

Abstract
We describe monozygotic twin girls with genetic variation at two separate loci resulting in a blended phenotype of Prader-Willi syndrome and Pitt-Hopkins syndrome. These girls were diagnosed in early infancy with Prader-Willi syndrome, but developed an atypical phenotype, with apparent intellectual deficiency and lack of obesity. Array-comparative genomic hybridization confirmed the apparently de novo paternal deletion of the 15q11.2q13 region and exome sequencing identified a second mutational event in both girls, which was a novel variant c.145+1G>A affecting a TCF4 canonical splicing site inherited from the mosaic mother. RNA studies showed that the splice site variant abolished the donor splicing site, which was accompanied by activation of an alternative non-canonical splicing-site which then predicts a premature stop codon in the following exon. Clinical re-evaluation of the twins showed that both variants are likely contributing to the more severe phenotypic presentation. Our data show that atypical clinical presentations may actually be the expression of blended clinical phenotypes arising from independent pathogenic events at two loci.
Keywords: atypical Prader-Willi Syndrome, blended phenotype, dual genetic diagnosis, mosaicism, PTHS
INTRODUCTION
Prader-Willi syndrome (PWS, MIM #176270) is an imprinting disorder caused by a paternal deletion of a 5–6 Mb region in 15q11.2q13 in 65–75% of patients and by maternal uniparental disomy in 20–30% of patients [Cassidy et al., 2012]. The PWS is characterized by severe hypotonia with a poor sucking reflex during early infancy followed by the development of early-onset obesity due to the lack of satiety during childhood. Most patients have mild to moderate intellectual disability (ID) [Cassidy et al., 2012]. There are few reports of patients severely affected by PWS that are explained by a (probably) larger deletion size[Reynolds et al., 1987], however, a correlation of deletion size and severity does not explain all variation in patients with atypical PWS [Calounova et al., 2008; Kim et al., 2012]. Therefore, additional genetic factors segregating in the affected patients may contribute to modulate severity and trait expression of such patients. Here we describe twin girls with an atypically severe PWS phenotype on whom combined analysis of the clinical features together with molecular studies identified a blended phenotype likely explained by a dual molecular diagnosis.
CLINICAL REPORT
The twin girls (BAB8466 and BAB8467) were the only children born to young and healthy non-consanguineous parents, with an unremarkable family history. The monozygotic twins were born by caesarean delivery after an uneventful pregnancy at 37 weeks gestation. Birth parameters were within normal range (Supplementary table I). Both were reported to have a weak cry, axial hypotonia, and to be hypoactive neonatally. Due to sucking difficulties both required nasogastric feeding up to 30 days of life. At seven months both had global hypotonia, small labia minora, small hands and feet, and high arched palate. Convulsions were reported for BAB8467 and EEG detected poorly organizing basal tracing, delta waves in anterior regions and multifocal spike/sharpen waves. Cerebral MRI of both twins showed ex-vacuo ventricular dilatation and hippocampal hypoplasia. With development, growth parameters showed normal weight and a decrease in height and OFC (Supplementary Table I and Supplementary Fig.1). Examination at 12 years of age and 13 years 10 months of age (Fig. 1A) showed both to have neurodevelopmental delay, never being able to stand or walk. Their vocabulary was limited to less than five words. Both were agitated and had continuous repetitive body and hand movements. They indicated hunger by signing or making sounds; however, the parents did not note a voracious appetite or lack of satiety. The BMIs were 22.5 and 20.9 for BAB8466 and BAB8467, respectively. Both girls had normal pubertal development.
Fig. 1.
