ferrari M. Multistage vector (MSV) therapeutics. J Control Release. 2015;219:406–15. doi: 10.1016/j.jconrel.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Godin B, Gu J, Serda RE, Ferrati S, Liu X, Chiappini C, et al. Multistage Mesoporous Silicon-based Nanocarriers: Biocompatibility with Immune Cells and Controlled Degradation in Physiological Fluids. Controll Release Newsl. 2008;25(4):9–11. [PMC free article] [PubMed] [Google Scholar]
- 28.Blanco E, Sangai T, Hsiao A, Ferrati S, Bai L, Liu X, et al. Multistage delivery of chemotherapeutic nanoparticles for breast cancer treatment. Cancer Lett. 2013;334(2):245–52. doi: 10.1016/j.canlet.2012.07.027. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka T, Mangala LS, Vivas-Mejia PE, Nieves-Alicea R, Mann AP, Mora E, et al. Sustained small interfering RNA delivery by mesoporous silicon particles. Cancer Res. 2010;70(9):3687–96. doi: 10.1158/0008-5472.CAN-09-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tasciotti E, Liu X, Bhavane R, Plant K, Leonard AD, Price BK, et al. Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nat Nanotechnol. 2008;3(3):151–7. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- 31.Tanei T, Leonard F, Liu X, Alexander JF, Saito Y, Ferrari M, et al. Redirecting Transport of Nanoparticle Albumin-Bound Paclitaxel to Macrophages Enhances Therapeutic Efficacy against Liver Metastases. Cancer Res. 2016;76(2):429–39. doi: 10.1158/0008-5472.CAN-15-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu R, Zhang G, Mai J, Deng X, Segura-Ibarra V, Wu S, et al. An injectable nanoparticle generator enhances delivery of cancer therapeutics. Nat Biotechnol. 2016;34(4):414–8. doi: 10.1038/nbt.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Godin B, Chiappini C, Srinivasan S, Alexander JF, Yokoi K, Ferrari M, et al. Discoidal Porous Silicon Particles: Fabrication and Biodistribution in Breast Cancer Bearing Mice. Adv Funct Mater. 2012;22(20):4225–35. doi: 10.1002/adfm.201200869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Godin B, Gu J, Serda RE, Bhavane R, Tasciotti E, Chiappini C, et al. Tailoring the degradation kinetics of mesoporous silicon structures through PEGylation. J Biomed Mater Res A. 2010;94(4):1236–43. doi: 10.1002/jbm.a.32807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brieu N, Pauly O, Zimmermann J, Binnig G, Schmidt G. Slide specific models for segmentation of differently stained digital histopathology whole slide images. Proc SPIE 9784, Medical Imaging: Image Processing. 2016;9784:978410. [Google Scholar]
- 36.Yokoi K, Hawke D, Oborn CJ, Jang JY, Nishioka Y, Fan D, et al. Identification and validation of SRC and phospho-SRC family proteins in circulating mononuclear cells as novel biomarkers for pancreatic cancer. Transl Oncol. 2011;4(2):83–91. doi: 10.1593/tlo.10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engelman JA, Zhang XL, Razani B, Pestell RG, Lisanti MP. p42/44 MAP kinase-dependent and -independent signaling pathways regulate caveolin-1 gene expression. Activation of Ras-MAP kinase and protein kinase a signaling cascades transcriptionally down-regulates caveolin-1 promoter activity. J Biol Chem. 1999;274(45):32333–41. doi: 10.1074/jbc.274.45.32333. [DOI] [PubMed] [Google Scholar]
- 38.Koleske AJ, Baltimore D, Lisanti MP. Reduction of caveolin and caveolae in oncogenically transformed cells. Proc Natl Acad Sci U S A. 1995;92(5):1381–5. doi: 10.1073/pnas.92.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sherif ZA, Sultan AS. Divergent control of Cav-1 expression in non-cancerous Li-Fraumeni syndrome and human cancer cell lines. Cancer Biol Ther. 2013;14(1):29–38. doi: 10.4161/cbt.22621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu D, Terrian DM. Regulation of caveolin-1 expression and secretion by a protein kinase cepsilon signaling pathway in human prostate cancer cells. J Biol Chem. 2002;277(43):40449–55. doi: 10.1074/jbc.M206270200. [DOI] [PubMed] [Google Scholar]
