

Abstract
Glycoproteins such as growth factor receptors and extracellular matrix have well-known functions in development and cancer progression, however, the glycans at sites of modification are often heterogeneous molecular populations which makes their functional characterization challenging. Here we provide evidence for a specific, discrete, well-defined glycan modification and regulation of a stage-specific cell migration in Caenorhabditis elegans. We show that a chain-terminating, putative null mutation in the gene encoding a predicted β1,4-N-acetylgalactosaminyltransferase, named ngat-1, causes a maternally rescued temperature sensitive (ts) defect in the second phase of the three phase migration pattern of the posterior, but not the anterior, hermaphrodite Distal Tip Cell (DTC). An amino-terminal partial deletion of ngat-1 causes a similar but lower penetrance ts phenotype. The existence of multiple ts alleles with distinctly different molecular DNA lesions, neither of which is likely to encode a ts protein, indicates that NGAT-1 normally prevents innate temperature sensitivity for phase 2 DTC pathfinding. Temperature shift analyses indicate that the ts period for the ngat-1 mutant defect ends by the beginning of post-embryonic development–nearly 3 full larval stages prior to the defective phase 2 migration affected by ngat-1 mutations. NGAT-1 homologs generate glycan-terminal GalNAc-β1-4GlcNAc, referred to as LacdiNAc modifications, on glycoproteins and glycolipids. We also found that the absence of the GnT1/Mgat1 activity [UDP-N-acetyl-D-glucosamine:α-3-D-mannoside β-1,2-N-acetylglucosaminyltransferase 1 (encoded by C. elegans gly-12, gly-13, and gly-14 and homologous to vertebrate GnT1/Mgat1)], causes a similar spectrum of DTC phenotypes as ngat-1 mutations–primarily affecting posterior DTC phase 2 migration and preventing manifestation of the same innate ts period as ngat-1. GnT1/Mgat1 is a medial Golgi enzyme known to modify mannose residues and initiate N-glycan branching, an essential step in the biosynthesis of hybrid, paucimannose and complex-type N-glycans. Quadruple mutant animals bearing putative null mutations in ngat-1 and the three GnT genes (gly-12, gly-13, gly-14) were not enhanced for DTC migration defects, suggesting NGAT-1 and GnT1 act in the same pathway. These findings suggest that GnTI generates an N-glycan substrate for NGAT-1 modification, which is required at restrictive temperature (25°C) to prevent, stabilize, reverse or compensate a perinatal thermo-labile process (or structure) causing late larval stage DTC phase 2 migration errors.
Introduction
The molecular mechanisms that regulate and guide cell and axon migrations have relevance to a variety of human neurological and mental health disorders as well as metastatic cancer. In the past, we discovered that mutants of UNC-6/Netrin signaling affect both axon guidance and the migration of two cells in C. elegans called distal tip cells (DTCs). These cells migrate at the leading distal tip of each of the two independently extending hermaphrodite gonad arms to shape these arms during post-embryonic development [1]. The DTCs are born post-embryonically in the ventral mid-body and migrate in 3 phases (see Fig 1A): (1) longitudinally in opposite directions away from the mid-body—the anterior DTC along the right and the posterior DTC along the left ventral body wall muscles (BWMs), then (2) from ventral to dorsal BWMs across the lateral epidermis on the same side as the phase 1 migration, then (3) back to the mid-body along dorsal BWMs. The migratory path of the DTCs determines the final shape of the adult gonad arms, thus in the wild type the gonad arms are each U-shaped and mirror image when the trajectory is viewed from a lateral perspective. In the studies reported here, the DTC migration paths were deduced from the shape of the hermaphrodite gonad arms in 4th larval stage (L4) animals and young adults (as in Fig 1A).
Fig 1. Common posterior Distal Tip Cell (DTC) trajectories in ngat-1(ev821) mutant animals.

Lateral view of the posterior gonad arm in L4 hermaphrodite animals by D.I.C. microscopy. In all panels, anterior is left, dorsal is up. (A) The 2 DTCs are born in ventral mid-body (in position of future vulva marked by triangle) and normally migrate post-embryonically in 3 phases [(1) thru (3)] to shape the developing U-shaped gonad arms. (B) Some ngat-1(ev821) animals grown at 25°C have a normal posterior DTC migration as in panel A, while many have an unc-6 mutant-like phase 2 failure with phases 2 and 3 executed on the ventral side (panel B), while others (panel C) show a partial return to mid-body before migrating dorsally. To the right of panels B and C are the predicted corresponding ventral clear patch (VCP) phenotypes visible by low magnification stereomicroscopy (asterisk marks partial VCP in panel C). Scale bar in panel A (for A-C) is 50 μm.
We previously showed that the secreted laminin-related guidance cue UNC-6/netrin requires the UNC-5 receptor acting alone or in combination with the UNC-40/DCC receptor to induce apparent repulsion from ventral sources of UNC-6 to guide ventral-to-dorsal (V-to-D) motor axon and phase 2 DTC migrations [1, 2]. In netrin signalling mutants (unc-5, unc-6 and unc-40 mutants) and the mutant subjects of this report, the phase 2 migration frequently fails to execute, nevertheless, the DTCs, stuck on the ventral side, move back to the mid-body along the ventral BWMs with proper phase 3 timing. This phase 2 migration failure causes a ventrally reflexed gonad arm in which the distal portion of the arm comes to lie beside the proximal portion of the same arm (as in Fig 1B). The side-by-side proximal and distal portions on the ventral side push the opaque intestine dorsally, creating a ventral clear patch (VCP) visible by low power stereo-microscopy in pre-adults (L4 stage larvae) and adults [see [3]]. Other kinds of DTC navigation errors cause smaller regions of gonad arms to lay side-by-side which manifest as smaller clear patches (as in Fig 1C). We crudely categorized VCPs as full (as in Fig 1B), near-full (full, but with a small opaque interruption), and partial (as in Fig 1C), depending on the number and extent of the clear patch(es) associated with each gonad arm. Clear patches therefore greatly facilitate scoring of a variety of DTC navigation errors.
The UNC-6/Netrin guidance system represents an important mechanism involving physical interaction and molecular cross-talk between DTCs and the extracellular matrix (ECM) during the course of migration. Further evidence for the importance of the ECM in DTC migration is that a variety of ECM molecules, their receptors, and enzymes that create or modify glycans present on ECM glycoproteins are known to affect DTC migration and in some cases also axon migration [4, 5]. These include proteoglycans UNC-52/perlecan [6] and SDN-1/syndecan [7], SQV-5/chondroitin synthase [8], HST-2/ heparan sulfate modifying enzyme [9], plus integrin related proteins PAT-4/ILK [10] and PAT-3/beta-integrin [11].
As development proceeds, the ECM is modified to form an acceptable substratum for the guidance of DTC and axon migrations. Genetic analysis demonstrated that two ADAMTS family matrix metalloproteases, GON-1 and MIG-17 are required to regulate DTC migration [12, 13]. GON-1, expressed in DTCs and BWMs, is essential for a normal rate of DTC migration while MIG-17, expressed in BWMs and localized to the gonad and DTC basement membrane, is essential for normal phases 2 and 3 DTC pathfinding. These results indicate that mig-17, like unc-5, -6 and -40, is not required for DTC migration per se, but influences the navigation of phase 2 and phase 3 migrations—albeit in a different way than do the netrin signaling genes (see below).
It has been proposed that GON-1 and MIG-17 regulate DTC migration by remodeling the extracellular or basement membrane substrata used for migration [12, 13]. How metalloproteases like MIG-17 help to do this remains to be elucidated, however, secretion of MIG-17 from BWM and proper localization to the gonadal/DTC surface [12, 14] requires N-glycosylation of its pro-domain [14, 15] and on the basement membrane protein MIG-6/papilin [16]. Poorly glycosylated MIG-17, as predicted by its consequent mis-localization, causes defects in DTC migration [15]. Several genes required for normal glycosylation, including those encoding Golgi complex proteins COGC-1 to 8 [17] and MIG-23, a Golgi-resident NDPase [15, 17] are required to glycosylate MIG-17 and probably other basement membrane glycoproteins. Mutants of the above glycosylation genes have DTC defects resembling those of mig-17 loss-of-function mutants in that the DTCs often meander widely along the dorso-ventral axis as they return to mid-body during phase 3 –a phenotype largely different from most of the phase 2 navigation errors of the netrin signaling mutants. Several papers have identified many additional genes required for DTC migration [18–24], but few are characterized beyond describing the DTC phenotypes induced by gene disruptions.
Proteins translated in the secretory pathway are N-glycosylated by covalent bonding of glycans to Asn (N) at NXS/T(X≠P) sites during translation and extrusion into the lumen of the endoplasmic reticulum (ER) [25]. The glycan portion of Glc3Man9GlcNAc2-pp-dolichol is transferred and serves as a ligand for chaperones calnexin and calreticulin, which bind the Glc-Man epitope and promote folding of the N-glycosylated polypeptide [26]. Glycoproteins traffic from the ER to the cis-Golgi where the N-glycans are trimmed by alpha-mannosidases and then rebuilt by the branching N-acetylglucosaminyltransferases (GnT1/Mgat1, Mgat2, Mgat4a,b,c and Mgat5). C. elegans has homologues for GnT1/Mgat1 (GLY-12, GLY-13, GLY-14), Mgat2 (GLY-20) and Mgat5 (GLY-2). In contrast to core glycan synthesis, which is constitutive in most cell types, the addition of branching and terminal sugars is often regulated in a tissue- or cell lineage–specific manner [27, 28]. Changes in terminal glycan structure are also often associated with malignant transformation in cancer [29–31] and metastasis [25, 29]. Tissue- and/or lineage-specific regulation of outer-chain biosynthesis is regulated by expression of the relevant glycosyltransferases, supply of the sugar-nucleotide substrates, and in some cases, by competition between enzymes for a common acceptor, however, the majority of regulated terminal glycosylations observed do not yet have a defined function.
Here we identify and characterize mutations in C. elegans genes encoding 2 types of glycosyltransferases that cause aberrant phase 2 DTC migration. One gene, ngat-1, that was identified by a mutation that causes VCPs, was cloned and found to encode a predicted β-1,4-N-acetylgalactosaminyl transferase predicted to generate a LacdiNAc glyco-epitope near or at the distal termini of oligosaccharides that decorate one or more unknown glycoproteins or glycolipids. The ngat-1 mutant has an unusual DTC migration phenotype in that phase 2 migration failures are largely confined to the posterior DTC. The ngat-1 mutants are also temperature sensitive (ts) for the posterior DTC migration defects with a ts period that ends just prior to or early during the first larval stage. The phase 2 navigation defects, largely confined to the posterior DTC, and the temperature-sensitivity of the ngat-1 mutants are unusual phenotypes that we discovered are also shared with the triple null mutant of three genes (gly-12, gly-13 and gly-14). These genes each encode an enzymatically active UDP-N-acetyl-D-glucosamine:α-3-D-mannoside β-1,2-N-acetylglucosaminyltransferase I [32] (also known as GnT1 or Mgat1), and together encode all of this Golgi enzyme activity, which is essential for hybrid, paucimannose, and complex-type N-glycan synthesis. The temperature-sensitive period for the gly-14; gly -12 gly-13 triple GnT1 mutant posterior DTC migration defects, as for the ngat-1 mutant, ends by the first larval stage of development, which surprisingly is nearly three full larval stages prior to the phase 2 DTC migration affected by these mutations. This is an unusual finding for any gene known to regulate cell migration and supports the suggestion that NGAT-1 and GnT1 activities may modify the same N-glycoprotein target(s) required for normal phase 2 DTC migration—modifications that are required for robust adaptation in early development to temperature variation, as revealed by subsequent altered phase 2 DTC migration patterns.