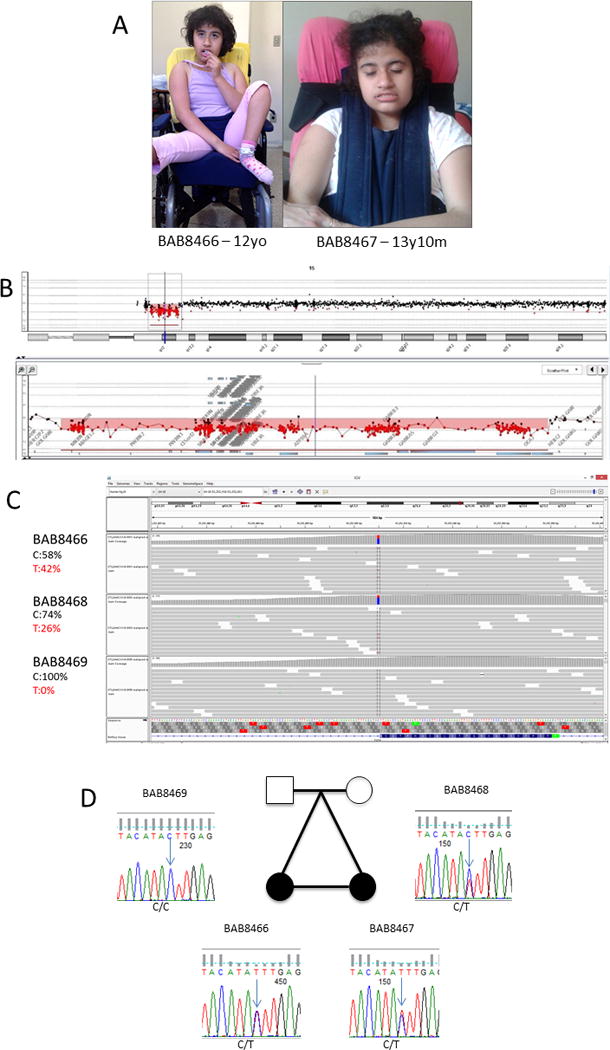
[A] Whole body picture of BAB8466 at 12 years of age and facial features of BAB8467 at 13 years 10 months of age showing deeply set eyes, protrusion of the mid and lower face, large nose, short philtrum, and thick, tented vermillion of the upper lip. [B] Chromosome-15 array-CGH profile from BAB8467 depicting a deletion of approximately 4.9 Mb at 15q11.2q13.1. The position of the deletion is indicated by the red line under the ideogram. Underneath, an enlargement of the 15q11.2q13.1 band shows a map of the deleted and non-deleted flanking probes. [C] The exome sequencing results for the trio BAB8466, and her parents showing a novel variant in (TCF4) present in one twin and the mother, and the estimated percentage of the wild (C) and mutant (T) allele in all individuals. [D] Sanger sequencing of segregation of the mutation in the family, confirming the exome results for the trio and showing the presence of the variant allele in BAB8467.
A diagnosis of PWS was suspected at 9 months of age based on hypotonia and feeding difficulties. Three markers in the 15q11.2q13 critical region (D15S11, D15S113, and GABRB3) indicated a paternal deletion in both twins. Human Genome CGH Microarray 60K (Agilent Technologies) further delineated an interstitial ~4.9 Mb deletion to arr[GRCh36] 15q11.2q13.1 (21258528-26193911)x1 dn, which is consistent with a typical type 2 deletion (Fig. 1B). No other potentially pathogenic genomic imbalances were observed. Methylation-sensitive polymerase chain reaction (MS-PCR) was performed for SNURF-SNRPN as described [Varela et al., 2005] which identified a methylated (maternal) allele but absence of the unmethylated (paternal) allele (results not shown). Although aforementioned results were consistent with PWS, genetic testing was resumed at 12 years of age due to their atypical phenotype. Methylation-sensitive high resolution melting (MS-HRM) [Rubatino et al., 2015], performed on peripheral blood DNA, confirmed methylation of the SNURF-SNRPN promoter in both twins (Supplementary Fig. 2). Taking into consideration the presence of some symptoms of Angelman Syndrome (AS, MIM #105830) such as apparent ID, microcephaly, lack of speech, autistic features, and epilepsy, it was hypothesized that the UBE3A gene on the non-deleted chromosome was potentially silenced by a mutation. Direct sequencing of all coding exons and intron/exons junctions of UBE3A in both girls using Sanger sequencing (accession number: NM_130838.1), did not identify any pathogenic variants (results not shown).
