

Abstract
The frequent occurrence of disease outbreaks in humans caused by group A Streptococcus (GAS) is an on-going public health threat. Conventional bacterial typing methods lack the discriminatory power to confidently confirm or refute outbreaks in hospital and community settings. Microbial whole genome sequencing (WGS) provides a potential solution to this, but, there has been limited population-based surveillance with accompanying sequence data. We performed retrospective genomic surveillance of 93 clinical GAS isolates from individuals in a defined geographic region. Detailed clinical information was obtained for closely related clusters of isolates. Genomic sequence data was contextualised through comparison with international data. We identified 18 different emm genotypes within our bacterial population, and revealed both highly diverse and closely related isolates. This high level of diversity was maintained even in the context of international sequence data. We also identified two emm1 clusters, and one emm3 cluster, of closely-related isolates that differed only by 1 to 4 single nucleotide polymorphisms. Analysis of clinical information identified no healthcare associated contact between patients, indicating cryptic community transmission. Our findings suggest that genomic surveillance of GAS would increase detection of transmission and highlight opportunities for intervention.
Introduction
The Lancefield group A Streptococcus (GAS; Streptococcus pyogenes) is a human pathogen capable of causing a wide spectrum of infections, ranging from self-limiting tonsillitis and pharyngitis to severe, and potentially lethal necrotising fasciitis and toxic shock syndrome. Globally, an estimated 663,000 cases of invasive GAS disease occur per year, resulting in approximately 163,000 deaths1. Epidemiological analysis suggests that the overall incidence rate of invasive GAS is increasing2, and sporadic increases in global and national prevalence are often associated with specific emm genotypes3–9. Whole genome sequencing (WGS) data has provided explanations for some of these epidemiological shifts, relating these to changes in gene content. For example, the acquisition of phage carrying novel virulence genes has been associated with a nationwide epidemic of emm3 GAS8, and homologous recombination of core genomic regions has been linked with a rise in prevalence of a new emm89 GAS variant in the UK, Europe and North America5, 6, 10.
GAS is also associated with outbreaks of disease. Prior to WGS, probable outbreaks were defined when two or more cases of GAS infection, related by person or place, occurred within a year and isolates shared the same subtypes based on molecular typing11, 12. Conventional bacterial typing methods may fail to confirm or refute an outbreak, because of insufficient discrimination between strains of the same lineage. WGS has been used successfully to investigate a small number of putative outbreaks, confirming a single causative strain that was distinct from the circulating population, which were otherwise indistinguishable by standard molecular typing methods12–14. Where epidemiological evidence for transmission is unclear or lacking, WGS data could potentially provide valuable supporting evidence to assist outbreak investigations15.
The use of WGS in routine practice will require access to contextual genome databases, to enable comparison of outbreak isolates with circulating lineages. This can be used to support decisions on transmission events and outbreaks, may provide more specific information on the abundance of variant strains and disease propensity, and identify cryptic disease clusters occurring in the community. There is currently a paucity of population-based genomic surveillance of GAS and associated sequence data. Here, we describe retrospective genomic surveillance of clinical GAS isolated from individuals in a circumscribed geographic region. This bacterial population contained 18 different emm genotypes, and both highly diverse and closely related isolates. The presence of clusters of highly similar isolates with no link to healthcare is indicative of cryptic community transmission.
Results
We conducted a retrospective observational cohort study at the Cambridge University Hospitals NHS Foundation Trust (CUH) in the UK, and identified 93 patients with at least one GAS isolate stored between 1st January 2006 and 31st December 2012 (Supplementary Table 1). Seventy isolates were from CUH patients, 14 from local district general and community hospitals (GCH), and 9 from general practice (GP). The median age of patients was 37 years (interquartile range; 17 to 67 years), and 47/93 (51%) were male. The majority of isolates were cultured from blood (n = 48) or skin and soft tissue samples (n = 24). The remainder were isolated from throat swabs (n = 7), respiratory secretions (n = 6), bone and joint specimens (n = 1), other sites (n = 1), or unspecified sites (n = 6). Seven patients died as a result of the infective episode within 30 days of the study sample.
Eighteen different emm-types were derived from sequence data for the 93 isolates (Fig. 1). The most common were emm1 (n = 25, 27%), emm28 (n = 13, 14%) and emm89 (n = 10, 11%), consistent with previous studies from the UK and elsewhere17–19. Although patient numbers were small, the mortality associated with emm3 was very high, as 3/5 invasive emm3 isolates (60%) were associated with attributable death, compared to 2/18 (11%) emm1, 1/6 (17%) emm12 and 1/8 (13%) emm89. Antimicrobial resistance to tetracycline, erythromycin and/or clindamycin was detected in 8 isolates, and was restricted to the less common emm-types (Supplementary Table 1). All three emm44 isolates and the single emm58 isolate carried tetM encoding tetracycline resistance, and ermTR encoding erythromycin resistance and inducible clindamycin resistance20. One of six emm75 isolates carried ermB and tetM and was erythromycin, clindamycin and tetracycline resistant. A further three isolates (emm5, emm43 and emm134) were resistant to tetracycline associated with tetM.
Figure 1.
