

ABSTRACT
The goal of this study was to develop effective and practical field sampling methods for quantification of aerial deposition of airborne conidia of Entomophaga maimaiga over space and time. This important fungal pathogen is a major cause of larval death in invasive gypsy moth (Lymantria dispar) populations in the United States. Airborne conidia of this pathogen are relatively large (similar in size to pollen), with unusual characteristics, and require specialized methods for collection and quantification. Initially, dry sampling (settling of spores from the air onto a dry surface) was used to confirm the detectability of E. maimaiga at field sites with L. dispar deaths caused by E. maimaiga, using quantitative PCR (qPCR) methods. We then measured the signal degradation of conidial DNA on dry surfaces under field conditions, ultimately rejecting dry sampling as a reliable method due to rapid DNA degradation. We modified a chamber-style trap commonly used in palynology to capture settling spores in buffer. We tested this wet-trapping method in a large-scale (137-km) spore-trapping survey across gypsy moth outbreak regions in Pennsylvania undergoing epizootics, in the summer of 2016. Using 4-day collection periods during the period of late instar and pupal development, we detected variable amounts of target DNA settling from the air. The amounts declined over the season and with distance from the nearest defoliated area, indicating airborne spore dispersal from outbreak areas.
IMPORTANCE We report on a method for trapping and quantifying airborne spores of Entomophaga maimaiga, an important fungal pathogen affecting gypsy moth (Lymantria dispar) populations. This method can be used to track dispersal of E. maimaiga from epizootic areas and ultimately to provide critical understanding of the spatial dynamics of gypsy moth-pathogen interactions.
KEYWORDS: airborne spores, convective transport, deposition, gypsy moth, insect pathogen, pathogen dispersal, population cycles, spatial synchrony
INTRODUCTION
The invasive gypsy moth, Lymantria dispar, is the most important forest defoliator in the eastern United States. Outbreaks cause severe defoliation, sometimes leading to extensive tree death and loss of aesthetic value in forest and residential settings. Economic analyses estimate an impact of around $3.2 billion per year in North America (1, 2), placing this species among the top five costliest invasive insects globally (3). Unsuccessful attempts were made to introduce the fungal pathogen Entomophaga maimaiga from Japan to North America, for biological control purposes, as early as 1910. Much later, between 1989 and 1992, the larval pathogen was detected in the northeastern United States (4). This pathogen spread rapidly through the gypsy moth's range in North America and is now common in most populations. It is often effective in suppressing outbreak populations, providing control both in the current year via infective airborne spores (conidia) and in subsequent years via long-lived azygospores (resting spores) in soil.
Insect-pathogen interactions can exhibit complex space-time relationships, and understanding these relationships may be key to understanding the role of pathogens in insect population dynamics (5). Dispersal of E. maimaiga conidia is likely to be a key determinant of spatial dynamics in this host-pathogen system. The gypsy moth and its pathogens exhibit spatial synchrony, or coincident changes in abundance across geographically disjunct populations (6), and we will gain understanding of this widespread phenomenon by studying the role of E. maimaiga dispersal. There is considerable evidence that dispersal of airborne conidia plays a critical role in the population dynamics of E. maimaiga (7, 8), but only limited information about the dispersal of airborne E. maimaiga conidia currently exists (9).
Entomophaga maimaiga belongs to the fungal order Entomophthorales, in which most species are obligate pathogens of arthropods. The majority of species produce two types of spores, i.e., long-lived resting spores, which are produced internally, fall to the soil, germinate, and produce infective conidia in the following year, and actively ejected conidia, which infect immediately (or produce another infective conidium). Entomophthoralean conidia can become airborne after being ejected from external conidiophores on dead insects, usually while attached to the tree. Some conidia fall slowly in still air (dry deposition) after being ejected (10). However, air movement (i.e., wind) can result in long-distance dispersal of the airborne conidia, with subsequent deposition via either dry settling or wet means (i.e., in precipitation). Leaf wetness was positively associated with conidial flux of E. maimaiga in the field, with time lags of 5 to 14 h (9), consistent with previous observations that higher humidity (11) or leaf wetness (12) is necessary for conidial discharge.
Airborne samples inspected microscopically have revealed spores of numerous species of Entomophthorales infecting aphids and lepidopteran larvae (13–17), including E. maimaiga, which is associated with gypsy moth larvae (9). Previous work on conidia dispersal in this pathogen has been based on visual counts of spores captured by sampling devices, which is very labor-intensive. DNA-based methods may give more accurate estimates of conidial deposition because of the instability of E. maimaiga conidia; conidia are relatively short-lived and may give rise to secondary and even tertiary spores within 1 or 2 days (4). These secondary and tertiary conidia are also actively ejected, raising the possibly of spores “escaping” from collecting surfaces; visual counting may possibly tally both empty primary and secondary conidia if the conidia do not travel far. The sensitivity of DNA-based methods also improves the detection of small numbers of spores, which are difficult to count at low densities. Finally, visual identification is based only on spore size and shape, and the presence and activity of closely related and similar-looking species, such as Entomophaga aulicae and Entomophaga grylli (18), could inflate counts. The use of a DNA-based assay specific to E. maimaiga prevents possible miscounts.