Materials and methods
Nematode culture
Standard procedures were used for the culture, maintenance and genetic analysis of C. elegans [33, 34]. The N2 Bristol strain was used as the standard wild-type strain. The CB4856 Hawaiian strain was used for one-step SNP/whole genome sequencing (WGS). Mutants used in this study were: Linkage group X (LGX): unc-6(ev400) [1], dpy-3(e27), gly-12(ok712), gly-13(id47) [35], dpy-3(e27), LG I: bre-4(ok3167), (dpy-5(e61), unc-40(ev543), LG II: dpy-10(e128), ngat-1(ev821), ngat-1(ev823), unc-52(e444), LG III: gly-14(id48) [35], LG V: him-5(e1490) [36]. Males from two transgenic lines were used: CF702 muIs32[mec-7::gfp; lin-15(+)]; him-5(e1490), and DE60 dnIs13[gly-18p::gfp; unc-119(+)]; him-5(e1490).
Strains not isolated in our laboratories were obtained from the Caenorhabditis elegans Genetics Center, courtesy of T. Stiernagle (University of Minnesota).
Genetic analyses
The ngat-1(ev821) and ngat-1(ev823) strains were backcrossed to N2 (wild type) at least two times before characterization. The gly-14; gly-12 gly-13 triple mutant was originally isolated and made available by one of us (HS) for characterization. The mutant strains characterized in the Results were derived as follows. We crossed ngat-1(ev821) males to the gly-14; gly-12 gly-13 triple to construct the ngat-1; gly-14; gly-12 gly-13 quadruple mutant. In this process, we isolated gly-14; gly-12 gly-13 triple and ngat-1(ev821) single mutant co-segregants of the quadruple, which should share, similar if not identical, genetic backgrounds with one another for purposes of comparison. For comparisons of penetrance for animals grown at the same temperature, the animals were grown in parallel, i.e., in the same incubator space to correct for variations in incubator temperature, which could account for somewhat different penetrance of defects observed following growth in the same incubator at different times. Differences in penetrance were also minimized by growing animals without starvation for at least two generations before scoring them for defects.
To separate ev821 from unc-40(ev543), we crossed double mutant hermaphrodites to males heterozygous for dpy-5(e61), a closely linked marker used as a balancer for unc-40(ev543), then cloned non-Unc F1s to identify clones segregating Dpy progeny. We failed to detect VCPs in the Dpy animals in the F2s, so we cloned 100 F2 Dpys and discovered several that produced F3s with significant penetrance of phase 2 DTC navigation errors–presumptive dpy-5; ev821 double mutants. We then eliminated the dpy-5 mutation by repeating the above procedure substituting N2 (wild type) males for dpy-5 heterozygous males in the cross and selecting for VCPs in the F3 to derive ev821 homozygotes lacking the dpy-5 mutation (Fig 1).
The genetic characteristics of ev821 were first assayed by crossing genetically marked males [DE60 dnIs13[gly-18p::gfp; unc-119(+)]; him-5(e1490) or CF702 muIs32[mec-7::gfp; lin-15(+)]; him-5(e1490)] to hermaphrodites homozygous for ev821 and analyzing subsequent F1 cross- and F2, F3 and F4 self-fertilization progeny. From a related cross of dpy-3 males to homozygous ev821 hermaphrodites, we also analyzed subsequent F1 cross- and F2, F3 and F4 self-fertilization progeny with results similar to those obtained from the CF702 and DE60 crosses.
WGS-SNP cloning of ngat-1
Mutant ev821 (N2 Bristol background) homozygous males were crossed with CB4856 (Hawaiian background) hermaphrodites. F1s were cloned and grown at 20°C. Self-fertilization progeny of cloned F2s were assayed for worms that displayed the ev821 ventral clear patch (VCP) phenotype. Forty-seven homozygous ev821 F3 lines were grown by self-fertilization and DNA was made from each line. DNAs were pooled for WGS at 30x coverage. The Cloudmap program [37] was used to localize the genomic regions enriched for Bristol (non-Hawaiian) strain DNA. Bristol DNA was found to be greatly enriched relative to Hawaiian DNA in only one region of the genome, which is on LG II. This region contained 4 Bristol genes that appeared carry mutations in open-reading-frame (ORFs) relative to a standard N2 wild type strain (see Results).
Molecular biology
Standard molecular biology methods [38] were used unless otherwise noted.
Transgenic constructs and germline transformation
The following constructs were newly designed and cloned in our lab. The sequences of plasmid constructs were verified by DNA sequence analysis.
ngat-1 rescue genomic fragment
A 3297 bp PCR fragment containing the ngat-1 gene was amplified from N2 genomic DNA. The fragment included 985 bps of sequence upstream of the initiator ATG and 633 bps of sequence downstream of the terminator TAA sequence.
myo-3 promoter-driven ngat-1 rescue fusion PCR (myo-3p::ngat-1(+))
The myo-3 promoter was amplified from pCFJ104 (Pmyo-3::mCherry::unc-54) (a gift from Erik Jorgensen) yielding a promoter fragment that included 2536 bp upstream of the initiator ATG sequence.
The ngat-1 coding fragment of 2344 bps was amplified from fosmid WRM0637aH09 DNA, spanning from the initiator ATG to 698 bps downstream of the terminator TAA codon. These two fragments were joined using fusion PCR such that the initiator ATG of the myo-3 gene substituted for the ATG of ngat-1.
lag-2 promoter-driven ngat-1 rescue fusion PCR (lag-2p::ngat-1(+))
The lag-2 promoter was amplified from pJK590 (gift from Judith Kimble) yielding a promoter fragment that included 3131 bps of lag-2 promoter DNA upstream of the initiator ATG. The ngat-1 coding sequence of 2344 bps was amplified from fosmid WRM0637aH09 DNA, spanning from the initiator ATG to 698 bps downstream of the terminator TAA codon. These two fragments were joined using fusion PCR such that the initiator ATG of the lag-2 gene substituted for the ATG of ngat-1.
ngat-1 promoter-driven GFP expression plasmid (ngat-1p::gfp)
Briefly, a 3181 bp ngat-1 genomic sequence including 1592 bps of DNA upstream of the initiator ATG was amplified from N2 genomic DNA and inserted into plasmid pCRII-TOPO (Invitrogen). The gfp sequence was amplified from plasmid pPD95.70 (Addgene plasmid # 1492, a gift from A. Fire) that included the SV-40 nuclear localization signal at the N-terminus and the 3’ untranslated sequence of unc-54. The gfp sequence was inserted into ngat-1 genomic sequence such that ngat-1 initiator ATG codon replaced the initiator ATG of the gfp gene to create construct ngat-1p::gfp used for transgenic analysis of ngat-1 gene expression.
dpy-7 promoter-driven ngat-1(+) rescue construct (dpy-7p::ngat-1(+))
The dpy-7 promoter was amplified from N2 genomic DNA yielding a promoter fragment that included 369 bps upstream of the initiator ATG sequence. The ngat-1 coding sequence of 2344 bps was amplified from fosmid WRM0637aH09 DNA, spanning from the initiator ATG to 698 bps downstream of the terminator TAA codon. These two fragments were joined using fusion PCR such that the initiator ATG of the dpy-7 gene substituted for the ATG of ngat-1.
elt-2 promoter-driven ngat-1(+) rescue construct (elt-2p::ngat-1(+))
A 2310 bp genomic segment of ngat-1 DNA sequence, that includes all six exons and the 3’ UTR, was inserted into plasmid pJM559 (58) downstream of the elt-2 intestine-specific promoter sequence (5049 bps upstream of ATG) using Gibson assembly methods. In this construct, the initiator ATG of the elt-2 gene replaces the initiator ATG of the ngat-1 gene
dpy-7p::mCherry epidermal reporter
To construct the dpy-7 promoter-driven epidermal reporter, mCherry DNA was amplified by PCR and sub-cloned into pPD96.41 dpy-7p::venus (gift from A. Fire), using AgeI and EcoRI sites.
odr-1p::reporter co-transformation reporter
In some cases an odr-1p::reporter DNA [39] was co-injected as a co-transformation reporter.
myo-2p::mCherry co-transformation reporter
In some cases addgene plasmid # 19328 was co-injected as a co-transformation reporter.
Germline transformation
Transgenic strains were generated by co-microinjection of the DNA mix into the distal gonad arms of hermaphrodites [40]. DNA mixes consisted of a test construct, a co-injection reporter, and carrier DNA to create a final DNA concentration of 100–150 ng/μl.
See S3 Table for details of DNA concentrations used for transgene injections.
Creation of a ngat-1::gfp expression strain
To examine ngat-1 expression, we injected a solution containing the following DNAs: 100 ng/μl ngat-1p::gfp [pTG2-#7], 50 ng/μl odr-1p::dsRed (as co-transformation marker) into wild-type strain N2 (WT). The extrachromosomal array formed by this injection was integrated into the genome by irradiation to create evIs460B[ngat-1p::gfp, odr-1p::ds red]. We next injected 60 ng/μl dpy-7p::mCherry as epidermal reporter, and 40 ng/μl PBS-KS(-) into the strain carrying evIs460B to create evEx481, which was then used for fluorescence microscopic examination of ngat-1 promoter driven expression of gfp relative to the epidermal dpy-7 promoter-driven mCherry.
CRISPR Cas9 gene editing
A CRISPR guide RNA was designed to target the sequence TAGCATTACATAGATTAGTG within the first exon of the ngat-1 gene. Plasmid pDD162 [41] expressing Cas9, the guide RNA and a visible co-injection marker were co-injected into the gonads of N2 worms and lines expressing the marker were recovered. Deletions and insertions in exon 1 were identified by polyacrylamide electrophoresis of PCR products and DNA sequence analysis.
Temperature shift experiments
To determine the ngat-1(ev821) mutant temperature-sensitive (ts) period, eggs were collected from mutant adults grown at 16°C or 25°C, and placed on 4 plates which were then incubated at 16°C or 25°C, respectively. One plate was grown continuously at 16°C or 25°C, whereas three plates were shifted to 25°C or 16°C, respectively, at different stages of the animal's development–one during the L1 stage, one during the L2 stage and one during the L3 stage—then grown to a mix of L4 and young adult stages when they were assayed by D.I.C. for their DTC migration phenotypes.
To determine the GnT1 triple knockout strain ts period, L1s were collected by settling in water from adults grown at 16°C or 25°C, and split into 2 portions for further growth—one portion was grown at the same temperature as the parents and the other shifted to the higher or lower temperature for subsequent growth to the L4 stage when they were scored for VCPs.
Statistical analysis
Ninety-five percent upper confidence limits in error bars are for a binomial distribution of the same sample size and the observed proportion as mean for DTC phenotypes. Statistical tests were carried out using a standard (one-tailed) comparison of two proportions (Moore and McCabe, 1998). All p values represent the probability that the measured penetrance of the phenotype is significantly different between two strains. A p value less than 0.05 is considered to be significant. All comparisons described as significant in the Results were based on this criterion.
Anatomical characterization
The ngat-1(ev821) mutant phenotype, a clear patch on the ventral side of the worm detected by low magnification stereo-microscopy, occurs as the result of DTC migration defects that affect the second phase of migration. The DTC defects were also assayed by D.I.C. microscopy to observe gonad arms of L4 stage and young adult animals. These stages were chosen because DTC migration is completed in the L4 stage and zygotes are not present in the uterus (even in young adults) to distort the shape of the gonad arms.