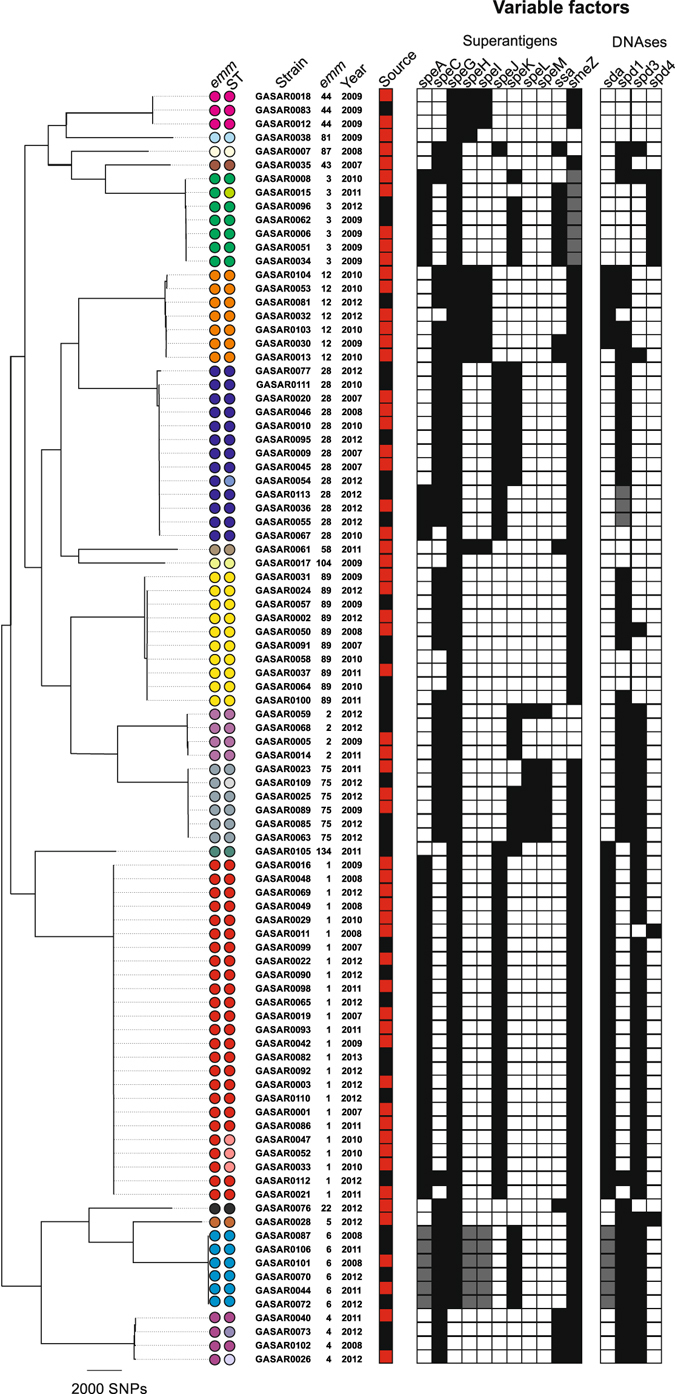
Core gene maximum likelihood phylogenetic analysis of all 93 isolates. All isolates clustered by emm-type and there was a broad association of variable factor complement with the emm-type. The superantigens speA, speH and speI are highlighted in grey in emm6 strains as these are atypical variants that may not be functional. Similarly, all emm3 strains were found to carry a non-functional smeZ gene, as previously described16. The DNase spd1 in three emm28 strains is highlighted grey to indicate that it is a different variant to that found in the other emm28 strains. Coloured circles represent emm-type/ST. The majority of emm-types were associated with a single ST type (colours of emm-type identical to colour of ST). Where an additional ST was associated with an emm-type this is represent by a different colour ST circle. emm1 (n = 25); ST28 (red/red) or ST785 (red/pink), emm2 (n = 4); ST55, emm3 (n = 7); ST15 (green/green) or ST315 (green/pale green), emm4 (n = 4); ST39 (purple/purple) or ST786 (purple/pale purple) or ST38 (purple/paler purple), emm6 (n = 6); ST382, emm12 (n = 7); ST36, emm28 (n = 13); ST52 (blue/blue) or ST787 (blue/pale blue), emm44 (n = 3); ST367, emm75 (n = 6); ST150 (grey/grey) or ST788 (grey/paler grey), emm89 (n = 10); ST101. Types represented by a single isolate comprised emm5; ST99, emm22; ST46, emm43; ST3 emm58; ST176, emm81; ST624, emm87; ST62, emm104; ST789, emm134; ST790. Source; Invasive (red) or non-invasive (black) site of isolation.
