

Abstract
In many human diseases, associated genetic changes tend to occur within non-coding regions, whose effect might be related to transcriptional control. A central goal in human genetics is to understand the function of such non-coding regions: Given a region that is statistically associated with changes in gene expression (expression Quantitative Trait Locus; eQTL), does it in fact play a regulatory role? And if so, how is this role “coded” in its sequence? These questions were the subject of the Critical Assessment of Genome Interpretation eQTL challenge. Participants were given a set of sequences that flank eQTLs in humans and were asked to predict whether these are capable of regulating transcription (as evaluated by massively parallel reporter assays), and whether this capability changes between alternative alleles. Here, we report lessons learned from this community effort. By inspecting predictive properties in isolation, and conducting meta-analysis over the competing methods, we find that using chromatin accessibility and transcription factor binding as features in an ensemble of classifiers or regression models leads to the most accurate results. We then characterize the loci that are harder to predict, putting the spotlight on areas of weakness, which we expect to be the subject of future studies.
Keywords: gene regulation, massive paralleled reporter assays, eQTLs, functional genomics
INTRODUCTION
Mapping genotype to phenotype has been the focus of many studies in the post genomic era, with an increasing focus on the non-coding genome (Farh, et al., 2015; Hindorff, et al., 2009; Maurano, et al., 2012; Weingarten-Gabbay and Segal, 2014; Welter, et al., 2014; Zhou, et al., 2013). Gene expression has been and is still one of the most well investigated phenotypes by such studies, starting with the pioneering work that modeled it as a function of sequence features of proximal promoter regions, focusing primarily on the occurrence, location, orientation, and cooperative interactions (Das, et al., 2004; Segal, et al., 2008) of transcription factor (TF) binding motifs, and K-mer frequencies (Beer and Tavazoie, 2004; Bussemaker, et al., 2001; Nguyen and D’Haeseleer, 2006). With the development of sequencing-based technologies for chromatin profiling (Thurman, et al., 2012), methods for prediction of gene expression advanced accordingly, now adding experimentally measured chromatin properties as features (e.g., TF binding or histone modifications using ChIP-seq; chromatin accessibility using DNAse-seq or ATAC-seq; (Dong, et al., 2012; Gonzalez, et al., 2015; Marstrand and Storey, 2014; Natarajan, et al., 2012; Wilczynski, et al., 2012)). With these new types of data, a number of related questions and challenges emerged. One set of studies aimed at annotating the state of the chromatin into broad categories (e.g., enhancers, insulators) based on observed chromatin features and/or DNA-sequence (Ernst and Kellis, 2010; Hoffman, et al., 2012) and then associate the resulting distal regulatory regions with the correct target gene (e.g., using chromatin conformation assays (Rao, et al., 2014) or computational inference (Gonzalez, et al., 2015)). Another set of studies aimed at predicting chromatin features (e.g., accessibility, TF binding) based on DNA sequences (Weingarten-Gabbay and Segal, 2014), and predict the dependence of epigenetic features on genetic variation (e.g., single nucleotide variants (Ernst and Kellis, 2010; Erwin, et al., 2014; Lee, et al., 2015)). Recent methods, some of which are applied here, “closed the loop” and use DNA sequence alterations to predict changes in epigenetic features, which are then used as features for predicting the pertaining effects on the expression of the putative target genes.
One of the major hurdles in advancing this field and characterizing the regulatory “code” of the genome has been the lack of a well-controlled and scalable experimental system, which allows to investigate the direct effect of any sequence alteration of interest. A substantial progress to this end was the development of Massively Parallel Reporter Assays (MPRA) - a cost effective, high-throughput activity screening of fully synthesized DNA regions (Figure 1; (Smith, et al., 2013; Weingarten-Gabbay and Segal, 2014)). In MPRA, a library of thousands of putative regulatory DNA elements (each about 150-nt in length) with coupled unique tags is synthesized and used to generate a pool of plasmids; this pool is then transfected into cells and the regulatory activity (as an enhancer or promoter) associated with the respective DNA element is assessed by sequencing the abundances of the expressed tags. Since oligoarrays can now be printed in a cost effective fashion, MPRA provides a feasible means (albeit only in an episomal context (Inoue, et al., 2017)) to systematically interrogate how regulatory activity is encoded in the DNA (Birnbaum, et al., 2014; Kheradpour, et al., 2013; Melnikov, et al., 2012; Mogno, et al., 2013; Patwardhan, et al., 2012; Sharon, et al., 2012; Smith, et al., 2013), and estimate the effects of sequence variants.
Figure 1.
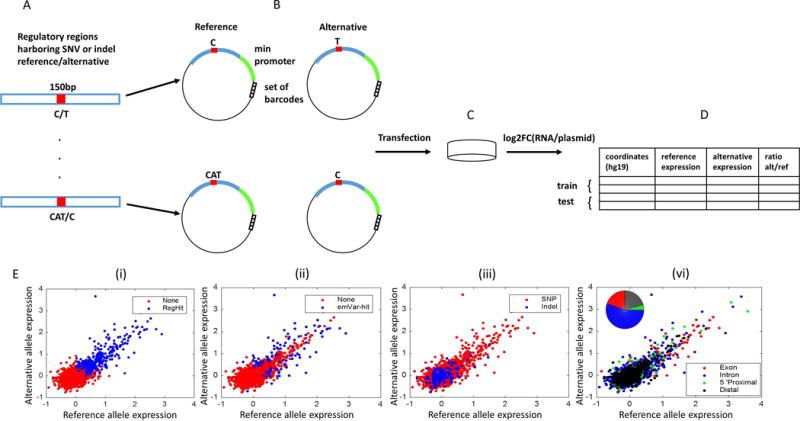
Experimental and challenge design: (A) Selection of regulatory regions that harbor a short polymorphism (SNV or indel). (B) Design of MPRA constructs for both reference and alternative alleles. (C) Transfection into two different LCLs. (D) Data provided for the challenge. (E) Alternative vs. reference allele expression for training set regions (i) regulatory hit/non-regulatory regions (ii) emVar hit/non- emVar regions (iii) indel/SNP regions are marked in blue/red respectively. (iv) exon, intron, promoter and distal regions are marked in red/blue/green/black respectively.
Since MPRA is still a nascent technology, computational methods that make effective use of it are still emerging (Gertz, et al., 2009). Specifically – how to leverage MPRA to build better and more accurate models for predicting whether a DNA region of interest plays a regulatory role, and if so, how does this its activity changes upon slight sequence variations (single nucleotide variants (SNV) of short insertions or deletions (indels)), commonly observed in human cohorts (Zhou, et al., 2013). The CAGI eQTL challenge is the first community effort aimed at advancing this type of studies. It is based on a comprehensive profiling of eQTLs observed in a subset of the Geuvadis database (Lappalainen, et al., 2013) with MPRA, culminating in over nine thousand regulatory sequences, in their reference genome form and their alternative (SNV or indel) form (Tewhey, et al., 2016). The goal of the challenge was two-fold – first, participants were asked to predict the regulatory activity of each regulatory sequence (reference or alternative allele) in isolation. Then, the participants were asked to predict the differences between each pair of alleles.