Pollen biologists, plant pathologists, and allergy epidemiologists have used many methods to quantify spore and pollen deposition. There are effective mechanical means of sampling (see, e.g., reference 19); however, it would be costly and impractical to deploy such equipment with adequate sampling replication over large areas of natural habitat. Selective media are often used to collect settling airborne spores of insect pathogenic hypocrealean fungi, followed by counting of colonies growing on plates (see, e.g., reference 20). Some investigations of fungal plant pathogens have used simple petri dishes for collection of airborne spores (21, 22), with an aqueous solution of 4× Tris-EDTA (TE) buffer as a wetting agent or dried preservative on pieces of filter paper. Our testing and observations of E. maimaiga conidia showered from infected insects indicated that none of these methods would be accurate for a replicated study. First, Entomophaga maimaiga is not easily cultured except on rich solid medium or in cell culture medium that simulates insect hemolymph; there are no selective media for this organism (4). Second, the conidia are relatively large (average size, 20.6 by 26.6 μm) (4), very sticky, and quick to germinate under the right conditions (3.4 to 9.0 h after ejection) (11). Finally, they have a tough outer wall that requires bead-beating for effective release of DNA during the extraction process, thus requiring transfer to a liquid medium.
Our goals were to develop and to optimize a method for collection of these conidia in the environment and then to apply quantitative PCR (qPCR) for quantification. First, we collected spores in the field on a dry surface and tested the ability to detect and to quantify them, but this was successful only over short periods. Next, we used laboratory-produced spores to test the persistence of spores on a dry surface, and we found rates of DNA degradation that were unacceptable for a seasonal field study. Finally, we modified standard pollen traps into a successful wet-cup collection method and tested it over one season.
RESULTS
Field sampling of settling E. maimaiga conidia on a dry surface.
Exposure of horizontal cellophane surfaces for 2 to 14 h at a field location with known active infections of L. dispar larvae resulted in positive readings for E. maimaiga. Fifteen of 22 samples contained E. maimaiga DNA, with threshold cycle (CT) values ranging from 24 to 39 cycles, thus confirming that we could detect conidial DNA in field-collected samples using qPCR.
Persistence of conidial DNA on dry surfaces in the field.
The persistence of conidia on polypropylene sheets in the field was relatively brief (Fig. 1). Within 48 h after initial exposure, the DNA signal had degraded significantly, in comparison to controls (fixed effect of treatment, F130 = 39.8; P < 0.0001) (the 12-h and 24-h values were not significantly different from control values). By 7 days, the DNA signal was not detectable. All qPCR plate efficiencies, which indicate relative rates of DNA copy production per cycle, were within the acceptable range (23) of 90 to 110% (actual range, 92 to 94%). Across 16 replicates, the average conidium count for controls (showered with conidia but not exposed to field conditions) was 869 conidia/cm2 (range, 149 to 2,743 conidia/cm2).
FIG 1.
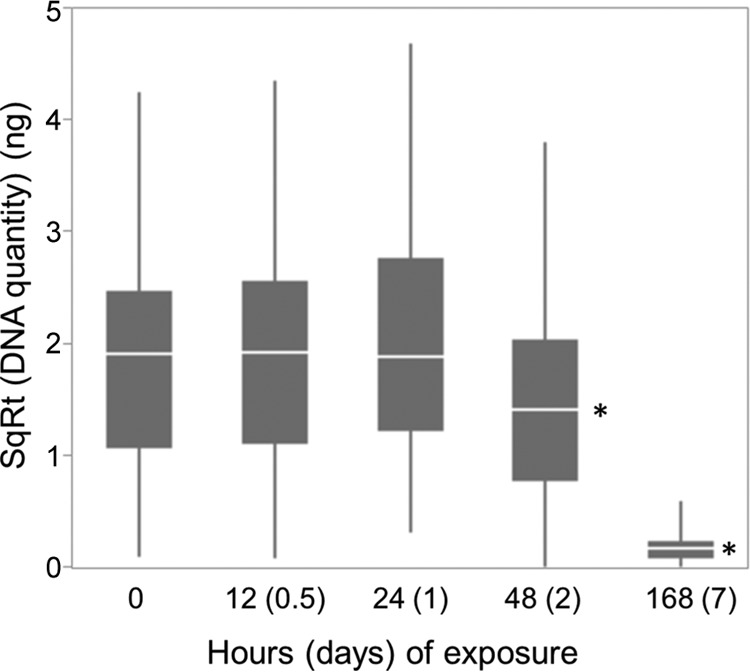
Boxplot of medians, interquartile ranges (IQRs) (25th quartile to 75th quartile), and outliers (up to 1.5 times the IQR), showing the persistence of DNA on dry polypropylene surfaces in the field. Asterisks indicate statistically significant differences from controls. SqRt, square root.
Wet-cup-modified Tauber traps.
Based on the findings of the dry surface experiments and in order to better collect and preserve conidia in the field, we modified a Tauber chamber-style trap originally designed for pollen collection (24, 25) with a slightly larger top opening and a wet cup containing buffer inside the trap (Fig. 2). We were able to detect the DNA of E. maimaiga from settling airborne conidia in most of the 20 traps deployed in Pennsylvania (Fig. 3). Overall, DNA quantity tended to be higher for early sampled traps, peaking on June 18, after which there was a steady decline through the season as larvae pupated; the last sighting of larvae was on June 30 (Fig. 4; with the data transformation, −6 on the y axis is equal to zero DNA).
FIG 2.

Photograph of the modified Tauber trap used in this study.
FIG 3.

Map of 2015 defoliation (shaded area) and 2016 trap locations (circles, eastern traps; triangles, central traps) in the state of Pennsylvania. The map is original (created with ArcGIS v10.1).
FIG 4.