The propensity for some strains to cause severe invasive disease has been attributed to loss-of-function mutations within virulence gene regulators, in particular the two component regulatory system CovRS, which negatively regulates a number of different virulence factors21–23. We extracted and compared the sequences of known transcriptional regulators, CovR, CovS, RocA (regulator of CovRS), RivR, Rgg1, Rgg2, Rgg3, Rgg4, and FasABCX (Supplementary Tables 2, 3 and 4). A single isolate (an invasive emm2) had a mutation in CovR, and 13 isolates, of varying emm-type, had mutations in CovS (10 invasive, 3 non-invasive isolates). Although mutations within CovRS have been associated with the virulence and invasive nature of GAS, particularly emm1, only two of the 18 invasive emm1 isolates had mutations in CovS; an amino acid change of Threonine to Proline at residue 214, and a premature stop codon after 390/500 residues. In addition, one emm1 strain (GASAR0033) carried a duplication mutation of 8 bp within rocA resulting in a premature stop codon after 32 amino acids, which is predicted to increase virulence factor expression through loss of CovRS repression24. None of the isolates from cases who died carried any additional gene regulatory mutations outside of those related to the emm-type lineage. Interestingly, 9/13 (69%) emm28 isolates carried a mutation in one of five gene regulators; CovS (2 isolates), RivR (1 isolate), Rgg1 (1 isolate), Rgg2 (2 isolates) and Rgg3 (3 isolates). This was a higher prevalence of regulatory gene mutations than other emm-types.
A maximum-likelihood tree based on 49,262 SNPs in the 1,295 conserved genes identified by pan-genome analysis identified clustering of isolates by emm-type (Fig. 1). Multilocus sequence type (MLST) derived from sequence data resolved 24 STs for the 93 isolates. Each ST contained a single emm-type, but five emm-types had multiple STs (Fig. 1). In each case the change in ST within an emm-type was associated with a single nucleotide base change within one of the seven MLST loci. We also derived emm-subtyping information (Supplementary Table 1) but all isolates within each emm-type were of the same emm-subtype. The exceptions were emm1, where the majority were emm1.0, but two isolates were emm1.52 and two isolates were emm1.6, and emm6, where the majority were emm6.0 but two isolates were emm6.4.
Pairwise comparison of SNPs in the core genome of isolates of the same emm-type determined a median SNP difference of 37 (range 0–370 SNPs), compared to 11,079 SNP differences (range 8149–13134 SNPs) between isolates of different emm-types. The range of zero to 370 SNP differences between isolates within emm-types suggest that both identical/very closely related isolates and diverse isolates are circulating within this local population.
The presence of variable accessory genes, which are often associated with mobile prophages, were mapped to the phylogenetic tree (Fig. 1). The pattern of types and subtypes of variable superantigen and DNase genes were predominantly associated with specific emm-types, although some were variably present in specific emm-type clusters. For example, four of 13 emm28 strains were positive for the superantigen speA but negative for the superantigen speK; both superantigens are associated with different prophages. Three of the same four emm28 isolates also had a variant of the DNase spd1, commonly found on the same prophage as the superantigen speC, that was different from the remaining emm28 isolates, and speC and spd1 were both absent in the fourth strain. This suggests that epidemiological studies using detection of these variable accessory genes for isolate discrimination, could be enhanced by including analysis of allelic variants.
Emm1
We confirmed the presence of two clusters of closely related emm1 isolates based on core gene analysis by mapping our emm1 population to an emm1 reference genome, MGAS500525 (Fig. 2). Cluster 1 contained three isolates, all CUH patients with invasive disease in 2010, and separated by only 1–2 SNPs. GASAR0033 and GASAR0052 were identical by core gene phylogeny (Fig. 1), but mapping to MGAS5005 resolved a unique non-synonymous SNP in GASAR0033 within a non-core gene (serine to leucine at amino acid residue 21 in M5005_Spy0853, short chain dehydrogenase). GASAR0033 also carried an insertion mutation in rocA, a regulator of CovRS. A single nucleotide base change in the MLST gene xpt of C194T in this cluster of isolates resulted in the unique ST785; other emm1 strains were ST28. Two of these patients initially presented with skin and soft tissue infections, and the third patient with bone and joint infection; all went on to develop bacteraemia and multi-organ failure. Patients with skin and soft tissue infections were admitted to the intensive care unit, and one patient died shortly after. GASAR0033 and GASAR0052 were isolated within the same month (22 days apart), although both were community-acquired infections, and GASAR0047 was isolated 7 months later. We were unable to identify any healthcare-related association between any of the three patients.
Figure 2.