In the following sections we describe the results of this challenge and the lessons that can be learned via a meta-analysis of the competing methods. We start by summarizing the properties that were used by the participants as predictive features, divide these properties into several categories, and inspect the predictive ability of representative features from each category in isolation. We then move to inspect the predictive algorithms used by the participants and evaluate their overall performance using a range of metrics. As expected, we find that the predictions for the first part of the challenge (i.e., predicting the regulatory activity of each allele separately) were much more accurate than the second part (predict the differences between alleles), reflecting the difficulty in modeling the effects of nuanced sequence modifications. Furthermore, we find that overall the ranking of the participants is stable across specific sub-tasks and performance metrics, with the most promising methods belonging to the ones that “close the loop” as stated above. These methods use the DNA sequence as a primary feature for predicting epigenetic properties. These properties, in turn, are used to train an ensemble of models (e.g., using different learning algorithms) to provide a robust prediction of transcriptional activity and its dependence on sequence variation.
Focusing on loci that showed strong transcriptional activity in the MPRA, and taking a meta-analysis approach, we find that there are two distinct subsets of loci – one whose activity is predicted accurately by all (or most) competing methods, and another whose activity is poorly predicted by all competitors. Expectedly, we find that the “hard-to-predict” regions are associated with “paradigm-violating” properties, such as lack of accessibility or no apparent TF binding (as inferred by DNA-seq and ChIP-seq, or predicted by sequence-based models). While this is probably to some extent a result of inspecting regulatory activity in episomal setting, it may also point to knowledge gaps in defining the predictive features (e.g., unknown TF binding preferences) or properly combining these features in a predictive model.
THE eQTL-CAUSAL VARIANT CHALLENGE
Tewhey and colleagues used MPRA as a tool for investigating genetic variants that are statistically associated with changes to gene expression, considering both SNVs and indels (Tewhey, et al., 2016). These expression Quantitative Trait Loci (eQTL) were inferred based on a collection of immortalized lymphoblastoid cell lines (LCLs) derived from a large set of individuals, where both genome sequences and transcription profiles are available (Tewhey, et al., 2016). The eQTL causal SNP CAGI challenge was based on a subset of these loci, and included 3,157 eQTLs inferred based on individuals of European ancestry (Genomes Project, et al., 2012; Lappalainen, et al., 2013). Each eQTL in this collection is in turn associated with one or more variants (with an average of 3 variants per eQTL) whose individual effects are statistically indistinguishable due to linkage disequilibrium (LD; considering all loci that are in perfect LD with the top associated variant), leading to an overall set of 9,116 variants (8,570 SNVs and 546 indels; Figure 1).
For each variant, a sequence of 150-bp was synthesized, which includes the surrounding genome sequence (using reference genome hg19) with the variant located at the central position (Figure 1A). Each SNV was associated with two sequences that are identical except for the respective variation in position 76. For indels, the longer of the two alleles was designed as a 150-nt oligonucleotide; the shorter allele was then designed with the same flanking sequences as the longer allele (e.g., for a single-nucleotide indel TC/C: X[TC]Y and X[T]Y, where X and Y are 74bp long DNA segments that flank the variant in the reference genome). To increase the accuracy and sensitivity of the assay, 20-nt barcodes were added to the oligos by emulsion PCR, such that each oligo is represented by an average of a thousand barcode tags within the plasmid pool (Figure 1B). The plasmid library was electroporated into two LCLs (NA12878 and NA19239), using five and three technical replicates respectively. Importantly, NA12878 is an ENCODE tier 1 cell line (Consortium, 2012) and thus a large number of genomic assays performed on this cell line are publically available. Twenty-four hours after transfection, the GFP reporter mRNA was captured by hybridization, and RNA sequencing of the 3′-UTR-adjacent barcodes was performed to quantify the influence of each 150-bp sequence on regulation of the reporter gene. RNA expression measured in barcode read counts was normalized relative to the input plasmid barcode counts determined by DNA sequencing, such that the reported MPRA output consisted of an estimate for the ratio between the number of transcripts (RNA-seq) and plasmids (DNA-seq) (Figure 1C).
The resulting MPRA dataset was divided into a training set, which was made available to the participants, and a test set, which was held off and used for evaluation by independent assessors. The complete data set, including training and test subsets is provided in Supp. Table S1. The training set (Figure 1D–E) consisted of MPRA results for 3,044 variants (2,874 SNVs and 170 indels) associated with 1,052 eQTLs. For each variant, the information available for training included: (1) the respective genomic coordinates (using the hg19 reference genome), and the position and type of the variant. (2) an estimated transcript to plasmid ratio for each allele (defined as log fold change (log2FC); averaged across replicates). (3) an indication whether or not at least one of the two alleles exhibits a significantly high ratio of transcripts to DNA (regulatory hit). Significance of differential abundance of transcripts vs. plasmid input was evaluated using DE-seq2 (Love, et al., 2014) with a false discovery rate (FDR) cutoff of 1% to call hits. (4) Comparison of transcriptional activity between the two alleles. As before, this included a quantitative field indicating the fold change between the transcriptional activity of the two alleles (alternative/reference; LogSkew); and a binary field, indicating whether the difference is statistically significant (expression-modulating variants; emVar hits); Significance of allelic skew was evaluated using a t-test on the log-transformed RNA-seq/plasmid ratios across replicates with a FDR cutoff of 5% to call hits. (5) the name of the eQTL associated gene and the association’s coefficient (beta), t-statistic, and p-value. As expected, hit regions have significantly higher expression (Figure 1E (i–ii)).
The challenge consisted of two parts, each with its own test set. In the first part, the participants were asked to predict the level of transcriptional activity (log2FC) for each allele, and determine for each variant whether at least one of the alleles is a regulatory hit. In addition, each prediction should have included a standard deviation, reflecting the confidence in the predicted values. The corresponding test set (Supp. Table S1) consisted of MPRA results for 3,006 variants (2,811 SNVs and 195 indels) associated with 1,050 eQTLs. In the second part, the participants were given variants that are confirmed regulatory hits and asked to predict the difference between the transcriptional activity of the two alleles, both quantitatively (LogSkew) and qualitatively (emVar hits). As before, each prediction should also include an estimate of statistical confidence. The corresponding test data consisted of MPRA results for 401 variants (370 SNVs and 31 indels) associated with 1,055 eQTLs (Supp. Table S1).