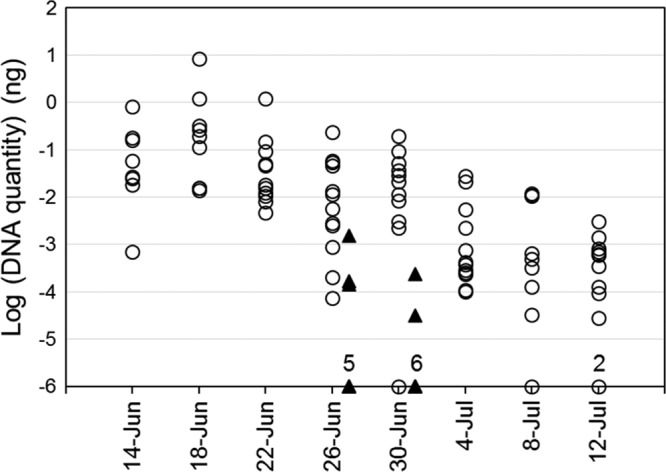
Transformed values, log(x + 10−6), of DNA quantities in all traps over the trapping period. Numbers above markers on the x axis indicate the number of superimposed points. The eastern group (circles) had 12 traps, and the central group (triangles) had 8 traps. Trap placement began on 10 June 2016, and samples were collected at 4-day intervals.
The standard threshold for all plates was 0.115 ΔRn, and the maximum quantity of DNA detected in a trap was 8.3 ng (equal to approximately 307 spores). The positive trap samples were all measured within the range of the standard dilutions; the smallest quantity detected in a trap sample was 2.7× 10−5 ng (CT = 37.0), while the lowest standard on that plate also exhibited a CT of 37.0. This is less than the amount expected in one multinucleate spore (estimated as 0.027 ng/spore for this standard), indicating possible degradation of spores during the trapping period or loss of DNA during sample processing.
For similar dates across all traps (eastern and central), DNA quantities were negatively correlated with the distance to the nearest defoliation, both with the previous year's (2015) defoliation data (r = −0.85; P < 0.00001) (Fig. 5a) and with the current year's (2016) (r = −0.79; P < 0.00001) (Fig. 5b). Across all dates (2015) for the 12 eastern traps, the pattern was similar (r = −0.61; P < 0.00001) (data not shown); we did not analyze the central traps separately due to small sample size.
FIG 5.
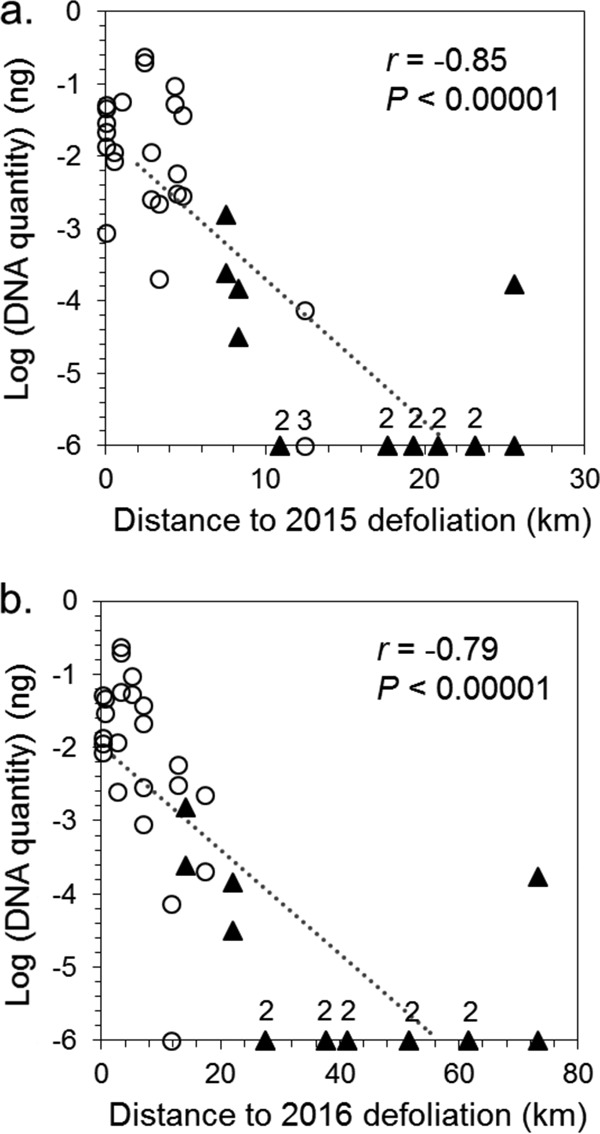
Transformed values, log(x + 10−6), of DNA quantities in eastern traps (circles) and central traps (triangles), from 26 June to 1 July 2016, versus the distance to the nearest defoliated area in 2015 (a) and 2016 (b). Numbers above markers on the x axis indicate the number of superimposed points. Note the scale difference in the x axes.
Across all samples, there was no overall correlation of DNA quantity with precipitation (i.e., sum of 5-day precipitation), although it appeared that rainfall on certain days in certain sites did correspond with an increase in conidial DNA quantity (data not shown). Traps were exposed to field conditions, and this affected the amounts and concentrations of buffer solution in the cups within the traps. Over all 105 trap collections, 18% of cups gained volume over the 4-day period, and the remaining cups had an average sample evaporative loss of approximately 50 ml (starting volume, 250 ml; mean final volume, 201 ml; median final volume, 185 ml [range, 50 to 460 ml]). None of the traps dried up completely or had cup overflow, and all concentrations remained at least 1× TE, because the volume of liquid did not double.
DISCUSSION
We combined and modified existing methods for a new application, i.e., quantifying densities of settling airborne conidia of an important entomopathogen, Entomophaga maimaiga. Our goals were to develop methods for trapping and quantification that would accurately measure the deposition of spores in natural habitats. We modified traps used in palynology (24), which are durable, reusable, easy to set up and to remove, nontoxic, and relatively unobtrusive in the landscape. This trapping method also appeared to be robust in the presence of at least moderate levels of precipitation during the sampling period. The qPCR assay proved to be repeatable and efficient. Perfect qPCR efficiency, i.e., doubling of copy number with each cycle, is 100%, and all of our efficiencies were within the range (90 to 110%) that is considered acceptable by researchers who use this technique (23).