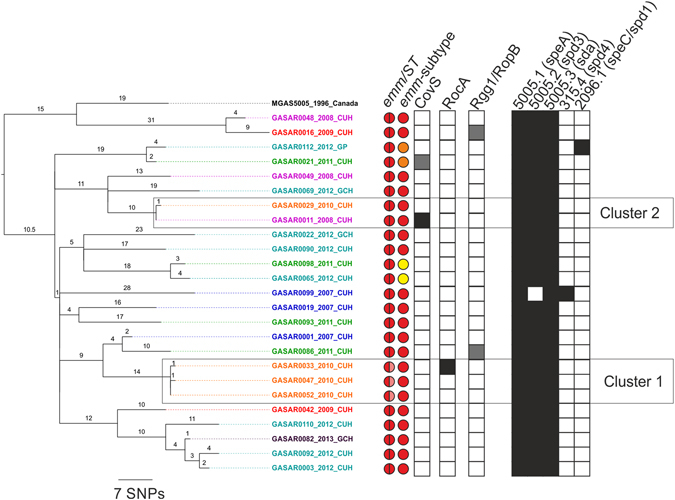
Identification of closely related clusters of emm1 isolates. Phylogenetic analysis of 25 emm1 isolates mapped to MGAS5005 (NC_007297). Two clusters were identified that were separated by only 1–2 SNPs. Two sequence types were identified: ST28 (red/red) or ST785 (red/pink). All were emm-subtype 1.0 except four isolates, of which two were subtype 1.52 (orange) and two were 1.6 (yellow) and the isolates belonging to these subtypes cluster within the phylogeny. Prophages are indicated on the right (presence indicated in black). The majority carried typical M1 prophages 5005.1 with the superantigen speA, 5005.2 with the DNase spd3 and 5005.3, with the DNase sda. GASAR0099 carried a prophage similar to 315.4 from the M3 strain MGAS315 with the DNase spd4, and GASAR0112 carried a prophage similar to 2096.1 from the M12 strain MGAS2096 with the superantigen speC and the DNase spd1. Five isolates had stop codon mutations (in black) or non-synonymous SNP mutations (grey) in three regulatory genes; covS, rocA and rgg1/ropB. Detailed in tip labels are isolate name, year of isolation and site of isolation; Cambridge University Hospital (CUH), Local or tertiary hospitals (GCH) or GP practices (GP). Branches are coloured based on year of isolation and branch numbers indicate SNPs.
Cluster 2 contained two invasive isolates from CUH, GASAR0011 (isolated in 2008) and GASAR0029 (isolated in 2010). They were only differentiated by a single non-synonymous mutation in rplJ in GASAR0029, but GASAR0011 also carried an insertion mutation in CovS of 25 bp after 1129/1503 bp truncating the protein at 390/500 amino acids, and a deletion mutation in vfr (M5005_0693), encoding for a potential virulence factor regulator, truncating the protein at 80/247 amino acids. As these were isolated three years apart, it seems unlikely that they represent direct or recent transmission, but may reflect persistence of this variant in the population.
In addition to Cluster 1 and Cluster 2, three isolate pairs were identified within our collection that were isolated 1–2 years apart and separated by only 6–7 SNPs (Fig. 2); GASAR00112 and GASAR00021, GASAR0098 and GASAR0065, GASAR0092 and GASAR0003. Based on a previously estimated substitution rate for emm1 GAS at 1.37 substitutions per core genome per year3, these isolate pairs are unlikely to represent recent transmission events, but may represent independent acquisitions of a lineage that is circulating in the community.
To contextualise our collection, we combined sequence data for the 25 study emm1 isolates with data for 1210 emm1 isolates from the USA, Canada, Denmark, Finland, Norway, Iceland and Sweden, collected between 2005–20133, and for 24 UK emm1 isolates from 2007–200814 (Fig. 3). Cluster 1 and Cluster 2 emm1 isolates remained clustered within this extended population. Our other emm1 isolates were interspersed with geographically distant isolates across the tree, indicating that the level of diversity in our local population is similar to that of a wider North American/European population. Of the 24 emm1 UK isolates sequenced by Turner et al.14, 15 isolates were part of an outbreak, and were previously determined to be identical to each other but distinct from emm1 isolates from other parts of the UK14. Incorporation of these 15 outbreak isolates into the extensive collection here supports this finding as they are clearly distinct from other isolates (Fig. 3).
Figure 3.
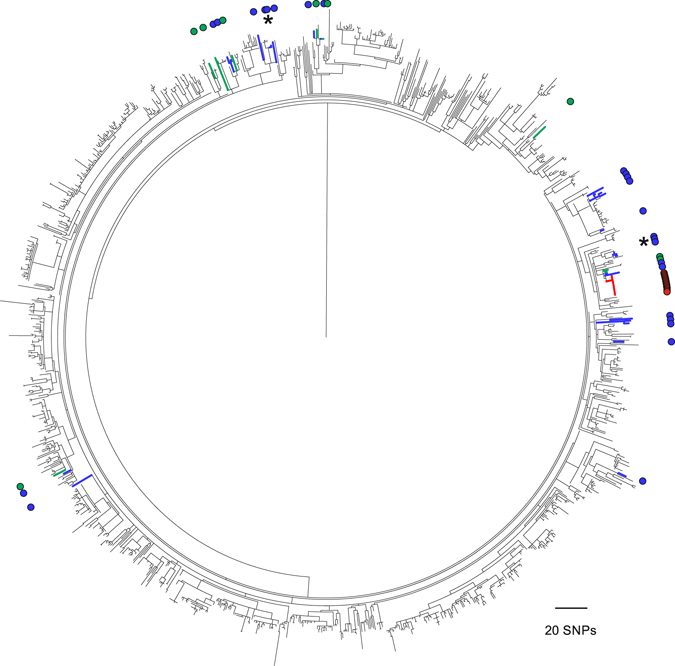
Global context of emm1 and outbreak identification. A maximum-likelihood phylogenetic tree was constructed using core genome SNPs (excluding prophages) identified in 1259 sequenced emm1 strains isolated 2005–2013 from USA (n = 381), Canada (n = 23), Denmark (n = 269), Finland (n = 204), Norway (n = 54), Iceland (n = 16), Sweden (n = 263)3 and the UK (n = 49)14 compared to MGAS5005. The UK isolates are identified by coloured branches and highlighted by coloured circles around the outside of the tree. They comprise 25 from the study collection (blue branches and circles), plus 9 from various locations in the UK (green branches and circles) and 15 emm1 outbreak isolates (red branches and circles) from a previous study14. Although the majority of UK isolates are dispersed across the tree, those associated with the outbreak and the two study local clusters (indicated by *) are clearly distinct from other emm1 isolates.