Exome Sequencing (ES) was performed on a trio (BAB8466 and the parents) at the Human Genome Sequencing Center (HGSC) at Baylor College of Medicine (BCM) through the Baylor Hopkins Center for Mendelian Genomics (BHCMG) initiative as described in the BCM-HGSC protocols. After a comprehensive analysis and rare variant filtering, two variants remained of interest. One was a maternally inherited, c.5546A>G (NM_004667), p.(Lys1849Arg) (NP_004658) missense variant in HERC2, a gene associated with an autosomal recessive intellectual disability syndrome (MRT38, MIM #615516) similar to AS [Puffenberger et al., 2012]. This variant was rare, present in 0.072% of individuals with European ancestry in dbSNP (rs201821203), and no homozygous variants were identified in ExAC. PolyPhen-2 [Adzhubei et al., 2010] and, SIFT [Sim et al., 2012] scores suggested that it is not damaging. The second candidate was a c.145+1G>A variant in intron 3 predicted to affect mRNA splicing of TCF4 (NM_001083962.1; NP_001077431.1; CCDS42438.1) (Fig. 1C) and not yet reported in any database. Sanger sequencing confirmed the presence of this TCF4 variant in both girls and their mother (Fig. 1D). Remarkably, ES data suggest that mother was mosaic as the number of variant reads was lower (49 reads - 26%) than the reference reads (142 reads - 74%), a finding exclusively observed in the maternal sample (Fig.1C). We were able to phase the splicing variant using a nearby SNP (rs599550) by Sanger sequencing of cloned TCF4 PCR products from the twins and the mother. Results showed that the mother has three distinct alleles for this amplicon, which confirms mosaicism (Supplementary Fig. 3). To determine the effect of the variant on splicing, cDNA was synthetized from RNA isolated from leukocytes of BAB8466 and the mother using the Invitrogen kit Superscript III First-Strand Synthesis SuperMix for qRT-PCR. The PCR products from exons 2 to 4 were amplified, cloned and sequenced, which showed that at least two distinct transcripts are being expressed (Supplementary Fig.4). Surprisingly, the c.145+1G>A variant does not use the canonical GT donor splicing site at the 5′ end of intron 3 and instead uses a non-canonical GC donor site within exon 3 while keeping the canonical AG acceptor at exon 4 splice junction. This splicing shift deletes the last 25 bases of exon 3 and changes the predicted reading frame to include a premature termination codon (PTC) at the beginning of exon 4 (Supplementary Fig.4).
This research was approved both by the Santa Casa Hospital ethics committee and by the Baylor Institutional Review Board (IRB) at BCM (protocol H-29697). Parents signed informed consents to participate on both studies and for data and photo publication.
DISCUSSION
We report clinical and genetic investigations of MZ twin girls molecularly diagnosed with PWS, but whom had an evolving atypical presentation, including apparent ID, lack of ambulation and speech, postnatal microcephaly, normal pubertal development, and absence of hyperphagia. After the exclusion of AS and other CNVs as the cause of the atypical PWS phenotype, we investigated whether they had an additional mutation by ES. The ES results identified two candidate variants for their phenotype, one at HERC2 and another at TCF4. Since HERC2 maps to the deleted 15q11.2q13 region, it could have unmasked this recessive trait. Prediction scores for amino acid changes do not support a role for the HERC2 change despite the fact that we cannot rule out that there is some contribution to the clinical phenotype.
