

Abstract
The efficient conversion of light energy into chemical energy is key for sustainable human development. Several photocatalytic systems based on photovoltaic electrolysis have been used to produce hydrogen via water reduction. However, in such devices, light harvesting and proton reduction are carried separately, showing quantum efficiency of about 10–12%. Here, we report a nano-hybrid photocatalytic assembly that enables concomitant reductive hydrogen production and pollutant oxidation with solar-to-fuel efficiencies up to 20%. The modular architecture of this plasmonic material allows the fine-tuning of its photocatalytic properties by simple manipulation of a reduced number of basic components.
Introduction
Artificial conversion of solar light into chemical bonds (aka artificial photosynthesis) has attracted wide interest because it yields storable energy deprived of greenhouse gas emission1, 2. The development of catalysts for photoinduced electron/hole transfer allows the production of molecular hydrogen and oxygen gases from water reduction and oxidation, respectively3. However, oxygen production relies on a difficult to achieve multi-step four-electron process, which yields a product with low-economic value, difficult to separate, and a direct competitor of protons for the photo-generated electrons. Strides have been made in the mitigation of the later problems by separating H2 and O2 evolution in space4 and time5 but fewer efforts are been done seeking for alternative substrates (e.g. pollutants), which can improve process safety and economic viability6. Moreover, H2 usages go beyond fuel applications7, 8, and include the synthesis of several bulk and fine chemicals, e.g. ammonia, methanol, fragrances and drugs. Depending on the approach, massive hydrogen production via water splitting may increase the concentration of oxygen in the atmosphere, an undesirable event that can affect profoundly the planet, as it did ca. 2.4 billion years ago9, 10.
Low-cost devices for the large-scale production of solar fuels can mitigate the use of fossil fuels. Developments based on large band gap semiconductors (>3 eV), such as TiO2 3, are hindered by the need of UV irradiation to induce charge separation. Efforts to shift their absorption to the visible range resulted in the decrease of both reaction scope and overall performance11. A promising strategy is semiconductor sensitization, as the case of dye-sensitized solar cells (DSSCs) concept12, where the sensitizer harvests sunlight and rapidly injects electrons (τinj < 1 ps) into the TiO2 conduction band (CB). Metallic nanoparticles (NPs) are attractive sensitizers due to their large optical cross-sections related to the excitation of localized surface plasmons (LSP). Gold group metals exhibit LSP resonances in the visible region, and their absorption maxima depend on nanoparticle morphology, enabling a good match with the solar spectrum. Additionally, their d 10 configuration bestows them chemical stability. Recently, the excitation of Au and Ag LSP resonances were shown to improve charge transfer in DSSCs13, photocurrents14, and photocatalytic oxidations15, 16.
Hallett-Tapley and coauthors17 suggested three LSP mediated reactive processes: thermal, electronic or antenna. The electronic process is the relevant for light conversion to chemical bonds, inferring hot electron creation18. Their generation upon LSP excitation was recently established via Au LIII-edge high-resolution X-ray absorption spectroscopy19. LSP excitation led to an upward shift in the threshold ionization energy (ca. 1.0 eV), and an increase of Au d-band hole population, consistent with hot electrons formation. The lifetime of hot electrons is too short (<100 fs) for any catalytic reaction to happen. However, transfer of hot electrons to the CB of TiO2 prolongs their lifetime to microseconds19, as confirmed by transient mid-IR spectroscopy20.
Another challenge to produce efficient solar fuels is the conciliation of light-induced single electron transfer with multi-electron catalysis. Systems must be able to accumulate/store charge particles provided by the fast light absorption process so the slow multi-electron catalysis can take place, i.e. accumulative charge separation concept 21. Metal NPs contain large amount of loosely bonded electrons that, combined with the capacity of LSP to redirect the light flow towards the NP (i.e. Poynting vector), makes multi-electron generation per single NP plausible. Small metal NPs are excellent hydrogen evolution catalysts, allowing charge accumulation and storage22. Gold NPs fabricated using top-down approaches have been extensively used for LSP-based conversion and storage of solar energy23. However, the bottom-up synthesis in solution of alternative silver NPs photocatalytic systems has been pursued both for practical and economic reasons.
Here, we present a modular nano-hybrid assembly fabricated using silver NPs produced via bottom-up synthesis. This chemical system is made up of a discrete number of components connected via covalent bonding and intermolecular interactions. In the present case, the subunits are silver metallic nanoparticles and n-type semiconductor TiO2 nanoparticles interconnected via a molecular linker. Our approach allows for the synthesis of a multitude of multifuctional materials from a relatively small number of basic units. The nano-hybrid architectures can accomplish long-lived charge separate states, enabling concomitant photocatalytic production of H2 and pollutant oxidation.
Results and Discussion
Figure 1 shows the blueprint of the nano-hybrid system, composed by natural product-stabilized Ag NPs, a molecular wire linker, TiO2 as a semiconductor and Ru NPs as a co-catalyst. Silver NPs stabilized with betanin and derivatives (Bts-Ag NPs) were synthesized in an automated microfluidic reactor using pure betanin extracted from beetroots24, as reducing and capping agent. A genetic optimization algorithm was used to ensure that particles size and light absorption are homogeneous25. Herein, we used monodispersed spherical Bts-Ag NPs with 28–30 nm, exhibiting a LSP resonance absorption maximum wavelength (λmax) at 405 nm; refer to the supporting information (SI) for DLS analysis (Fig. S2), SEM (Fig. S3), UV-Vis spectrum (Fig. S4), and AFM (Fig. S5).
Figure 1.

Nano-hybrid assembly synthesized using modular design approach. The architecture is comprised of light absorber and oxidation catalyst (Ag NPs), a molecular wire linker (pABA), a semiconductor (TiO2), a co-catalyst (Ru NPs) and regenerators (Bts).