Emm3
Our local collection of seven emm3 isolates were associated with high mortality (60%). Like emm1, this small population consisted of diverse isolates and a cluster of closely related isolates (Fig. 4). One isolate belonged to a recently identified lineage (Lineage C) associated with an epidemic upsurge in invasive disease in the UK8. Four isolates, all obtained in 2009 but isolated one, four and almost 12 months apart from the first isolate, were separated by only 1–4 SNPs (Fig. 4) and two of these isolates (GASAR0006 and GASAR0062) were associated with a fatal outcome. They differed from their nearest relative by two non-synonymous SNPs; threonine to alanine at amino acid residue 112 in rgpG, encoding a putative polysaccharide biosynthesis protein, and aspartic acid to asparagine at amino acid residue 588 in rpoB, a putative DNA-dependent RNA polymerase subunit β. GASAR0062 also carried a SNP within the −35 transcription box of the hasA hyaluronic acid capsule-synthesis gene promoter, which may impact on expression of the hyaluronic acid capsule. GASAR0034 carried a synonymous SNP in pbp2a and an 11 bp deletion in CovS, truncating the protein after 221/500 amino acids. Detailed clinical information from all four cases indicated that three patients presented with lower respiratory tract infections and the fourth with pharyngitis (isolate GASAR0034). Three patients developed bacteraemia (isolates GASAR0006, GASAR0034, GASAR0051) and one patient had a skin and soft tissue infection. The relatedness of the strains determined by WGS and the supporting clinical information provides evidence that this was a community outbreak and that it may have extended beyond our cohort.
Figure 4.
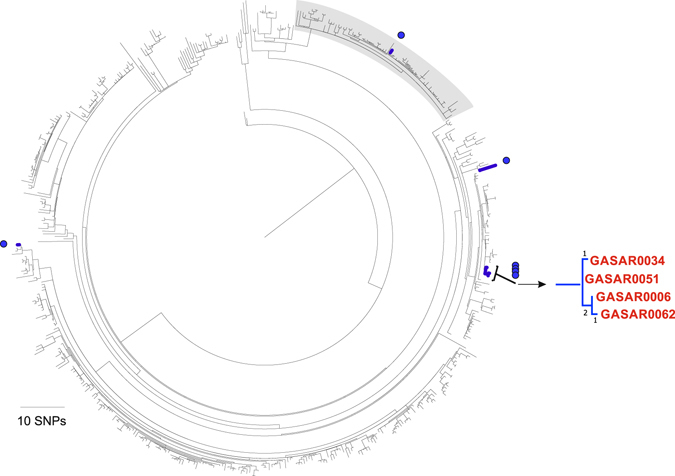
Diverse and closely related isolates within emm3. A maximum-likelihood phylogenetic tree was constructed using core genome SNPs (excluding prophages) identified in sequenced emm3 strains from our collection (n = 7), emm3 strains isolated 1992–2007 from Canada (n = 70)26, and emm3 strains isolated 2001–2013 from UK and Ireland (n = 442)8, compared to the reference emm3 genome strain MGAS315. Isolates from our collection are highlighted in blue branches and blue circles around the outside of the tree. One of our isolates resided within the previously identified Lineage C (lineage highlighted grey) associated with an increase in invasive disease in the UK8. This strain, like the majority of others within this lineage, carries the superantigen speC and the DNase spd1 but has lost the superantigen ssa. An expanded region of the main tree is indicated on the right hand side and highlights four 2009 isolates that are separated from each other by only 1–4 SNPs. Branch labels represent number of SNPs.
Emm12
A recent study into the outbreak of scarlet fever in Hong Kong identified four major clades of emm12, three of which were associated with scarlet fever isolates7. Our local collection of just seven emm12 contained isolates from each of the four major clades, suggesting that these clades are a common feature of the emm12 population (Supplementary Figure 1). Only two of our isolates carried the superantigen ssa which has been attributed to scarlet fever, both were associated with clade I but neither were associated with scarlet fever. Although there has been a recent increase in scarlet fever in the UK, emm12 has not yet been identified as a major contributor27.
Discussion
The limited amount of WGS data currently available for GAS means that relatively little is known about the circulating populations in the community. Population-based genomic surveillance of GAS and associated sequence data are crucial in supporting transmission and outbreak investigations. We have identified that even within localised populations there can be both diverse and closely related isolates, indicating both locally circulating clones and the introduction of new lineages.