Seven groups participated in the challenge. Each group was allowed to submit multiple predictions, resulting in overall 20 submissions for the first part, and 13 submissions for the second part. The submissions spanned a wide array of predictive features and prediction algorithms, as summarized in Table 1.
Table 1.
Summary of submissions. Methods and features used by each group for the two parts of the challenge. Features are divided into 4 classes: 1) experimentally measured epigenetic properties, 2) predicted epigenetic properties 3) other locus-specific properties 4) DNA k-mer frequencies. For the methods: R corresponds to the Regression tasks (predicting Log2FC in part I or LogSkew in part II); C corresponds to the classification tasks (predicting regulatory hits in part I or emVar hits in part II).
| Group | Features (feature classes 1–4) |
Methods (part I) (R: regression; C: classification) |
Methods (part II) (R: regression; C: classification) |
|---|---|---|---|
| 1 | Histone modifications in K562 cells (Consortium, 2012) (class 1); Evolutionary conservation (Siepel, et al., 2005) (class 3); k-mer frequencies (class 4); | Regularized regression (e.g., elastic net (Hui Zou, 2005); R, C), random forest (R, C), SVR (R), SVM (C) | Same as part I |
| 2 | Histone modifications, DHS, and TFBS in LCL (Consortium, 2012) (class 1); Predictions of DHS in 164 cell lines (Kelley, et al., 2016) (class 2); Predictions of TFBS (Cowper-Sallari, et al., 2012) and LCL-specific histone modifications and DHS based on (Consortium, 2012) (class 2). | Ensemble of gradient boosting models (Fabian Pedregosa, 2011). Each model trained on a different feature subset (R, C). | Same as part I |
| 3 | k-mers (class 4) | Linear SVR (R) and SVM (C) | Same as part I |
| 4 |
Part I: Segmentation of genomic regions based on histone modifications in LCL (Consortium, 2012; Ernst and Kellis, 2012) (class 1); Predictions of TFBS, DHS, and histone marks (using (Alipanahi, et al., 2015; Zhou and Troyanskaya, 2015), with data from (Consortium, 2012; Romanoski, et al., 2015); class 2). Part II: allele-specific activity level predicted by the models in part I |
Ensemble of models, using LASSO or Random Forest, and trained on different feature subsets (R). Ensemble of neural networks, trained different feature subsets (C) |
Difference between predicted alleles’ scores (R) Ensemble of classifiers (e.g., KNN; C) |
| 5 | Predictions of DHS (using (Ghandi, et al., 2014) with LCL data from (Consortium, 2012); class 2). | Predicted alleles’ DH scores are used directly (R,C) | Difference between DHS scores of the two alleles (Ghandi, et al., 2014) (R, C) |
| 6 |
Part I: Histone modifications, DHS, DNA-methylation, and TFBS in LCL (Consortium, 2012) (class 1); Predictions of TFBS, and protein binding sites in the transcribed RNA (using (Alipanahi, et al., 2015; Grant, et al., 2011; Hume, et al., 2015), with data from (Alipanahi, et al., 2015; Consortium, 2012); class 2). Part II: all of features form part I, plus allele-specific activity levels predicted by the models in part I. |
Random forest (R, C). The classifier used the results of the regression task as additional features. | Random forest (R, C) |
| 7 | Predictions of TFBS, DHS, and histone marks, using (Zhou and Troyanskaya, 2015) with data from (Consortium, 2012); class 2). 0/1 Indicator of leading variant and eQTL p-value (class 3). | Random forest | Same as part I |
References
Alipanahi B, Delong A, Weirauch MT, Frey BJ. 2015. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol 33(8):831–8.
Consortium EP. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414):57–74.
Cowper-Sal lari R, Zhang X, Wright JB, Bailey SD, Cole MD, Eeckhoute J, Moore JH, Lupien M. 2012. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat Genet 44(11):1191–8.
Ernst J, Kellis M. 2012. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9(3):215–6.
Fabian Pedregosa GV, Alexandre Gramfort, Vincent Michel, Bertrand Thirion, Olivier Grisel, Mathieu Blondel, Peter Prettenhofer, Ron Weiss, Vincent Dubourg, Jake Vanderplas, Alexandre Passos, David Cournapeau, Matthieu Brucher, Matthieu Perrot, Édouard Duchesnay. 2011. Scikit-learn: Machine Learning in Python. Journal of Machine Learning Research 12:2825–2830.
Ghandi M, Lee D, Mohammad-Noori M, Beer MA. 2014. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol 10(7):e1003711.
Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27(7):1017–8.
Hui Zou TH. 2005. Regularization and variable selection via the elastic net. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 67(2):301–320.
Hume MA, Barrera LA, Gisselbrecht SS, Bulyk ML. 2015. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res 43(Database issue):D117–22.
Kelley DR, Snoek J, Rinn J. 2016. Basset: Learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res.
Romanoski CE, Glass CK, Stunnenberg HG, Wilson L, Almouzni G. 2015. Epigenomics: Roadmap for regulation. Nature 518(7539):314–6.
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S and others. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15(8):1034–50.
Zhou J, Troyanskaya OG. 2015. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods 12(10):931–4.
RESULTS
PREDICTIVE FEATURES
The features that were used by the participating groups can be categorized into several classes: (1) Experimentally measured epigenetic properties, including Transcription factor (TF) binding sites (TFBS), histone marks, chromatin accessibility (primarily by identifying DNase-hypersensitivity sites; henceforth abbreviated as DHS), and DNA-methylation. To define these features, each reference allele is mapped to the reference human genome, and then queried against tracks of epigenetic properties (primarily from ENCODE (Consortium, 2012) and the Epigenome Road map (Romanoski, et al., 2015)), measured in LCLs and other cell lines. (2) Predicted epigenetic properties. This set of features covers similar properties as the experimentally-derived ones (e.g., TFBS or DHS). However, instead of being directly measured, the properties are inferred based on the DNA sequence of the respective MPRA construct, using models trained on experimental data (e.g., protein binding microarrays (Newburger and Bulyk, 2009) for TFBS, or DNase-seq (Consortium, 2012) for DHS). A wide array of models for prediction of epigenetic properties from sequence were used, from simple DNA-binding motif scoring (Grant, et al., 2011) to more recent supervised learning algorithms such as DeepBind (Alipanahi, et al., 2015), gkm-SVM (Ghandi, et al., 2014) and Basset (Kelley, et al., 2016). (3) Other locus-specific properties, including Variant information (e.g., indication if a variant is defined as a leading SNP (Tewhey, et al., 2016)), and evolutionary conservation; and (4) DNA k-mer frequencies.
Notably, the sequence-based features (feature classes 2 and 4), associate the reference and alternative alleles with different values (reflecting the differences in their respective DNA sequences). Conversely, in features that are based on direct characterization of the loci in the reference genome (feature classes 1 and 3), the two alleles are associated with the same value. Features of the former classes may therefore be more directly applicable for distinguishing between the two alleles at the second part of this challenge.