Heterozygous TCF4 mutations cause Pitt-Hopkins syndrome (PTHS, OMIM #610954) in an autosomal dominant manner. The PTHS comprises severe intellectual disability and speech impairment with distinctive facial features, seizures, typical behavior and intermittent hyperventilation and postnatal onset of microcephaly [de Winter et al., 2016; Peippo & Ignatius, 2012]. Of interest, PTHS, it is an important differential diagnosis for AS [Tan et al., 2014]. The molecular diagnosis identified by ES is a novel splice variant, that causes the abolishment of the donor in intron 3 resulting in the use of a non-canonical donor site in exon 3. This TCF4 aberrant splicing predicts deletion of 25 bases from exon 3 and shifts the mRNA coding frame, introducing a (PTC) in exon 4 (Supplementary Fig. 4). Introduction of a PTC may lead to mRNA degradation due to nonsense-mediated RNA decay (NMD) and/or to the translation of a truncated protein lacking most of the C-terminal domains of the protein. It has been proposed that TCF4 transcripts code for 18 N-terminally distinct protein isoforms, and while all isoforms contain the transactivation domain AD2 (encoded by exons 14–16) and the bHLH domain (encoded by exon 19), many do not include the entire transactivation domain AD1 (encoded by exons 3–6) [Sepp et al., 2011]. As the mutation herein reported introduces a PTC in exon 4, this transcript probably lacks all long isoforms that include AD1. The fact that we identified two different transcripts, both in the mother and in the twin’s samples, provides evidence that not all mutated mRNA is being degraded by NMD, however, it is reasonable to anticipate that the truncated protein would not be functional. Our results are consistent with the hypothesis from [Sepp et al., 2012] that suggests that the deficit of isoforms containing AD1 and/or NLS might not be compensated by the remaining isoforms and that partial loss-of-function is sufficient to cause PTHS.
Both girls inherited the TCF4 variant from their mosaic mother who reported a convulsion in adolescence and a panic disorder in the last 10 years. De Pontual and colleagues [de Pontual et al., 2009] also reported a woman mosaic for a TCF4 variant with chronic depression and epilepsy. This raises the question whether the lower levels of wild-type TCF4 or the presence of a variant form could cause mild adult-onset phenotypes such as depression or epilepsy.
Our results suggest that both girls have a blended phenotype from dual molecular diagnoses of PWS and PTHS. Individuals with more than one independently segregating molecular diagnosis can present with clinical features that represent a blending of two or more disease phenotypes that may initially suggest an apparent novel, atypical, or broader presentation of one of the diseases [Boycott & Innes, 2017; Posey et al., 2017]. A complete comparison of the twins phenotype and the most common clinical symptoms of PTHS can be found in Supplementary Table II. Reports of multilocus genetic diagnoses contributing to clinical phenotypes are increasing with the use of ES and genome sequencing and might not be as rare as previously thought [Posey et al., 2017]. This report illustrates the challenges of reaching a final diagnosis in complex clinical presentations when the genetic results do not completely explain the clinical phenotype observed or match the anticipated clinical presentation.
Supplementary Material
Acknowledgments
We thank the patients and their parents for the willingness in participating in this research. We thank Drs. V. Reid Sutton and Jennifer E. Posey for thoughtful comments and discussions. This work was supported by the Brazilian Agencies CAPES, CNPq, FAPEMIG, FAPESP and the US National Human Genome Research Institute-National Heart Lung and Blood Institute grant to the BHCMG [UM1 HG006542]. J.J.W. was funded in part by the Smith-Magenis Syndrome Research Foundation. J.R.L. has stock ownership in 23 and Me and Lasergen, is a paid consultant for Regeneron Pharmaceuticals, and is a co-inventor on multiple patents related to molecular diagnostics for inherited neuropathies, eye diseases and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at derives revenue from the genomic testing offered in the Baylor Genetics Laboratory.
Footnotes
Other authors have no disclosures relevant to the manuscript.