Worms were placed on a 2% agarose pad and immobilized with 5–10 μL of 25 μM sodium azide. The samples were examined using a Leica DMRA2 microscope set to DIC microscopy. Images were captured using a Hamamatsu ORCA-ER digital camera mounted to the microscope.
Microscopy
Strains that carried a gfp reporter were viewed using a Leica DMRXA microscope. In some cases, confocal microscopy was performed using a Leica DMFLS laser confocal microscope equipped with a 63 PC APO CS lens (1.40–0.60) or with a Nikon CSi laser confocal microscope. Confocal images were analyzed by processing confocal z-axis series using Volocity (Quorum Technologies) or ImageJ software (NCBI).
Results
ev821 isolation and phenotypic characterization
Using a low-magnification stereo-microscope to screen for ventral clear patch (VCP) enhancers of DTC defects of a weak hypomorphic unc-40(ev543) allele, we discovered several mutations, one of which, ev821, we have characterized genetically and phenotypically. We first backcrossed the enhanced double mutant to isolate ev821 homozygotes separated from unc-40(ev543) and propagated the ev821 homozygotes (more readily identified in the F3 generation—see below) by several generations of self-fertilization. These homozygotes produced a substantial frequency of self-fertilization progeny with ventral clear patches (VCPs) of unc-6 mutant-like phase 2 DTC migration failures (as in Fig 1B). As self-fertilizing hermaphrodites grown in parallel at room temperature (approximately 23°C) for several generations, the penetrance of the full plus near-full VCP phenotype (observed for posterior DTCs only and not including partial VCPs) in the unc-40(ev543) single mutant, the unc-40(ev543); ev821 double mutant and ev821 single mutant were 23% (n = 244), 55% (n = 268) and 38% (n = 418), respectively.
The penetrance of posterior VCPs in ev821 single mutants grown at 25°C was somewhat higher than for mutant animals grown at room temperature and generally of 2 major types that occurred almost exclusively in the posterior gonad arm (Fig 1). Thirty-nine percent (n = 115) of mutant animals had full or near-full VCPs in which the ventral clear patch extended from the vulva posteriorly to two-thirds of the distance from the vulva to the rectum (Fig 1B), and nearly as many (35%) had a partial clear patch (Fig 1C) that started some distance posterior to the vulva and extended a short distance toward the rectum.
When gonad arms of ev821 mutant animals grown at 25°C were analyzed by higher magnification differential interference contrast (D.I.C.) microscopy, we found that 47% (n = 261) had a phase 2 DTC migration failure consistent with a full or near-full VCP (as in Fig 1B and Fig 2 i), and that 25% had a morphology consistent with a partial clear patch (Fig 2 ii-iv). Those consistent with a partial or near-full clear patch included animals in which the phase 2–3 migration started toward mid-body by migrating along the ventral BWMs, then migrated on a diagonal path to the dorsal BWMs (10.7%) (Fig 2 ii), meandered ventrally along the dorso-ventral axis back toward mid-body (4.2%) (Fig 2 iii), or changed direction frequently making multiple short excursions between turns (6.1%) (Fig 2 iv). It is important to note that some of the animals scored as unc-6 mutant-like phase 2 failures by D.I.C. microscopy sometimes had a DTC that made a dorsal excursion near the end of their phase 3 migration along the ventral side that might have appeared as partial VCPs by low power stereo- microscopy–this could readily account for the difference between the scores observed using stereo-microscopy compared with D.I.C. microscopy. Wild-type N2 animals grown at 25°C were not totally devoid of phase 2 DTC migration errors as 6 of 211 animals had phase 2 migration errors.
Fig 2. DTC navigational and ventral clear patch (VCP) phenotypes of ngat-1(ev821) animals.

The number (#) and percentage (%) of ngat-1(ev821) and GnT1 triple (see below) animals grown at 25°C with a given diagrammed posterior DTC abnormal (i-iv) or wild-type-like (WT = vi) trajectory (deduced from posterior gonad arm shapes) are shown. Diagrams are based on D.I.C. microscopy of gonad arms from a lateral perspective. Predicted corresponding ventral clear patch (VCP) phenotypes are indicated in the right-most column.
Nearly all of the observed phase 2 DTC migration errors occurred in the posterior gonad arm; the anterior arm was largely normal with fewer than 10% of animals showing anterior gonad arms with various abnormal morphologies, few of which resembled the phase 2 failures of the posterior DTC. In addition to the phase 2 posterior DTC migration errors, there were also a variety of DTC defects observed in the ev821 mutant grown at 25°C, all of which were also observed to roughly the same extent in N2 wild-type animals grown at 25°C. There were frequent defects in phase 1 and phase 3 navigation. Anterior and posterior DTC phase 1 navigation defects were largely limited to gonad arms that migrated prematurely to the dorsal side during phase 1 (5 of 211 posterior DTCs and 5 of 199 anterior DTCs). Phase 3-specific navigation defects included distal gonad arms that returned to the ventral side following a normal phase 2 (see Fig 2v), reversed direction on the dorsal side (2 of 199 anterior DTCs), or much more frequently migrated to the opposite side of the animal (switched sides) before stopping or continuing to migrate toward mid-body on the dorsal side. In rarer cases an entire gonad arm was found on the wrong side of the animal (e.g., 3 of 211 and 1 of 69 posterior DTCs in N2 and ev821 animals, respectively). Many of the 25°C-generated anterior and posterior gonad arms, including ones exhibiting normal navigation, had a grossly enlarged phase 3-generated distal portion—fewer had a grossly enlarged phase 1-generated proximal portion. All of the above phases 1 and 3 phenotypes were also observed with roughly equal ferequencies in N2 wild-type animals grown at 25°C, suggesting they do not result from the ngat-1 mutation, rather from growth at 25°C.
Genetic characterization of ngat-1(ev821)
The genetic characteristics of ev821 were first assayed by crossing genetically marked males (see Materials and methods) to hermaphrodites homozygous for ev821. The F1 cross progeny and subsequent generations of self-fertilization progeny were grown at 20°C. None of more than 1,000 F1s and only approximately 0.5% of the F2 animals from each cross had a full or near-full (not including partial) VCP phenotype (n = 2,071), compared to 29% for the starting ev821 parent strain propagated for several generations at 20°C, suggesting the possibility of maternal rescue for the ev821 gene (see below). From two of the above crosses, we cloned a total of 158 F2s and found that 36 of them (23%) segregated F3s with obvious VCPs, which is roughly the expected Mendelian frequency (25%) of ev821 homozygotes among the F2s. From 3 additional reciprocal crosses of ev821 males to genetically marked hermaphrodites, no F1s (0/200) or their self-fertilization F2 progeny (0/178) exhibited VCPs. From 178 cloned wild-type F2s, 50 (28%) segregated F3s with visible VCPs.
The relative absence of the VCP phenotype in the F1 and F2 generations and its subsequent reappearance in the F3 from the above crosses suggests a maternal rescue of the ev821 gene phase 2 DTC migration defect. This was evident when the ev821 allele in the F1came from the hermaphrodite or the male parent. These results, when considered together, suggest that ev821 can be largely or totally rescued by a maternally or a zygotically functioning wild-type gene.
Evidence for temperature-sensitivity in ev821
In order to determine if different growth temperatures affect the penetrance of VCPs or other aspects of the ev821 phenotype, ev821 worms were grown for a minimum of two generations without starvation at 16°C, 20°C, and 25°C. We observed that ev821 animals grown at 16°C, 20°C and 25°C exhibited 9% (n = 200), 51% (n = 100) and 74% (n = 100) penetrance, respectively, for all 3 of the VCP phenotypes (including full, near-full, and partial VCPs) (Panel A in S1 Fig).
These results prompted us to repeat the first ev821 inheritance experiment to determine if the maternal-effect is still apparent when ev821 animals are crossed and grown at 25°C. In this experiment, none of 300 F1 worms had DTC defects, however, 14 of 15 cloned F1s segregated DTC migration defective (full or near-full VCP) F2 animals, which represented 2% (n = 1254) of the F2s—significantly higher (p<0.001 by one-tail z-test for difference between 2 proportions) than the 0.5% penetrance (n = 2071) among the F2 progeny grown at 20°C. Furthermore, 20 of the F2s with phase 2 DTC migration errors were cloned and all segregated substantial numbers of DTC defective VCP F3s. This shows that the degree of maternal rescue is reduced as the defect among the F2s becomes more penetrant at the higher temperature.
ev821 has a temperature-sensitive period for phase 2 DTC migration that ends near or soon after completion of embryogenesis
We also determined an approximate ts period for the ev821 phase 2 DTC migration errors (detected using D.I.C. microscopy) by growing the animals at 16°C or 25°C for several generations then shifting to 25°C or 16°C, respectively, at various stages of development (Fig 3). The results shown in Fig 3demonstrate that embryonic growth at 25°C causes a high penetrance of phase 2 posterior DTC guidance defects (categorized in S2 Fig) approximately equal to the penetrance of defects that occurs if shifted back to 16°C during or after the first larval (L1) phase. This means that growth during the ts period (late embryogenesis) at 25°C is almost completely causal for the increased penetrance of these defects relative to growth at 16°C in the ngat-1(ev821) mutant. In contrast, ngat-1(ev821) mutant animals grown continuously at 16°C (including throughout the ts period) have a lower penetrance of phase 2 DTC defects, but this penetrance is slightly increased (from 13% to approximately 27%) by shifting to growth at 25°C during one of the L1 to L3 larval stages. The finding that some increased penetrance of the mutant phenotype can occur following a shift to growth at 25°C during early (e.g., L1) and even later larval stages suggests that growth at 16°C during the ts period does not entirely protect against mutant phase 2 DTC migration defects following a shift to 25°C post-embryonically, whereas, growth at 25°C during the ts period is nearly completely causal for the increased penetrance of these defects relative to growth at 16°C.
Fig 3. Approximate ts period for the ev821 phase 2 DTC navigation errors.

ngat-1(ev821) animals were grown at 16°C or 25°C then shifted to test temperature (25°C or 16°C, respectively) at various larval stages of development (L1, L2, and L3). Control animals were maintained at 16°C or 25°C test temperature throughout the experiment. Animals were scored by D.I.C. microscopy in the 4th larval stage (L4) or as young adults for phase 2 posterior DTC migration errors. A temperature-sensitive period was readily identified for these defects, which was developmentally much earlier than manifestation of the late L3 stage phase 2 DTC migration defect. Error bars represent z-test based 95% confidence intervals.
It is easy to lose sight of the fact that ngat-1(+) function in an animal does not just prevent the temperature sensitivity of the phase 2 DTC migration, but it also prevents the phase 2 DTC migration defect that occurs following growth at the permissive temperature. This realization raises the possibility that NGAT-1 does not necessarily directly, or even indirectly, affect the ts process (or structure) per se to prevent its thermo-lability and subsequent effect on DTC phase 2 migration, but rather may act by a completely different, possibly temperature-independent mechanism to somehow bypass or compensate for the innate ts process (or structure). Further experiments to determine the migration-relevant glycoprotein target(s) of NGAT-1 and GnT1 activities should help distinguish between these possibilities.
It is worth noting that mutants of gale-1 which encodes UDP-glactosamine-4-epimerase, (interconverts UDP-galactose and UDP-glucose and in some animals UDP-N-acetylgalactosamine and UDP-N-acetylglucosamine), as well as sqv-7, which encodes a transporter of UDP- glucuronic acid, UDP-galactose, and UDP-N-acetylgalactosamine into the Golgi—also exhibit DTC migration defects that have not been extensively characterized [42]. Also of interest, an L1 period sensitive to rapidly alternating temperature changes, but not sensitive to a constant temperature, has been observed in miRNA mir-34 and mir-83 mutants at the time of DTC birth, but these mutations affect phases 1 and 3 of DTC migration [43]. The relationship of this form of temperature sensitivity to the temperature sensitivity of ngat-1 and other mutants (see below) for phase 2 posterior DTC migration remains to be determined.