Betanin (Bn) and its derivatives from alkaline hydrolysis or oxidation, betalamic acid (HBt), cyclo-DOPA 5-O-glucoside (cDOPA), and neobetanin (neoBn), are expected to retard electron/hole recombination and are used as regenerators. TiO2 semiconductor was used to enhance electron lifetime19 and ruthenium NPs as hydrogen evolution catalyst26, which were prepared via chemical reduction (see SI).
A molecular linker (4-aminobenzoic acid, pABA) was used to enhance the electronic coupling between the electron donor (Ag NPs) and the electron acceptor (TiO2). Electron transfer through a π-conjugated molecular wire is essential to a multitude of biologic systems and molecular electronics27 but has seldom usage in the synthesis of nano-based artificial photosynthesis systems. The linker pABA provides an excellent electronic coupling because of its good π-conjugated, rigidity and planarity28. The connectivity between electron donor and acceptor parts via molecular linker was accessed via Raman spectroscopy (Fig. S6). Due to the similarities in functional groups between betanin derivatives and pABA, there are not many characteristic functionalities that can be firmly used for the assignment. There is, however, a peak at 1727 cm−1 in the silver containing samples that is absent in the pure linker, assigned to C=O terminal group of betalamic acid29. Addition of the linker to Bts-Ag NPs led to the appearance of a small peak at 1605 cm−1, ascribed to secondary amine N–H bend, a broad peak from 1620 to 1800 cm−1, associated to aromatic ring combination band (1660–2000 cm−1) and carboxylic acid (1700–1725 cm−1)29, suggesting the anchoring of the amino group of the linker to the Bts-Ag NPs. Addition of TiO2-Ru led to a decrease in the ratio between the broad peak and 1605 cm−1, suggesting the anchoring of the carboxylic group of the linker to TiO2.
Both the molecular wire linker and the regenerator are important for photocatalytic hydrogen production (Fig. 2a) and methylene blue (model pollutant) depletion (Fig. 2b). Experiments were carried out using a flow photoreactor (Fig. S1)30. The results show a noteworthy increase in hydrogen evolution when the systems are connected by a molecular wire. Such increase was not observed using PVP-Ag NPs, prepared in the absence of betanin via the polyol method (Fig. 2a) confirming the active role of the capping layer in the process, as rationalized below. Transient absorption infrared spectroscopy measurements performed on complete system with PVP-Ag NPs, revealed very fast charge recombination, which justifies the low activity.
Figure 2.
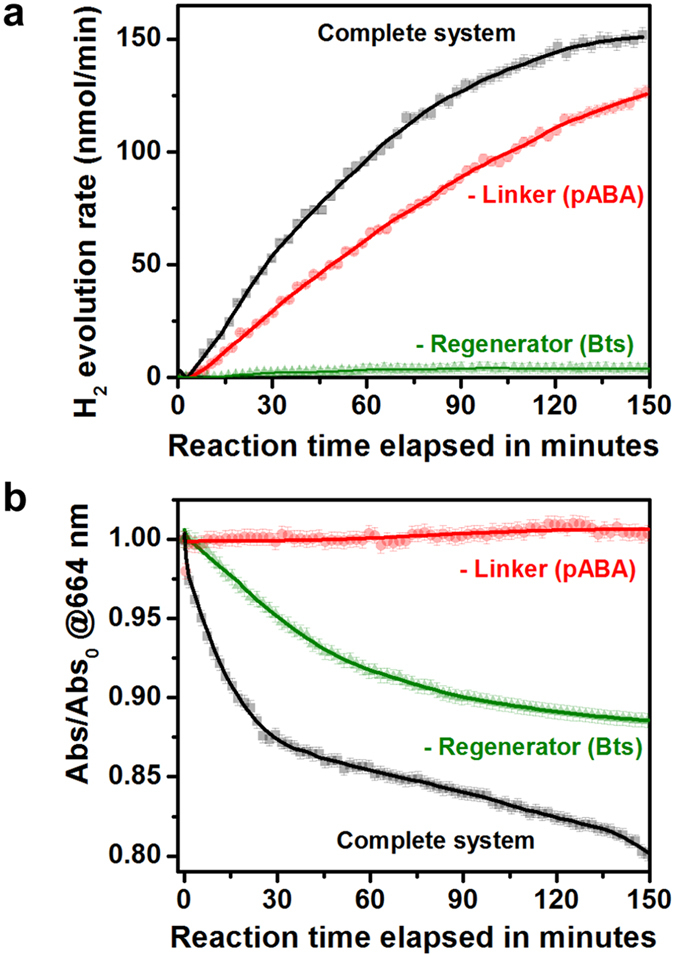
Effect of components in hydrogen production and methylene blue (MB+) oxidation. (a) Hydrogen production and (b) methylene blue (MB+) oxidation from the complete silver based nano-hybrid assembly (Bts-Ag NP–linker–TiO2–Ru NPs), without the regeneration layer (–Regenerator (Bts)) and without the molecular wire linker (–pABA), upon monochromatic excitation at 405 nm.
A noticeable rise in the multi-electron photo-oxidation of methylene blue was detected for the complete system, contrasting with the low activity of the unconnected system (Fig. 2b) in which electron transfer occurs via random collision between Bts-Ag NPs (electron donor) and TiO2:Ru NPs (electron acceptor). Individual components show no photocatalytic activity (control experiments are shown in Fig. S7). There is however evidences for methylene blue adsorption on the control experiment using only TiO2:Ru NPs, reflected in absorption signal drop and large oscillation in the date, which is to be expected due to TiO2 large surface area. Preliminary tests show that the complete system is stable for at least 5 h on stream and recyclable, since the second run of photocatalytic test show virtually no change in photoactivity. Raman and AFM measurements performed after the photocatalytic tests revealed no significant differences between fresh, used in the photocatalytic tests and aged for 3 weeks samples.