Sampling airborne conidia of Entomophaga maimaiga presents some challenges, including their large size and strong adhesion to surfaces. Entomophaga maimaiga conidia actively ejected from dead larvae (i.e., primary conidia) also have a short life span and can begin developing and ejecting secondary conidia within 1 day (4). We showed that, over short periods (<1 day), we could detect E. maimaiga DNA deposited on dry cellophane. However, undislodged conidia remained on the cellophane, which collapsed during washing, so we sought a different material for dry spore capture. We found that conidia could be easily dislodged from small dry pieces of flexible polypropylene plastic using a weak detergent solution (0.05 to 0.1% Tween). However, when conidia on polypropylene were subjected to field conditions, DNA detection dropped off within 48 h of field exposure, perhaps because secondary conidia were ejected, rainfall washed off the spores, or the conidia were dislodged, covered, or decomposed because of other organisms falling on the surface. This result led us to abandon sampling on dry surfaces and pointed toward trapping conidia in liquid buffer as a better choice for extending trapping periods. The trapping method described does not require daily visitation of the traps, although optimization of trap timing for different research goals is possible.
As a field trial application of the wet sampling device, 20 traps were monitored for collection of naturally deposited spores over a 1-month period in June and July 2016, when an outbreak of L. dispar and a natural epizootic of E. maimaiga occurred over a large geographic area in northeastern Pennsylvania. Although larval infection rates for E. maimaiga reached 86% in some of the sampling areas (T. D. Bittner, unpublished data), many gypsy moth populations persisted at outbreak levels throughout the season, and areas of northeastern Pennsylvania suffered defoliation. We found that conidial DNA was negatively related to the distance from defoliated areas, which are putative sources for populations of airborne conidia. The pattern of steeply declining densities of settling conidia with increasing distances (ranging from 10 to 20 km) from defoliation (Fig. 5) suggests a diffusive pattern of conidial dispersal. However, one trap captured conidia over 70 km from defoliation, suggesting the possibility of occasional long-distance convective transport.
We sampled during a relatively dry period; June precipitation in the eastern region (Allentown, PA) was 6.2 cm below normal, and that in central Pennsylvania (State College, PA) was 5.5 cm below normal (Northeast Regional Climate Center) (http://www.nrcc.cornell.edu/wxstation/nowdata.html). However, we found that conidial DNA was deposited in many of our traps over the sampling period, and conidial densities declined throughout the season, as would be expected due to host death and completion of gypsy moth larval development. The observed pattern was strongly influenced by the eastern group of traps; the central group was operated over a shorter period, included fewer traps, and had many zeroes.
Available data on the rates of E. maimaiga invasion into new areas suggest that they vary widely, from 350 m season−1 to more than 100 km season−1, and they seemed to be correlated with differences in precipitation (4). Modeling studies suggested that different processes are important for short-distance and long-distance spread of E. maimaiga. For example, a model by Dwyer et al. (7) showed that a randomly diffusing conidial dispersal pattern could account for the lower range of observed spread rates but, when Weseloh (8) used a model incorporating a nonnormal (leptokurtic) probability function for conidial dispersal, which allowed for occasional long-distance spread, it could account for the observed rate of 100 km season−1. For a gypsy moth invasion front moving at 12.2 km year−1, E. maimaiga followed with a 3-year lag but a similar rate of spread (26).
Interestingly, we did not find a spatial association of spore deposition with local rainfall. Rainfall is known to increase both sporulation rates at the source sites (via leaf wetness and humidity) and deposition rates at the deposition sites directly (via wet deposition) (16, 27). Although spatial variation in rainfall can be expected to affect variation in conidial production among sporulation sites, the spatial variation may not be reflected among deposition sites after conidia have dispersed.
The gypsy moth and its pathogens exhibit spatial synchrony, or coincident changes in abundance, across geographically disjunct populations (5, 28). In many organisms, outbreaks and subsequent population crashes occur cyclically over large ranges, but the causes of this synchrony are difficult to disentangle. The mobility of predators has been shown to synchronize prey population crashes (29), and possibly a mobile pathogen could have a similar effect. If precipitation is an underlying driver of synchrony in this system, as supported by Haynes et al. (28), then the mechanism could be via direct increases in the production, survival, and mobility of E. maimaiga spores over long distances. Future spatially detailed measurements of conidial DNA over several seasons, in combination with larval densities and infection rates, would give a better picture of the variability in conidial densities and their relationship to spatial synchrony. In addition, viability tests of deposited conidia will be needed to demonstrate conidial survival during movement.
Early prediction of an epizootic of E. maimaiga could have economic and environmental benefits if pest managers are able to target or to reduce pesticide spray programs. Future work may improve predictive models to forecast the success of E. maimaiga in controlling the gypsy moth in a given year or from one year to the next; this would require integrating information about the early presence and abundance of pathogenic conidia with stochastic environmental factors, such as spring storms, that could be driving epizootics (8). An epizootic may serve to suppress L. dispar outbreaks over larger areas, and dispersal of conidia is a probable mechanism. These and other questions can now be explored with the methodology presented here.
MATERIALS AND METHODS
Production of conidia for testing.
Conidia were produced for testing of persistence on dry surfaces, assessment of buffers in wet cups, and determination of standard curves for qPCR. To generate conidia in the laboratory, we obtained first-instar L. dispar from a colony maintained by the USDA Animal and Plant Health Inspection Service and reared larvae to fourth instars at 25°C, with 10 larvae per 200-ml cup of artificial diet (Southland Products, Inc.), using a 14-h light/10-h dark regimen. On a daily basis, larvae that had reached the fourth instar were transferred to storage (maximum of 7 days) at 10°C until use.