Mapping and cloning of the ngat-1 gene identified by the ev821 mutation
The ev821 mutation was identified by one-step SNP mapping/whole genome sequencing (WGS) [44]. SNP-WGS mapping of the ev821 cross to CB4856 Hawaiian strain placed the ev821 mutation within a chromosome II region between 11 and 11.5 Mb corresponding to around 3.5 cM (sqt-1 is at 11.3 megabases at 3.44 cM). Comparison of the DNA sequence of the ev821 and wild-type strains in this region indicated potential missense or predicted chain terminating nonsense mutations in four genes: ptp-3/ protein tyrosine phosphatase, rsf-1/ras association domain protein, D1043.1/ transcription regulatory protein, and W02B12.11/glycosyltransferase enzyme. To identify the ev821 gene among these candidates, we carried out phenotypic rescue studies of the ev821 mutant animals grown at 25°C, starting with W02B12.11 (S3 Fig). Fosmid WRM0637aH09 (30 kB) spanning the W02B12.11 gene significantly rescued the ev821 DTC phenotype. A small (4.3kB) fragment of genomic DNA within this fosmid that included the W02B12.11 gene, 0.9 kB of sequence upstream of the predicted transcription start site, plus 1.3 kB of sequence downstream of the predicted transcription termination site was amplified and found to also efficiently rescue the ev821 posterior DTC phenotype—reducing the migration defects from 53% to 11% and strongly supporting the identification of W02B12.11 as responsible for the ev821 mutant DTC migration defect.
W02B12.11 encodes a 387-residue protein (Figs 4 and 5). The DNA sequence data indicates that the ev821 mutant strain carries a T for C substitution creating a chain terminating stop codon for codon 209 in exon IV of the W02B12.11 gene. To confirm the identification of ev821, we generated a second allele of the W02B12.11 gene using CRISPR-Cas9 gene editing [41] to target a deletion in the first exon of ngat-1. Candidate insertion/deletion strains were identified and a 96 base pair deletion (ev823) was verified that removed the sequence encoding nucleotides -2 to +94 with the A of ATG as position +1 of exon 1 (Fig 5). Thus the ev823 mutant is predicted to encode a protein missing the normal initiator codon and the N-terminal 31 residues of W02B12.11. The next in-frame ATG codon is codon 89 within the Stem domain. Mutant ngat-1(ev823) animals grown at 25°C and analyzed by D.I.C. microscopy manifested posterior DTC phase 2 migration defects similar to those observed in ngat-1(ev821), but at lower penetrance of 33% (n = 268) of animals, compared with 68% (n = 123) for ev821 grown at 25°C. As with ev821, this phenotype was temperature sensitive, with the penetrance of VCPs reduced to 16% (n = 168) following growth at 20°C. These results strongly support the identification of W02B12.11 as causal for the ev821 and ev823 phase 2 DTC migration defects. The finding that both an apparently strong, possibly null allele like ev821, and a weaker, possibly hypomorphic allele like ev823 cause the same spectrum of phase 2 defects and are both ts for these phenotypes suggests that any reduction of ngat-1/W02B12.11 gene activity reveals an underlying ts process or structure required for normal phase 2 DTC navigation.
Fig 4. Comparison of galactosyl transferases from C. elegans, D. melanogaster (fly), and humans.

Alignments were made of the most closely-related C. elegans (wormbase), fly (FBgn0027538, FBgn0031495) and human (Q8NCL4, Q10472) galactosyltransferases using CLUSTAL [47]. The ngat-1(ev821) nonsense mutation is indicated by blue triangles and the position of the Leu (or in this case Ile) residue, which specifies N-acetylgalactosamine transfer (vs Tyr or Phe, which do not) [48] is indicated by red triangles. Metal ion (red underline), acceptor (blue underline), and donor (green underline) binding sites as predicted from human galactosyl transferases [45]. Identical residues are shaded black and similar residues are shaded grey.
Fig 5. Organization of ngat-1 coding sequence and encoded protein.

Boxes represent cDNA exons whose approximate codon size is indicated within. Exon boundaries and the positions of the ev821 point mutation (red) and the ev823 deletion (red) are indicated above the exon boxes. The corresponding protein domain structure [48] is indicated below the exons (Cyto = Cytoplasmic domain, TM = transmembrane domain). Color coding for binding sites is as in Fig 4.
The W02B12.11 gene encodes a protein with sequence homology to the ß1-4- galactosyltransferase gene family (Fig 4). In these Golgi localized enzymes, a key residue in the catalytic domain determines if the enzyme utilizes UDP-galactose or UDP-N-acetylgalactosamine for the transfer reaction—with tyrosine and phenylalanine specifying galactose, and leucine or isoleucine specifying N-acetylgalactosaminyl transfer [45]. This key residue in W02B12.11 is isoleucine (Fig 4)—characteristic of enzymes that transfer N-acetylgalactosamine; thus, we have renamed the gene ngat-1 for ß1,4-N-acetylgalactosaminyltransferase.
The ev821 chain-termination nonsense mutation at codon 209 predicts deletion of the C-terminal half of the catalytic domain (Fig 5), including key residues for metal ion binding, sugar donor and sugar acceptor interactions [45]. This mutation is therefore likely to be a null mutation. Only one other C. elegans gene, bre-4, encodes an enzyme with the same predicted activity as NGAT-1, but it functions in the intestine [46] and does not cause posterior DTC migration defects, nor does it enhance the DTC defects of ngat-1(ev821) (S1 Table).
Loss of complex-type N-glycans causes DTC migration defects
The evidence that ngat-1 is required for normal DTC migration indicates that carbohydrate modifications play an important role in guided DTC migration. A possible role for the NGAT-1 enzyme may involve addition of N-acetylgalactosamine to complex N-glycans on one or more migration-relevant glycoproteins required for phase 2 DTC migration. If so, mutations that disrupt early steps in the elaboration of core glycans could block the synthesis of NGAT-1 dependent substrate structures and cause migration defects similar to those observed for ev821. C. elegans has three genes (gly-12,-13, and -14) encoding functional β-1,2-N-acetylglucosaminyltransferase enzymes (also known as GnTI or Mgat1) that catalyze the first step of N-glycan branching in the Golgi. Deletion of GnT1 activity in mice causes mid-gestation embryonic lethality [49, 50]. It was reported previously that a gly-14(id48)III; gly-12(id47) gly-13(ok712)X triple null mutant (putative GnT1/Mgat1 null) of C. elegans has no obvious developmental defects (Zhu et al. 2004); however, careful examination of the gonad arms of triple mutant hermaphrodites grown at 25°C revealed a normal phase 1 trajectory with phase 2 migration errors in 114 of 130 (88%) of posterior DTCs examined. Among 56 of these animals that were also scored for anterior DTC migration errors, there were only 4 anterior DTC migration defects, 2 of which were phase 2 specific and one each phase 1 or phase 3 specific. The posterior DTC phase 2 errors in the triple mutant often manifested somewhat differently from the phase 2 errors of the ngat-1(ev821) mutant. In the ngat-1 mutant, most gonad arms project back toward mid-body usually along the proximal arm of the gonad on the ventral body wall, however, in the GnT1 triple mutant, more often than in the ngat-1(ev821) mutant, the posterior DTC at the end of phase 1 projected a short distance toward mid-body, then moved away from the ventral side making several short migrations (rarely oriented away from mid-body) separated by abrupt changes in direction before stopping at some distance from the mid-body region (Fig 6A and 6B). Changes in direction were usually accompanied by a bulbous protrusion in the girth of the gonad arm and occasional changes in sidedness of the DTC migration during phase 3. The migration back toward mid-body along the proximal gonad arm on the ventral side did not occur as frequently or to the same extent in the GnT1 triple null mutant as in the ngat-1 mutants (Fig 6C), thereby causing a lower penetrance (approximately 30%) of visible VCPs compared to the nearly 75% penetrance of VCPs in the ngat-1 mutant animals. Furthermore, the phase 2 posterior DTC migration errors in GnT1 triple null mutant animals grown at 25°C were more frequent (48 of 56) than those grown at 20°C (30 of 563), which were more frequent than those grown at 16°C (8 of 62) (Panel B in S1 Fig). Similar phase 2 migration errors, the same temperature-sensitivity, and similar differences in penetrance of posterior vs anterior DTC migration defects raise the possibility that NGAT-1 and the GnT1 enzymes act sequentially on the same N-glycoprotein target(s) to prevent posterior DTC migration errors, with the ngat-1 defect being perhaps a slightly milder version of the GnT1 triple mutant defects, which are more penetrant.
Fig 6. Common posterior gonad arm phenotypes of the GnT1/Mgat1 triple null mutant.

(A-C) Diagram iv errors of Fig 2are of various trajectories in ngat-1 and the GnT1 triple mutants following normal phase 1 navigation, three of which are shown here by D.I.C. microscopy. In these, following a normal phase 1, the posterior DTC changes direction often and seemingly randomly, but rarely away from mid-body, although the distal arm in these cases does not return to the mid-body region. Panel C shows a posterior gonad arm like that of diagram ii. The return toward mid-body on the ventral side is usually shorter in the GnT1 triple compared to the ngat-1(ev821) mutant. Premature phase 1 ventral-to-dorsal migrations were observed for 7 anterior DTCs of 130 GnT1 triple mutant animals examined. Scale bar in panel A (for A-C) is 50 μm.
Temperature sensitive period of GnT1 triple null mutant
To further examine the idea that GnT1 and NGAT-1 may affect the same target(s) for glycosylation required for phase 2 DTC migration, we determined the approximate ts period of the GnT1 triple knockout mutant by scoring posterior VCPs. From Fig 7it is clear that the GnT1 triple null mutant strain cannot reverse a phase 2 DTC migration failure induced by embryonic growth at 25°C if shifted back to 16°C during the first larval L1 phase, whereas embryonic growth at 16°C leads to wild-type/normal DTC migration that can be only partially prevented by a shift to 25°C during the L1 stage. This shows that the ts period for phase 2 DTC migration revealed by the GnT1 triple mutant probably ends during late embryogenesis or during the early L1 phase—just as the ts period revealed by the ngat-1(ev821) mutant does. This unusual phenotype suggests that both NGAT-1 and GnT1/Mgat1 acting in series may directly prevent, stabilize, reverse, or indirectly compensate for the same early developmental thermo-labile process (or structure) that leads to the much later 3rd larval stage phase 2 navigation defects observed in both kinds of mutants, possibly by modifying glycan(s) on the same target glycoprotein(s) relevant to phase 2 DTC migration.
Fig 7. Temperature-sensitive period for GnT1 triple mutant phase 2 DTC migration errors.