While the competing groups combined multiple feature sets using learning algorithms, we first wanted to explore the predictive capacity of each feature in isolation. To this end, we assembled a representative set from each feature class and measured its accuracy when applied on the test data sets (Supp. Table S1, Figure 2). For class 1, we include twenty epigenetic properties, derived from experimental profiling of LCLs by the ENCODE consortium (Consortium, 2012). These features include DHS (using DNase-seq), multiple histone modification, and TFBS (using ChIP-seq). These profiles were interpreted as binary, where a value of 1 indicates that the respective region overlaps with a peak of the respective signal (provided by the ENCODE unified pipeline (Consortium, 2012)). For class 2, we included the number of predicted TFBS based on the presence of DNA binding motifs form the ENCODE collection (Grant, et al., 2011; Kheradpour and Kellis, 2014), or using a Neural network model trained on protein-binding microarrays (Alipanahi, et al., 2015). We also included the distance between the transcription start site of the MPRA construct and the nearest motif hit, and several sequence-based properties related to the DNA structure, including: length of polyA/T sub-sequence representing nucleosome disfavoring sequences, GC content, and predicted DNA shape features including: minor groove width (MGW), roll, propeller twist (PROT), and helix twist (HELT) (Zhou, et al., 2013). For class 3, we included evolutionary conservation scores, predicted by phastCons (Siepel, et al., 2005). We did not include class 4 features (k-mers) in this analysis.
Figure 2.
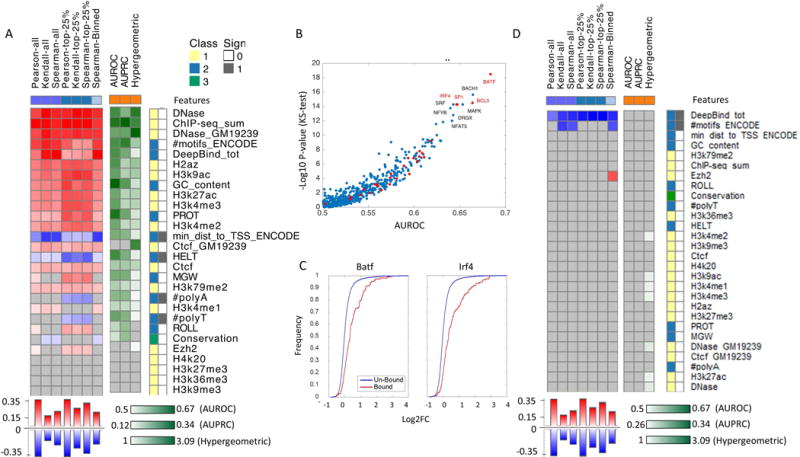
Individual feature accuracy using standard statistical tests. Features classes are divided to four categories (1) experimentally measured epigenetic properties (i.e., DHS, multiple histone modification and TFBS using ChIP-seq). (2) Predicted epigenetic properties (i.e., TFBS predictions: #motifs_ENCODE - number of predicted TFBS based on the presence of DNA binding motifs form ENCODE), or DeepBind_tot - using a Neural network model trained on protein-binding microarrays. min_dist_to_TSS_ENCODE - the distance between the transcription start site of the MPRA construct and the nearest motif hit, #PolyA, #PolyT - length of polyA/T sub-sequence, GC content and DNA shape features: minor groove width (MGW), roll, propeller twist (PROT), and helix twist (HELT)). DeepBind_tot feature was derived by marking the regions that score at the top 90% as hits for every TF, and for every region, count the number of TFs for which it has a hit. The aggregation method we use for these features is log fold between the alternate and reference allele and subtraction for DeepBind_tot. (3) locus-specific properties (i.e., evolutionary conservation scores) (4) k-mer frequencies (not included in this analysis). For regression tasks we applied several correlation measures (Person, Spearman, Kendall), considering either the entire test data (purple squares); variants at the top 25% of quantitative measurements (blue squares); or a binning of the data (light blue squares). For the binary predictions we record the AUROC and AUPRC (orange squares). For both regression and classification tasks we applied a hypergeometric test. The features are ranked based on the median performance across all tests and presented sorted from the most to the least predictive. Non-significant correlations are marked in grey, high positive/high negative/low correlation is marked in red/blue/white for regression and dark green/light green/white respectively for classification. Features categories 1/2/3/4 (only the first 3 presented in this figure) are denoted by yellow/blue/green/pink and the sign of the correlation positive/negative is marked with white/grey. (A) Part I: regression tasks include the expression of the reference and alternate allele, classification task includes regulatory hit prediction. (B) Contribution of individual TFs for predicting regulatory activity of MPRA constructs measured by the minus log p-value of the ks-test for AUROC per factor (C) Cumulative distribution of regulatory activity for regions that are bound/un bound by two of the most predictive factors (BATF and IRF4). (D) Part II: regression tasks includes allelic skew and classification task includes emVar hit prediction.
We used a number of tests to evaluate the accuracy of each feature (Supp. Note S1). For the regression tasks, i.e. predicting the expression of the reference and alternate allele (log2FC) and their ratio (LogSkew), we applied several correlation measures (Person, Spearman, Kendall), considering either the entire test data; variants at the top 25% transcriptional activity (defined as the maximum log2FC of the two alleles) or absolute allelic skew; or a discretization of the data (predicted and observed) into quintiles. For the binary predictions (i.e., Regulatory hit and emVar hit) we record the area under the Receiver Operating Characteristic (ROC) and Precision Recall (PR) curves (AUC). To better account for the binary predicted values (i.e., class 1 features), we also applied a fold enrichment test by examining the overlap between the set of true positives (Regulatory hits or emVar hits) and the predicted positives. For quantitative features (classes 1, 3) the predicted positives were defined as regions with value higher than the mean. The significance of each test was evaluated by the respective statistical test (correlation p-values for the regression tasks; Kolmogorov–Smirnov (KS) test for ROC and PR; hypergeometric p-value for the enrichment test). All p-values were corrected using the Benjamini–Hochberg procedure, and only associations below a false discovery rate (FDR) of 5% are presented.
Consistent with the previous literature (Erwin, et al., 2014; Kwasnieski, et al., 2014; Smith, et al., 2013), the most highly predictive features for the absolute expression levels (part I) are those related to TF binding (number of bound TF, inferred either computationally or experimentally) and chromatin accessibility (Figure 2A). We note that there is an overall high correlation within features categories (Supp. Figure S1) and negligible correlation between features and eQTL statistics (Supp. Figure S2). Notably, the set of all MPRA regions is significantly enriched with majority of histone marks with respect to the entire genome (Supp. Table S2), as expected (Francois Aguet, 2017; Fromer, et al., 2016). However, within those regions, we find the histone marks to have a low ability to predict eQTL strength (Supp. Figure S2).