INTERNET RESOURCES
Baylor exome methods:
https://www.hgsc.bcm.edu/content/protocols-sequencing-library-construction
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott KM, Innes AM. When One Diagnosis Is Not Enough. N Engl J Med. 2017;376:83–85. doi: 10.1056/NEJMe1614384. [DOI] [PubMed] [Google Scholar]
- Calounova G, Hedvicakova P, Silhanova E, Kreckova G, Sedlacek Z. Molecular and clinical characterization of two patients with Prader-Willi syndrome and atypical deletions of proximal chromosome 15q. Am J Med Genet A. 2008;146A:1955–1962. doi: 10.1002/ajmg.a.32416. [DOI] [PubMed] [Google Scholar]
- Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14:10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- de Pontual L, Mathieu Y, Golzio C, Rio M, Malan V, Boddaert N, Soufflet C, Picard C, Durandy A, Dobbie A, Heron D, Isidor B, Motte J, Newburry-Ecob R, Pasquier L, Tardieu M, Viot G, Jaubert F, Munnich A, Colleaux L, Vekemans M, Etchevers H, Lyonnet S, Amiel J. Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum Mutat. 2009;30:669–676. doi: 10.1002/humu.20935. [DOI] [PubMed] [Google Scholar]
- de Winter CF, Baas M, Bijlsma EK, van Heukelingen J, Routledge S, Hennekam RC. Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J Rare Dis. 2016;11:37. doi: 10.1186/s13023-016-0422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Miller JL, Kuipers PJ, German JR, Beaudet AL, Sahoo T, Driscoll DJ. Unique and atypical deletions in Prader-Willi syndrome reveal distinct phenotypes. Eur J Hum Genet. 2012;20:283–290. doi: 10.1038/ejhg.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peippo M, Ignatius J. Pitt-Hopkins Syndrome. Mol Syndromol. 2012;2:171–180. doi: 10.1159/000335287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, Walkiewicz M, Bi W, Xiao R, Ding Y, Xia F, Beaudet AL, Muzny DM, Gibbs RA, Boerwinkle E, Eng CM, Sutton VR, Shaw CA, Plon SE, Yang Y, Lupski JR. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N Engl J Med. 2017;376:21–31. doi: 10.1056/NEJMoa1516767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puffenberger EG, Jinks RN, Wang H, Xin B, Fiorentini C, Sherman EA, Degrazio D, Shaw C, Sougnez C, Cibulskis K, Gabriel S, Kelley RI, Morton DH, Strauss KA. A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder. Hum Mutat. 2012;33:1639–1646. doi: 10.1002/humu.22237. [DOI] [PubMed] [Google Scholar]
- Reynolds JF, Daniel A, FitzGerald J. Atypical phenotype associated with deletion (15) (pter––q11::q13––qter) Am J Med Genet. 1987;28:55–58. doi: 10.1002/ajmg.1320280108. [DOI] [PubMed] [Google Scholar]
- Rubatino FV, Carobin NV, Freitas ML, de Oliveira VT, Pietra RX, Oliveira PP, Bosco AA, Jehee FS. Manipulation of primer affinity improves high-resolution melting accuracy for imprinted genes. Genet Mol Res. 2015;14:7864–7872. doi: 10.4238/2015.July.14.12. [DOI] [PubMed] [Google Scholar]
- Sepp M, Kannike K, Eesmaa A, Urb M, Timmusk T. Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5′ exon usage and splicing. PLoS One. 2011;6:e22138. doi: 10.1371/journal.pone.0022138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepp M, Pruunsild P, Timmusk T. Pitt-Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum Mol Genet. 2012;21:2873–2888. doi: 10.1093/hmg/dds112. [DOI] [PubMed] [Google Scholar]
- Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–457. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan WH, Bird LM, Thibert RL, Williams CA. If not Angelman, what is it? A review of Angelman-like syndromes. Am J Med Genet A. 2014;164A:975–992. doi: 10.1002/ajmg.a.36416. [DOI] [PubMed] [Google Scholar]
- Varela MC, Kok F, Setian N, Kim CA, Koiffmann CP. Impact of molecular mechanisms, including deletion size, on Prader-Willi syndrome phenotype: study of 75 patients. Clin Genet. 2005;67:47–52. doi: 10.1111/j.1399-0004.2005.00377.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.