

Abstract
Somatic embryogenesis (SE) is a critical transition from vegetative to embryogenic growth in higher plants; however, few studies have investigated the mechanism that regulates SE initial differentiation. Most cotton varieties have not undergone regeneration by SE, so only a few varieties can be used in genetic engineering. Here, two varieties of cotton with different SE capabilities (HD, higher differentiation and LD, lower differentiation) were analyzed by high throughout RNA-Seq at the pre-induction stage (0h) and two induction stages (3h and 3d) under callus-induction medium (CIM). About 1150 million clean reads were obtained from 98.21% raw data. Transcriptomic analysis revealed that “protein kinase activity” and “oxidoreductase activity” were highly represented GO terms during the same and different treatment stages among HD and LD. Moreover, several stress-related transcription factors might play important roles in SE initiation. The SE-related regulation genes (SERKs) showed different expression patterns between HD and LD. Furthermore, the complex auxin and ethylene signaling pathway contributes to initiation of differentiation in SE. Thus, our RNA-sequencing of comparative transcriptome analysis will lay a foundation for future studies to better define early somatic formation in cotton with different SE capabilities.
Introduction
Somatic embryogenesis (SE) resulting in regeneration of new plants is an important step in the Agrobacterium-mediated method. This process includes two stages. First, the cell reverts back to its own lineage to re-enter the cell cycle and transform into a dedifferentiated cell state. Callus formation is frequently considered the manifestation of the dedifferentiated cellular state. Second, calli can regenerate somatic embryos and new plants by redifferentiation1, 2. Dedifferentiation is frequently considered the manifestation of the stem cell-like state to switch fate preceding the commitment for proliferation3. Following that, dedifferentiation is an important biological phenomenon in whole SE process.
Various molecular biological technologies have been described to investigate molecular mechanisms during SE in different plant species4, including carrot5, Arabidopsis 2, soybean6 and potato7. These studies have revealed valuable information regarding the physiological and biochemical changes that occur during SE8. In addition, many studies have investigated the initiation of SE in various plant species. Plant hormones and stress are the two most important factors involved in stimulation of SE initiation9. However, the regulatory mechanism of vegetative-to-embryogenic triggered remains elusive10. For example, numerous candidate genes associated with hormonal regulation, oxidative stress and response to stress were shown to be specifically activated or exhibit differential expression11, 12.
Cotton is a major raw material used by the textile industry and a source of oil. Transgenic technologies have been widely applied to cotton molecular breeding through Agrobacterium-mediated transformation via SE. However, few varieties have been successfully used in genetic engineering and regeneration in vitro 13. The majority of the desirable cotton cultivars are incapable of regeneration via SE14, and many factors can effect SE, including culture conditions and tissue background, furthermore, the key factor is a genotype-dependent response15. Several recent studies have been conducted to investigate the molecular mechanism and identify genes critical for SE16. More recently, RNA deep-sequencing technology has provided a platform for analysis of differences in gene expression17. There are reports demonstrating that hormonal signaling pathways such as auxin and cytokinin16, as well as stress-responsive pathways are critical during cotton SE development18. However, the initial SE molecular mechanisms have not been thoroughly studied and are still unclear in different dedifferentiated cotton cultivars. Here, we investigated early events of SE during the without induction stage (0h) and induction stage (3h and 3 d) by comparing cotton of different SE capability (HD and LD) using RNA-seq profiling combined with differentially expressed genes (DEG) and GO analysis. This study provides a comprehensive analysis of gene expression of different cotton types during the initial stages of somatic embryogenesis. Furthermore, the data presented herein provides an abundant gene catalog that should be a useful resource for future studies of molecular and developmental mechanisms of SE.
Results
Differentiation efficiency of various cotton species
Four Gossypium hirsutum L cultivars (YZ1, R15, X33 and X42) were sampled at three stages, the pre-induction stage (0h) and two induction stages (3h and 3d) in the CIM. YZ1 and R15 (HD) were selected because they exhibited good SE potential and were the main transgenic materials. Although X33 and X42 (LD) are the major commercial cultivars in Xinjiang, China, they have a low rate of differentiation during SE relative to YZ1 and R15. The high differentiation lines and the recalcitrant lines showed diverse morphology during the callus induction stage. The early calli of HD were friable, loose, and primarily whitish (Fig. 1a–d and i–l), while those of LD were firmer and less friable than HD (Fig. 1e–g and m–p). The significant differences in calli may be due to differences in gene expression during SE initiation; therefore, cDNA libraries were constructed from three time stages between HD and LD and two biological replicates were performed.
Figure 1.

Four-cotton cultivars showing different differentiation at 10d and 1 month in the CIM. (a–g) after 10d of culture in CIM. (i–o) after 1M of culture in CIM. Four cotton cultivars of different induction stages were sampled, YZ1 (a,b,i and j), R15 (c,d,k and l), X33 (e,f,m and n) and X42 (g,h,o and p).
Illumina HiSeq mRNA sequencing
High-throughput RNA-Seq sequencing generated 1171 million raw reads from 24 RNA samples using the Illumina HiSeqTM 2000 platform, approximately 48 million reads for each line. After discarding adapters, poly-N and low quality reads from raw data, a total of 1150 million clean reads were obtained from the total raw reads (98.26%). Two biological replicates were evaluated for each line, and the results indicated good agreement of the RNA-seq data (0.958 < R2 < 0.986). Overall, 86.31%–88.94% of the high-quality reads were mapped to the Gossypium hirsutum reference genome using TopHat29. Of the mapped reads, 84.3–89.4% were distributed in exon regions, while 1.7–2.4% were located in introns and 8.0–13.2% were in intergenic regions (Table 1). For all sequence data, the average Q20, Q30, and GC contents were 97.03%, 92.64%, and 43.82%, respectively. The error rate of all samples was 0.01%–0.02% (Table. S1). Of which three categories of mapped data: 1) multiple mapped (7.63–9.11%) and unique mapped reads (77.75–81.06%), 2) forward mapped (38.83–40.53%) and reverse mapped reads (38.88–40.54%), and 3) non-splice reads (48.35–53.15%) and splice reads (26.87–29.64%) (Table S1).
Table 1.
Summary of transcriptome sequencing data.
| Sample name | Raw reads | Clean reads | Clean bases | Total mapped | Exon (%) | Intron (%) | Intergenic (%) |
|---|---|---|---|---|---|---|---|
| R15_0h_1 | 45813740 | 44974414 | 6.75 G | 38815533 | 84.3 | 2.4 | 13.2 |
| R15_0h_2 | 44436266 | 43634922 | 6.55 G | 38509898 | 85.7 | 2.2 | 12 |
| R15_3h_1 | 46339512 | 45602268 | 6.84 G | 40352472 | 87.6 | 1.8 | 10.6 |
| R15_3h_2 | 55664292 | 54795000 | 8.22 G | 48396413 | 88.2 | 1.8 | 10 |
| R15_3d_1 | 58297408 | 57185824 | 8.58 G | 49594358 | 87.2 | 1.9 | 10.9 |
| R15_3d_2 | 45504132 | 44718654 | 6.71 G | 39092086 | 86.4 | 1.8 | 11.8 |
| YZ1_0h_1 | 54411212 | 53434048 | 8.02 G | 46862756 | 87 | 2 | 11 |
| YZ1_0h_2 | 42332338 | 41472822 | 6.22 G | 36244862 | 87.2 | 2 | 10.8 |
| YZ1_3h_1 | 48182224 | 47414878 | 7.11 G | 41720626 | 88.2 | 1.8 | 10 |
| YZ1_3h_2 | 51910688 | 51103964 | 7.67 G | 45108556 | 89.4 | 1.7 | 8 |
| YZ1_3d_1 | 41334482 | 40551474 | 6.08 G | 35085465 | 88 | 1.8 | 10,2 |
| YZ1_3d_2 | 53111840 | 51931008 | 7.79 G | 45087397 | 87.9 | 1.7 | 10.3 |
| X33_0h_1 | 55528574 | 54606234 | 8.19 G | 48091726 | 85.1 | 2.3 | 12.5 |
| X33_0h_2 | 48595188 | 47799278 | 7.17 G | 42052352 | 86.5 | 2 | 11.5 |
| X33_3h_1 | 51455830 | 50559176 | 7.58 G | 44749580 | 87.9 | 1.8 | 10.2 |
| X33_3h_2 | 47675942 | 46858792 | 7.03 G | 41408584 | 87.8 | 1.7 | 10.5 |
| X33_3d_1 | 44247472 | 43462830 | 6.52 G | 38118346 | 87.7 | 1.7 | 10.6 |
| X33_3d_2 | 45522790 | 44759502 | 6.71 G | 39403962 | 86.8 | 1.8 | 11.3 |
| X42_0h_1 | 40809328 | 40079772 | 6.01 G | 35489334 | 86.9 | 1.9 | 11.2 |
| X42_0h_2 | 49641394 | 48808912 | 7.32 G | 43159067 | 86.6 | 2.1 | 11.3 |
| X42_3h_1 | 49429384 | 48648396 | 7.3 G | 43221078 | 88.5 | 1.9 | 9.7 |
| X42_3h_2 | 45973326 | 45219188 | 6.78 G | 40215890 | 87.3 | 2 | 10.7 |
| X42_3d_1 | 55203894 | 54213944 | 8.13 G | 47493700 | 86.3 | 2 | 11.7 |
| X42_3d_2 | 49845162 | 49014372 | 7.35 G | 43117216 | 87 | 1.9 | 11.1 |
DEGs and GO terms during same-stage comparisons
Significant differences in gene expression were examined in three same-stage comparisons of the two difference species (HD and LD). Using a P-value < 0.05 and |log2FoldChange| > 2 in one sampling point as the higher criteria. Our date shows a heatmap of DEGs and Venn diagrams showing details regarding the number of DEGs at each pairwise comparison (Fig. 2). The results revealed 140 DEGs at 0h (Fig. 2b), 143 DEGs at 3h (Fig. 2c) and 123 DEGs at 3d (Fig. 2d). Analysis of these DEGs revealed that protein kinase-related genes were differentially expressed. The 0h comparison led to identification of one G-type lectin S-receptor-like Serine/Threonine-kinase and three putative homologs of the protein kinase superfamily protein. Additionally, two homologs of the G-type lectin S-receptor-like Serine/Threonine-kinase, two protein kinase superfamily proteins and one MAP kinase kinase 2 were involved at 3h. In addition, the G-type lectin S-receptor-like Serine/Threonine-kinase, protein kinase superfamily protein and MAP kinase kinase 2 were significantly down-regulated at 3d (Table 2).