We also find a significant positive relationship in various histone modifications that are associated with active regulatory regions (e.g., H3K27Ac). Considering the contribution of individual TFs, we find several regulators whose predicted binding sites are particularly predictive of regulatory activity of MPRA constructs (Figure 2B). Interestingly, among the top TFs are Batf and Irf4 (supported by ChIP-seq data as well; Figure 2C), which are highly expressed in LCL and are known to form a heterodimer that performs pioneer functions (i.e., recruitment of chromatin remodeling machinery) in T cell development (Ciofani, et al., 2012). For part II, we do not observe any individual features that are significantly predictive, considering different ways to aggregate the scores of the two alleles in feature class 2 [max, min, difference], and using a lenient cutoff of FDR<0.1 (Figure 2D). The only exception was a weak positive signal from the differences in number of predicted TFBS, attesting to the complexity of predicting differences that hinge on a single or few nucleotide difference. Since MPRA is subject to experimental error, as most other assays, we have tested the robustness of these results by resampling, and observed overall consistent results (Supp. Table S3).
COMBINING PREDICTIVE FEATURES WITH LEARNING ALGORITHMS
The prediction algorithms for the first part of this challenge included a wide array of standard machine learning techniques, both for the classification task (regulatory hits; e.g., support vector machine (SVM), random forest, neural networks), and the regression tasks (Log2FC; e.g., support vector regression (SVR), regularized linear models, regression trees), with random forests being the most widely-used method (Table 1). Some of the groups further used an ensemble of predictors, by varying either the type of prediction algorithm (e.g., combining linear models and random forest) or the set of predictive features (e.g., different classifiers, each using epigenetic features predicted by a different algorithm (Alipanahi, et al., 2015; Zhou and Troyanskaya, 2015)). For instance, group #2 applied gradient boosting for classification of regulatory hits, and reported an aggregate over the predicted scores of eight different models, trained on eight distinct feature sets, with each set including either experimentally-derived epigenetic properties (e.g., ChIP-seq signal over the respective loci in LCL from ENCODE), or computationally-derived ones (e.g., DNase hypersensitivity predictions in 164 cell lines using Basset (Kelley, et al., 2016)).
In part II, we observed two main strategies: the first was to treat this part independently from part I, and build predictive models using the feature categories summarized above. The alternative strategy was to use the individual activity of each allele, as predicted in part I, to infer their differential activity (quantitatively - LogSkew; and qualitatively - emVar hits). For instance, using a similar strategy as in the deepSEA framework (Zhou and Troyanskaya, 2015), group #4 (Haoyang Zeng, 2016) built a two-step classifier: in the first level it applies the classifiers from part I to predict the transcriptional activity of each allele; in the second part it uses the predicted activity of each allele and the difference between them as features for predicting emVar hits using an ensemble of classifiers, including regularized logistic regression, random forest, SVM, and K nearest neighbors (KNN).
PERFORMANCE EVALUATION
The groups submitted their predictions for the held-out test data sets, which we then used for evaluation. We used similar performance tests as above to rank the groups, and then derived an overall ranking by taking the median across tests (Figure 3). Not surprisingly, we observe that the relative performance within each part is overall consistent across the different tasks; for instance, the accuracy of predicting transcriptional activities (Log2FC) is highly indicative of the performance in the related classification task (regulatory hit). The consistency between the two parts was less substantial (even though the top ranking group was similar), for instance with group #5 performing well in part II, but less so in part I. Interestingly, this group based its predictions on feature class 2 only (sequence-based predictions of epigenetic properties). While their method was previously shown to be predictive of the effects of subtle sequence changes (mostly SNV) on chromatin accessibility (Ghandi, et al., 2014), the lack of direct experimental measurements as features (i.e., class 1) might be the cause for the less favorable performance in part I compared to groups that used class 1 features. More generally, we observe a substantially worse performance in part II vs. part I, which is consistent with the single feature analysis above, and reflects the difficulty of predicting the effects of nuanced sequence modifications.
Figure 3.
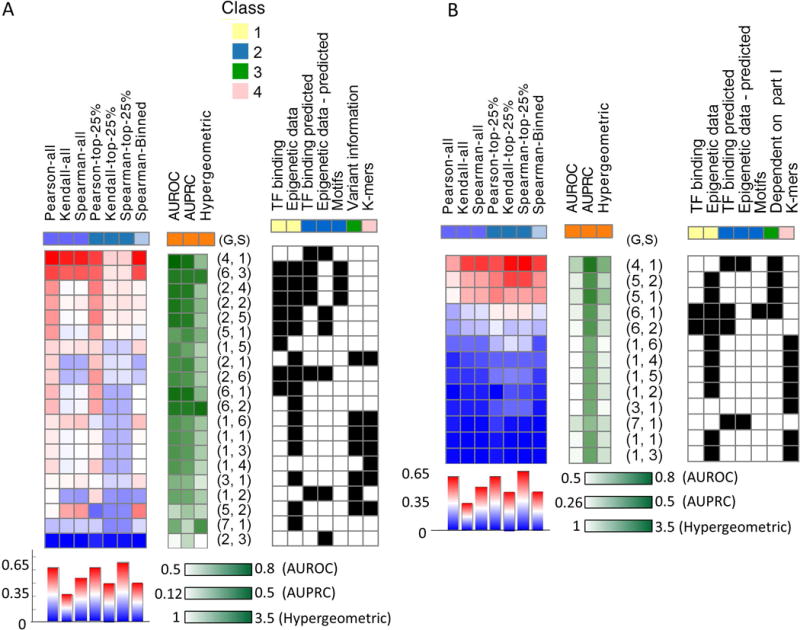
Summary of performance and features used per submission. The statistical tests used and features examined are similar to Figure 2. High/low performance is marked in red/blue and dark/light green respectively. Group number and submission are denoted by (G,S), features use is indicated by black(1)/white(0) heat maps. The submissions are ranked based on the median performance across all tests and presented sorted from high to low performance. (A) Part I. (B) Part II. The third class of features for part II indicates if the model is dependent on predictions from part I.
Next, we wanted to assess if there are specific classes of models or combinations of features that are associated with better performance. To address this, we record for each submission the types of models and features that were used (Table 1 and Figure 3). We note that ensemble methods were generally better performing, highlighting the need for robust inference methodologies, and consistently with other applications of machine learning in biology (Marbach, et al., 2012). Furthermore, it is clear that non-linear methods perform better - an expected results given the plausibility of non-linear (Das, et al., 2004) and combinatorial (Spitz and Furlong, 2012) effects of the features. For part I, we observe that, generally, including TFBS as features (either predicted or experimentally-derived), leads to better performance, which is consistent with the individual feature analysis (Figure 3A). For part II, we find that relying on models trained in part I (i.e., using the predicted allele activity levels as features) leads to improved performance (Wilcoxon ranksum test p-value 0.0028). (Figure 3B).