We used isolates of E. maimaiga (ARSEF 6212 and ARSEF 6625) to grow liquid cultures of protoplasts in Grace's insect medium (BioWhittaker/Lonza, Walkersville, MD) containing 5% heat-inactivated fetal bovine serum (Invitrogen/Gibco, Grand Island, NY). Larvae were injected intrahemocoelically, at the base of a proleg, with 1 × 105 protoplasts per insect, using a manual microinjector fitted with a 3-ml syringe and a 23-gauge needle. Injected larvae were held at 16°C and checked daily for death. Cadavers were transferred to 2% water agar, held in darkness at 16°C, and checked daily for production of conidia. Conidia were collected using the “showering” method (30, 31).
Field sampling of settling E. maimaiga conidia on a dry surface.
In preliminary observations, we found that conidial adhesion to hard surfaces such as glass and plastic petri dishes was too strong, and the conidia could not be dislodged for DNA extraction. Therefore, we tested small dry pieces of flexible materials that could be transferred and washed inside a tube. We started by testing cellophane. In June 2015, we set out 22 open 10-cm petri dishes near Beaver Springs, Pennsylvania, at the edge of a forest with a moderately dense population of late-instar L. dispar dying from E. maimaiga (evidenced by fresh cadavers hanging on trees). Each dish held a rectangle (5 by 7 cm) of cellophane, and the dishes were placed horizontally on short tables (0.33 m [width] by 0.45 m [length] by 0.25 m [height]) on the forest floor, on stainless steel trays covered with netting. The tables were protected by a cage made of hardware cloth, to prevent animals from disturbing the dishes. Dishes were open to aerial deposition for 2 to 14 h. At the end of a collection period, each cellophane rectangle was transferred to a 50-ml sterile plastic tube and kept dry and cool (4 to 10°C) until it was returned to the laboratory for inspection and processing (see “Sample Preparation,” “DNA Extraction,” and “Quantitative PCR” below).
Persistence of conidial DNA on dry surfaces.
Cellophane proved too thin and water absorbent for our needs, so we used plastic rectangles cut from standard page-protector sleeves (Staples), which are made of polypropylene. Between 10 and 24 sporulating cadavers were suspended above a rotating platform, as described in reference 30, inside a humid plastic box to shower conidia onto 10 flexible plastic rectangles (2.5 by 3.5 cm). All pieces within a showering box were arranged in a circle to rotate along the same path, relative to the cadavers above, and thus be uniformly showered. Showering boxes were operated for approximately 4 h each, in the dark, at 21°C and 70 to 80% humidity.
A total of 16 replicate showers, conducted on 9 (nonconsecutive) days, were analyzed. The 10 showered plastic rectangles per box were allocated to the following treatments (2 rectangles from each box per field exposure): no (control), 12-h, 24-h, 48-h, and 7-day field exposure.
The 2 control rectangles were used to determine the average starting conidial count and starting DNA quantity. To verify that adequate densities of conidia were present, we counted 5 random grids per control rectangle at ×100 magnification, with an ocular micrometer (0.951 mm2 per grid), and averaged the counts across the 2 control rectangles from the same shower. Using a total area of 875 mm2 per rectangle, we calculated the average number of conidia per rectangle (these counts were not used in the statistical analysis). Immediately after counting, control rectangles were rolled with the showered side facing in, placed in 2-ml screw-cap tubes with 500 μl of 0.05% Tween, and frozen for later quantification of E. maimaiga DNA, as described below (see “Sample Preparation,” “DNA Extraction,” and “Quantitative PCR”).
Field exposure was conducted by transferring the remaining 8 rectangles at random, with the showered side up, to open halves of 6-cm-diameter plastic petri dishes. The natural exposure site was on the north edge of a small mixed-hardwood woodlot, and caged tables were placed under the canopy about 3 m from the forest edge, inside the shrubby edge vegetation zone. Conidial samples were placed on the tables within the 1-h period before sunset, and groups of dishes were removed at the appropriate intervals. Upon removal from the dishes, rectangles were stored as described for controls. Field exposures were conducted after the season of gypsy moth larval development, from 14 August to 29 September 2015, in Ithaca, New York, where there had been no outbreak. Thus, there was no possibility of contamination from aerially deposited E. maimaiga conidia.
Wet-cup-modified Tauber traps.
Traps were made of polyvinyl chloride (PVC) pipe (diameter, 15.2 cm) and two endcaps with a slightly domed shape (Fig. 2). The body was a 33.0-cm length of pipe with the bottom end pounded into an endcap, which fit tightly so that adhesives were not required. The outside surface of the upper trap body was sanded down to allow easier removal of the lid endcap, which was drilled at the center of the dome, using a circular bit (diameter, 11.4 cm) on a drill press, to create the trap opening. A steel mesh screen (6.4-mm grid) was attached across the opening using four small screws, to prevent entry of larger material. The assembled outer width and height of the traps were approximately 16 cm and 35 cm, respectively. Traps were deployed resting upright on the ground and attached firmly to a 1.2-m metal fence post using cable ties (zip ties). We used a 739-ml clear plastic cup (diameter, 14.6 cm; height, 7 cm), which fit closely inside the trap body, to hold 250 ml of buffer. Based on references 21 and 22 and preliminary testing (T. D. Bittner, unpublished data), we used 2× TE buffer with 0.05% Tween. This combination maintained the normal appearance of conidia (i.e., they did not wrinkle or lyse) and prevented germination. Before installing the trap lid, we sprayed the outer upper PVC edges liberally with Tangle-Trap sticky insect trap coating (Tanglefoot Co., Grand Rapids, MI), which prevented the entry of most slugs and captured many small insects that might otherwise have fallen into the buffer.