Figure 2.
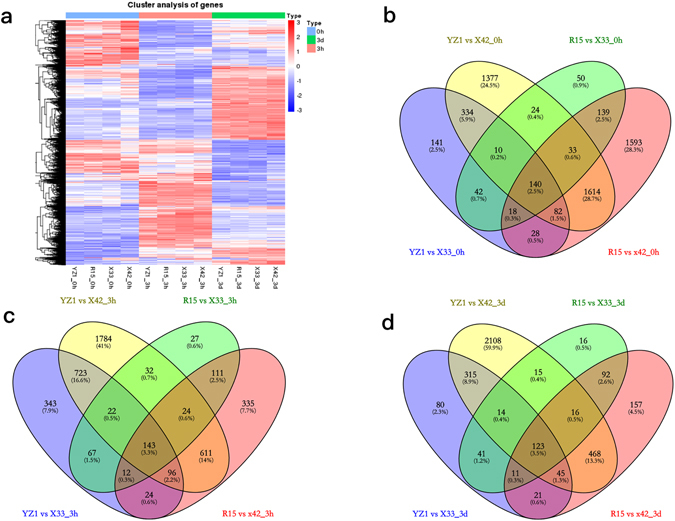
DEGs in three same-stage comparisons between HD and LD. (a) Heatmap of DEGs (P-value < 0.05, |log2FoldChange| > 2 at one sampling point); (b–d) Venn analysis of the DEGs in two types of cotton during three time stages.
Table 2.
Protein kinase-related genes differentially expressed in the same-stage.
| Gene id | log2 Fold Change | Gene description | ||||
|---|---|---|---|---|---|---|
| R15 vs X33 | R15 vs X42 | YZ1 vs X33 | YZ1 vs X42 | |||
| 0h | Gh_A06G1786 | −3.6825 | −3.2886 | −3.9907 | −3.5972 | G-type lectin S-receptor-like Serine/Threonine-kinase |
| Gh_A03G1310 | 2.0274 | 1.1129 | 1.989 | 1.0746 | Protein kinase superfamily protein | |
| Gh_A06G1617 | −3.2889 | −2.0637 | −2.715 | −1.489 | ||
| Gh_A11G2003 | 1.3657 | 1.7908 | 1.5901 | 2.0144 | ||
| Gh_A02G1701 | 1.2525 | 1.3822 | 1.3948 | 1.5242 | ||
| 3h | Gh_A06G1786 | −2.6061 | −2.6257 | −3.5518 | −3.5623 | G-type lectin S-receptor-like Serine/Threonine-kinase |
| Gh_D10G0162 | 2.2204 | 2.5952 | 2.4067 | 2.7904 | ||
| Gh_A06G1617 | −5.1736 | −3.7709 | −4.0353 | −2.6227 | Protein kinase superfamily protein | |
| Gh_A11G2002 | −2.3912 | −2.7061 | −1.3735 | −1.6803 | ||
| Gh_D07G2384 | −2.0518 | −2.1876 | −1.6726 | −1.7997 | MAP kinase kinase 2 | |
| 3d | Gh_A06G1786 | −2.8407 | −3.143 | −2.859 | −3.1611 | G-type lectin S-receptor-like Serine/Threonine-kinase |
| Gh_A06G1617 | −2.5474 | −1.322 | −2.6999 | −1.4741 | Protein kinase superfamily protein | |
| Gh_D07G2384 | −2.7462 | −2.4769 | −1.8776 | −1.6079 | MAP kinase kinase 2 | |
Comparison of DEGs and GO terms at different-stages
DEGs and three main GO terms were identified by comparisons of two induction stages (3h and 3d) and the pre-induction stage (0h). Comparison of 3h vs 0h revealed 7175 and 10,718 DEGs in the HD and LD cultivars, respectively, and more than half (50.8%) of the DEGs were present in all cotton cultivars at 3h vs 0h (Fig. 3a and b). Upon GO classification based on biological processes, “oxidation-reduction process”, “protein phosphorylation” and “regulation of transcription, DNA-dependent” were found to be associated with a higher number of DEGs in HD and LD (Fig. 3d). The six top molecular functions were “protein binding”, “ATP binding”, “protein kinase activity”, “protein tyrosine kinase activity”, “protein serine/threonine kinase activity” and “oxidoreductase activity” (Fig. 3d). Among cellular components, “membrane”, “integral component of membrane” and “nucleus” were highly represented in both cotton types (Fig. 3d).
Figure 3.
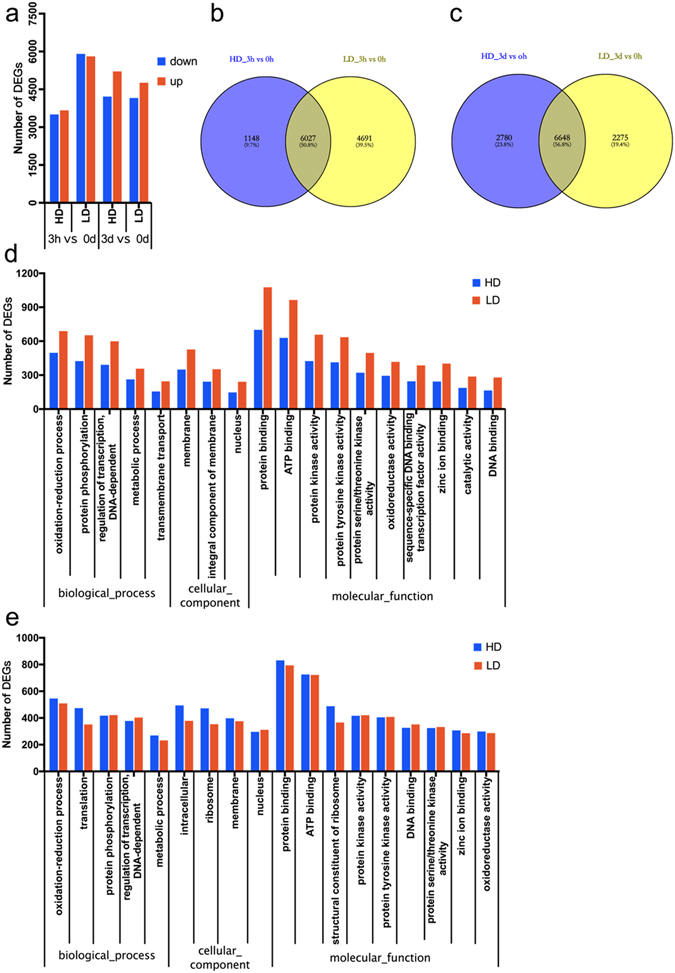
DEGs in four cotton cultivars during different-stage comparisons. (a) Number of DEGs at 3h vs 0h and 3d vs 0h; (b,c) Venn analysis of the DEGs in two types of cotton during different comparisons. (d,e) Go function analysis of DEGs at 3h vs 0h and 3d vs 0h, respectively.