Analysis of the population core genome identified closely related clusters within the emm1 and emm3 genotypes that differed only by 0–1 SNPs, indicative of cryptic community transmission. By mapping to a completed reference genome of the same emm-type, further differences between isolates were resolved. This suggests that disease clusters can be identified through pan-genome analysis without an emm-type specific reference, which is necessary as, although reference genomes have been proven useful, not all emm-types are represented. Where disease-clusters are associated with common emm-types sharing typical subtypes (MLST, emm-subtype, resistance genes), WGS can provide irrefutable evidence of relatedness and identify any unique features that could underpin a specific disease phenotype. In our study, we did not have sufficient numbers to directly link variant isolates or transcriptional regulatory gene mutations with specific clinical markers or disease outcomes. Unusually, however, our emm28 isolates did show a higher level of regulatory gene mutations than other emm-types, although the relevance of these mutations could not be resolved in our study. With further study and if WGS becomes more routine, is may be possible to tailor treatments towards the specific infection variant.
The majority of invasive disease cases were associated with emm1 and mortality was particularly high with emm3, making the detection and prevention of disease clusters with these emm-types critical. All patients with Cluster 1 emm1 isolates went on to develop severe disease, with two requiring ICU admission and one patient died, suggesting this variant may have propensity towards severe invasive disease. Similarly, two of the four closely-related cluster emm3 isolates were associated with mortality. These patients with emm3 presented with lower respiratory tract infection or pharyngitis, and went on to develop bacteraemia or, in one case, skin and soft tissue infection. Cluster 1 emm1 isolates carried a unique MLST and therefore may have been identified through routine typing. The emm3 cluster, however, lacked distinguishing conventional typing features (emm, MLST, variable accessory factors) suggesting that without whole genome sequencing it would not have been identified. The clinical reports for three of the emm3 patients indicated possible contact with other GAS patients outside of this cohort. Although we could not confirm this, it is possible that this cluster extended beyond the four cases that we identified.
There is limited data on GAS transmission and possible infection prevention strategies in the community. Two recent studies identified a 731–2000 fold increased risk of invasive GAS disease among household contacts of a GAS index case compared to the general population, indicating a potential benefit of antibiotic prophylaxis within households28, 29. With the rarity of index cases, the number of cases where house-hold or community transmission has been confirmed is very small. Real-time genomic surveillance may provide alerts to outbreak or transmission events and could support the introduction and assessment of community interventions.
We have demonstrated that WGS of isolates within a local population can identify cryptic community transmission and disease clusters. This supports previous findings that WGS of GAS populations can identify disease clusters within the community as well as the spread of unusual GAS genotypes entering the population or the emergence of new GAS variants that may lead to disease upsurges4–6, 8, 9, 30, 31. By monitoring local bacterial populations with WGS, informed by circulating national and international genomic data isolates, community transmission could be rapidly identified and interventions put in place. It may also provide early indication of new and potentially harmful variants entering the population that can rapidly become epidemic.
Methods
Study Setting, Design and Participants
A retrospective observational cohort study was conducted at the Cambridge University Hospitals NHS Foundation Trust (CUH) in the United Kingdom (UK), a tertiary referral centre in the United Kingdom with 1,170 beds. We identified samples that were culture positive for GAS at the on-site Public Health England Clinical Microbiology and Public Health Laboratory (CMPHL) between 1st January 2006 and 31st December 2012. This diagnostic laboratory receives samples from three hospitals and 75 general practice (GP) surgeries in Cambridgeshire. Isolates were cross-referenced with patient information to identify cases with at least one stored GAS isolate (n = 93). The first stored isolate for each patient was recovered from the laboratory freezer archive for sequencing. Phenotypic antibiotic susceptibility data were obtained from the CMPHL laboratory database. Isolates had previously been tested for susceptibility to penicillin, tetracycline, erythromycin, vancomycin, teicoplanin and clindamycin, using the disc diffusion method (British Society for Antimicrobial Chemotherapy; BSAC methods for Antimicrobial Susceptibility Testing; Version 14 January 2015). Inducible clindamycin resistance was detected using the D-test method.
Clinical data on all 93 cases was collected from paper and electronic patient records, and consisted of date of sample collection, gender, age, infection type/site and 30-day mortality. Seventy isolates were CUH patients, 14 from local district general and community hospitals (GCH), and 9 from general practice (GP). Invasive GAS disease was defined as the isolation of GAS from a normally sterile site or from a wound in a patient with necrotizing fasciitis or streptococcal toxic shock syndrome32. Where we suspected clusters of related cases the healthcare records were retrieved and available clinical data was reviewed.