Traps were placed individually at each of 20 sites in eastern or central Pennsylvania in June 2016, during a regional gypsy moth outbreak (Fig. 3). Traps were deployed in grassy open areas for better collection of samples from the open air, rather than under a tree canopy, where settling conidia might have originated largely from local infected larvae. The buffer and cup were replaced every 4 days. Collected buffer was transported back to the laboratory in 500-ml sample bottles, on ice. Empty cups were also returned to the laboratory and filled with 50 to 100 ml of double-distilled water; this rinsate remained stored in the cup with the sample bottles at 4°C until sample preparation. The purpose of rinsing the cups was 2-fold, i.e., to dislodge any conidia left behind in the cup and to wash away the buffer salts during the filtering step (see “Sample Preparation”).
In the eastern part of the state, 12 traps spanning 75.5 km were operated for up to 32 days (8 collections); the average distance between traps was 13.7 km (range, 3.2 to 37.7 km). In the central part of the state, 8 traps spanning 61.3 km were operated for 8 days (2 collections); the average distance between traps was 10.7 km (range, 5.7 to 14.9 km).
Sample preparation.
Sample preparation prior to DNA extraction depended on the collection technique used. For dry collections (field detection and persistence experiments), the flexible surfaces (cellophane or polypropylene) were washed in sterile tubes by vortex-mixing for 1 min with 0.05% Tween. For cellophane sheets (5 by 7 cm), we used 50-ml tubes and, after vortex-mixing, we pelleted material by centrifugation in a swinging bucket rotor at 1,300 × g for 20 min. For persistence polypropylene sheets (2.5 by 3.5 cm), we used 2-ml screw-cap tubes and, after vortex-mixing, we pelleted material in a microcentrifuge at 14,100 × g for 10 min. Sheets were observed under the microscope to confirm the removal of conidia and then were discarded. In the dry surface field experiment, the pellet material was transferred from the 50-ml tubes to 2-ml bead-beating tubes prior to extraction; we diluted the pellet with 400 μl of lysis buffer from the extraction kit (see below) and then transferred the liquid.
For wet-cup trap collections, we took note of the buffer sample condition, including color, contaminants, insects, and other small invertebrates. To collect conidia from the buffer, a two-stage Swinnex filter system (diameter, 2.5 cm; Millipore International) with a 60-ml Luer-lock syringe (BD) was used. The top filter, with a 60-μm net, captured unwanted large material, and the second filter, with a 10-μm net, captured spores. The total volume of sample liquid was measured during filtering, and then the rinse water from the matching cup was pushed through the syringe to rinse the sample. Finally, air (2 or 3 puffs) was pushed through the filter system to remove excess moisture from the nets.
We transferred each 10-μm net to a 2-ml bead-beating tube with 1 ml of molecular grade water. Each sample was vortex-mixed for 1 min, and then the nets were removed and discarded. Samples were centrifuged for 10 min at 14,100 × g, and water was removed from the pellet. We immediately added 400 μl of the extraction kit buffer to each pellet (see below) and froze the samples for later extraction.
DNA extraction.
For all experiments, we used DNeasy Plant minikits (Qiagen) and 0.5 g of 0.7-mm-diameter zirconium beads (BioSpec Products). DNA extraction proceeded according to the kit instructions after bead-beating of each sample for 60 s at 48 krpm in a Mini-BeadBeater (BioSpec Products). All extractions were performed by one person, using consistent quantities of reagents added and transferred at each step, to minimize variations. The final elution volume for all samples was 50 μl.
Quantitative PCR.
We performed quantitative PCR to quantify the conidia present in each sample by comparison to a standard curve of DNA extracted from known concentrations of conidia. To produce DNA for the standard curve, conidia were showered from infected cadavers over a dish containing 0.1% Tween in water. Conidia were filtered from the Tween using 10-μm nylon nets in the Swinnex filter system (described above). The resulting conidial suspension was quantified at a magnification of ×100, using a hemocytometer, and then was centrifuged and extracted as described above. The DNA concentration was measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific). For each plate, new dilutions of the DNA standard were made to generate the standard curve. From a starting quantity of 17.5 ng, we made 1:10 serial dilutions (at least 6 and up to 8 dilutions per plate).
Each 96-well plate for qPCR contained triplicates of standard dilutions, samples, and negative controls. Reactions with iTaq Universal Probes supermix (Bio-Rad Laboratories, Hercules, CA) were performed in 25-μl volumes (containing 5 μl of template), with 40 cycles of qPCR on an Applied Biosystems ViiA7 system, at the Cornell University Biotechnology Resource Center. Detection was achieved using TaqMan chemistry with a 6-carboxyfluorescein (FAM) reporter, primers, and probe from reference 32.
Data analysis.
qPCR results were visualized and analyzed using QuantStudio v1.2 (Applied Biosystems). If one of the triplicate reactions was an outlier from the other two, then the outlier was omitted and the plate was reanalyzed. Within an experiment, if multiple plates were needed to fit all of the samples, then the threshold value for each run was adjusted to the mean of all threshold values originally determined by the software for all plates in the experiment. DNA quantities (as generated by QuantStudio) from the triplicate reactions were averaged and used for further analyses.
In the dry persistence study, estimates of DNA quantities were square root transformed to improve normality and were used in a mixed-model analysis of variance (ANOVA) (with showers as a random factor and hours of exposure as a fixed factor) to test for differences among hours of exposure, with Dunnett's adjustment for multiple comparisons (33), in JMP (SAS, Cary, NC).