Comparison of the 3d vs 0h samples revealed 9428 DEGs in the HD species and 8923 DEGs in the LD cultivars (Fig. 3a and c). “Oxidation-reduction process”, “translation” and “protein phosphorylation” were the main GO terms identified among differentially expressed genes corresponding to biological processes. Among molecular functions, the highly represented GO terms were “protein binding”, “ATP binding”, “structural constituent of ribosome”, “protein kinase activity”, “protein tyrosine kinase activity” and “DNA binding”. “Intracellular”, “ribosome” and “membrane” were grouped into cellular components in HD and LD (Fig. 3e).
The number of DEGs identified in LD was higher than in HD at 3h vs 0h, while the number of DEG detected at 3d vs 0h showed the opposite pattern. With the exception of the comparisons of LD at 3h vs 0h, the number of up-regulated DEGs was slightly higher than that of down-regulated DEGs in both comparisons (Fig. 3a). The cellular component was the most overrepresented GO term in different-stage comparisons between HD and LD.
Analysis of TF during SE initiation
Transcription factors (TFs) recognize DNA in a sequence-specific manner to regulate gene expression. Based on the |log2FoldChange| > 2 at one sampling point, a total of 1590 specifically different transcription factors were annotated in 69 TF families between two cultivars. Most TFs were involved in response to pathogen challenge, including AP2-EREBP (211), MYB (155), bHLH (128), WRKY (91), NAC (89), HB (73), C2H2 (72), GRAS (47), bZIP (43), AUX/IAA (42), C2C2-Dof (38), CH3 (33) and CCAAT (24) (Fig. 4a). Putative homologs of SE differentiation related genes were identified in two cotton cultivars (Fig. 4b–d). UPBEAT1 (UPB1), belongs to bHLH TF family, three UPB1 homologues were same expression model and were up-regulated during at 3h, while then the expression level reduced to very low (FPKM < 1) at 3d. However, another two bHLH transcription factors, MYC1 and GL3, showed higher expression levels at 3d. The expression of MYC1 homologues increased with induction. GL3 decreased at 3h, then increased at 3d. The MYB transcription factors CPC and ETC1 showed different expression patterns during three time stages. GAI is a subfamily of the GRAS family, the value of FPKM was decreased with induction. bZIP transcription factors, bZIP1, bZIP44 and acid responsive elements-binding factor, ABF2 and ABF3, most of those TFs showed higher FPKM at the 3h, while bZIP44 (Gh_A06G1847) was generally opposite expression pattern.
Figure 4.
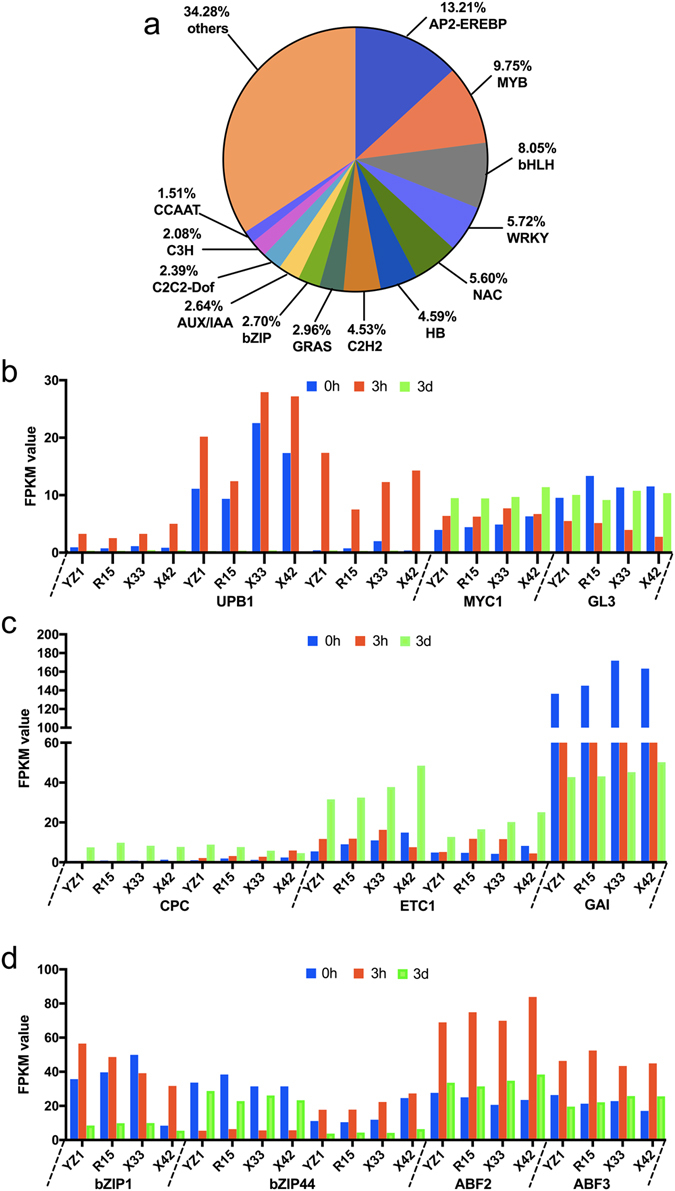
(a) TF genes and classification of TF families. TF genes were classified into TF families using iTAK. A total of 1590 different transcription factors were annotated in 69 TF families. Numbers represent the percentage of TF genes. (b–d) Detailed expression profiles of SE differentiation related TF genes. The x-axis indicates the distribution of SE differentiation related TF genes in the four cotton species, and the y-axis represents the value of FPKM in each TF gene.
Analysis of SE-related functional genes
A number of embryogenic-regulating genes that were differentially expressed during early SE (|log2FoldChange| > 2 at one sampling point) were selectively analyzed. The expression patterns of SE-related genes are shown in Table 3 and Fig. 5. WUSCHEL-related homeobox genes were involved, including WOX5 (1) and WOX11 (2). Overall, WOX homeobox genes were significantly up-regulated in the two cotton cultivars at 3d vs 0h, while there was no significant difference in expression between 3h and 0h. WOX5 and WOX11 exhibited similar expression patterns at 3d vs 0h, which were just significant up-regulated in HD and LD. In the present study, SERK1 (somatic embryogenesis receptor kinase 1), SERK2 (somatic embryogenesis receptor kinase 2), BAK1 (SERK3/BRI1-associated kinase 1), TDR/PXY (TDIF receptor/phloem intercalated with xylem) and BAM1 were identified in two cotton types. SERK1 showed a similar expression pattern at 3h vs 0h between HD and LD. SERK2 and one BAK1 were up-regulated at 3d vs 0h, while BAK1 displayed the opposite expression pattern in LD. Four homologs of BAM1 were found to be down-regulated at 3h vs 0h, while just one BAK1 (Gh_D12G0758) was down-regulated at 3d vs 0h among all cotton cultivars. The CLAVATA3/ESR-RELATED (CLE) family of seven genes was showed, these genes involving CLE1, CLE5, CLE27, CLE44 and TDIF. At 3h vs 0h, CLE1 and CLE27 showed the opposite expression in LD cotton, while CLE44 and two TDIF were down-regulated between HD and LD. At 3d vs 0h, CLE44 and one TDIF were down-regulated in both cultivars, while CLE5 (Gh_D01G0413) and TDIF (Gh_A07G1469) were up-regulated in LD and HD, respectively. Five homologous genes of EBE were significantly upregulated at 3h compared to 0h in all cotton cultivars. With the exception of Gh_A11G0359 and Gh_A12G2541, the three EBE homologous genes were also up-regulated at 3d vs 0h in the two cotton cultivars, while one EBE (Gh_A11G0309) was up regulated at 3d vs 0h in the HD. One YUC4, four YUC8 and one YUC10 were confirmed to show differential expression patterns between cotton types. Only YUC4 was up-regulated in the HD cultivars at 3h vs 0h. At 3d vs 0h, YUC8 and YUC10 showed the opposite expression patterns in the two types.
Table 3.