Whole genome sequencing and analysis
DNA was extracted using the QIAxtractor instrument (QIAgen, Hilden, Germany). DNA library preparation was conducted according to the Illumina protocol and sequencing was performed on an Illumina HiSeq. 2000 with 100-cycle paired-end runs. Sequence data have been submitted to the European Nucleotide Archive (ENA) (www.ebi.ac.uk/ena) under the accession numbers listed in Supplementary Table 5. Genomes were de novo assembled using Velvet33 with the pipeline and improvements found at https://github.com/sanger-pathogens/vr-codebase and https://github.com/sanger-pathogens/assembly_improvement (assembly statistics in Supplementary Table 5). Genome assemblies were annotated using Prokka and a pan-genome estimated using Roary34. A maximum-likelihood tree was created based on 49,262 single nucleotide polymorphisms (SNPs) in the 1,295 conserved genes (present in 100% of isolates with greater than 95% shared identity) using RAxML35, a midpoint root and 100 bootstraps. Multilocus sequence types (MLSTs) were identified from the sequence data using the MLST database (pubmlst.org/spyogenes) and an in-house script (https://github.com/sanger-pathogens/mlst_check). Emm-types were derived from sequence data and the presence of the 11 known variable chromosomal and prophage-encoded superantigen genes (speA, speC, speG-M, ssa, smeZ) and four prophage-encoded DNAse genes (sda, spd1, spd3, spd4) determined by BLAST analysis. The sequences of key virulence gene regulators (covR, covS, rocA, rgg1, rgg2, rgg3, rgg4, fasA, fasB, fasC, fasX, rivR) were extracted from the assembled genomes and compared to all available reference genomes to identify variant alleles. Within the gene regulators, some genetic variation was identified that was related to emm-type, suggesting lineage specific variation and not spontaneous non-functional mutants. We therefore only defined alleles as potential non-functional mutant variants when non-synonymous SNPs occurred in addition to emm-type lineage specific variations.
To provide genetic context, sequence data for 1210 emm1 isolates from USA/Canada/Europe3 and 24 UK emm1 isolates14 were downloaded from the ENA and combined with the 25 study emm1 genomes and mapped to the genome of GAS emm1 strain MGAS5005 (ENA accession number NC_007297). Sequence data for 442 emm3 GAS isolates from the UK and Ireland8 and 70 Canadian emm3 isolates26 were downloaded from ENA and mapped with the 7 study emm3 genomes to emm3 strain MGAS315 (ENA accession number NC_004070). 132 emm12 isolates from Hong Kong collected between 2005–20117 were downloaded from the ENA and combined with the 7 study emm12 isolates and mapped to emm12 strain HKU16 (ENA accession number AFRY01000001). All mapping was performed using SMALT (http://www.sanger.ac.uk/resources/software/smalt/), and SNPs in the core genome (excluding prophages) were used to construct a maximum-likelihood tree using RAxML35.
Ethics approval
The study was approved by the National Research Ethics Service (ref: 12/EE/0439) the Cambridge University Hospitals NHS Foundation Trust Research and Development Department (ref: A092428) and we performed the study in accordance with the guidelines and regulations.
Data availability
Sequence data have been submitted to the European Nucleotide Archive (ENA) (www.ebi.ac.uk/ena) under the accession numbers listed in Supplementary Table 5.
Electronic supplementary material
Acknowledgements
We thank Elizabeth Blane for laboratory assistance, and the library construction, sequencing and core informatics teams at the Wellcome Trust Sanger Institute. This publication presents independent research supported by the Health Innovation Challenge Fund (HICF-T5-342 and WT098600), a parallel funding partnership between the UK Department of Health and Wellcome Trust. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health, Public Health England or the Wellcome Trust. MET is a Clinician Scientist Fellow, supported by the Academy of Medical Sciences, the Health Foundation and the NIHR Cambridge Biomedical Research Centre.
Author Contributions
C.E.T. and L.B. performed the analysis, K.J. provided laboratory assistance, N.M.B. provided the bacterial isolates and access to clinical information, M.E.T. assisted with protocols and ethical approvals, J.P. provided resources, C.E.T., L.B. and S.J.P. wrote the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Claire E. Turner and Luke Bedford contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-08914-x
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–94. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 2.Sims Sanyahumbi, A., Colquhoun, S., Wyber, R. & Carapetis, J. R. In: Ferretti, J. J., Stevens, D. L. & Fischetti, V. A. editors. Streptococcus pyogenes: Basic Biology to Clinical Manifestations. Oklahoma City (OK); 2016.
- 3.Nasser W, et al. Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc Natl Acad Sci USA. 2014;111:E1768–76. doi: 10.1073/pnas.1403138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu L, et al. A molecular trigger for intercontinental epidemics of group A Streptococcus. J Clin Invest. 2015;125:3545–59. doi: 10.1172/JCI82478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turner CE, et al. Emergence of a new highly successful acapsular group A Streptococcus clade of genotype emm89 in the United Kingdom. MBio. 2015;6 doi: 10.1128/mBio.00622-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friaes A, et al. Emergence of the same successful clade among distinct populations of emm89 Streptococcus pyogenes in multiple geographic regions. MBio. 2015;6:e01780–15. doi: 10.1128/mBio.01780-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies MR, et al. Emergence of scarlet fever Streptococcus pyogenes emm12 clones in Hong Kong is associated with toxin acquisition and multidrug resistance. Nat Genet. 2015;47:84–7. doi: 10.1038/ng.3147. [DOI] [PubMed] [Google Scholar]