In the wet-cup trap study, estimates of DNA quantity were transformed via log(x + 10−6). For each location where wet-cup traps were deployed, we obtained estimates of daily total precipitation from the PRISM Climate Group database (Oregon State University) (http://prism.oregonstate.edu). The daily time frame for this database is the 24-hour period ending at 7 a.m. EST. We used these estimates to calculate the total precipitation at each trapping site for 5-day intervals, beginning 1 day before and including the 4-day trapping period. Due to the data cutoff, any precipitation that occurred during the day on which the trap buffers were changed (between 9 a.m. and 5 p.m.) was not included.
In order to explore the relationship between settling conidial densities and distances to source gypsy moth outbreaks, we calculated the distance of each trap from the nearest defoliated forest. Defoliation is mapped annually via aerial sketch mapping by the Pennsylvania Bureau of Forestry, and data are compiled by US Forest Service Forest Health Protection staff in a geographical information system database (https://www.fs.fed.us/foresthealth/technology/ads_standards.shtml). Using these defoliation data for Pennsylvania, we measured the distance (in kilometers) from each spore trap to the nearest defoliation polygon for both the previous year (2015) and the current year (2016). We then correlated transformed DNA quantities with distances to defoliation.
ACKNOWLEDGMENTS
Direct funding for this project came from the USDA Forest Service Forest Health Technology Enterprise Team and the Gypsy Moth Slow the Spread Foundation.
We thank Kendra McLauchlan for her modified Tauber trap design, Jacob Henry for assistance in building Tauber traps, Elson Shields for equipment, Jacob Henry, Gino Luzader, Alyssa Espinoza, and Austin Cody for assistance in the field and the laboratory, Gino Luzader for compiling PRISM data and maps, and Françoise Vermeylen for statistical consulting. We thank Tim Marasco and Scott Stitzer of the Pennsylvania Bureau of Forestry for defoliation data and assistance in monitoring gypsy moth populations and/or finding sites and Craig Bingman of the Snyder County Conservation District for connecting us with landowners. We also thank the various private property owners who granted access to their properties for the placement of spore traps or the collection of insects for determination of infection rates.
REFERENCES
- 1.Aukema JE, Leung B, Kovacs K, Chiver C, Britton KO, Englin J, Frankel SJ, Haight RG, Holmes TP, Liebhold AM, McCullough DG, Von Holle B. 2011. Economic impacts of non-native forest insects in the continental United States. PLoS One 6:e24587. doi: 10.1371/journal.pone.0024587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colautti RI, Bailey SA, van Overdijk CDA, Amundsen K, MacIsaac HJ. 2006. Characterised and projected costs of nonindigenous species in Canada. Biol Invasions 8:1–11. doi: 10.1007/s10530-005-0236-y. [DOI] [Google Scholar]
- 3.Bradshaw CJA, Leroy B, Bellard C, Roiz D, Albert C, Fournier A, Barbet-Massin M, Salles J-M, Simard F, Courchamp F. 2016. Massive yet grossly underestimated global costs of invasive insects. Nat Commun 7:12986. doi: 10.1038/ncomms12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hajek AE. 1999. Pathology and epizootiology of Entomophaga maimaiga infections in forest Lepidoptera. Microbiol Mol Biol Rev 63:814–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dwyer G. 1994. Density dependence and spatial structure in the dynamics of insect pathogens. Am Nat 143:533–562. doi: 10.1086/285619. [DOI] [Google Scholar]
- 6.Liebhold A, Koenig WD, Bjørnstad ON. 2004. Spatial synchrony in population dynamics. Annu Rev Ecol Evol Syst 35:467–490. doi: 10.1146/annurev.ecolsys.34.011802.132516. [DOI] [Google Scholar]
- 7.Dwyer G, Elkinton JS, Hajek AE. 1998. Spatial scale and the spread of a fungal pathogen of gypsy moth. Am Nat 152:485–494. doi: 10.1086/286185. [DOI] [PubMed] [Google Scholar]
- 8.Weseloh RM. 2003. Short and long range dispersal in the gypsy moth (Lepidoptera: Lymantriidae) fungal pathogen, Entomophaga maimaiga (Zygomycetes: Entomophthorales). Environ Entomol 32:111–122. doi: 10.1603/0046-225X-32.1.111. [DOI] [Google Scholar]
- 9.Hajek AE, Olsen C, Elkinton JS. 1999. Dynamics of airborne conidia of the gypsy moth (Lepidoptera: Lymantriidae) fungal pathogen Entomophaga maimaiga (Zygomycetes: Entomophthorales). Biol Control 16:111–117. doi: 10.1006/bcon.1999.0740. [DOI] [Google Scholar]
- 10.Sawyer AJ, Griggs MH, Wayne R. 1994. Dimensions, density, and settling velocity of entomophthoralean conidia: implications for aerial dissemination of spores. J Invertebr Pathol 63:43–55. doi: 10.1006/jipa.1994.1008. [DOI] [Google Scholar]
- 11.Hajek AE, Carruthers RI, Soper RS. 1990. Temperature and moisture relations of sporulation and germination of Entomophaga maimaiga (Zygomycetes: Entomopthoraceae), a fungal pathogen of Lymantria dispar. Environ Entomol 19:85–90. doi: 10.1093/ee/19.1.85. [DOI] [Google Scholar]