The log2 fold change of SE-related genes in different-stage comparisons.
| Gene name | Gene ID | log2 Fold Change | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 3h vs 0h | 3d vs 0h | ||||||||
| YZ1 | R15 | X33 | X42 | YZ1 | R15 | X33 | X42 | ||
| WOX5 | Gh_A10G2087 | None | None | None | None | 3.08 | Inf | 5.45 | Inf |
| WOX11 | Gh_A13G1402 | None | None | None | None | Inf | Inf | Inf | Inf |
| WOX11 | Gh_D13G1717 | None | None | None | None | Inf | Inf | Inf | 6.17 |
| SERK1 | Gh_A01G0158 | −3.69 | −4.36 | −4.09 | −4.51 | None | None | None | None |
| SERK2 | Gh_Sca006973G01 | None | None | 2.11 | 2.63 | None | None | None | None |
| BAK1 | Gh_D13G0548 | None | None | 2.32 | 2.56 | None | None | None | None |
| BAK1 | Gh_D13G0550 | None | None | −2.35 | −2.35 | None | None | None | None |
| TDR/PXY | Gh_A06G0865 | −2.79 | −2.44 | −2.64 | −3.21 | None | None | None | None |
| BAM1 | Gh_A11G1901 | −4.28 | −3.49 | −4.30 | −4.65 | None | None | None | None |
| BAM1 | Gh_D12G0758 | −3.23 | −2.99 | −3.29 | −2.97 | −2.38 | −2.18 | −2.60 | −2.70 |
| BAM1 | Gh_D11G2071 | −2.69 | −2.14 | −2.06 | −2.57 | None | None | None | None |
| BAM1 | Gh_A03G1912 | −3.61 | −3.49 | −3.75 | −3.07 | None | None | None | None |
| TDIF | Gh_A07G1469 | None | None | None | None | 3.38 | 3.40 | None | None |
| TDIF | Gh_D07G1565 | −2.27 | −2.04 | −2.11 | −2.66 | None | None | None | None |
| TDIF | Gh_D11G1411 | −3.22 | −3.78 | −3.88 | −4.79 | −3.76 | −3.66 | −3.82 | −2.67 |
| CLE1 | Gh_A04G0317 | None | None | 3.25 | 5.10 | None | None | None | None |
| CLE5 | Gh_D01G0413 | None | None | None | None | None | None | 4.18 | 4.54 |
| CLE27 | Gh_D02G1729 | None | None | −4.83 | −2.35 | None | None | None | None |
| CLE44 | Gh_A11G1262 | −6.00 | −4.66 | −5.76 | −4.44 | −2.19 | −2.75 | −3.33 | −2.78 |
| YUC4 | Gh_D03G1753 | 5.06 | Inf | None | None | None | None | None | None |
| YUC8 | Gh_A07G1853 | None | None | None | None | −3.90 | −3.95 | None | None |
| YUC8 | Gh_D07G1540 | None | None | None | None | None | None | −4.91 | −4.53 |
| YUC8 | Gh_A11G1241 | None | None | None | None | None | None | −4.99 | −3.36 |
| YUC8 | Gh_D11G1388 | None | None | None | None | None | None | −4.26 | −5.63 |
| YUC10 | Gh_A08G0657 | None | None | None | None | 2.27 | 3.60 | 3.06 | 2.35 |
| EBE | Gh_D12G0658 | 6.08 | 7.37 | 5.89 | 7.27 | None | None | None | None |
| EBE | Gh_D11G0418 | 3.98 | 3.97 | 3.84 | 3.03 | 5.21 | 5.02 | 4.52 | 3.20 |
| EBE | Gh_A12G1761 | 5.71 | 6.26 | 7.90 | 6.84 | 2.52 | 3.16 | 4.41 | 3.83 |
| EBE | Gh_A12G2541 | 3.66 | 6.14 | 6.07 | 5.97 | None | None | None | None |
| EBE | Gh_D12G1915 | 6.19 | 6.33 | 6.03 | 6.25 | 2.33 | 2.97 | 2.45 | 2.81 |
| EBE | Gh_A11G0359 | None | None | None | None | 3.00 | 2.88 | None | None |
(Note: Among the 3d vs 0h, Inf indicates that the FPKM of WOX5 was close to 0 at 3d).
Figure 5.
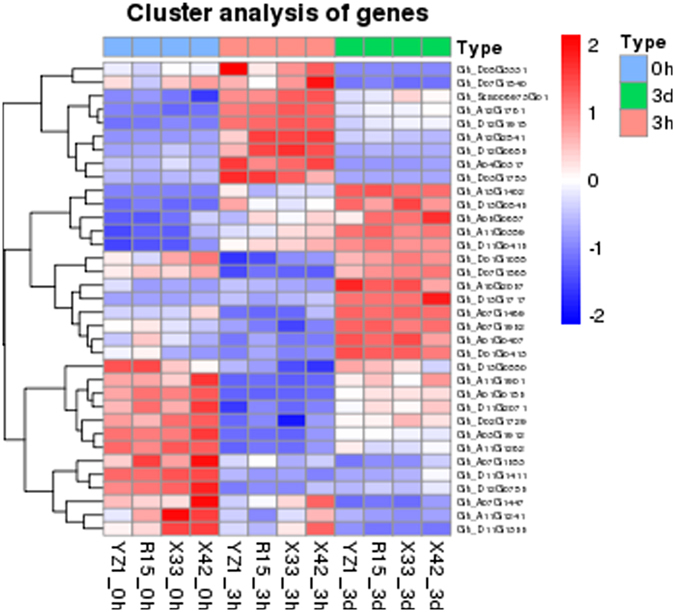
Heatmap of SE-related functional genes (P-value < 0.05, |log2FoldChange| >2 at one sampling point).
Validation of RNA-seq data by qRT-PCR
To further validate the RNA-seq data, 11 DEGs were randomly selected along with their specific primers for qRT-PCR analysis. The cotton ERF1α (NCBI Reference Sequence: XM_016892582.1) was used for relative gene expression normalization. In addition, RT-qPCR data were analyzed using the Origin 8 software. As shown in Fig. 6, there were strong positive correlations (R2 = 0.98–0.96) between RNA-seq data and qRT-PCR data, demonstrating the reliability of the RNA-seq data.
Figure 6.
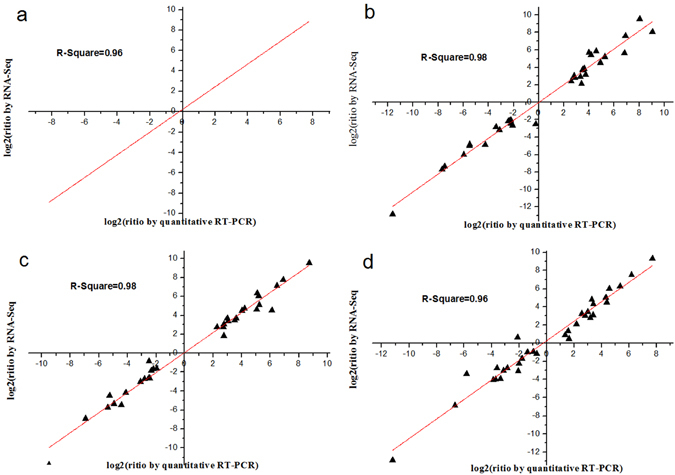
Comparison of expression profile by RNA-Seq and qRT-PCR. (a–d) Comparison of expression profile of 11 randomly selected DEGs by RNA-Seq and qRT-PCR at YZ1, R15, X33 and X42 (3h vs 0h, 3d vs 0h and 3d vs 3h).
Discussion
Transcriptome analysis of SE initial dedifferentiation in two cotton cultivars
Gossypium hirsutum is one of the most recalcitrant cultivars for in vitro plant regeneration through somatic embryogenesis. Moreover, only a few cultivars can produce SE. The number of cotton totipotent cells differentiating into calli is key to manipulating the SE process. Despite a few studies conducted to determine the molecular mechanisms regulating SE initial dedifferentiation16, different cultivars for SE initial dedifferentiation have been not well elucidated. In the present study, gene expression changes during SE initiation and formation were analyzed between the cultivars in which it was easy or difficult to produce SE. Our analyses showed that the number of DEGs and GO identified in similar types of cotton at different induction stages were generally higher than those observed during same-stage comparisons between HD and LD, indicating that HD and LD of different differentiation ability may be not only its varieties of differences, the more important be the response diversity for induction.