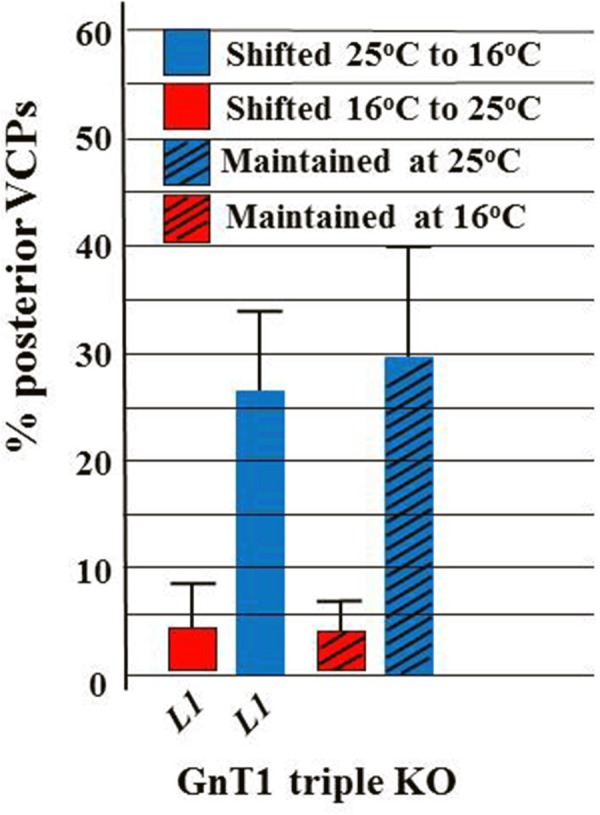
A temperature shift experiment similar to that of Fig 3, but limited to shifting only L1s (based on the Fig 3ev821 results) was performed for the GnT1 triple null mutant scored for all 3 kinds of VCPs by low magnification stereomicroscopy. Visible VCPs are not as frequent as phase 2 defects scored by D.I.C. in the GnT1 triple mutant, probably because the distal arm return along the proximal arm is typically much shorter (creating a much smaller VCP) in the GnT1 triple than in ngat-1(ev821). Neither the ngat-1(ev821) nor the GnT1 triple null mutant can prevent, reverse, or compensate a phase 2 DTC migration failure induced by embryonic growth at 25°C if shifted back to 16°C during the first larval (L1) phase, whereas embryonic growth at 16°C leads to largely normal DTC migration that cannot be reversed by a shift to 25°C during the first larval (L1) stage.
We decided to further examine the possibility that GnT1 and NGAT-1 act in the same pathway to prevent the temperature sensitivity of phase 2 DTC migration To do this, we generated the ngat-1; gly-14; gly-12 gly-13 quadruple null mutant and compared its DTC migration defects to those of the gly-14; gly-12 gly-13 triple null mutant. We expected an enhancement of the phase 2 DTC migration defects following growth at 20°C if the enzymes act in different pathways (e.g., via glycosylation of different targets) and no enhancement if they act in the same pathway (e.g., via glycosylation of the same target(s)). We found that the quadruple null mutant had nearly the same penetrance of posterior DTC phase 2 migration errors (42%, n = 65, measured by D.I.C.) as the triple null mutant (40%, n = 86) following growth in parallel at 20°C, while the ngat-1(ev821) segregant (see Materials and methods) grown in parallel manifested 46% (n = 63) phase 2 posterior DTC migration defects (S2 Table). These results suggest that NGAT-1 and GnT1 enzyme activities function in the same pathway to prevent the early developmental temperature sensitivity of the posterior DTC phase 2 migration. Glycolipids in C. elegans can be modified by the NGAT-1 homolog BRE-4 [46], however, these quadruple mutant results effectively exclude glycolipids as relevant to the DTC migration phenotype observed in this study.
ngat-1::gfp expression pattern
An ngat-1p::gfp transcriptional reporter was made comprising 1592 bps of the putative 5’ upstream promoter region of ngat-1 driving the gfp gene and a 3’ UTR of unc-54. Embryonic expression begins in the last third of embryonic development somewhat prior to the comma stage of embryogenesis (Fig 8). Embryonic expression was observed in many cells, many of which also express a dpy-7 promoter-driven epidermal mCherry reporter, in presumptive BWMs, in intestinal precursors, in several unidentified accessory cells in the head, and in several additional unidentified embryonic cell types. GFP expression was also observed in the lateral epidermis, intestine, and body wall muscles of ngat-1::gfp larvae and adults (Fig 8). There was no obvious embryonic or post-embryonic expression in the gonad or DTCs, however, germline-specific expression is highly dependent on specific 3’ UTR binding proteins [51], so our use of the unc-54 3’ UTR in the above expression construct may prevent germline expression
Fig 8. ngat-1p::gfp transcriptional reporter expression pattern.

A chromosomally integrated ngat-1p::gfp transcriptional reporter strain was made as described in Materials and methods. A dpy-7p::mCherry transcriptional reporter was injected into the ngat-1::gfp integrated line to create a non-integrated marker for epidermal cells (see Materials and methods), which may not mark all epidermal cells in a given animal because of potential mitotic loss of the extrachromosomal array. (A-B) merge of gfp and mCherry signals showing presumptive mCherry-expressing epidermal cells (thick arrows) that co-express gfp (as indicated by orange tinge) plus presumptive muscle cells (thin arrows), and presumptive intestinal cells (star) that express only ngat-1p::gfp in a comma stage embryo. (C-E) The ngat-1p::gfp reporter is expressed in body wall muscles (arrows in C and E) and in lateral epidermis (asterisks in D and E) in all post-embryonic larval stages. Some unidentified elongated cells in the head also express gfp and at least one (arrowhead) also expresses mCherry. (Dark oval-shaped areas in the lateral epidermis are lateral seam cells). rf = red fluorescence channel; gf = green fluorescence channel. Scale bar in panel A (for A and B) is 12.5 μm. Scale bar in panel C (for C-E) is 100 μm.
Transcriptional reporters for the GnT1 encoding genes gly-12, gly-13 and gly-14 were reported by Chen et al. [32]. The gly-12 reporter was expressed in all stages of development in many tissues including intestine, muscle, epidermis, other epithelial cells, and in ganglia in the head and tail region. The gly-13 reporter was expressed from late embryogenesis to adulthood and was confined to intestinal cells in L1 larvae, but later roughly mimicked gly-12 expression. The gly-14 reporter was observed only in the intestine in animals ranging from L1 to adulthood. Expression was not detected in embryos, although significant levels of gly-14 embryonic mRNA were detected.
Cell/tissue-specific rescue
For cell type specific rescue experiments, we made animals transgenic for constructs in which cell type-specific heterologous promoters drive expression of ngat-1(+)—encoded by genomic DNA that includes all 6 exons and 698 bps of sequence downstream of the translational terminator codon. The ev821 mutant DTC defects following growth at 25°C were largely rescued when ngat-1(+) expression was driven by the putatively muscle-specific myo-3 promoter or by the putatively DTC-specific lag-2 promoter (S3 Fig, S3 Table). This suggests that body wall muscle expression and DTC expression of ngat-1(+) are each sufficient to rescue the mutant DTC migration defects (however, see caveats in Discussion).
ngat-1(ev821) mutant animals transgenic for a construct of ngat-1(+) regulated by the intestine-specific elt-2 promoter, were not significantly rescued their phase 2 DTC migration defects (S3 Fig, S3 Table). To examine the ability of hypodermal/epidermal expression to rescue, we used the dpy-7 epidermis-specific promoter to drive ngat-1(+) expression, but were unable to recover viable transgenic progeny in several attempts at making them.
NGAT-1 functions in parallel with the UNC-6/netrin pathway
To determine if NGAT-1 might function exclusively in the UNC-6/netrin signaling pathway required to prevent phase 2 DTC migration failures, we made an ngat-1(ev821); unc-6(ev400) double mutant and quantified its VCP phenotype following growth at 25°C to compare with unc-6(ev400) and ngat-1(ev821) single mutant animals grown in parallel (S4 Table). In this experiment the ev821 homozygotes exhibited 39% netrin mutant-like phase 2 DTC migration failures identified by a full ventral clear patch extending from the vulva toward the tail (n = 105). There was an additional 35% penetrance of a partial clear patch posterior to and separate from the clear patch of the vulva. The unc-6(ev400) mutant grown in parallel under identical conditions exhibited a penetrance of 78% for these two kinds of VCPs (65% full and 13% partial VCPs, n = 101). The unc-6(ev400); ngat-1(ev821) double mutant grown under identical conditions exhibited a penetrance of 95% VCPs (84% full and 11% partial, n = 110), which is just statistically significant (p = 0.0423 by one-tail z-test for difference between 2 proportions) and almost exactly what is expected [78% + (74% x 22%) = 94%] if NGAT-1 has functions in parallel to the UNC-6/netrin signaling pathway that mediates phase 2 DTC migration. There was no apparent enhancement of the anterior phase 2 DTC VCPs of the unc-6(ev400) mutant by ngat-1(ev821) (S4 Table)—a result that is consistent with the finding that ngat-1(ev821) has little apparent effect on anterior DTC migration.
Discussion
Phase 2 ventral-to-dorsal DTC migration across the lateral epidermis occurs during the late 3rd larval stage of development and this migration frequently fails in unc-6/netrin mutants and mutants of the netrin receptor encoding genes unc-40 and unc-5. Our identification of weak alleles of unc-40 in a screen for suppressors of axon guidance defects induced by ectopic expression of UNC-5 [52], supplied a sensitized genetic background in which to generate enhancer mutations that identify other genes required for DTC migration. The ev821 mutation was identified as such an enhancer, but when isolated away from the weak unc-40(ev543) allele was found to induce DTC migration defects on its own. We cloned the ev821 mutant gene by one step SNP/WGS and the ev821 mutation was found to create a premature stop codon in the ORF for a putative N-acetyl-galactosaminyltransferase encoding gene we named ngat-1. Many, but not all of the ngat-1(ev821) mutant DTC migration defects were phase 2 DTC migration failures, nearly half of which were similar to those previously observed in mutants of unc-5, unc-40 and unc-6, while other phase 2 migration errors varied in migration pattern, including those that began migrating toward mid-body on the ventral BWMs then migrated to the dorsal BWMs before reaching mid-body. Other relatively infrequent defects in ev821 mutant DTC migration included DTCs meandering along the D/V axis on the ventral side as they migrate back toward the mid-body during the equivalent of phases 2 and 3 of migration. The DTC migration defects in ngat-1(ev821) (and GnT1/Mgat1 triple) mutants differ in several additional ways from the netrin mutant DTC migration defects. First, they appear to affect primarily posterior DTC migration and have few effects on anterior migration. This is somewhat unexpected, except that for unknown reasons, even in unc-5, unc-6 and unc-40 mutants the phase 2 navigational errors of the posterior DTC are more penetrant than the anterior DTC. However, anterior DTC migrations in the ngat-1 and GnT1 mutants might simply fall below some threshold for ventral to dorsal migration activity that is not present in the posterior DTC and not encountered in the netrin mutants. This seems unlikely, however, because one would have expected that any effect of ngat-1 mutation on the phase 2 anterior DTC migration would have manifested in a genetic background sensitized by the absence of unc-6 function, yet ngat-1(ev821) failed to enhance the anterior DTC defects (VCPs) of the unc-6(ev400) mutant. This suggests that ngat-1, unlike unc-6, has little if any role in anterior DTC migration (or is redundant with another unknown gene) and its function largely in posterior DTC migration may begin to account for known differences in the sensitivity of the two DTCs to mutations in other migration genes.
A second difference between the netrin mutants and ngat-1(ev821) is that the ngat-1 mutant DTC migration defects are maternally rescued, that is, the homozygous mutant self-fertilization progeny of a heterozygous hermaphrodite parent rarely manifest phase 2 DTC navigation errors, whereas the self-fertilization progeny of these homozygotes do manifest a significant penetrance of these errors. Maternal rescue is known to occur for the majority of mutations in genes required for embryonic viability, which suggests that products of most maternally rescued genes are present when their function is needed during embryogenesis. Unlike most essential embryonic genes showing maternal rescue, the ngat-1(+) gene is required for a much later (post-embryonic) process leading to phase 2 DTC migration. A third difference between ev821 and netrin mutants—the severe temperature sensitivity of the former and not the latter (S5 Table)–allowed us to estimate the developmental period in which subsequent phase 2 DTC migration is sensitive to temperature in the absence of NGAT-1 or, as we later showed, GnT1 function. Surprisingly, the ts period for ngat-1 and GnT1 mutants ends by the beginning of post-embryonic development or slightly thereafter in the first post-embryonic larval stage to allow a migration that occurs nearly 3 full larval stages later.