- 12.Hajek AE, Soper RS. 1992. Temporal dynamics of Entomophaga maimaiga after death of gypsy moth (Lepidoptera: Lymantriidae) larval hosts. Environ Entomol 21:129–135. doi: 10.1093/ee/21.1.129. [DOI] [Google Scholar]
- 13.Wilding N. 1970. Entomophthora conidia in the air-spora. J Gen Microbiol 62:149–157. doi: 10.1099/00221287-62-2-149. [DOI] [PubMed] [Google Scholar]
- 14.Harper JD, Herbert DA, Moore RE. 1984. Trapping patterns of Entomophthora gammae (Weiser) (Entomophthorales: Entomophthoraceae) conidia in a soybean field infested with the soybean looper, Pseudoplusia includens (Walker) (Lepidoptera: Noctuidae). Environ Entomol 13:1186–1190. doi: 10.1093/ee/13.5.1186. [DOI] [Google Scholar]
- 15.Steinkraus DC, Hollingsworth RG, Boys GO. 1996. Aerial spores of Neozygites fresenii (Entomophthorales: Neozygitaceae): density, periodicity, and potential role in cotton aphid (Homoptera: Aphididae) epizootics. Environ Entomol 25:48–57. doi: 10.1093/ee/25.1.48. [DOI] [Google Scholar]
- 16.Hemmati F, Pell JK, McCartney HA, Deadman ML. 2001. Airborne concentrations of conidia of Erynia neoaphidis above cereal fields. Mycol Res 105:485–489. doi: 10.1017/S0953756201003537. [DOI] [Google Scholar]
- 17.Hemmati F, Pell JK, McCartney HA, Clark SJ, Deadman ML. 2001. Conidial discharge in the aphid pathogen Erynia neoaphidis. Mycol Res 105:715–722. doi: 10.1017/S0953756201004014. [DOI] [Google Scholar]
- 18.Bałazy S. 1993. Flora of Poland: fungi (mycota), vol. 24. Entomophthorales. Polish Academy of Sciences, W. Szafer Institute of Botany, Cracow, Poland. [Google Scholar]
- 19.West JS, Atkins SD, Emberlin J, Fitt BDL. 2008. PCR to predict risk of airborne disease. Trends Microbiol 16:380–387. doi: 10.1016/j.tim.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Shimazu M, Sato H, Maehara N. 2002. Density of the entomopathogenic fungus, Beauveria bassiana Vuillemin (Deuteromycotina: Hyphomycetes) in forest air and soil. Appl Entomol Zool 37:19–26. doi: 10.1303/aez.2002.19. [DOI] [Google Scholar]
- 21.Luchi N, Ghelardini L, Belbahri L, Quartier M, Santini A. 2013. Rapid detection of Ceratocystis platani inoculum by quantitative real-time PCR. Appl Environ Microbiol 79:5394–5404. doi: 10.1128/AEM.01484-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schweigkofler W, O'Donnell K, Garbelotto M. 2004. Detection and quantification of airborne conidia of Fusarium circinatum, the causal agent of pine pitch canker, from two California sites by using real-time PCR approach combined with a simple spore trapping method. Appl Environ Microbiol 70:3512–3520. doi: 10.1128/AEM.70.6.3512-3520.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Life Technologies Corp. 2012. Real-time PCR handbook. Life Technologies Corp., Carlsbad, CA. [Google Scholar]
- 24.Tauber H. 1974. A static non-overload pollen collector. New Phytol 73:359–369. doi: 10.1111/j.1469-8137.1974.tb04770.x. [DOI] [Google Scholar]
- 25.Commerford JL, McLauchlan KK, Minckley TA. 2016. High dissimilarity within a multiyear annual record of pollen assemblages from a North American tallgrass prairie. Ecol Evol 6:5273–5289. doi: 10.1002/ece3.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hajek AE, Tobin PC. 2011. Introduced pathogens follow the invasion front of a spreading alien host. J Anim Ecol 80:1217–1226. doi: 10.1111/j.1365-2656.2011.01870.x. [DOI] [PubMed] [Google Scholar]
- 27.Reilly JR, Hajek AE, Liebhold AM, Plymale R. 2014. Impact of Entomophaga maimaiga (Entomophthorales: Entomophthoraceae) on outbreak gypsy moth populations (Lepidoptera: Erebidae): the role of weather. Environ Entomol 43:632–641. doi: 10.1603/EN13194. [DOI] [PubMed] [Google Scholar]
- 28.Haynes KJ, Bjørnstad ON, Allstadt AJ, Liebhold AM. 2013. Geographical variation in the spatial synchrony of a forest-defoliating insect: isolation of environmental and spatial drivers. Proc Biol Sci 280:20122373. doi: 10.1098/rspb.2012.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ims RA, Steen H. 1990. Geographical synchrony in microtine population cycles: a theoretical evaluation of the role of nomadic avian predators. Oikos 57:381–387. doi: 10.2307/3565968. [DOI] [Google Scholar]
- 30.Filotas MJ, Hajek AE, Humber RA. 2003. Prevalence and biology of Furia gastropachae (Zygomycetes: Entomophthorales) in populations of forest tent caterpillars (Lepidoptera: Lasiocampidae). Can Entomol 135:359–378. doi: 10.4039/n02-004. [DOI] [Google Scholar]
- 31.Hajek AE, Papierok B, Elienberg J. 2012. Methods for study of the Entomophthorales, p 285–316. In Lacey LA. (ed), Manual of techniques in invertebrate pathology, 2nd ed Academic Press, London, United Kingdom. [Google Scholar]
- 32.Castrillo LA, Thomsen L, Juneja P, Hajek AE. 2007. Detection and quantification of Entomophaga maimaiga resting spores in forest soil using real-time PCR. Mycol Res 111:324–331. doi: 10.1016/j.mycres.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Dunnett CW. 1955. A multiple comparison procedure for comparing several treatments with a control. J Am Stat Assoc 50:1096–1121. doi: 10.1080/01621459.1955.10501294. [DOI] [Google Scholar]