The DEGs and GO groups showed many differences in the 3h vs 0h and 3d vs 0h comparisons, indicating that 3h and 3d of induction may be the main regulation phase in the SE initiation. Previous studies have shown that hypocotyls cultured for 3h had no morphological changes compared to non-induction hypocotyls16. In tobacco and Arabidopsis, cellular dedifferentiation and initiation of cell division by protoplasts occurs within 48–72 h. Moreover, histological observations have shown that cotton somatic cells undergo initial differentiation within 72 h19. Furthermore, GO enrichment analysis revealed that protein kinase activity, oxidation-reduction processes and protein/ATP binding were associated with a higher number of DEGs in different and same time stages between HD and LD. Therefore, we inferred that these GO terms would be more likely to play a role in cells differentiating during the SE process.
Stress-related transcription factors regulate differentiation of cotton SE
Transcription factors play regulatory roles in the embryogenesis processes in different plant species20. In the present study, 624 TFs were identified, among which the TF families involved in diverse stress responses as well as in SE processes such as MYB, bHLH, bZIP and GRAS are of particular interest21, 22. In Arabidopsis SE initiation, half of the stress-related transcription factors can be induced by 2, 4-D23. Stress and differentiation are strongly interrelated, overlapping processes24, with certain cells being competent to switch cell fate25. In our study, most of the TF families displayed diverse expression profiles that have complex regulatory functions during SE in cotton. Furthermore, the most differentially expressed TFs were identified in the greater the difference differentiation rate between the varieties.
In Arabidopsis, bHLH transcription factors play a pivotal role in cell fate determination22. UPBEAT1 (UPB1), a bHLH transcription factor, regulated the balance between cell proliferation and differentiation by directly regulating the expression of a set of peroxidases26. Three UPB1 homologues were expressed at higher levels at 3h, and the characterized UPB1 might indicate that cell fate was changed from cellular proliferation to initial dedifferentiation at 3h. MYC1 and GL3 influence cell differentiation for root formation27. MYB proteins work together with BHLH proteins in a variety of cellular processes including control of cell fate determination and regulation of the cell cycle28. The MYB transcription factors, CPC and ETC1, are central regulators of cell differentiation in Arabidopsis thaliana, and ETC1 has enhancer functions with CPC 29, 30. In the present study, the expression patterns of CPC and ETC1 were consistent with those during three time stages. Finally, lower expression levels were observed in the CPC gene than in the two ETC1 (Fig. 4b,c).
GAI is a highly homologous DELLA protein31. DELLA proteins are also GA signaling repressors that are responsive to GA-induced degradation by the ubiquitin-proteasome pathway32, as well as activation of GA-mediated physiological and developmental processes33. GA is a negative regulator of SE in Arabidopsis34. As revealed in this study, GAI decreased with increasing induction times. In plants, the bZIP transcription factors regulate diverse functions, such as plant development and stress response35. In the present study, bZIP1, ABF2 and ABF3 showed the same expression pattern. GmbZIP1 was most closely related to AtABF2 36. Cross-talk between bZIP1 and ABA occurred, potentially leading to improved tolerance to abiotic stresses36. In contrast, GmbZIP44 is a negative regulator of ABA signaling37 that showed the opposite expression model compared to bZIP1. In the present study, bZIP homologues showed diverse expression profiles, which suggests a complex regulated by bZIP proteins as an ABA-dependent transcription factor in cotton SE. In addition, low ABA:GA ratios acted synergistically to stimulate SE in Medicago truncatula34. Our results suggest that these TFs interactions and crosstalk regulate stress response and influence the initiation of SE differentiation. However, further research is required to determine how TFs affect differentiation of cotton SE.
Reactive oxygen species (ROS) regulated differentiation of SE initiation
According to our transcriptomic data, the oxidation-reduction process was significantly enriched in most comparisons. Redox homeostasis is essential to sustain metabolism and growth. In this study, some oxidative phosphorylation related genes also showed differential expression. The strength and duration of signaling regulated reactive oxygen species (ROS) via the redox-dependent signal transduction pathway38. NADPH oxidase (RBOH protein) is an important generator of ROS in the M. truncatula SE induction period34. The major NADPH oxidase catalytic subunits of homologues AtRBOHD and AtRBOHF are required for accumulation of ROS intermediates in the plant defense response in Arabidopsis39. In our study, the same expression model was observed in two RBOHD homologous genes, with relatively high expression occurring at 3h compared to 0h and 3d (Fig. 7a,b). In addition, RBOHA, RBOHB and RBOHC were found to have different expression patterns. Most studies of the interaction between ROS and other genes have been well documented. AtRBOHD and AtrRBOHF mediate ABA-induced ROS production in Arabidopsis 39. The UPBEAT1 gene of the bHLH TF family controls the transition from cellular proliferation to differentiation by directly regulating the expression of ROS26. DELLA activity is regulated by environmental variability40, and DELLAs then regulate plant growth and stress tolerance through modulation of ROS levels40. Our results suggest that synergistic interactions between ROS and other genes may be essential for the differentiation of SE initialization.
Figure 7.
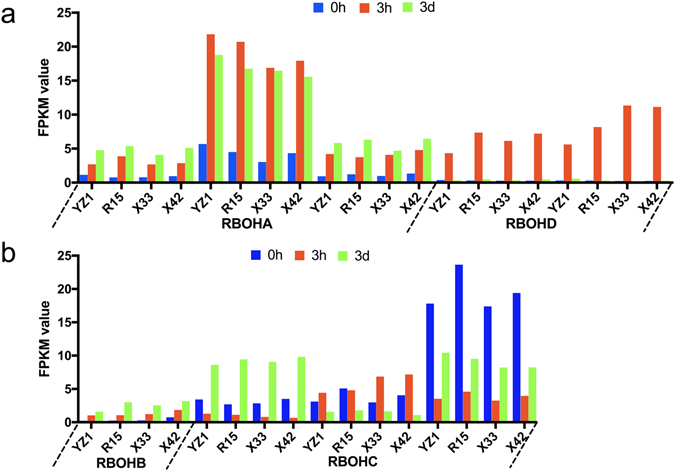
Expression of ROS genes.
Crosstalk among auxin and ethylene contributes to initiation of SE differentiation
Auxin and ethylene are critical plant growth regulators (PGRs) for initiating differentiation in the induction of SE. The differentiation of cotton is correlated with sharp changes in endogenous anxin levels, which may be one of the first steps regulating SE41. ARFs and AUX/IAAs are the key transcription factors involved in regulating the expression of auxin-responsive genes. ARFs bind auxin response promoter elements, mediate transcriptional responses to auxin and regulate auxin-mediated transcriptional activation/repression together with Aux/IAA 42. ARF19 and ARF TF family members43, 44 were down-regulated during two induction stages (Fig. 8a). The function of ARF19 may be negatively regulated by IAA14 and other Aux/IAAs 44. In addition, auxin induces degradation of Aux/IAAs, which release ARFs to regulate transcription of their target genes, thus establishing a negative feedback loop. In the present study, two IAA14 (Fig. 8b) displayed different expression patterns, with one (Gh_Sca006585G01) showing the same pattern as ARF19. Gh_A09G1947 was up-regulated at 3h, then down-regulated at 3d. YUCCA (YUC) is an auxin biosynthetic gene45, and YUC10 is essential for embryogenesis and leaf formation in Arabidopsis 45. In the present study, YUC10 increased continuously during the three time stages (Fig. 8a).
Figure 8.