WHERE DO WE FAIL?
We next wanted to characterize the regions that were proven to be hard to predict. We start by ranking the MPRA constructs by their observed activity levels (Log2FC). For each competing submission and each MPRA construct, we then define the respective accuracy as the absolute difference between the observed and predicted rank, scaled by the difference expected by a random ranking (which becomes smaller the closer we are to the average). We note that this measure is more robust than taking the difference of the original (non-rank transformed) values, which due to the scaling of variance with the mean (as expected), leads to strong bias for highly active constructs (Supp. Figure S3).
We then assess how consistent different groups are in their performance within a region. To this end, we record the Spearman correlation coefficient of region performance between every pair of groups (taking the maximal correlation among all pairs of submissions). The cumulative distribution of these coefficients (Figure 4A) suggests that there is an overall agreement in regions performance for the predictions in part I across the different submissions (Figure 4B left panel) and less coherent agreement for part II (Figure 4C left panel). Similarly, we observe an overall agreement in regions performance for the predictions in part I, based on the 10 most predictive features individually (Figure 4B right panel) but not for part II (Figure 4C right panel).
Figure 4.
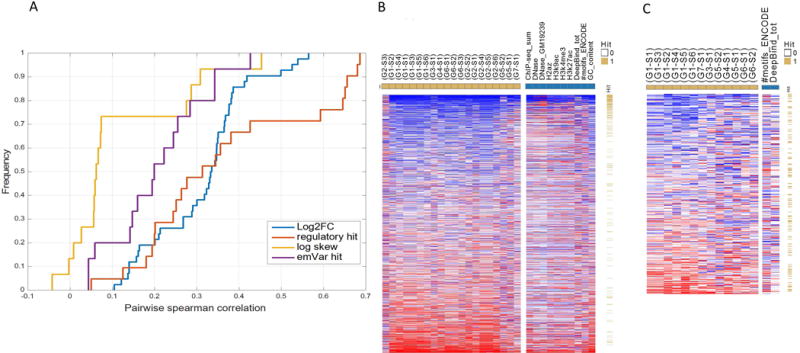
Regions hardness. Respective accuracy per region and submission is defined as the absolute difference between the observed and predicted rank, scaled by the expected difference (using random ranking). The “region hardness to predict” per part is defined as the mean rank across all tasks. Hard/easy to predict regions are denoted by red/blue respectively and sorted from easy to hard. (A) Cumulative distribution of the Spearman correlation coefficient when comparing each pair of groups (taking the maximum over all possible pairs of submissions) for their regions accuracy per prediction (log2FC, regulatory hit, log skew, emVar hit). (B) Left panel: heat-map of regions hardness for part I when using the predictions from all groups (yellow squares). The regions are sorted by their rank and denoted if they are regulatory hits (yellow/white). Right panel - heat-map of regions hardness when using the top 10 features as predictors (blue squares). (C) Left panel: heat-map of regions hardness for part II when using the predictions from all groups (yellow squares). The regions are sorted by their rank and denoted if they are emVar hits (yellow/white). Right panel - heat-map of regions hardness when using the top 2 features as predictors (blue squares).
Given the consistency of submissions for part I, we focus our analysis on pinpointing which genomic and epigenetic features are associated with the ability to predict the activity level (Log2FC) of a region (i.e. region “hardness”). Considering all the variants in the test set of part I, we find that regions that are accurately predicted by all or most competing submissions, are highly enriched with regulatory hits (Figure 4B left panel). This observation, that the activity of truly active regions is generally easier to predict, is expected since the activity level of clear non-hits is likely to fall within the regimen of noise. To gain a better understanding of what makes a region hard to predict, we focused our attention on regulatory hits. We used similar performance tests as above to evaluate the extent to which the different feature classes (Figure 2) and other properties (Figure 5A) is indicative for the difficulty in predicting the activity of a regulatory hit. First, we find that the measurement noise (evaluated based on reproducibility of the MPRA assay (Supp. Figure S3) does not discriminate between hard and easy to predict regions. Second, we find that hard-to-predict regions are associated with a lower transcriptional activity (as expected), however, this association (AUROC = 0.54) is not as strong as that observed for other features (Figure 5A). Next, considering the contribution of individual TFs, we find several regulators whose binding sites are enriched in either hard or easy regions (Figure 5B). Evidently, the TFs that are enriched in hard regions tend to have more binding sites across the genome (based on ChIP-seq in LCL; Figure 5C), which –from the machine learning perspective- naturally makes them less powerful in discriminating active from inactive regions. More globally, we observe that hard-to-predict regions tend to have less TF binding sites and reduced association with open chromatin and active chromatin marks in the genome, as well as lower GC content. These results reflect our overall conception of what characterizes an active region (Figure 2A) and are in line with the previous literature, for instance that the expression of genes whose promoters has a low GC content is more difficult to predict (Dong, et al., 2012). Indeed, looking at individual cases, we find a number of regions that are highly-active in the MPRA assay, but are not associated with any TFBS or accessible chromatin in LCL (Figure 5D). While these apparent discrepancies may be related to the episomal nature of the MPRA assay, close investigation of such regions may be valuable for identifying genetic or epigenetic properties that are predictive of transcriptional activity, in addition to those employed in this challenge.
Figure 5.
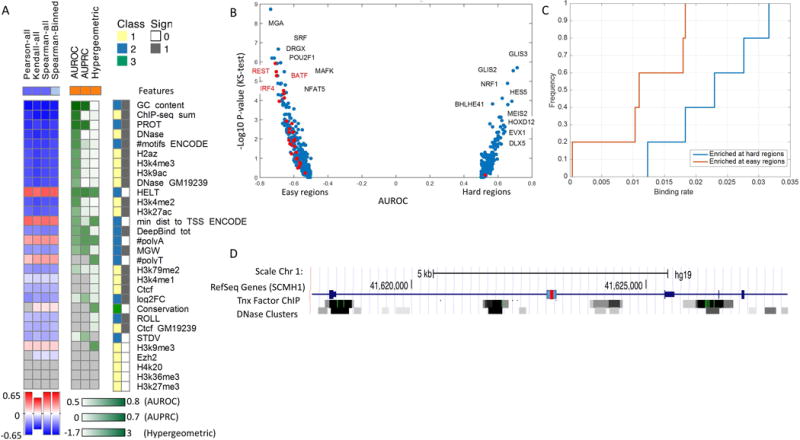
Features correlation with region hardness for regulatory hit regions in part I.