- 8.Al-Shahib, A. et al. Emergence of a novel lineage containing a prophage in emm/M3 group A Streptococcus associated with upsurge in invasive disease in the UK. mGen. 2 (2016). [DOI] [PMC free article] [PubMed]
- 9.Fittipaldi N, et al. Full-genome dissection of an epidemic of severe invasive disease caused by ahypervirulent, recently emerged clone of group AStreptococcus. Am J Path. 2012;180:1522–32. doi: 10.1016/j.ajpath.2011.12.037. [DOI] [PubMed] [Google Scholar]
- 10.Zhu L, Olsen RJ, Nasser W, de la Riva Morales I, Musser JM. Trading capsule for increased cytotoxin production: contribution to virulence of a newly emerged clade of emm89 Streptococcus pyogenes. MBio. 2015;6:e01378–15. doi: 10.1128/mBio.01378-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steer JA, et al. Guidelines for prevention and control of group A streptococcal infection in acute healthcare and maternity settings in the UK. J Infect. 2012;64:1–18. doi: 10.1016/j.jinf.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Chalker VJ, et al. Integration of genomic and other epidemiologic data to investigate and control a cross-institutional outbreak of Streptococcus pyogenes. Emerg Infect Dis. 2016;22:973–80. doi: 10.3201/eid2206.142050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowen AC, et al. Whole genome sequencing reveals extensive community-level transmission of group A Streptococcus in remote communities. Epidemiol Infect. 2016;144:1991–8. doi: 10.1017/S095026881500326X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turner CE, et al. Molecular analysis of an outbreak of lethal postpartum sepsis caused by Streptococcus pyogenes. J Clin Microbiol. 2013;51:2089–95. doi: 10.1128/JCM.00679-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galloway-Pena J, et al. Application of whole-genome sequencing to an unusual outbreak of invasive group A streptococcal disease. Open Forum Infect Dis. 2016;3 doi: 10.1093/ofid/ofw042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turner CE, et al. Superantigenic activity of emm3 Streptococcus pyogenes is abrogated by a conserved, naturally occurring smeZ mutation. PLoS One. 2012;7 doi: 10.1371/journal.pone.0046376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luca-Harari B, et al. Clinical and microbiological characteristics of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol. 2009;47:1155–65. doi: 10.1128/JCM.02155-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. Global emm-type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect Dis. 2009;9:611–6. doi: 10.1016/S1473-3099(09)70178-1. [DOI] [PubMed] [Google Scholar]
- 19.Lamagni TL, et al. Epidemiology of severe Streptococcus pyogenes disease in Europe. J Clin Microbiol. 2008;46:2359–67. doi: 10.1128/JCM.00422-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seppala H, Skurnik M, Soini H, Roberts MC, Huovinen P. A novel erythromycin resistance methylase gene (ermTR) in Streptococcus pyogenes. Antimicrob Agents Chemother. 1998;42:257–62. doi: 10.1093/jac/42.2.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turner CE, Kurupati P, Jones MD, Edwards RJ, Sriskandan S. Emerging role of the interleukin-8 cleaving enzyme SpyCEP in clinical Streptococcus pyogenes infection. J Infect Dis. 2009;200:555–63. doi: 10.1086/603541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. Genome-wide analysis of group a streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2006;2 doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin JN, Chang LL, Lai CH, Lin HH, Chen YH. Association between polymorphisms in the csrRS two-component regulatory system and invasive group A streptococcal infection. Eur J Clin Microbiol Infect Dis. 2014;33:735–43. doi: 10.1007/s10096-013-2005-7. [DOI] [PubMed] [Google Scholar]
- 24.Lynskey NN, et al. RocA Truncation Underpins Hyper-Encapsulation, Carriage Longevity and Transmissibility of Serotype M18 Group A Streptococci. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sumby, P. et al. Evolution and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J Infect Dis.192, 771–782 (2005). [DOI] [PubMed]
- 26.Beres SB, et al. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc Natl Acad Sci USA. 2010;107:4371–6. doi: 10.1073/pnas.0911295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turner CE, et al. Scarlet fever upsurge in England and molecular-genetic analysis in North-West London, 2014. Emerg Infect Dis. 2016;22:1075–8. doi: 10.3201/eid2206.151726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carapetis JR, et al. Effectiveness of clindamycin and intravenous immunoglobulin, and risk of disease in contacts, in invasive group A streptococcal infections. Clin Infect Dis. 2014;59:358–65. doi: 10.1093/cid/ciu304. [DOI] [PubMed] [Google Scholar]
- 29.Lamagni, T. L., Oliver, I. & Stuart, J. M. Global assessment of invasive group A Streptococcus infection risk in household contacts. Clin Infect Dis.60, 166–7 (2015). [DOI] [PubMed]
- 30.Fittipaldi N, et al. Integrated whole-genome sequencing and temporospatial analysis of a continuing group A Streptococcus epidemic. Emerg Microbes Infect. 2013;2 doi: 10.1038/emi.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Athey TBT, et al. High incidence of invasive group A Streptococcus disease caused by strains of uncommon emm types in Thunder Bay, Ontario, Canada. J Clin Micro. 2016;54:83–92. doi: 10.1128/JCM.02201-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson GE, et al. Epidemiology of invasive group A streptococcal infections in the United States, 2005–2012. Clin Infect Dis. 2016;63:478–86. doi: 10.1093/cid/ciw248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Page AJ, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rokas, A. Phylogenetic analysis of protein sequence data using the Randomized Axelerated Maximum Likelihood (RAXML) Program. Curr Protoc Mol Biol. Chapter 19 (2011). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data have been submitted to the European Nucleotide Archive (ENA) (www.ebi.ac.uk/ena) under the accession numbers listed in Supplementary Table 5.