The solar-to-fuel efficiency (quantum yield) of the nano-hybrid system was measured directly, assuming the steady-state amount of H2 produced (150 nmol min−1) at a known photon flux. A quantum yield of 19.9 ± 0.5% was measured assuming that every photon absorbed has an equal probability of generating a hot electron and a reaction stoichiometry of two electrons for each H2 molecule formed. Currently, the best performing systems based on photovoltaic electrolysis reach 10–12% conversion efficiency. For example, Luo and coworkers31 reported recently 12.3% water photolysis efficiency via perovskite photovoltaics and earth-abundant catalysts. Jacobsson and collaborators32 described a monolithic water splitting device based on series interconnected thin film absorbers capable of attaining 10% solar-to-hydrogen efficiency. Kim and coworkers33 developed a dual photoanodes-silicon solar cell, reaching an unbiased water splitting efficiency of 7.7%. Finally, Nocera’s artificial leaf model without precious metals water-splitting catalysts can attain a 10% solar-to-fuels efficiency34. Apart of superseding the current efficiencies, the reported value was obtained without component ratio optimization, leaving large room for further improvement.
The mechanism of electron transport from donor to acceptor site by the molecular wire linker was investigated by performing ultrafast transient infrared absorption measurements. Infrared spectroscopy is a very sensitive technique to evaluate the arrival of electrons to TiO2 conduction band because photoinduced charge carriers in semiconductor conduction band have mid-IR absorption originated from the quasi-metallic state formed20. Figure 3 shows the kinetic trace extracted at 2081 cm−1 from transient absorption infrared spectroscopy measurements upon excitation at 405 nm for the Bts-Ag NP–linker–TiO2 (linked system) and Bts-Ag NP–TiO2 (unlinked system). Analysis of signal intensity shows that the presence of the organic linker increased by roughly 3-4-fold the transference of electrons from Ag NPs to the TiO2 conduction band comparatively to the unlinked system, which relies on collisional electron transfer. Kinetic fitting of the transient signal revealed a rising edge time component of ca. 600–700 fs that is significantly higher than the instrument response function (ca. 200 fs), assigned to the time necessary for electron transfer from Bts-Ag NPs to TiO2. The signal was fitted with two exponential decay functions with τ1 = 5 ps (93%) and τ2 = 80 ps (7%) (<τ> = 10 ps), which endorses ultrafast recombination dynamics and accordingly good connectivity between two sites. Note that roughly 30% of the initial signal is still present after 5 ns, our longest time delay line, suggesting electron stabilization and consequent increase in charge separation, necessary for photocatalysis to occur.
Figure 3.
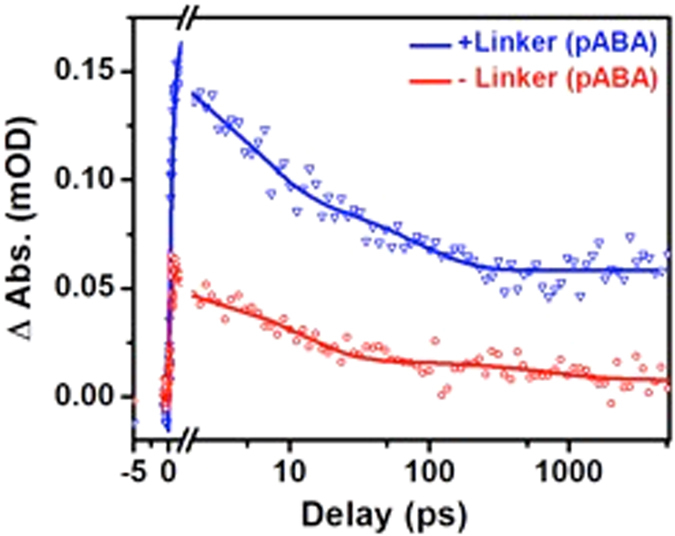
Effect of linking Ag NPs to TiO2 with a molecular linker to the ultrafast transient infrared absorption signal. Kinetic traces extracted at 2081 cm−1 from transient absorption infrared spectroscopy measurements upon excitation at 405 nm for the Bts-Ag NP–linker–TiO2 (linked system, + linker (pABA)) and Bts-Ag NP–TiO2 (unlinked system, – linker (pABA)).
The measurement was repeated during the self-assembling of the nano-hybrid system and control experiments were carried out using unlinked nanoparticles, i.e. in the absence of the molecular linker pABA. The intensity of the transient absorption infrared spectroscopy signal increases with time when in the presence of the molecular linker, but remains unchanged for the unlinked system (Fig. 4a,b). This is particularly noticeable in the inserts, where infrared intensity extracted upon completion of charge transfer (1.5 ps) is plotted over time. There is a clear increase in intensity until the process of linking donor and acceptor is completed, which takes up to 90 min after joining all the components. Again, there is virtually no signal change in the absence of pABA (Fig. 4b and insert).
Figure 4.