- 41.Engelman JA, Zhang XL, Lisanti MP. Sequence and detailed organization of the human caveolin-1 and -2 genes located near the D7S522 locus (7q31.1). Methylation of a CpG island in the 5′ promoter region of the caveolin-1 gene in human breast cancer cell lines. FEBS Lett. 1999;448(2–3):221–30. doi: 10.1016/s0014-5793(99)00365-8. [DOI] [PubMed] [Google Scholar]
- 42.Chen ST, Lin SY, Yeh KT, Kuo SJ, Chan WL, Chu YP, et al. Mutational, epigenetic and expressional analyses of caveolin-1 gene in breast cancers. Int J Mol Med. 2004;14(4):577–82. [PubMed] [Google Scholar]
- 43.Cui J, Rohr LR, Swanson G, Speights VO, Maxwell T, Brothman AR. Hypermethylation of the caveolin-1 gene promoter in prostate cancer. Prostate. 2001;46(3):249–56. doi: 10.1002/1097-0045(20010215)46:3<249::aid-pros1030>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 44.Kunze E, Von Bonin F, Werner C, Wendt M, Schlott T. Transitional cell carcinomas and nonurothelial carcinomas of the urinary bladder differ in the promoter methylation status of the caveolin-1, hDAB2IP and p53 genes, but not in the global methylation of Alu elements. Int J Mol Med. 2006;17(1):3–13. [PubMed] [Google Scholar]
- 45.Bai L, Deng X, Li Q, Wang M, An W, Deli A, et al. Down-regulation of the cavin family proteins in breast cancer. J Cell Biochem. 2012;113(1):322–8. doi: 10.1002/jcb.23358. [DOI] [PubMed] [Google Scholar]
- 46.Syeed N, Hussain F, Husain SA, Siddiqi MA. 5′-CpG island promoter hypermethylation of the CAV-1 gene in breast cancer patients of Kashmir. Asian Pac J Cancer Prev. 2012;13(1):371–5. doi: 10.7314/apjcp.2012.13.1.371. [DOI] [PubMed] [Google Scholar]
- 47.Gray SG, Baird AM, O’Kelly F, Nikolaidis G, Almgren M, Meunier A, et al. Gemcitabine reactivates epigenetically silenced genes and functions as a DNA methyltransferase inhibitor. Int J Mol Med. 2012;30(6):1505–11. doi: 10.3892/ijmm.2012.1138. [DOI] [PubMed] [Google Scholar]
- 48.Yong KJ, Milenic DE, Baidoo KE, Brechbiel MW. Cell Killing Mechanisms and Impact on Gene Expression by Gemcitabine and 212Pb-Trastuzumab Treatment in a Disseminated i.p. Tumor Model. PLoS One. 2016;11(7):e0159904. doi: 10.1371/journal.pone.0159904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mai J, Huang Y, Mu C, Zhang G, Xu R, Guo X, et al. Bone marrow endothelium-targeted therapeutics for metastatic breast cancer. J Control Release. 2014;187:22–9. doi: 10.1016/j.jconrel.2014.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu R, Huang Y, Mai J, Zhang G, Guo X, Xia X, et al. Multistage vectored siRNA targeting ataxia-telangiectasia mutated for breast cancer therapy. Small. 2013;9(9–10):1799–808. doi: 10.1002/smll.201201510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shen H, Rodriguez-Aguayo C, Xu R, Gonzalez-Villasana V, Mai J, Huang Y, et al. Enhancing chemotherapy response with sustained EphA2 silencing using multistage vector delivery. Clin Cancer Res. 2013;19(7):1806–15. doi: 10.1158/1078-0432.CCR-12-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen J, Wu X, Lee Y, Wolfram J, Yang Z, Mao ZW, et al. Porous silicon microparticles for delivery of siRNA therapeutics. J Vis Exp. 2015;95:52075. doi: 10.3791/52075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5(3):161–71. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 54.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–51. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leonard F, Curtis LT, Yesantharao P, Tanei T, Alexander JF, Wu M, et al. Enhanced performance of macrophage-encapsulated nanoparticle albumin-bound-paclitaxel in hypo-perfused cancer lesions. Nanoscale. 