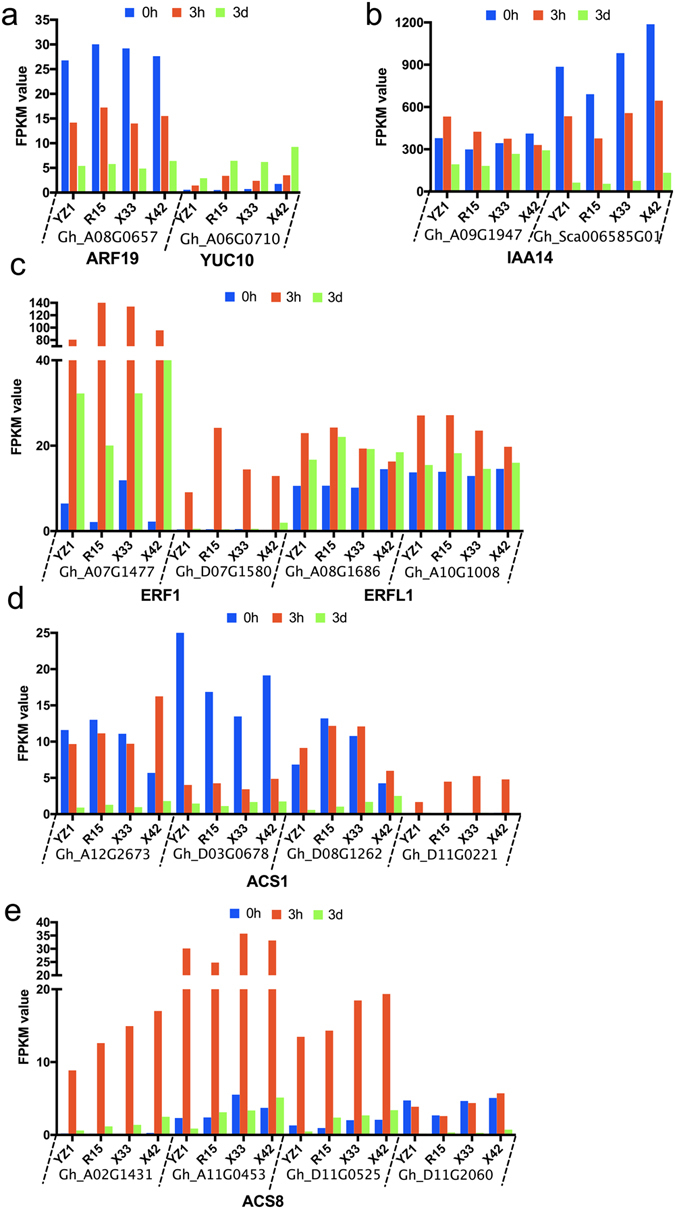
Expression of auxin (a–c) and ethylene (d–h) related genes at three time stages, 0h, 3h and 3d, in the two cultivars, HD and LD.
AP2-EREBP is crucial for ethylene-regulated developmental processes, and ethylene may play a positive role in SE. ERFs contain an AP2 DNA-binding domain of transcriptional factors46. ERF1 (ETHYLENE RESPONSE FACTOR1) is a key integrator of the ethylene responses in developmental processes47. In our study, two ERF1 and two ERFL1 were identified (Fig. 8c), with the highest expression level being observed in 3h. Ethylene also interferes with auxin during the initiation of embryogenesis1. Previous research showed that auxin could increase ethylene levels via activation of the expression of ACC (1-aminocyclopropane-1-carboxylic acid) synthase genes, leading to induction of the expression of ARF19 to regulate downstream gene expression. ARF19 serves as a cross talk point between auxin and ethylene43. In the present study, the FPKM of ACS1 and ACS8 showed the lowest level at 3d (Fig. 8d,e). Both auxin related genes and ethylene related genes were differentially expressed in the two cotton cultivars. Overall, auxin related genes and ethylene related genes showed complex expression profiles, and the results indicate that they may crosstalk with each other during SE dedifferentiation in different cultivars.
Complex regulation of LRR-RLKs in cotton SE initiation
In the present study, many genes were classified into “protein kinase” and “protein/ATP binding” terms, such as LRR-RLKs (leucine-rich receptor-like protein kinase family protein), which have been shown to play important roles in cell differentiation and embryo pattern formation48. SERKs, TDR/PXY and BAM1 belong to the LRR-RLK family of proteins. In Arabidopsis, BAM1 plays an important regulatory role in stem cell maintenance49. In the present study, BAM1 showed significant differences at 3h vs 0h and 3d vs 0h (Table 3). SERK genes play an essential role in determining embryogenic competence. In Arabidopsis, SERK proteins included two distinct groups, one group containing SERK1 and SERK2, BAK1, SERK4, and SERK5 are clustered to another group50. Our results showed that SERK1, SERK2, BAK1 and SERK1 displayed similar expression patterns between HD and LD, while SERK2 and BAK1 expression differed significantly in LD at 3h vs 0h. BAK1 cannot replace SERK1 in male fertility50. TDR/PXY also belongs to the LRR-RLK subclass51. SERKs serve as co-receptors in TDIF that interact with PXY to regulate plant development51. CLE (CLAVATA3/EMBRYO SURROUNDING REGION-RELATED) contains a conserved CLE domain at the C-terminal region and a hydrophobic signal peptide at the N-terminal region (CLAVATA3/EMBRYO)52, 53. Based on bioactivity and receptor specificity, R-type CLE and H-type CLE can be recognized in the CLE peptide family54. CLV3 (CLAVATA3) belongs to R-type CLE, while the H-type CLE includes TDIF, and CLE peptides play important roles in regulating differentiation of stem cells and maintenance of shoot apical meristems in Arabidopsis 52, 55, 56. When compared to HD and LD, CLE proteins showed more differences in LD species at different time stages. When combined with the results of previous studies, our results indicate that LRR-RLKs play a role in the SE process and regulate the SE differentiation rate.
Materials and Methods
Plant materials and tissue culture conditions
Four cultivars of Gossypium hirsutum, YZ1, R15, X33 and X42, were investigated, among which YZ1 and R15 have a relatively high SE differentiation rate and therefore comprise the main transgenic material57. Although X33 and X42 are the major commercial cultivars in Xinjiang, China, they have a low rate of differentiation during SE compared with YZ1 and R15. Specimens of all cultivars have been conserved in our laboratory.
Four cotton seeds were sterilized with 0.1% (w/v) Hg2Cl2 for 10 min, then rinsed five times with sterilized distilled water. The sterilized seeds were germinated on ½ MS for hypocotyl induction58, then incubated at 28 °C in the dark for 6 d. Next, hypocotyls were excised from sterile seedlings, cut into 1 cm segments and placed on callus-induction medium (CIM; MS medium plus B5 vitamins, supplemented with 0.05 mg l–1 IAA, 0.05 mg l–1 kinetin, 0.05 mg l–1 2,4-D, pH 5.8)14. Induction cultures were conducted under 16 h light: 8 h dark conditions at 28 °C. Different stages of explants at 0h, 3h and 3d were used for transcription analysis.
RNA and library preparation for transcriptome sequencing
Total RNA was extracted from each sample using the PurelinkTM RNA Mini Kit (Life Technologies, Carlsbad, CA, USA) following the manufacturer’s protocol. We collected 24 samples from two biological replicates of each sample and monitored RNA degradation and contamination on 1% agarose gels. The RNA purity was checked using a NanoPhotometer® spectrophotometer (IMPLEN, CA, USA), while the RNA concentration and integrity were measured using a Qubit® RNA Assay Kit and a Qubit® 2.0 Flurometer (Life Technologies, CA, USA) and the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). RNA samples were stored at −80 °C until further processing. For each sample, a total of 3 µg RNA was used as the input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext® Ultra™ RNA Library Prep Kits for Illumina® (NEB, USA) according to the manufacturer’s instructions, and index codes were added to attribute sequences to each sample. Clustering of the index-coded samples was performed on a cBot Cluster Generation System using the TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s recommendations. Finally, paired-end reads of 125 bp/150 bp were generated via an Illumina Hiseq platform.
Bioinformatics analysis of RNA-Seq data
Quality control is first step, clean reads were obtained by removing adaptor tags, reads containing ploy-N and low quality reads from the raw reads of fastq format. In the addition, the clean reads of Q20, Q30 and GC were calculated, and high quality clean reads were used in all downstream analyses. For annotation, all reads were mapped to the reference sequences. An index of the reference genome was constructed using Bowtie v2.2.3 and paired-end clean reads were aligned to the reference genome using TopHat v2.0.12. To quantify the gene expression level, we employed HTSeq v0.6.1 to calculate the numbers of mapped reads and then normalized the results to the FPKM (expected number of Fragments Per Kilobase of transcript sequence per millions of base pairs sequenced), which is the most commonly used normalized method for estimating gene expression levels that considers the effects of sequencing depth and gene length for the reads at the same time59. We used the DESeq R package (1.18.0) to confirm the differential expression analysis. The resulting P-values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. We used a threshold value of an adjusted P-value (padj) ≤0.05 and a log2FoldChange ≥ 2 in at least one of these stages.