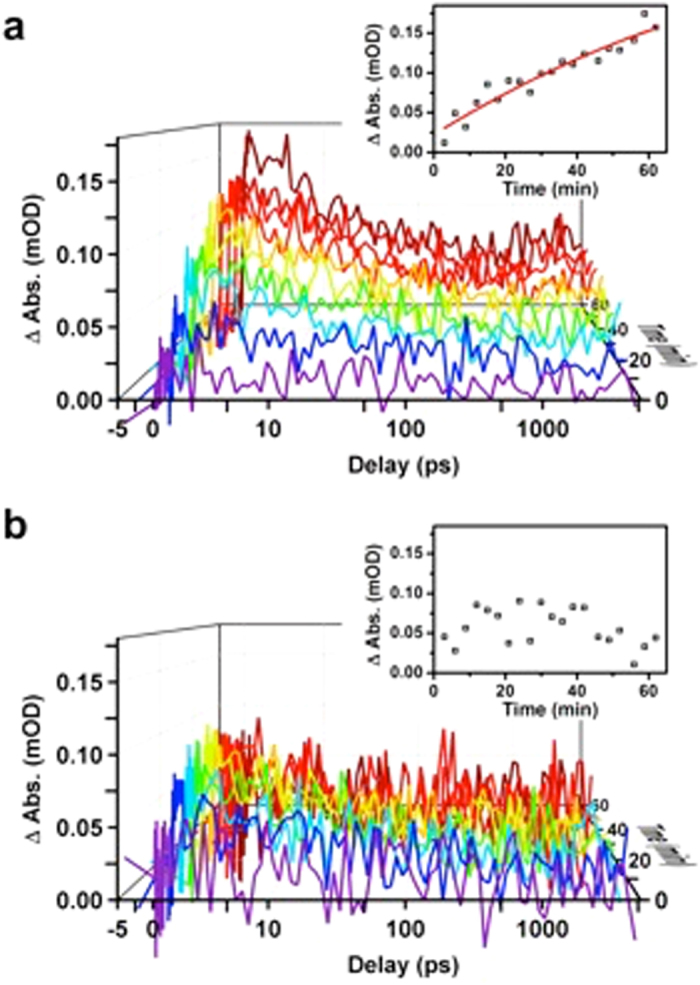
Evolution of ultrafast transient infrared absorption signal as Ag NPs connect to TiO2 with a molecular linker. Temporal evolution of the kinetic traces extracted at 2081 cm−1 from transient absorption infrared spectroscopy measurements upon excitation at 405 nm for the (a) Bts-Ag NP–linker–TiO2 (linked system), and (b) Bts-Ag NP–TiO2 (unlinked system). Inserts show the infrared absorption intensity over time extracted at 1.5 ps.
Ultrafast transient infrared absorption measurements show that the molecular linker improve the electronic coupling between electron donor and electron acceptor units of the nano-hybrid assembly. However, electrons must be trapped at the surface of TiO2 to be transferred to Ru NPs and conversely participate in a photocatalytic reaction. Ultrafast transient infrared absorption usually does not allow for distinction of bulk and surface-trapped electrons due to characteristic narrow spectral range of laboratory setups. However, surface-trapped electrons on TiO2 yield an absorption band at 795 nm35 that can be monitored by ultrafast transient absorption measurements (Fig. 5). Experiments were carried out either using the complete nano-hybrid system or in the absence of Ru NPs. In both cases, the signal has a rising edge time component of ca. 800 fs (without Ru NPs 800 fs, and complete system 820 fs) that was assigned to the time necessary for the electron transfer from Bts-Ag NPs to TiO2 and filling of its surface trap states. Since the rising edge component only differs by roughly 100–200 fs from the one extracted from transient infrared measurements, one can conclude that electrons fill the surface trap states rather quickly.
Figure 5.
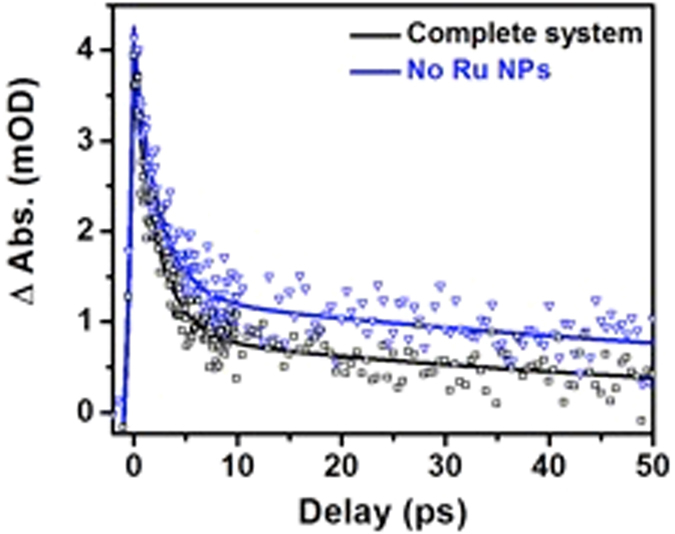
Effect of adding the catalyst to the ultrafast transient absorption signal. Kinetic traces extracted at 795 nm from transient absorption spectroscopy measurements upon excitation at 405 nm for the complete system and in the absence of Ru NPs (hydrog en evolution catalyst). The instrument response function is ca. 200 fs.
Conversely, good connectivity between the two sites should result in an intensification of the undesired recombination path, characterized by decays in the ultrafast domain. The signal was fitted with two exponential decay functions with τ1 = 2.6 ps (65%) and τ2 = 92 ps (35%) (<τ> = 34 ps), which endorses ultrafast recombination dynamics and accordingly good connectivity between two sites. By adding Ru NPs one expects a suppression of the recombination pathway as electrons are expected to transfer from TiO2 to the Ru 4d empty states, leading to an increase of the charge separation state lifetime. Transient absorption measurements showed faster decay when Ru NPs were present (τ1 = 2.6 ps (75%) and τ2 = 67 ps (25%), (<τ> = 19 ps)) upholding the suggestion that hot electrons are transferred to the Ru NPs after reaching TiO2 CB.
This observation contributes for the understanding of the increase in hydrogen evolution in the complete system, but leaves behind rationalization concerning the increase in methylene blue degradation comparatively to the unconnected system and even more so to the system prepared with Ag NPs with PVP termination. The most logical explanation for the rise in the photo-oxidation turnover is further increase in charge separation state lifetime, leading to the accumulation of reactive holes necessary for the multi-electron photo-oxidation process. The proposed hole stabilization mechanism is sensitive to Ag NPs termination, thus must involve the molecular structures on Ag NPs. A negative absorption signal was detected during the reaction with Bts-Ag NPs_pABA_TiO2:Ru NPs that is virtually absent for all the other materials tested (Fig. 6) and can be assigned to an emission resulting from a photophysical process occurring during the photocatalysis. The emission is centered at around 515 nm with a shoulder at 475 nm and is different from the emission of pure betalamic acid upon excitation at 405 nm (Fig. S8)36 but compatible with the photoluminesce profile of betanin submitted to alkaline hydrolysis (Fig. 6b). The lifetime of the emission emanated from the molecular structures on Ag NPs is 15.8 ps, estimated from time-resolved fluorescence (Fig. 6c). Poor light absorption of betanin at 405 nm and its low fluorescence quantum yield37 suggests that the direct excitation of surface betanin has little or no contribution to the emission observed during photocatalytic reaction. However, the molar attenuation coefficient of betalamic acid and neobetanin at 405 nm are higher than 20,000 L∙mol−1∙cm−1 and fluorescence of both compounds in solution is centered at around 500 nm38.