2016;8(25):12544–52. doi: 10.1039/c5nr07796f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanei T, Leonard F, Liu X, Alexander JF, Saito Y, Ferrari M, et al. Redirecting Transport of Nanoparticle Albumin-Bound Paclitaxel to Macrophages Enhances Therapeutic Efficacy against Liver Metastases. Cancer Res. 2016;76(2):429–39. doi: 10.1158/0008-5472.CAN-15-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martinez JO, Chiappini C, Ziemys A, Faust AM, Kojic M, Liu X, et al. Engineering multi-stage nanovectors for controlled degradation and tunable release kinetics. Biomaterials. 2013;34(33):8469–77. doi: 10.1016/j.biomaterials.2013.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jurkic LM, Cepanec I, Pavelic SK, Pavelic K. Biological and therapeutic effects of ortho-silicic acid and some ortho-silicic acid-releasing compounds: New perspectives for therapy. Nutr Metab (Lond) 2013;10(1):2. doi: 10.1186/1743-7075-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen J, Xu R, Mai J, Kim HC, Guo X, Qin G, et al. High capacity nanoporous silicon carrier for systemic delivery of gene silencing therapeutics. ACS Nano. 2013;7(11):9867–80. doi: 10.1021/nn4035316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang M, Xu R, Xia X, Yang Y, Gu J, Qin G, et al. Polycation-functionalized nanoporous silicon particles for gene silencing on breast cancer cells. Biomaterials. 2014;35(1):423–31. doi: 10.1016/j.biomaterials.2013.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanaka T, Godin B, Bhavane R, Nieves-Alicea R, Gu J, Liu X, et al. In vivo evaluation of safety of nanoporous silicon carriers following single and multiple dose intravenous administrations in mice. Int J Pharm. 2010;402(1–2):190–7. doi: 10.1016/j.ijpharm.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Godin B, Gu J, Serda RE, Ferrati S, Liu X, Chiappini C, et al. Multistage Mesoporous Silicon-based Nanocarriers: Biocompatibility with Immune Cells and Controlled Degradation in Physiological Fluids. Controll Release Newsl. 2008;25(4):9–11. [PMC free article] [PubMed] [Google Scholar]
- 63.Tanaka T, Decuzzi P, Cristofanilli M, Sakamoto JH, Tasciotti E, Robertson FM, et al. Nanotechnology for breast cancer therapy. Biomed Microdevices. 2009;11(1):49–63. doi: 10.1007/s10544-008-9209-0. [DOI] [PubMed] [Google Scholar]
- 64.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55(17):3752–6. [PubMed] [Google Scholar]
- 65.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–92. [PubMed] [Google Scholar]
- 66.Hidalgo M, Plaza C, Musteanu M, Illei P, Brachmann CB, Heise C, et al. SPARC Expression Did Not Predict Efficacy of nab-Paclitaxel plus Gemcitabine or Gemcitabine Alone for Metastatic Pancreatic Cancer in an Exploratory Analysis of the Phase III MPACT Trial. Clin Cancer Res. 2015;21(21):4811–8. doi: 10.1158/1078-0432.CCR-14-3222. [DOI] [PubMed] [Google Scholar]
- 67.Bertino EM, Williams TM, Nana-Sinkam SP, Shilo K, Chatterjee M, Mo X, et al. Stromal Caveolin-1 Is Associated With Response and Survival in a Phase II Trial of nab-Paclitaxel With Carboplatin for Advanced NSCLC Patients. Clin Lung Cancer. 2015;16(6):466–74. doi: 10.1016/j.cllc.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary table I. Characteristics of patients with PDAC.
Supplementary figure 1. Presence of tumor stroma in mouse PDAC samples. (A) Alpha-smooth muscle actin staining (B) Hematoxylin and eosin staining. Scalebar: 200 μm.
Supplementary figure 2. Effect of treatment with nAb-PTX + gem with or without MSV delivery in nude mice bearing L3.6pl pancreatic cancer: A) Animal body weight. B) Image of pancreatic tumors.
Supplementary figure 3. Reduction of tumor proliferation and increase of tumor apoptosis in response to nAb-PTX + gem with or without MSV delivery in nude mice bearing L3.6pl pancreatic cancer. Confocal images of pancreatic tumors with Ki67 staining and TUNEL assay to determine tumor proliferation and tumor apoptosis, respectively. Scalebar: 50 μm.