How can one reconcile the apparent early ts period with the late effect on DTC migration of missing NGAT-1 and GnT1 function? In principle, ngat-1(ev821) (and possibly one or more of the gly gene mutants) is temperature sensitive for synthesis of the corresponding gene product (which normally occurs during the ts period) or creates a ts NGAT-1 protein. However, the possibility of either of these scenarios for NGAT-1 or GnT1 is remote, given the high likelihood that both ngat-1(ev821) and the triple gly-14; gly-12 gly-13 mutants are null for their respective NGAT-1 and GnT1 activities. This consideration suggests that NGAT-1 and GnT1 activities somehow prevent the innate temperature sensitivity of the phase 2 DTC migration, which has a ts period that ends by the L1 stage. NGAT-1 and GnT1 could do this by acting directly in a ts process or structure to prevent or reverse its thermolability (in which case the ts period could represent the period of NGAT-1 and GnT1 activity) or it could act in a separate process that bypasses or compensates for the ts deficiency in some other unknown way (in which case the ts period may not represent the time of NGAT-1 and GnT1 action). However, the great majority of maternally rescued genes with few exceptions [53] function during embryogenesis or perinatally, therefore it seems likely that NGAT-1 and GnT1 function during the innate ts period rather than closer to the time of the phase 2 migration they regulate. Distinguishing between these possibilities will require—as a first step—the identification and characterization of the target(s) of NGAT-1 and GnT1activity relevant to phase 2 DTC migration.
NGAT-1 and GnT1 are predicted by their enzymatic activity to modify one or more secreted glycoproteins. The ability of a lag-2 promoter-driven ngat-1(+) construct to rescue mutant DTC migration defects suggests that a DTC-expressed glycoprotein can be modified by GnT1 and NGAT-1 to rescue the innate temperature sensitivity of the phase 2 DTC migration. The ability of body wall muscle (BWM) expressed NGAT-1 to rescue the ngat-1(ev821) DTC migration defects—even though BWM is not the substratum for the phase 2 migration–also suggests that a glycoconjugate target (possibly the same as the DTC-expressed target) of NGAT-1 activity can be secreted by BWMs to promote phase 2 migration at restrictive temperature, probably by incorporating into the gonad/DTC basement membrane. Precedent for this kind of autonomous and non-autonomous activity that promotes phase 2 DTC migration is found in MIG-17, which is made by body wall muscles and incorporates into the gonad basement membrane where it functions, but can also rescue a mig-17 mutant DTC defects when expressed in the DTCs [12, 14]. An important caveat to the above interpretations is that lag-2 and myo-3 reporters are not entirely specific to DTCs and muscle, respectively. For example, myo-3 is also expressed by the gonad sheath cells [54], which help create the gonad/DTC basement membrane, and may conceivably also be expressed below limits of detection in the DTCs and lag-2 is expressed in the ventral nerve cord and vulval precursor cells in the L3 stage [55]. Nonetheless, it is possible (as occurs for mig-17) that non-autonomous rescue by BWM and perhaps other tissue-specific expression of NGAT-1 does occur and possible that cell autonomous rescue can also occur.
One of our immediate goals is to identify potential targets for LacdiNAc modification by NGAT-1 in C. elegans and by related galactosyl transferases in mammals to begin to illuminate the ngat-1 requirement for cell migration. When we started, we considered possible targets for NGAT-1 mediated glycosylation included N- or O- glycoproteins or glycolipids. The finding that gly-14;gly-12 gly-13 triple mutant animals have DTC migration defects similar to those of ngat-1 mutants (temperature-sensitive phase 2 errors largely limited to posterior DTCs with a similar early ts period) suggests that the migration-relevant NGAT-1 target(s) is the same N-glycoprotein(s) as the GnT1 target(s). The nearly identical penetrance of phase 2 DTC migration defects in the gly-14; gly-12 gly-13 triple null and the ngat-1; gly-14, gly-12 gly-13 quadruple null mutant suggests that the GnT1 activity encoded by the 3 gly genes and the activity encoded by ngat-1 function in the same glycosylation pathway, which strongly supports the idea that GnT1 activity creates a N-glycan modification that serves as a substrate for NGAT-1-mediated LacdiNAc modification. Mass spectrometry analysis may be required to confirm this possibility, which if true would suggest that it is not the absence of GnT1 derived core glycan structures per se that cause the phase 2 DTC migration defects, but more likely the absence of the terminal LacdiNAc modification dependent upon these core glycan structures that is largely, but not totally, responsible for these defects.
The ngat-1 null mutant DTC migration phenotype differs from other glycosylation- associated gene mutants
Other glycogene mutants affecting DTC migration like cogc-1, cogc-3, mig-22 and mig-23 are likely to cause severe but unpredictable alterations of glycan chains of many proteins. These all appear to cause a meandering back to mid-body DTC migration pattern following an apparently normal phase 1 migration. The ngat-1 mutations on the other hand are predicted to cause a very specific alteration in glycan structure (absence of a glycan-terminal LacdiNAc modification) which in this case leads to a fairly specific defect in DTC migration–phase 2-specific migration errors, which are frequent in the ngat-1 mutants but not an obvious frequent phenotype observed in other glycogene mutants. This phenotype in ngat-1 mutants does resemble the DTC phase 2 errors that are characteristic of netrin signaling mutants, however, there are some variants, especially DTCs that following phase 1 appear to return partially toward mid-body along the ventral BWMs before turning dorsally. The double mutant analysis indicates that NGAT-1 (and by inference GnT1) has navigation functions independent of the netrin signaling pathway as might be expected of a gene affecting the substratum for phase 2 DTC migration (the lateral epidermis for phase 2 migration), which should be necessary for both netrin dependent and netrin independent migration.
Potential vertebrate roles
The ngat-1 gene and its targets represent a potential causal link between a specific glyco-epitope (LacdiNAc) and cell migration. The discovery of glycogene mutants with cell migration defects predicted to cause deficits in LacdiNAc modification of glycoproteins has an interesting parallel to reports that the LacdiNAc epitope in mammals plays a role in cancer progression and metastasis [56–60]. Furthermore, one of us (JWD) has reported previously that deficiencies in one or more vertebrate enzymes of the N-glycan branching pathway (comprising Mgat1/GnT1, alpha-mannosidase II/ManII, Mgat2, Mgat4, and Mgat5 activity—see [61]) modifies tumor growth and reduces cell migration, invasion and metastasis [29, 61–63].
The predicted absence of a specific glyco-epitope (LacdiNAc) in ngat-1 and Mgat1/GnT1 (gly12, -13, -14) mutants that causes a specific cell migration defect and the existence of reagents that detect this epitope [64, 65] suggest it should be possible to biochemically identify the target(s) of NGAT-1 and GnT1 mediated glycosylation required for normal DTC phase 2 pathfinding, which are presumably present in both BWMs and DTCs as determined by cell type specific rescue experiments of the ngat-1 mutant. The link between LacdiNAc modification and tumor progression further suggests that a genetic dissection of this mechanism in C. elegans and analysis of mammalian homologs of the relevant target genes identified in this genetic model could have translational potential. The maternal effect and later manifestation of a cell migration phenotype also suggests the possibility that NGAT-1 related enzymes in mammals might mediate a previously unrecognized perinatal function that contributes to later onset human health issues.
Supporting information
Bars indicate the penetrance of DTC phase 2 errors when grown >2 generations at the indicated temperature. (A) ngat-1(ev821) posterior DTC migration defects were scored as VCPs by stereomicroscopy, whereas, (B) GnT1/gly-14; gly-12 gly-13 mutant DTC defects were scored by D.I.C. microscopy. Error bars represent z-test based 95% confidence intervals for a proportion.
(TIF)
(A) Bars shows the penetrance of phase 2 posterior DTC migration failures of the types (DTC-1 to DTC-6) represented diagrammatically in panel B. L1-L4 = larval stages 1–4. Note that in a different plot of these data, shown in Fig 3, the DTC-5 category is not included as phase 2 errors because many similar trajectories were later found in wild-type animals grown at 25°C.
(TIF)
Constructs carrying ngat-1(+) driven by different regulatory regions were made as described in Materials and methods. These include 1) ngat-1(+) in fosmid WRM0637aH09, 2) a 4.3 PCR product encompassing W02B12.11, 3) ngat-1(+) driven by body wall muscle specific promoter myo-3p, 4) DTC-specific promoter lag-2, and intestine-specific promoter elt-2 (see Materials and methods). These results show that W02B12.11/ngat-1(+) expression in body wall muscles or in DTCs (see caveat in Discussion) is largely sufficient to rescue the ngat-1(ev821) mutant DTC migration defects of animals grown at 25°C, whereas, expression in embryonic intestine by the elt-2 promoter does not rescue these defects. Error bars represent z-test based 95% confidence intervals for a proportion.
(TIF)
1An ngat-1(ev821); bre-4(ok3167) double mutant—constructed by standard methods—was grown in parallel with both single mutants for two generations at 23°C before monitoring VCPs in L4 animals. Shown are number (#) of gonad arms scored and percentage (%) of animals with given VCP phenotype.
(TIF)
1Number (#) and percentage (%) of animals with phase 2 posterior DTC migration defects.
2Two of the 3 anterior DTC navigation errors (in the triple and quadruple mutants) were premature phase 1 migration toward the dorsal side and 1 was a phase 2 error.
3One anterior DTC migrated along a normal 3 phase trajectory except on the left instead of the right side epidermis in the triple mutant. There was 1, 1 and 3 otherwise normally guided but wrong-sided posterior DTC migrations in the triple, quadruple, and single mutant, respectively.
(TIF)
(TIF)
1An ngat-1(ev821); unc-6(ev400) double mutant—constructed by standard methods—was grown in parallel with both single mutants for two generations at 25°C before scoring VCPs in L4 animals. Ant. = Anterior DTC; Post. = Posterior DTC; Full = full and near-full, Part. = Partial; VCP = Ventral Clear Patch, # = number of gonad arms scored. % = percentage of animals with a given VCP phenotype.
(TIF)
1Number (#) of gonad arms scored following growth for two generations at 16°C or 25°C and percentage (%) of animals with a given VCP phenotype.
2 p = 0.66 by 2-tailed z-test.
3 p = 0.47 by 2-tailed z-test.
(TIF)
Acknowledgments
The authors want to thank Louise Brown for help with microscopy, Kin Chan for whole genome sequencing, and Aldis Krizus for technical assistance with the GnT1 triple mutant.
Data Availability
All relevant data are in the paper and its Supporting Information files. Sequencing data are uploaded to WormBase. The URL for W02B12.11 (= ngat-1) is www.wormbase.org/species/c_elegans/gene/WBGene00012206#0b-f9e4a56g3d8ci1-10.
Funding Statement
This work was supported by Natural Sciences and Engineering and Research Council of Canada, Grant number RGPIN-20160o6644 UT to JGC (URL: http://www.nserc-crsng.gc.ca/onlineservices-servicesenligne/index_eng.asp); and Canadian Institutes of Health Research, Grant number MOP 77722 to JGC (URL: http://cihr-irsc.gc.ca/e/193.html).