Figure 6.
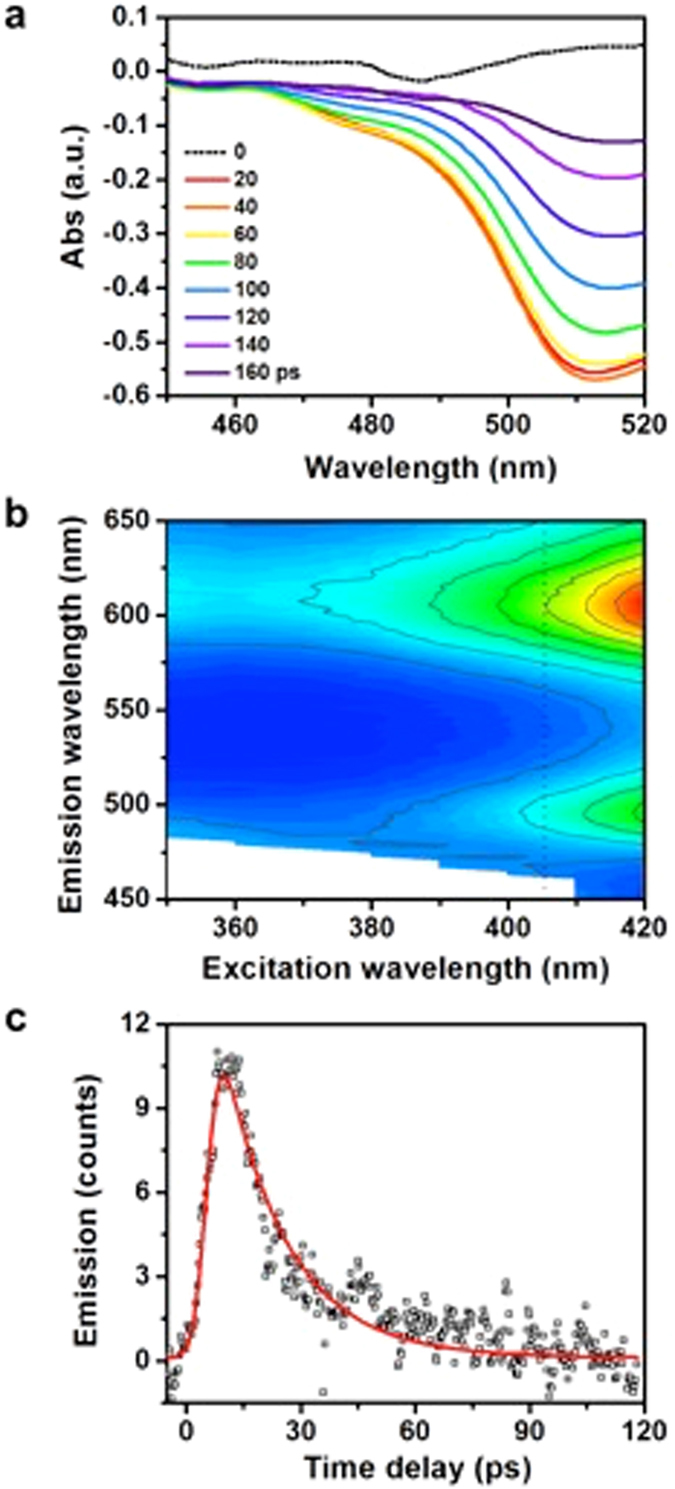
Photoluminescence signal emanated from Ag NPs regenerating layer. (a) Steady-state photoluminescence emanated from the complete system during photocatalysis. (b) Steady-state fluorescence profile of betanin in water (10 ppm AcOEt) at pH = 12. (c) Kinetic trace extracted at 515 nm with streak camera time-resolved fluorescence upon excitation at 390 nm of Bts-Ag NPs_pABA_TiO2:Ru NPs in aqueous solution.
One possible explanation for the stabilization of the hole and increase of electron-hole pair lifetime is that upon electron transfer from Ag NPs to TiO2, the Bts moieties transfer electrons to Ag NPs mediated by a proton release from Bts resulting in hole decay via a radiative reversible electron transfer, i.e. hole stabilization via proton-couple electron transfer, which is an essential process in chemical and biological catalysis, for example in water splitting, oxygen activation, proton reduction, nitrogen fixation, and ribonucleotide reductase reactions44–46. Another possibility is the concurrence between electron/hole recombination, electron transfer from Ag NPs to TiO2 and electron transfer from Ag NPs to the HOMO (highest occupied molecular orbital) of excited Bts. The latter process results in a stabilized anion-radical of Bts at the surface of the Ag NPs, increasing the persistence of holes. Next, electron transfer from Bts− to h+-Ag NP is coupled to the radiative deactivation of the resulting excited state of Bts (Fig. 7). In both scenarios, the resulting accumulation of reactive holes in silver increases methylene blue abatement.
Figure 7.

Charge particles pathways and reaction intermediates in the nano-hybrid assembly leading to a long-lived charge separated state. Electronic excitation is represented as dashed red arrows; this scheme does not necessarily represent the physical location where events take place. Energy levels were taken from the literature39–43.