(A) The statistical tests used and features examined are similar to Figure 2. The features are sorted from the most predictive (blue/red) to the least predictive (white) to regions hardness, based on the mean rank across all methods across the three tests in part I. The positive class is defined as the top 100 hardest regions. Two additional features are included: noise (log(STD/Mean)) across replicates and log2FC. (B) Contribution of individual TFs to the prediction of hard and easy regions, measured by the minus log p-value of the ks-test for AUROC per factor. (C) Frequency of binding sites across the genome for TFs that are enriched in hard and easy regions. (D) The genomic loci of a specific regulatory hit region with high expression levels that is hard to predict across all submissions. Gene annotations, DHS, and transcription factor ChIP tracks from ENCODE are shown. The 150bp region is marked with a blue rectangular with a red mark in the middle indicating the SNP.
DISCUSSION
The outcome of the Critical Assessment of Genome Interpretation eQTL challenge serves two main purposes. The first is providing a benchmark and encouraging the development of methods for predicting transcriptional activity of DNA-regions, thus improving our understanding of the individual genetic and epigenetic properties that make up the regulatory code, and the appropriate way to model their inter-dependence in a predictive mathematical model. The second purpose takes a translational point of view – a given eQTL variant is usually associated with multiple loci that cannot be discriminated due to LD. The methods developed here thus join and enhance the published cohort of computational studies (e.g., (Alipanahi, et al., 2015; Ghandi, et al., 2014; Kelley, et al., 2016; Kircher, et al., 2014; Ritchie, et al., 2014; Zeng, et al., 2016; Zhou and Troyanskaya, 2015)) that prioritize likely causal variants in an LD block, based on the predicted allelic shift in chromatin state.
As opposed to existing body of computational studies, the task of identifying causal variants in high throughput was tackled by Tewhey and colleagues experimentally - using a combined pipeline of eQTL analysis followed by MPRA of the identified loci (Tewhey, et al., 2016). Evidently, the resulting MPRA data proved valuable for the identification of key loci in the original study (Tewhey, et al., 2016) and for development of predictive methods in this challenge. However, it is important to bear in mind that MPRA is conducted outside of the natural context of the chromatin and the cell’s regulatory network, thus potentially leading to inaccuracies. Indeed, it is still not clear whether the MPRA constructs (Figure 1) are capable of acquiring physiological chromatin in a manner comparable to endogenous loci (Inoue, et al., 2017). While some studies successfully used MPRA to model interactions between TFs (Kwasnieski, et al., 2014; Smith, et al., 2013), other studies suggest that episomal assays may in some cases fail to reflect cooperative TF activity, e.g., due to differences in histone H1 stoichiometry and nucleosome positioning (Archer, et al., 1992; Hebbar and Archer, 2007; Hebbar and Archer, 2008; Smith and Hager, 1997). Future novel experimental approaches, including lentivirus based MPRAs that can allow for integration into the genome (Inoue, et al., 2017), will shed more light on the features that determine regions functionality. Future challenges should focus on finer annotation of TFBS and epigenetic assays which seem to encompass the majority of information regarding regions regulatory activity. Specifically, considering cellular context (e.g., pioneer factors as shown in Figure 2, or RNA-seq data) when prioritizing features may improve prediction, as opposed to treating all TFs equally. Lastly, we note this challenge is based on MPRA in regions that harbor an eQTL, the results in Figure 1E indicate that distal regions (i.e., regions that do not intersect with promoters, introns or exons) show lower transcriptional activity, which might be a result of an error in the eQTL association (which, naturally, tends to be more error prone for distal sites due to statistical burden).
In conclusion, while the task of predicting expression-phenotype from genotype is immensely complex, this challenge has seen some promising methodologies. Development of such methods and pinpointing which genetic and epigenetic features contribute to regions functionality is essential to the study of human disease.
Supplementary Material
Acknowledgments
The CAGI experiment coordination is supported by NIH U41 HG007446 and the CAGI conference by NIH R13 HG006650. NY and AK were supported by NIH grant U01HG007910.
The answer key, predictions, and assessment are available on the CAGI website: https://genomeinterpretation.org/content/4-eQTL-causal_SNPs
References
- Alipanahi B, Delong A, Weirauch MT, Frey BJ. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol. 2015;33(8):831–8. doi: 10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- Archer TK, Lefebvre P, Wolford RG, Hager GL. Transcription factor loading on the MMTV promoter: a bimodal mechanism for promoter activation. Science. 1992;255(5051):1573–6. doi: 10.1126/science.1347958. [DOI] [PubMed] [Google Scholar]
- Beer MA, Tavazoie S. Predicting gene expression from sequence. Cell. 2004;117(2):185–98. doi: 10.1016/s0092-8674(04)00304-6. [DOI] [PubMed] [Google Scholar]
- Birnbaum RY, Patwardhan RP, Kim MJ, Findlay GM, Martin B, Zhao J, Bell RJ, Smith RP, Ku AA, Shendure J, et al. Systematic dissection of coding exons at single nucleotide resolution supports an additional role in cell-specific transcriptional regulation. PLoS Genet. 2014;10(10):e1004592. doi: 10.1371/journal.pgen.1004592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussemaker HJ, Li H, Siggia ED. Regulatory element detection using correlation with expression. Nat Genet. 2001;27(2):167–71. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M, et al. A validated regulatory network for Th17 cell specification. Cell. 2012;151(2):289–303. doi: 10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das D, Banerjee N, Zhang MQ. Interacting models of cooperative gene regulation. Proc Natl Acad Sci U S A. 2004;101(46):16234–9. doi: 10.1073/pnas.0407365101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Greven MC, Kundaje A, Djebali S, Brown JB, Cheng C, Gingeras TR, Gerstein M, Guigo R, Birney E, et al. Modeling gene expression using chromatin features in various cellular contexts. Genome Biol. 2012;13(9):R53. doi: 10.1186/gb-2012-13-9-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28(8):817–25. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin GD, Oksenberg N, Truty RM, Kostka D, Murphy KK, Ahituv N, Pollard KS, Capra JA. Integrating diverse datasets improves developmental enhancer prediction. PLoS Comput Biol. 2014;10(6):e1003677. doi: 10.1371/journal.pcbi.1003677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJ, Shishkin AA, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518(7539):337–43. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois Aguet AAB, Castel Stephane, Davis Joe R, Mohammadi Pejman, Segre Ayellet V, Zappala Zachary, Abell Nathan S, Fresard Laure, Gamazon Eric R, Gelfand Ellen, Gloudemans Machael J, He Yuan, Hormozdiari Farhad, Li Xiao, Li Xin, Liu Boxiang, Garrido-Martin Diego, Ongen Halit, Palowitch John J, Park YoSon, Peterson Christine B, Quon Gerald, Ripke Stephan, Shabalin Andrey A, Shimko Tyler C, Strober Benjamin J, Sullivan Timothy J, Teran Nicole A, Tsang Emily K, Zhang Hailei, Zhou Yi-Hui, Battle Alexis, Bustamonte Carlos D, Cox Nancy J, Engelhardt Barbara E, Eskin Eleazar, Getz Gad, Kellis Manolis, Li Gen, MacArthur Daniel G, Nobel Andrew B, Sabbati Chiara, Wen Xiaoquan, Wright Fred A, Consortium GTEx. Lappalainen Tuuli, Ardlie Kristin G, Dermitzakis Emmanouil T, View ORCID Profile. Christopher Brown D, Montgomery Stephen B. Local genetic effects on gene expression across 44 human tissues. BioRxiv 2017 [Google Scholar]
- Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, Ruderfer DM, Oh EC, Topol A, Shah HR, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 2016;19(11):1442–1453. doi: 10.1038/nn.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz J, Siggia ED, Cohen BA. Analysis of combinatorial cis-regulation in synthetic and genomic promoters. Nature. 2009;457(7226):215–8. doi: 10.1038/nature07521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Lee D, Mohammad-Noori M, Beer MA. Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Comput Biol. 2014;10(7):e1003711. doi: 10.1371/journal.pcbi.1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez AJ, Setty M, Leslie CS. Early enhancer establishment and regulatory locus complexity shape transcriptional programs in hematopoietic differentiation. Nat Genet. 2015;47(11):1249–59. doi: 10.1038/ng.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27(7):1017–8. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haoyang Zeng MDE, Yuchun Guo, Gifford David K. Accurate eQTL prioritization with an ensemble-based framework. BioRxiv. 2016 doi: 10.1002/humu.23198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbar PB, Archer TK. Chromatin-dependent cooperativity between site-specific transcription factors in vivo. J Biol Chem. 2007;282(11):8284–91. doi: 10.1074/jbc.M610554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbar PB, Archer TK. Altered histone H1 stoichiometry and an absence of nucleosome positioning on transfected DNA. J Biol Chem. 2008;283(8):4595–601. doi: 10.1074/jbc.M709121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106(23):9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods. 2012;9(5):473–6. doi: 10.1038/nmeth.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue F, Kircher M, Martin B, Cooper GM, Witten DM, McManus MT, Ahituv N, Shendure J. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 2017;27(1):38–52. doi: 10.1101/gr.212092.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Snoek J, Rinn J. Basset: Learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016 doi: 10.1101/gr.200535.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpour P, Ernst J, Melnikov A, Rogov P, Wang L, Zhang X, Alston J, Mikkelsen TS, Kellis M. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Res. 2013;23(5):800–11. doi: 10.1101/gr.144899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradpour P, Kellis M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 2014;42(5):2976–87. doi: 10.1093/nar/gkt1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnieski JC, Fiore C, Chaudhari HG, Cohen BA. High-throughput functional testing of ENCODE segmentation predictions. Genome Res. 2014;24(10):1595–602. doi: 10.1101/gr.173518.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedlander MR, t Hoen PA, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–11. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Gorkin DU, Baker M, Strober BJ, Asoni AL, McCallion AS, Beer MA. A method to predict the impact of regulatory variants from DNA sequence. Nat Genet. 2015;47(8):955–61. doi: 10.1038/ng.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Costello JC, Kuffner R, Vega NM, Prill RJ, Camacho DM, Allison KR, Consortium D. Kellis M, Collins JJ, et al. Wisdom of crowds for robust gene network inference. Nat Methods. 2012;9(8):796–804. doi: 10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marstrand TT, Storey JD. Identifying and mapping cell-type-specific chromatin programming of gene expression. Proc Natl Acad Sci U S A. 2014;111(6):E645–54. doi: 10.1073/pnas.1312523111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG, Jr, Kinney JB, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol. 2012;30(3):271–7. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogno I, Kwasnieski JC, Cohen BA. Massively parallel synthetic promoter assays reveal the in vivo effects of binding site variants. Genome Res. 2013;23(11):1908–15. doi: 10.1101/gr.157891.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan A, Yardimci GG, Sheffield NC, Crawford GE, Ohler U. Predicting cell-type-specific gene expression from regions of open chromatin. Genome Res. 2012;22(9):1711–22. doi: 10.1101/gr.135129.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburger DE, Bulyk ML. UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2009;37:D77–82. doi: 10.1093/nar/gkn660. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DH, D’Haeseleer P. Deciphering principles of transcription regulation in eukaryotic genomes. Mol Syst Biol. 2006;2:20060012. doi: 10.1038/msb4100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, Lee C, Andrie JM, Lee SI, Cooper GM, et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol. 2012;30(3):265–70. doi: 10.1038/nbt.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–80. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie GR, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nat Methods. 2014;11(3):294–6. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanoski CE, Glass CK, Stunnenberg HG, Wilson L, Almouzni G. Epigenomics: Roadmap for regulation. Nature. 2015;518(7539):314–6. doi: 10.1038/518314a. [DOI] [PubMed] [Google Scholar]
- Segal E, Raveh-Sadka T, Schroeder M, Unnerstall U, Gaul U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 2008;451(7178):535–40. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- Sharon E, Kalma Y, Sharp A, Raveh-Sadka T, Levo M, Zeevi D, Keren L, Yakhini Z, Weinberger A, Segal E. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat Biotechnol. 2012;30(6):521–30. doi: 10.1038/nbt.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15(8):1034–50. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Hager GL. Transcriptional regulation of mammalian genes in vivo. A tale of two templates. J Biol Chem. 1997;272(44):27493–6. doi: 10.1074/jbc.272.44.27493. [DOI] [PubMed] [Google Scholar]
- Smith RP, Taher L, Patwardhan RP, Kim MJ, Inoue F, Shendure J, Ovcharenko I, Ahituv N. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat Genet. 2013;45(9):1021–8. doi: 10.1038/ng.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet. 2012;13(9):613–26. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell. 2016;165(6):1519–29. doi: 10.1016/j.cell.2016.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten-Gabbay S, Segal E. The grammar of transcriptional regulation. Hum Genet. 2014;133(6):701–11. doi: 10.1007/s00439-013-1413-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–6. doi: 10.1093/nar/gkt1229. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilczynski B, Liu YH, Yeo ZX, Furlong EE. Predicting spatial and temporal gene expression using an integrative model of transcription factor occupancy and chromatin state. PLoS Comput Biol. 2012;8(12):e1002798. doi: 10.1371/journal.pcbi.1002798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Hashimoto T, Kang DD, Gifford DK. GERV: a statistical method for generative evaluation of regulatory variants for transcription factor binding. Bioinformatics. 2016;32(4):490–6. doi: 10.1093/bioinformatics/btv565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods. 2015;12(10):931–4. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Yang L, Lu Y, Dror I, Dantas Machado AC, Ghane T, Di Felice R, Rohs R. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res. 2013;41:W56–62. doi: 10.1093/nar/gkt437. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.