References
- 1.Hedgecock EM, Culotti JG, Hall DH. The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron. 1990;4(1):61–85. Epub 1990/01/01. . [DOI] [PubMed] [Google Scholar]
- 2.Merz DC, Zheng H, Killeen MT, Krizus A, Culotti JG. Multiple signaling mechanisms of the UNC-6/netrin receptors UNC-5 and UNC-40/DCC in vivo. Genetics. 2001;158(3):1071–80. Epub 2001/07/17. ; PubMed Central PMCID: PMC1461735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Su M, Merz DC, Killeen MT, Zhou Y, Zheng H, Kramer JM, et al. Regulation of the UNC-5 netrin receptor initiates the first reorientation of migrating distal tip cells in Caenorhabditis elegans. Development. 2000;127(3):585–94. Epub 2000/01/13. . [DOI] [PubMed] [Google Scholar]
- 4.Saied-Santiago K, Townley RA, Attonito JD, da Cunha DS, Diaz-Balzac CA, Tecle E, et al. Coordination of Heparan Sulfate Proteoglycans with Wnt Signaling To Control Cellular Migrations and Positioning in Caenorhabditis elegans. Genetics. 2017. Epub 2017/06/04. doi: 10.1534/genetics.116.198739 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diaz-Balzac CA, Lazaro-Pena MI, Tecle E, Gomez N, Bulow HE. Complex cooperative functions of heparan sulfate proteoglycans shape nervous system development in Caenorhabditis elegans. G3 (Bethesda). 2014;4(10):1859–70. Epub 2014/08/08. doi: 10.1534/g3.114.012591 ; PubMed Central PMCID: PMC4199693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Merz DC, Alves G, Kawano T, Zheng H, Culotti JG. UNC-52/perlecan affects gonadal leader cell migrations in C. elegans hermaphrodites through alterations in growth factor signaling. Dev Biol. 2003;256(1):173–86. Epub 2003/03/26. . [DOI] [PubMed] [Google Scholar]
- 7.Schwabiuk M, Coudiere L, Merz DC. SDN-1/syndecan regulates growth factor signaling in distal tip cell migrations in C. elegans. Dev Biol. 2009;334(1):235–42. Epub 2009/07/28. doi: 10.1016/j.ydbio.2009.07.020 . [DOI] [PubMed] [Google Scholar]
- 8.Suzuki N, Toyoda H, Sano M, Nishiwaki K. Chondroitin acts in the guidance of gonadal distal tip cells in C. elegans. Dev Biol. 2006;300(2):635–46. Epub 2006/09/20. doi: 10.1016/j.ydbio.2006.08.037 . [DOI] [PubMed] [Google Scholar]
- 9.Kinnunen T, Huang Z, Townsend J, Gatdula MM, Brown JR, Esko JD, et al. Heparan 2-O-sulfotransferase, hst-2, is essential for normal cell migration in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2005;102(5):1507–12. Epub 2005/01/27. doi: 10.1073/pnas.0401591102 ; PubMed Central PMCID: PMC547812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu X, Rongali SC, Miles JP, Lee KD, Lee M. pat-4/ILK and unc-112/Mig-2 are required for gonad function in Caenorhabditis elegans. Exp Cell Res. 2006;312(9):1475–83. Epub 2006/02/16. doi: 10.1016/j.yexcr.2006.01.006 . [DOI] [PubMed] [Google Scholar]
- 11.Lee M, Cram EJ, Shen B, Schwarzbauer JE. Roles for beta(pat-3) integrins in development and function of Caenorhabditis elegans muscles and gonads. J Biol Chem. 2001;276(39):36404–10. Epub 2001/07/27. doi: 10.1074/jbc.M105795200 . [DOI] [PubMed] [Google Scholar]
- 12.Nishiwaki K, Hisamoto N, Matsumoto K. A metalloprotease disintegrin that controls cell migration in Caenorhabditis elegans. Science. 2000;288(5474):2205–8. Epub 2000/06/24. . [DOI] [PubMed] [Google Scholar]
- 13.Blelloch R, Anna-Arriola SS, Gao D, Li Y, Hodgkin J, Kimble J. The gon-1 gene is required for gonadal morphogenesis in Caenorhabditis elegans. Dev Biol. 1999;216(1):382–93. Epub 1999/12/10. doi: 10.1006/dbio.1999.9491 . [DOI] [PubMed] [Google Scholar]
- 14.Ihara S, Nishiwaki K. Prodomain-dependent tissue targeting of an ADAMTS protease controls cell migration in Caenorhabditis elegans. EMBO J. 2007;26(11):2607–20. Epub 2007/05/12. doi: 10.1038/sj.emboj.7601718 ; PubMed Central PMCID: PMC1888677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishiwaki K, Kubota Y, Chigira Y, Roy SK, Suzuki M, Schvarzstein M, et al. An NDPase links ADAM protease glycosylation with organ morphogenesis in C. elegans. Nat Cell Biol. 2004;6(1):31–7. Epub 2003/12/23. doi: 10.1038/ncb1079 . [DOI] [PubMed] [Google Scholar]
- 16.Kawano T, Zheng H, Merz DC, Kohara Y, Tamai KK, Nishiwaki K, et al. C. elegans mig-6 encodes papilin isoforms that affect distinct aspects of DTC migration, and interacts genetically with mig-17 and collagen IV. Development. 2009;136(9):1433–42. Epub 2009/03/20. doi: 10.1242/dev.028472 ; PubMed Central PMCID: PMC2674254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kubota Y, Sano M, Goda S, Suzuki N, Nishiwaki K. The conserved oligomeric Golgi complex acts in organ morphogenesis via glycosylation of an ADAM protease in C. elegans. Development. 2006;133(2):263–73. Epub 2005/12/16. doi: 10.1242/dev.02195 . [DOI] [PubMed] [Google Scholar]
- 18.Levy-Strumpf N, Culotti JG. Netrins and Wnts function redundantly to regulate antero-posterior and dorso-ventral guidance in C. elegans. PLoS Genet. 2014;10(6):e1004381 Epub 2014/06/06. doi: 10.1371/journal.pgen.1004381 ; PubMed Central PMCID: PMC4046927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itoh B, Hirose T, Takata N, Nishiwaki K, Koga M, Ohshima Y, et al. SRC-1, a non-receptor type of protein tyrosine kinase, controls the direction of cell and growth cone migration in C. elegans. Development. 2005;132(23):5161–72. Epub 2005/10/28. doi: 10.1242/dev.02103 . [DOI] [PubMed] [Google Scholar]
- 20.Lucanic M, Kiley M, Ashcroft N, L'Etoile N, Cheng HJ. The Caenorhabditis elegans P21-activated kinases are differentially required for UNC-6/netrin-mediated commissural motor axon guidance. Development. 2006;133(22):4549–59. Epub 2006/10/20. doi: 10.1242/dev.02648 . [DOI] [PubMed] [Google Scholar]
- 21.Lundquist EA, Reddien PW, Hartwieg E, Horvitz HR, Bargmann CI. Three C. elegans Rac proteins and several alternative Rac regulators control axon guidance, cell migration and apoptotic cell phagocytosis. Development. 2001;128(22):4475–88. Epub 2001/11/21. . [DOI] [PubMed] [Google Scholar]
- 22.Soto MC, Qadota H, Kasuya K, Inoue M, Tsuboi D, Mello CC, et al. The GEX-2 and GEX-3 proteins are required for tissue morphogenesis and cell migrations in C. elegans. Genes Dev. 2002;16(5):620–32. Epub 2002/03/06. doi: 10.1101/gad.955702 ; PubMed Central PMCID: PMC155352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabello J, Neukomm LJ, Gunesdogan U, Burkart K, Charette SJ, Lochnit G, et al. The Wnt pathway controls cell death engulfment, spindle orientation, and migration through CED-10/Rac. PLoS Biol. 2010;8(2):e1000297 Epub 2010/02/04. doi: 10.1371/journal.pbio.1000297 ; PubMed Central PMCID: PMC2814829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cram EJ, Shang H, Schwarzbauer JE. A systematic RNA interference screen reveals a cell migration gene network in C. elegans. J Cell Sci. 2006;119(Pt 23):4811–8. Epub 2006/11/09. doi: 10.1242/jcs.03274 . [DOI] [PubMed] [Google Scholar]
- 25.Varki A, Kannagi R, Toole BP. Glycosylation Changes in Cancer. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, et al. , editors. Essentials of Glycobiology. 2nd ed. Cold Spring Harbor; (NY: )2009. [PubMed] [Google Scholar]
- 26.Deprez P, Gautschi M, Helenius A. More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol Cell. 2005;19(2):183–95. Epub 2005/07/26. doi: 10.1016/j.molcel.2005.05.029 . [DOI] [PubMed] [Google Scholar]
- 27.Warren CE, Krizus A, Dennis JW. Complementary expression patterns of six nonessential Caenorhabditis elegans core 2/I N-acetylglucosaminyltransferase homologues. Glycobiology. 2001;11(11):979–88. Epub 2001/12/18. . [DOI] [PubMed] [Google Scholar]
- 28.Warren CE, Krizus A, Roy PJ, Culotti JG, Dennis JW. The Caenorhabditis elegans gene, gly-2, can rescue the N-acetylglucosaminyltransferase V mutation of Lec4 cells. J Biol Chem. 2002;277(25):22829–38. Epub 2002/04/09. doi: 10.1074/jbc.M201390200 . [DOI] [PubMed] [Google Scholar]
- 29.Lau KS, Dennis JW. N-Glycans in cancer progression. Glycobiology. 2008;18(10):750–60. Epub 2008/08/15. doi: 10.1093/glycob/cwn071 . [DOI] [PubMed] [Google Scholar]
- 30.Contessa JN, Bhojani MS, Freeze HH, Ross BD, Rehemtulla A, Lawrence TS. Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin Cancer Res. 2010;16(12):3205–14. Epub 2010/04/24. doi: 10.1158/1078-0432.CCR-09-3331 ; PubMed Central PMCID: PMC3413408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126(5):855–67. Epub 2006/09/09. doi: 10.1016/j.cell.2006.08.019 . [DOI] [PubMed] [Google Scholar]
- 32.Chen S, Zhou S, Sarkar M, Spence AM, Schachter H. Expression of three Caenorhabditis elegans N-acetylglucosaminyltransferase I genes during development. J Biol Chem. 1999;274(1):288–97. Epub 1998/12/29. . [DOI] [PubMed] [Google Scholar]
- 33.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77(1):71–94. Epub 1974/05/01. ; PubMed Central PMCID: PMC1213120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wood WB. The Nematode Caenorhabditis elegans. Cold Spring Harbour, NY: Cold Spring Harbor Laboratory Press; 1988. [Google Scholar]
- 35.Zhu S, Hanneman A, Reinhold VN, Spence AM, Schachter H. Caenorhabditis elegans triple null mutant lacking UDP-N-acetyl-D-glucosamine:alpha-3-D-mannoside beta1,2-N-acetylglucosaminyltransferase I. Biochem J. 2004;382(Pt 3):995–1001. Epub 2004/07/02. doi: 10.1042/BJ20040793 ; PubMed Central PMCID: PMC1133976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hodgkin J, Horvitz HR, Brenner S. Nondisjunction Mutants of the Nematode CAENORHABDITIS ELEGANS. Genetics. 1979;91(1):67–94. Epub 1979/01/01. ; PubMed Central PMCID: PMC1213932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minevich G, Park DS, Blankenberg D, Poole RJ, Hobert O. CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics. 2012;192(4):1249–69. Epub 2012/10/12. doi: 10.1534/genetics.112.144204 ; PubMed Central PMCID: PMC3512137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sambrook J, Fristsch E. F., and Maniatis T. Molecular Cloning: A Laboratory Manual: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 39.L'Etoile ND, Bargmann CI. Olfaction and odor discrimination are mediated by the C. elegans guanylyl cyclase ODR-1. Neuron. 2000;25(3):575–86. Epub 2000/04/25. . [DOI] [PubMed] [Google Scholar]
- 40.Mello C, Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–82. Epub 1995/01/01. . [PubMed] [Google Scholar]