Functional classification of differentially expressed genes (DEGs)
Functional analysis of DEGs including Gene Ontology (GO) and KEGG was performed. GO enrichment analysis of DEGs was implemented by the GOseq R package. We used the KOBAS software to test the statistical enrichment of differential expression genes in the KEGG database. GO terms and KEGG pathways with corrected P-values ≤ 0.05 were considered the thresholds to determine significant enrichment by differentially expressed genes.
qRT-PCR analysis
Eleven DEGs were selected to estimate the validation of RNA-seq data by qRT-PCR. Gene primers (Table S2) were designed using NCBI/Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome) and synthesized by BGITECH. The cDNA was synthesized from 1 μL of total RNA (200–500 ng) using the PrimerScript RT reagent Kit (Takara, Dalian, China) in a 10 μL reaction mixture according to the manufacturer’s instructions. qRT-PCR was performed in 10 μL reactions on a LightCycler® 480 (Roche) using 1 μL of first-strand cDNA as the template, 5 μL of 2 × SYBR Premix Ex Taq II (TLi RanseH Plus) (Takara, Dalian, China), 0.4 μL each of 10 μM forward and reverse gene-specific primers and 3.2 μL of ddH2O in 96-well plates. The cotton ERF1α (NCBI Reference Sequence: XM_016892582.1) was used as an internal standard. The qRT-PCR conditions were as follows: pre-incubation at 95 °C for 30 s, followed by amplification by 45 cycles at 95 °C for 10 s, 60 °C for 10 s and 72 °C for 10 s. qRT-PCR analysis was conducted in the three biological replicates. Data were analyzed using the Origin 8 software.
Electronic supplementary material
Acknowledgements
This work was supported by Breeding of Transgenic Short-season Cotton New Varieties in Exceptional Early Mature Cotton Region [2014ZX08005002-004].
Author Contributions
A.C., Y.Z., B.C., and Y.Y. conceived and designed the experiments. A.C., D.S., X.W. and L.Y. performed the experiments. A.C. and B.M. analyzed the data. A.C., Y.Z. and Y.Y. contributed reagents/materials/analysis tools. A.C. wrote the paper. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-08763-8
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jiménez VM. Involvement of Plant Hormones and Plant Growth Regulators on in vitro Somatic Embryogenesis. Plant Growth Regulation. 2005;47:91–110. doi: 10.1007/s10725-005-3478-x. [DOI] [Google Scholar]
- 2.Kurczyńska EU, Mazur E. Histological analysis of direct somatic embryogenesis in Arabidopsis thaliana (L.) Heynh. Planta. 2007;226:619–628. doi: 10.1007/s00425-007-0510-6. [DOI] [PubMed] [Google Scholar]
- 3.Fehér A, Pasternak TP, Dudits D. Transition of somatic plant cells to an embryogenic state. Plant Cell, Tissue and Organ Culture (PCTOC) 2003;74:201–228. doi: 10.1023/A:1024033216561. [DOI] [Google Scholar]
- 4.Grafi G. How cells dedifferentiate: a lesson from plants. Developmental Biology. 2004;268:1–6. doi: 10.1016/j.ydbio.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 5.Toonen MAJ, et al. Description of somatic-embryo-forming single cells in carrot suspension cultures employing video cell tracking. Planta. 1994;194:565–572. doi: 10.1007/BF00714471. [DOI] [Google Scholar]
- 6.Thibaud-Nissen F, Shealy RT, Khanna A, Vodkin LO. Clustering of Microarray Data Reveals Transcript Patterns Associated with Somatic Embryogenesis in Soybean. Plant Physiology. 2003;132:118–136. doi: 10.1104/pp.103.019968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma SK, Millam S, Hedley PE, Mcnicol J, Bryan GJ. Molecular regulation of somatic embryogenesis in potato: an auxin led perspective. Plant Molecular Biology. 2008;68:185–201. doi: 10.1007/s11103-008-9360-2. [DOI] [PubMed] [Google Scholar]
- 8.Zhenzhen X, C. Z, et al. Transcriptome Profiling Reveals Auxin and Cytokinin Regulating Somatic Embryogenesis in Different Sister Lines of Cotton Cultivar CCRI24 †. Journal of Integrative Plant Biology. 2013;55:631–642. doi: 10.1111/jipb.12073. [DOI] [PubMed] [Google Scholar]
- 9.Karami O, Saidi A. The molecular basis for stress-induced acquisition of somatic embryogenesis. Molecular Biology Reports. 2010;37:2493–2507. doi: 10.1007/s11033-009-9764-3. [DOI] [PubMed] [Google Scholar]
- 10.Karami O, Aghavaisi B, Mahmoudi PA. Molecular aspects of somatic-to-embryogenic transition in plants. Journal of Chemical Biology. 2009;2:177–190. doi: 10.1007/s12154-009-0028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeng F, et al. Isolation and characterization of genes associated to cotton somatic embryogenesis by suppression subtractive hybridization and macroarray. Plant Molecular Biology. 2006;60:167–183. doi: 10.1007/s11103-005-3381-x. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X. Regulation of Somatic Embryogenesis in Higher Plants. Critical Reviews in Plant Sciences. 2010;29:36–57. doi: 10.1080/07352680903436291. [DOI] [Google Scholar]
- 13.Jin S, et al. Identification of a novel elite genotype for in vitro culture and genetic transformation of cotton. Biologia Plantarum. 2006;50:519–524. doi: 10.1007/s10535-006-0082-5. [DOI] [Google Scholar]
- 14.Zheng W, et al. AtWuschel Promotes Formation of the Embryogenic Callus in Gossypium hirsutum. Plos One. 2014;9:e87502–e87502. doi: 10.1371/journal.pone.0087502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumria R, et al. High-frequency somatic embryo production and maturation into normal plants in cotton (Gossypium hirsutum) through metabolic stress. Plant Cell Reports. 2003;21:635–639. doi: 10.1007/s00299-002-0554-9. [DOI] [PubMed] [Google Scholar]
- 16.Yang X, et al. Transcript profiling reveals complex auxin signalling pathway and transcription regulation involved in dedifferentiation and redifferentiation during somatic embryogenesis in cotton. BMC Plant Biology. 2012;12:1–19. doi: 10.1186/1471-2229-12-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang XW, et al. De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. BMC Genomics. 2010;11:326–333. doi: 10.1186/1471-2164-11-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng WH, et al. De novo transcriptome analysis reveals insights into dynamic homeostasis regulation of somatic embryogenesis in upland cotton (G. hirsutum L.) Plant Molecular Biology. 2016;92:279–292. doi: 10.1007/s11103-016-0511-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu HG, et al. Analysis of genes differentially expressed during initial cellular dedifferentiation in cotton. Science Bulletin. 2008;53:3666–3676. doi: 10.1007/s11434-008-0468-1. [DOI] [Google Scholar]
- 20.El OS, et al. Control of somatic embryogenesis and embryo development by AP2 transcription factors. Plant Molecular Biology. 2010;74:313–326(314). doi: 10.1007/s11103-010-9674-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Llorca CM. M. P., Ulrike Zentgraf. bZIPs and WRKYs: two large transcription factor families executing two different functional strategies. Frontiers in Plant Science. 2014;5:169–169. doi: 10.3389/fpls.2014.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao H, Li X, Ma L. Basic helix-loop-helix transcription factors and epidermal cell fate determination in Arabidopsis. Plant Signaling & Behavior. 2012;7:1556–1560. doi: 10.4161/psb.22404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gliwicka M, Nowak K, Balazadeh S, Muellerroeber B, Gaj MD. Extensive Modulation of the Transcription Factor Transcriptome during Somatic Embryogenesis in Arabidopsis thaliana. Plos One. 2013;8 doi: 10.1371/journal.pone.0069261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grafi G, et al. Plant response to stress meets dedifferentiation. Planta. 2011;233:433–438. doi: 10.1007/s00425-011-1366-3. [DOI] [PubMed] [Google Scholar]
- 25.Grafi G, Florentin A, Ransbotyn V, Morgenstern Y. The stem cell state in plant development and in response to stress. Frontiers in Plant Science. 2011;2:1085–1091. doi: 10.3389/fpls.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsukagoshi H, Busch W, Benfey PN. Transcriptional regulation of ROS controls transition from proliferation to differentiation in the root. Cell. 2011;143:606–616. doi: 10.1016/j.cell.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 27.Bernhardt C, Lee MA, Zhang F, Lloyd A, Schiefelbein J. The bHLH genes GLABRA3 (GL3) andENHANCER OF GLABRA3 (EGL3) specify epidermal cell fate in the Arabidopsis root. Development. 2004;130:6431–6439. doi: 10.1242/dev.00880. [DOI] [PubMed] [Google Scholar]