In conclusion, a silver based nano-hybrid assembly was designed combining natural pigments and molecular wire linkers used in DSSC and LSP-based photoconversion devices. The dramatic increase in the charge separation state lifetime compared to conventionally synthesize silver systems allowed the simultaneous production of hydrogen and oxidative pollutant abatement under visible light irradiation. This bottom-up modular architecture can be tuned to fit desired applications by changing the identity of a small number basic units, as well as the ratio between them. The plasmonic structure, the linker, the semiconductor and the metal co-catalyst can be permuted and combined in almost infinite ways, in strong parallel to metallic organic frameworks.
Methods
Materials synthesis
All reagents were purchased from commercial vendors (Sigma Aldrich) and used as received. Ag NPs (nanoparticles) decorated with betalamic acid were synthesized via base hydrolysis of betanin extracted from beetroot in an automated microfluidic reactor with an optimization algorithm that ensures the particles size and light absorption is homogeneous25. AgNO3 was used a metal precursor and betanin works as reducting and capping agent. The Ag NPs decorated with PVP were prepared with the same procedure and reactor but using PVP and NaBH4 as capping and reducing agent, respectively.
Ruthenium NPs on TiO2 (TiO2_Ru, H2 evolution catalyst) was prepared by an impregnation-reduction method reported by Mishra et al.47. For the of Ru NPs on TiO2 with 3 nm nominal size, RuCl3· × H2O (0.02 M) was dissolved in dry ethanol (15 mL) in a round bottle flask with a mechanical stirrer and an argon inlet. The mixture was stirred at room temperature under argon for 24 h. Then, 0.2 M solution of NaBH4 in ethanol was added dropwise to the mixture under argon and continuously stirring. The solution turned dark grey within minutes but was left to stir for additional 24 h at room temperature to ensure complete reduction. Catalyst was recovered from solution after removing the solvent via centrifugation, and cleaned via a few cycles of washing/centrifugation with ethanol and water. The material was then dried at room temperature, yielding a grey powder. The TiO2 nanoparticles were supplied by Sachtleben, Hombikat UV 100 with a nominal size of 3 nm and anatase as crystal structure.
For the preparation of silver nano-hybrid architecture, Ag NPs decorated with betalamic acid or PVP (4.9 × 10−3 nM) were mixed with water solution of 4-aminobensoic acid (5 × 10−6 M) and stirred under argon for 15 min. TiO2_Ru suspension in water (0.15 mg) was added afterwards. The elemental units were combined using a 1:1:1 ratio, i.e., each Ag NPs is linked via a single organic linker molecule to TiO2_Ru.
Standard characterization
SEM (scanning electron microscopy), DLS (dynamic light scatterring) and UV-Vis spectroscopy were used to monitor the Ag NPs homogeneity, average size and optical properties. UV-Vis spectra were acquired on a setup equipped with a deuterium-tungsten-halogen light source (DH-2000-BAL, Ocean Optics) and a USB4000 spectrometer (Ocean Optics). The size a distribution index of the particles was determined using DLS Malvern NanoS. SEM images of Ag NPs were acquired on a Leo 1550 SEM (Zeiss, Germany) instrument. The samples were supported on a silicon substrate and mounted in an aluminum stub. Prior to the measurements, samples were sputtered for 40 s with Au/Pd using a plasma current of 30 mA to minimize charging. Non-resonant Raman spectra were collected in a RENISHAW Raman microscope with excitation at 532 nm, objective of 20×, laser power at 100%, exposure time 10 s and five acquisitions. Liquid samples were mounted directly into a microscope glass slide.
Photocatalytic studies
Ag NPs decorated with betalamic acid was prepared as described above. In the case of sample without linker, the set involving adding 4-aminobenzoic acid was replaced by simply adding an equivalent amount of pure water. Finally, methylene blue (1.3 × 10−5 M) and 1 drop of trifluoracetic acid were added to the reaction mixture. A schematic representation of the photocatalytic reactor is presented in Fig. S1. A 405 nm CW laser was used to excite selectively the Ag NPs LSP and the gaseous products were extracted via a carrier gas (argon) and analyzed in flow mode with a QMS (quadrupole mass-spectrometer), while methylene blue depletion and emission from the sample was monitored by UV-Vis spectroscopy in batch mode.
Transient infrared absorption spectroscopy
Experiments were carried out in a femtosecond transient absorption spectrometer (Helios IR, Ultrafast Systems LLC) at room temperature. A one-box Ti:Sapphire based amplifier with integrated oscillator and pump lasers (800 nm, 40 fs, 3 kHz, Libra LHE, Coherent Inc.) was used to pump two TOPAS Primes coupled with frequency mixers (Light Conversion Ltd) to produce the depolarized visible excitation pulses ranging (405 nm) and the broad mid-IR probe spectrum. The samples were compacted between two CaF2 windows. Experiments were performed with humid samples, conferring conditions similar to what occurs during photocatalytic reaction. This approach also ensures good signal-to-noise ratio for pump-probe infrared measurements in transmission.
Transient absorption spectroscopy
Experiments were carried out on laser based spectroscopy, with laser powers equating to less than one photon absorption per particle. Samples for transient absorption experiments were measured as suspensions in a liquid cell with quartz windows (1 mm). A Coherent Legend Ti:Sapphire amplifier (800 nm, 100 fs pulse length, 1 kHz repetition rate) was used. The output is split to pump and probe beams. Excitation pulses at the wavelength of 405 nm were acquired using an optical parametric amplifier (Topas C, Light Conversion). The probe pulses (a broad supercontinuum spectrum) were generated from the 800 nm pulses in a CaF2 crystal and split by a beam splitter into a probe pulse and a reference pulse. The probe pulse and the reference pulse were dispersed in a spectrograph and detected by a diode array. Instrumental response time is ~100 fs. Polarization of the pump was set at magic angle, 54.7° relative to the probe beam. The kinetic traces were extracted at 795 nm.