- 41.Dickinson DJ, Ward JD, Reiner DJ, Goldstein B. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods. 2013;10(10):1028–34. Epub 2013/09/03. doi: 10.1038/nmeth.2641 ; PubMed Central PMCID: PMC3905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brokate-Llanos AM, Monje JM, Murdoch Pdel S, Munoz MJ. Developmental defects in a Caenorhabditis elegans model for type III galactosemia. Genetics. 2014;198(4):1559–69. Epub 2014/10/10. doi: 10.1534/genetics.114.170084 ; PubMed Central PMCID: PMC4256771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burke SL, Hammell M, Ambros V. Robust Distal Tip Cell Pathfinding in the Face of Temperature Stress Is Ensured by Two Conserved microRNAS in Caenorhabditis elegans. Genetics. 2015;200(4):1201–18. Epub 2015/06/17. doi: 10.1534/genetics.115.179184 ; PubMed Central PMCID: PMC4574240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doitsidou M, Poole RJ, Sarin S, Bigelow H, Hobert O. C. elegans mutant identification with a one-step whole-genome-sequencing and SNP mapping strategy. PLoS One. 2010;5(11):e15435 Epub 2010/11/17. doi: 10.1371/journal.pone.0015435 ; PubMed Central PMCID: PMC2975709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramakrishnan B, Qasba PK. Role of a single amino acid in the evolution of glycans of invertebrates and vertebrates. J Mol Biol. 2007;365(3):570–6. Epub 2006/11/07. doi: 10.1016/j.jmb.2006.10.034 ; PubMed Central PMCID: PMC1850938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Griffitts JS, Huffman DL, Whitacre JL, Barrows BD, Marroquin LD, Muller R, et al. Resistance to a bacterial toxin is mediated by removal of a conserved glycosylation pathway required for toxin-host interactions. J Biol Chem. 2003;278(46):45594–602. Epub 2003/08/29. doi: 10.1074/jbc.M308142200 . [DOI] [PubMed] [Google Scholar]
- 47.McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, et al. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013;41(Web Server issue):W597–600. Epub 2013/05/15. doi: 10.1093/nar/gkt376 ; PubMed Central PMCID: PMC3692137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qasba PK, Ramakrishnan B, Boeggeman E. Structure and function of beta -1,4-galactosyltransferase. Curr Drug Targets. 2008;9(4):292–309. Epub 2008/04/09. ; PubMed Central PMCID: PMC2365515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ioffe E, Stanley P. Mice lacking N-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proc Natl Acad Sci U S A. 1994;91(2):728–32. Epub 1994/01/18. ; PubMed Central PMCID: PMC43022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Metzler M, Gertz A, Sarkar M, Schachter H, Schrader JW, Marth JD. Complex asparagine-linked oligosaccharides are required for morphogenic events during post-implantation development. EMBO J. 1994;13(9):2056–65. Epub 1994/05/01. ; PubMed Central PMCID: PMC395055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pushpa K, Kumar GA, Subramaniam K. Translational Control of Germ Cell Decisions. Results Probl Cell Differ. 2017;59:175–200. Epub 2017/03/02. doi: 10.1007/978-3-319-44820-6_6 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colavita A, Culotti JG. Suppressors of ectopic UNC-5 growth cone steering identify eight genes involved in axon guidance in Caenorhabditis elegans. Dev Biol. 1998;194(1):72–85. Epub 1998/03/14. doi: 10.1006/dbio.1997.8790 . [DOI] [PubMed] [Google Scholar]
- 53.Hekimi S, Boutis P, Lakowski B. Viable maternal-effect mutations that affect the development of the nematode Caenorhabditis elegans. Genetics. 1995;141(4):1351–64. Epub 1995/12/01. ; PubMed Central PMCID: PMC1206872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ono K, Ono S. Two distinct myosin II populations coordinate ovulatory contraction of the myoepithelial sheath in the Caenorhabditis elegans somatic gonad. Mol Biol Cell. 2016;27(7):1131–42. Epub 2016/02/13. doi: 10.1091/mbc.E15-09-0648 ; PubMed Central PMCID: PMC4814220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen N, Greenwald I. The lateral signal for LIN-12/Notch in C. elegans vulval development comprises redundant secreted and transmembrane DSL proteins. Dev Cell. 2004;6(2):183–92. Epub 2004/02/13. . [DOI] [PubMed] [Google Scholar]
- 56.Huang J, Liang JT, Huang HC, Shen TL, Chen HY, Lin NY, et al. Beta1,4-N-acetylgalactosaminyltransferase III enhances malignant phenotypes of colon cancer cells. Mol Cancer Res. 2007;5(6):543–52. Epub 2007/06/21. doi: 10.1158/1541-7786.MCR-06-0431 . [DOI] [PubMed] [Google Scholar]
- 57.Che MI, Huang J, Hung JS, Lin YC, Huang MJ, Lai HS, et al. beta1, 4-N-acetylgalactosaminyltransferase III modulates cancer stemness through EGFR signaling pathway in colon cancer cells. Oncotarget. 2014;5(11):3673–84. Epub 2014/07/09. doi: 10.18632/oncotarget.1981 ; PubMed Central PMCID: PMC4116512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hirano K, Matsuda A, Shirai T, Furukawa K. Expression of LacdiNAc groups on N-glycans among human tumors is complex. Biomed Res Int. 2014;2014:981627 Epub 2014/07/09. doi: 10.1155/2014/981627 ; PubMed Central PMCID: PMC4066867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsu WM, Che MI, Liao YF, Chang HH, Chen CH, Huang YM, et al. B4GALNT3 expression predicts a favorable prognosis and suppresses cell migration and invasion via beta(1) integrin signaling in neuroblastoma. Am J Pathol. 2011;179(3):1394–404. Epub 2011/07/12. doi: 10.1016/j.ajpath.2011.05.025 ; PubMed Central PMCID: PMC3157223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Machado E, Kandzia S, Carilho R, Altevogt P, Conradt HS, Costa J. N-Glycosylation of total cellular glycoproteins from the human ovarian carcinoma SKOV3 cell line and of recombinantly expressed human erythropoietin. Glycobiology. 2011;21(3):376–86. Epub 2010/10/30. doi: 10.1093/glycob/cwq170 . [DOI] [PubMed] [Google Scholar]
- 61.Beheshti Zavareh R, Sukhai MA, Hurren R, Gronda M, Wang X, Simpson CD, et al. Suppression of cancer progression by MGAT1 shRNA knockdown. PLoS One. 2012;7(9):e43721 Epub 2012/09/08. doi: 10.1371/journal.pone.0043721 ; PubMed Central PMCID: PMC3434202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dennis JW, Laferte S, Waghorne C, Breitman ML, Kerbel RS. Beta 1–6 branching of Asn-linked oligosaccharides is directly associated with metastasis. Science. 1987;236(4801):582–5. Epub 1987/05/01. . [DOI] [PubMed] [Google Scholar]
- 63.Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med. 2000;6(3):306–12. Epub 2000/03/04. doi: 10.1038/73163 . [DOI] [PubMed] [Google Scholar]
- 64.Nyame AK, Leppanen AM, DeBose-Boyd R, Cummings RD. Mice infected with Schistosoma mansoni generate antibodies to LacdiNAc (GalNAc beta 1—>4GlcNAc) determinants. Glycobiology. 1999;9(10):1029–35. Epub 1999/10/16. . [DOI] [PubMed] [Google Scholar]
- 65.van Remoortere A, Hokke CH, van Dam GJ, van Die I, Deelder AM, van den Eijnden DH. Various stages of schistosoma express Lewis(x), LacdiNAc, GalNAcbeta1-4 (Fucalpha1-3)GlcNAc and GalNAcbeta1-4(Fucalpha1-2Fucalpha1-3)GlcNAc carbohydrate epitopes: detection with monoclonal antibodies that are characterized by enzymatically synthesized neoglycoproteins. Glycobiology. 2000;10(6):601–9. Epub 2000/05/18. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bars indicate the penetrance of DTC phase 2 errors when grown >2 generations at the indicated temperature. (A) ngat-1(ev821) posterior DTC migration defects were scored as VCPs by stereomicroscopy, whereas, (B) GnT1/gly-14; gly-12 gly-13 mutant DTC defects were scored by D.I.C. microscopy. Error bars represent z-test based 95% confidence intervals for a proportion.
(TIF)
(A) Bars shows the penetrance of phase 2 posterior DTC migration failures of the types (DTC-1 to DTC-6) represented diagrammatically in panel B. L1-L4 = larval stages 1–4. Note that in a different plot of these data, shown in Fig 3, the DTC-5 category is not included as phase 2 errors because many similar trajectories were later found in wild-type animals grown at 25°C.
(TIF)
Constructs carrying ngat-1(+) driven by different regulatory regions were made as described in Materials and methods. These include 1) ngat-1(+) in fosmid WRM0637aH09, 2) a 4.3 PCR product encompassing W02B12.11, 3) ngat-1(+) driven by body wall muscle specific promoter myo-3p, 4) DTC-specific promoter lag-2, and intestine-specific promoter elt-2 (see Materials and methods). These results show that W02B12.11/ngat-1(+) expression in body wall muscles or in DTCs (see caveat in Discussion) is largely sufficient to rescue the ngat-1(ev821) mutant DTC migration defects of animals grown at 25°C, whereas, expression in embryonic intestine by the elt-2 promoter does not rescue these defects. Error bars represent z-test based 95% confidence intervals for a proportion.
(TIF)
1An ngat-1(ev821); bre-4(ok3167) double mutant—constructed by standard methods—was grown in parallel with both single mutants for two generations at 23°C before monitoring VCPs in L4 animals. Shown are number (#) of gonad arms scored and percentage (%) of animals with given VCP phenotype.
(TIF)
1Number (#) and percentage (%) of animals with phase 2 posterior DTC migration defects.
2Two of the 3 anterior DTC navigation errors (in the triple and quadruple mutants) were premature phase 1 migration toward the dorsal side and 1 was a phase 2 error.
3One anterior DTC migrated along a normal 3 phase trajectory except on the left instead of the right side epidermis in the triple mutant. There was 1, 1 and 3 otherwise normally guided but wrong-sided posterior DTC migrations in the triple, quadruple, and single mutant, respectively.
(TIF)
(TIF)
1An ngat-1(ev821); unc-6(ev400) double mutant—constructed by standard methods—was grown in parallel with both single mutants for two generations at 25°C before scoring VCPs in L4 animals. Ant. = Anterior DTC; Post. = Posterior DTC; Full = full and near-full, Part. = Partial; VCP = Ventral Clear Patch, # = number of gonad arms scored. % = percentage of animals with a given VCP phenotype.
(TIF)
1Number (#) of gonad arms scored following growth for two generations at 16°C or 25°C and percentage (%) of animals with a given VCP phenotype.
2 p = 0.66 by 2-tailed z-test.
3 p = 0.47 by 2-tailed z-test.
(TIF)
Data Availability Statement
All relevant data are in the paper and its Supporting Information files. Sequencing data are uploaded to WormBase. The URL for W02B12.11 (= ngat-1) is www.wormbase.org/species/c_elegans/gene/WBGene00012206#0b-f9e4a56g3d8ci1-10.