- 28.Zhang F, Gonzalez A, Zhao M, Payne CT, Lloyd A. A network of redundant bHLH proteins functions in all TTG1-dependent pathways of Arabidopsis. Development. 1991;130:4859–4869. doi: 10.1242/dev.00681. [DOI] [PubMed] [Google Scholar]
- 29.Kirik V, Simon M, Wester K, Schiefelbein J, Hulskamp M. ENHANCER of TRY and CPC 2 (ETC2) reveals redundancy in the region-specific control of trichome development of Arabidopsis. Plant Molecular Biology. 2004;55:389–398. doi: 10.1007/s11103-004-0893-8. [DOI] [PubMed] [Google Scholar]
- 30.Kirik, V., Simon, M., Huelskamp, M. et al. The ENHANCER OF TRY AND CPC1 gene acts redundantly with TRIPTYCHON and CAPRICE in trichome and root hair cell patterning in Arabidopsis. Developmental Biology268, 506–513 (2004). [DOI] [PubMed]
- 31.Peng J, et al. The Arabidopsis GAI gene defines a signaling pathway that negatively regulates gibberellin responses. Genes & Development. 1998;11:3194–3205. doi: 10.1101/gad.11.23.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itoh H, Ueguchitanaka M, Sato Y, Ashikari M, Matsuoka M. The gibberellin signaling pathway is regulated by the appearance and disappearance of SLENDER RICE1 in nuclei. Plant Cell. 2002;14:57–70. doi: 10.1105/tpc.010319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards DE, King KE, Tahar Aitali A, Harberd NP. How Gibberellin Regulates Plant Growth and Development: A Molecular Genetic Analysis of Gibberellin Signaling. Plant Biology. 2001;52:67–88. doi: 10.1146/annurev.arplant.52.1.67. [DOI] [PubMed] [Google Scholar]
- 34.Nolan KimE, Siyang Liao YS, Saeed NasirA, Zhang Xiyi, Rose RayJ. An Unusual Abscisic Acid and Gibberellic Acid Synergism Increases Somatic Embryogenesis, Facilitates Its Genetic Analysis and Improves Transformation in Medicago truncatula. Plos One. 2014;9 doi: 10.1371/journal.pone.0099908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ying S, et al. Cloning and characterization of a maize bZIP transcription factor, ZmbZIP72, confers drought and salt tolerance in transgenic Arabidopsis. Planta. 2012;235:253–266. doi: 10.1007/s00425-011-1496-7. [DOI] [PubMed] [Google Scholar]
- 36.Gao SQ, et al. The soybean GmbZIP1 transcription factor enhances multiple abiotic stress tolerances in transgenic plants. Plant Molecular Biology. 2011;75:537–553. doi: 10.1007/s11103-011-9738-4. [DOI] [PubMed] [Google Scholar]
- 37.Liao Y, et al. Soybean GmbZIP44, GmbZIP62 and GmbZIP78 genes function as negative regulator of ABA signaling and confer salt and freezing tolerance in transgenic Arabidopsis. Planta. 2008;228:225–240. doi: 10.1007/s00425-008-0731-3. [DOI] [PubMed] [Google Scholar]
- 38.Burhans WC, Heintz NH. The cell cycle is a redox cycle: linking phase-specific targets to cell fate. Free Radical Biology & Medicine. 2009;47:1282–1293. doi: 10.1016/j.freeradbiomed.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 39.Torres MA, Dangl JL, Jones JD. Arabidopsis gp91phox homologues AtrbohD and AtrbohF are required for accumulation of reactive oxygen intermediates in the plant defense response. Proceedings of the National Academy of Sciences. 2002;99:517–522. doi: 10.1073/pnas.012452499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Achard P, Renou JP, Berthomé R, Harberd NP, Genschik P. Plant DELLAs restrain growth and promote survival of adversity by reducing the levels of reactive oxygen species. Current Biology. 2008;18:656–660. doi: 10.1016/j.cub.2008.04.034. [DOI] [PubMed] [Google Scholar]
- 41.Thomas C, et al. Immuno-cytochemical localization of indole-3-acetic acid during induction of somatic embryogenesis in cultured sunflower embryos. Planta. 2002;215:577–583. doi: 10.1007/s00425-002-0791-8. [DOI] [PubMed] [Google Scholar]
- 42.Xiao-Yi Z, Li S, Hong-Wei X. Brassinosteroids Regulate the Differential Growth of Arabidopsis Hypocotyls through Auxin Signaling Components IAA19 and ARF7. Molecular Plant. 2013;6:887–904. doi: 10.1093/mp/sss123. [DOI] [PubMed] [Google Scholar]
- 43.Li J, Dai X, Zhao Y. A role for auxin response factor 19 in auxin and ethylene signaling in Arabidopsis. Plant Physiology. 2006;140:899–908. doi: 10.1104/pp.105.070987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okushima Y, et al. Functional genomic analysis of the AUXIN RESPONSE FACTOR gene family members in Arabidopsis thaliana: unique and overlapping functions of ARF7 and ARF19. Plant Cell. 2005;17:444–463. doi: 10.1105/tpc.104.028316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Youfa Cheng XD. Yunde Zhao. Auxin Synthesized by the YUCCA Flavin Monooxygenases Is Essential for Embryogenesis and Leaf Formation in Arabidopsis. Plant Cell. 2007;19:2430–2439. doi: 10.1105/tpc.107.053009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mao, J. L. et al. Arabidopsis ERF1 Mediates Cross-Talk between Ethylene and Auxin Biosynthesis during Primary Root Elongation by Regulating ASA1 Expression. Plos Genetics12 (2016). [DOI] [PMC free article] [PubMed]
- 47.Lorenzo O, Piqueras R, Sánchezserrano JJ, Solano R. ETHYLENE RESPONSE FACTOR1 integrates signals from ethylene and jasmonate pathways in plant defense. Plant Cell. 2003;15:165–178. doi: 10.1105/tpc.007468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J. Multi-tasking of somatic embryogenesis receptor-like protein kinases. Current Opinion in Plant Biology. 2010;13 doi: 10.1016/j.pbi.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 49.Deyoung BJ, et al. The CLAVATA1-related BAM1, BAM2 and BAM3 receptor kinase-like proteins are required for meristem function in Arabidopsis. Plant Journal. 2006;45:1–16. doi: 10.1111/j.1365-313X.2005.02592.x. [DOI] [PubMed] [Google Scholar]
- 50.Aandentoorn M, Albrecht C, Devries S. On the Origin of SERKs: Bioinformatics Analysis of the Somatic Embryogenesis Receptor Kinases. Molecular Plant. 2015;8:762–782. doi: 10.1016/j.molp.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 51.Zhang H, et al. SERK Family Receptor-Like Kinases Function as a Co-receptor with PXY for Plant Vascular Development. Molecular Plant. 2016;9:1406–1414. doi: 10.1016/j.molp.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 52.Ito Y, et al. Dodeca-CLE peptides as suppressors of plant stem cell differentiation. Science. 2006;313:842–845. doi: 10.1126/science.1128436. [DOI] [PubMed] [Google Scholar]
- 53.Xu TT, Song XF, Ren SC, Liu CM. The sequence flanking the N-terminus of the CLV3 peptide is critical for its cleavage and activity in stem cell regulation in Arabidopsis. BMC Plant Biology. 2013;13:1–12. doi: 10.1186/1471-2229-13-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kinoshita A, et al. Gain-of-function phenotypes of chemically synthetic CLAVATA3/ESR-related (CLE) peptides in Arabidopsis thaliana and Oryza sativa. Plant and Cell Physiology. 2008;48:1821–1825. doi: 10.1093/pcp/pcm154. [DOI] [PubMed] [Google Scholar]
- 55.Huang S, Ness LA. The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell. 2000;100:635–644. doi: 10.1016/S0092-8674(00)80700-X. [DOI] [PubMed] [Google Scholar]
- 56.Hirakawa Y, Kondo Y, Fukuda H. TDIF peptide signaling regulates vascular stem cell proliferation via the WOX4 homeobox gene in Arabidopsis. Plant Cell. 2010;22:2618–2629. doi: 10.1105/tpc.110.076083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin S, Zhang X, Y., Guo X, Liang S, Zhu H. Identification of a novel elite genotype for in vitro culture and genetic transformation of cotton. Biologia Plantarum. 2006;50:519–524. doi: 10.1007/s10535-006-0082-5. [DOI] [Google Scholar]
- 58.Murashige T, Skoog F. A Revised Medium for Rapid Growth and Bio Assays with Tobacco Tissue Cultures. Physiologia Plantarum. 1962;15:473–497. doi: 10.1111/j.1399-3054.1962.tb08052.x. [DOI] [Google Scholar]
- 59.C T, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.