Electronic supplementary material
Acknowledgements
The authors would like to thank Uppsala University, ÅForsk (grant no. 15–455), STINT (grant no. IB2015-6474), São Paulo Research Foundation – FAPESP (grant 2014/14866-2) and Swedish Research Council (grant no. 2015-03764) for the financial support.
Author Contributions
J.S. proposed the concept and directed the research. M.V.P. and M.A. designed the experiments. M.V.P., A.B.F., M.A., D.L.A.F., C.O.M., I.R., Y.H. and C.P. carried out the experiments. M.V.P. and E.L.B. prepared the figures. J.S. and E.L.B. wrote the main manuscript text. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Mariia V. Pavliuk, Arthur B. Fernandes and Mohamed Abdellah contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-09261-7
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Erick L. Bastos, Email: elbastos@iq.usp.br
Jacinto Sá, Email: jacinto.sa@kemi.uu.se.
References
- 1.Gust D, et al. Solar fuels via artificial photosynthesis. Acc. Chem. Res. 2009;42:19890–1898. doi: 10.1021/ar900209b. [DOI] [PubMed] [Google Scholar]
- 2.Tachibana Y, et al. Artificial photosynthesis for solar water-splitting. Nat. Photonics. 2012;6:511–518. doi: 10.1038/nphoton.2012.175. [DOI] [Google Scholar]
- 3.Fujishima A, Honda K. Electrochemical photolysis of water at a semiconductor electrode. Nature. 1972;238:37–38. doi: 10.1038/238037a0. [DOI] [PubMed] [Google Scholar]
- 4.Reece SY, et al. Wireless solar water splitting using silicon-based semiconductors and earth-abundant catalysts. Science. 2011;334:645–648. doi: 10.1126/science.1209816. [DOI] [PubMed] [Google Scholar]
- 5.Rausch B, et al. Decoupled catalytic hydrogen evolution from a molecular metal oxide redox mediator in water splitting. Science. 2014;345:1326–1330. doi: 10.1126/science.1257443. [DOI] [PubMed] [Google Scholar]
- 6.Wu B, et al. Anisotropic growth of TiO2 onto gold nanorods for plasmon-enhanced hydrogen production from water reduction. J. Am. Chem. Soc. 2016;138:1114–1117. doi: 10.1021/jacs.5b11341. [DOI] [PubMed] [Google Scholar]
- 7.Lewis NS, Nocera DG. Powering the planet: chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA. 2006;103:15729–15735. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Goff A, et al. From hydrogenases to noble metal-free catalytic nanomaterials for H2 production and uptake. Science. 2009;326:1384–1387. doi: 10.1126/science.1179773. [DOI] [PubMed] [Google Scholar]
- 9.Lyons TW, et al. The rise of oxygen in Earth’s early ocean and atmosphere. Nature. 2014;506:307–315. doi: 10.1038/nature13068. [DOI] [PubMed] [Google Scholar]
- 10.Schwarz G, et al. Molybdenum cofactors, enzymes and pathways. Nature. 2009;460:839–847. doi: 10.1038/nature08302. [DOI] [PubMed] [Google Scholar]
- 11.Asahi R, et al. Visible-light photocatalysis in nitrogen-doped titanium oxides. Science. 2001;293:269–271. doi: 10.1126/science.1061051. [DOI] [PubMed] [Google Scholar]
- 12.O’Regan B, Grätzel M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 film. Nature. 1991;353:737–740. doi: 10.1038/353737a0. [DOI] [Google Scholar]
- 13.Brown MD, et al. Plasmonic dye-sensitized solar cells using core−shell metal−insulator nanoparticles. Nano Lett. 2011;11:438–445. doi: 10.1021/nl1031106. [DOI] [PubMed] [Google Scholar]
- 14.Tian Y, Tatsuma T. Mechanisms and applications of plasmon-induced charge separation at TiO2 films loaded with gold nanoparticles. J. Am. Chem. Soc. 2005;127:7632–7637. doi: 10.1021/ja042192u. [DOI] [PubMed] [Google Scholar]
- 15.Christopher P, et al. Visible-light-enhanced catalytic oxidation reactions on plasmonic silver nanostructures. Nat. Chem. 2011;3:467–472. doi: 10.1038/nchem.1032. [DOI] [PubMed] [Google Scholar]
- 16.Linic S, et al. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 2011;10:911–921. doi: 10.1038/nmat3151. [DOI] [PubMed] [Google Scholar]
- 17.Hallett-Tapley GL, et al. Plasmon-mediated catalytic oxidation of sec-phenethyl and benzyl alcohols. J. Phys. Chem. C. 2011;115:10784–10790. doi: 10.1021/jp202769a. [DOI] [Google Scholar]
- 18.Brongersma ML, et al. Plasmon-induced hot carrier science and technology. Nat. Nanotechnol. 2015;10:25–34. doi: 10.1038/nnano.2014.311. [DOI] [PubMed] [Google Scholar]
- 19.Sá J, et al. Direct observation of charge separation on Au localized surface plasmons. Energy Environ. Sci. 2013;6:3584–3588. doi: 10.1039/c3ee42731e. [DOI] [Google Scholar]
- 20.Antila LA, et al. Hunting for the elusive shallow traps in TiO2 anatase. Chem. Commun. 2015;51:10914–10916. doi: 10.1039/C5CC02876K. [DOI] [PubMed] [Google Scholar]
- 21.Hammarström L. Accumulative charge separation for solar fuels production: coupling light-induced single electron transfer to multielectron catalysis. Acc. Chem. Res. 2015;48:840–850. doi: 10.1021/ar500386x. [DOI] [PubMed] [Google Scholar]
- 22.Sá J, et al. Photoformed electron transfer from TiO2 to metal clusters. Catal. Commun. 2008;9:1991–1995. doi: 10.1016/j.catcom.2008.03.041. [DOI] [Google Scholar]
- 23.Mubeen S, et al. An autonomous photosynthetic device in which all charge carriers derive from surface plasmon. Nat. Nanotechnol. 2013;8:247–251. doi: 10.1038/nnano.2013.18. [DOI] [PubMed] [Google Scholar]
- 24.Gonçalves LCP, et al. A comparative study of the purification of betanin. Food Chem. 2012;131:231–238. doi: 10.1016/j.foodchem.2011.08.067. [DOI] [Google Scholar]
- 25.Fernandes DLA, et al. Green microfluidic synthesis of monodisperse silver nanoparticles via genetic algorithm optimization. RSC Adv. 2016;6:95693–95697. doi: 10.1039/C6RA20877K. [DOI] [Google Scholar]
- 26.Yamada Y, et al. Photocatalytic hydrogen evolution under highly basic conditions by using Ru nanoparticles and 2-phenyl-4-(1-naphthyl)quinolinium ion. J. Am. Chem. Soc. 2011;133:16136–16145. doi: 10.1021/ja206079e. [DOI] [PubMed] [Google Scholar]
- 27.Caroll RL, Gorman CB. The genesis of molecular electronics. Angew. Chem. Int. Ed. 2002;41:4378–4400. doi: 10.1002/1521-3773(20021202)41:23<4378::AID-ANIE4378>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 28.Sukegawa J, et al. Electron transfer through rigid organic molecular wires enhanced by electronic and electron-vibration coupling. Nat. Chem. 2014;6:899–905. doi: 10.1038/nchem.2026. [DOI] [PubMed] [Google Scholar]
- 29.Coates, J. ‘Interpretation of infrared spectra, a practical approach’, Encyclopedia of Analytical Chemistry, R. A. Meyers (Ed.), John Wiley & Sons Ltd.
- 30.Fernandes DLA, et al. Novel photo-reactor for fast screening of photo-catalytic systems. J. Photochem. Photobiol. A. 2017;335:36–39. doi: 10.1016/j.jphotochem.2016.11.015. [DOI] [Google Scholar]
- 31.Jingshan L, et al. Water photolysis at 12.3% efficiency via perovskite photovoltaics and Earth-abundant catalysts. Science. 2014;345:1593–1596. doi: 10.1126/science.1258307. [DOI] [PubMed] [Google Scholar]
- 32.Jacobsson TJ, et al. A monolithic device for solar water splitting based on series interconnected thin film absorbers reaching over 10% solar-to-hydrogen efficiency. Energy Environ. Sci. 2013;6:3676–3683. doi: 10.1039/c3ee42519c. [DOI] [Google Scholar]
- 33.Kim JH, et al. Hetero-type dial photoanodes unbiased solar water splitting with extended light harvesting. Nat. Commun. 2016;7 doi: 10.1038/ncomms13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cox CR, et al. Ten-percent solar-to-fuel conversion with nonprecious materials. Proc. Natl. Acad. Sci. 2014;111:14057–14061. doi: 10.1073/pnas.1414290111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takami Y, et al. Dynamics of efficient electron–hole separation in TiO2 nanoparticles revealed by femtosecond transient absorption spectroscopy under the weak-excitation condition. Phys. Chem. Chem. Phys. 2007;9:1453–1460. doi: 10.1039/B617552J. [DOI] [PubMed] [Google Scholar]
- 36.Cabanes J, et al. Fluorescent bioinspired protein labeling with betalamic acid. Derivatization and characterization of novel protein-betaxanthins. Dyes & Pigments. 2016;133:458–466. doi: 10.1016/j.dyepig.2016.06.037. [DOI] [Google Scholar]
- 37.Bartoloni FH, et al. Photophysics and hydrolytic stability of betalains in aqueous trifluoroethanol. Monatsh. Chem. 2013;144:567–571. doi: 10.1007/s00706-012-0883-5. [DOI] [Google Scholar]
- 38.Wendel M, et al. Ultrafast internal conversion in neobetanin in comparison to betacyanins. J. Photochem. Photobiol. A Chem. 2017;332:602–610. doi: 10.1016/j.jphotochem.2016.09.027. [DOI] [Google Scholar]
- 39.Lim A, et al. Co-dominant effect of selected natural dye sensitizers in DSSC performance. Spectrochim. Acta A. 2016;167:26–31. doi: 10.1016/j.saa.2016.05.024. [DOI] [PubMed] [Google Scholar]
- 40.Oprea CI, et al. A combined experimental and theoretical study of natural betalain pigments used in dye-sensitized solar cells. J. Photochem. Photobiol. A Chem. 2012;240:5–13. doi: 10.1016/j.jphotochem.2012.05.003. [DOI] [Google Scholar]
- 41.Lee JS, et al. Highly photoactive, low bandgap TiO2 nanoparticles wrapped by grapheme. Adv. Mater. 2012;24:1084–1088. doi: 10.1002/adma.201104110. [DOI] [PubMed] [Google Scholar]
- 42.Clavero C. Plasmon-induced hot-electron generation at nanoparticle/metal-oxide interfaces for photovoltaic and photocatalytic devices. Nat. Photonics. 2014;8:95–103. doi: 10.1038/nphoton.2013.238. [DOI] [Google Scholar]
- 43.Thomas AG, et al. Adsorption studies of p-aminobenzoic acid on the anatase TiO2 (101) surface. Langmuir. 2014;30:12306–12314. doi: 10.1021/la5032619. [DOI] [PubMed] [Google Scholar]
- 44.Hoganson CW, Babcock GT. A metalloradical mechanism for the generation of oxygen from water in photosynthesis. Science. 1997;277:1953–1956. doi: 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
- 45.Babcock GT, Wikström M. Oxygen activation and the conservation of energy in cell respiration. Nature. 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 46.Peters JW, et al. X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science. 1998;282:1853–1858. doi: 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]
- 47.Mishra DK, et al. Ruthenium nanoparticles supported on zeolite Y as an efficient catalyst for selective hydrogenation of xylose to xylitol. J. Molec. Catal. A: Chem. 2013;376:63–70. doi: 10.1016/j.molcata.2013.04.011. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.