

ABSTRACT
The genomes of most bacteria contain mobile DNA elements that can contribute to undesirable genetic instability in engineered cells. In particular, transposable insertion sequence (IS) elements can rapidly inactivate genes that are important for a designed function. We deleted all six copies of IS1236 from the genome of the naturally transformable bacterium Acinetobacter baylyi ADP1. The natural competence of ADP1 made it possible to rapidly repair deleterious point mutations that arose during strain construction. In the resulting ADP1-ISx strain, the rates of mutations inactivating a reporter gene were reduced by 7- to 21-fold. This reduction was higher than expected from the incidence of new IS1236 insertions found during a 300-day mutation accumulation experiment with wild-type ADP1 that was used to estimate spontaneous mutation rates in the strain. The extra improvement appears to be due in part to eliminating large deletions caused by IS1236 activity, as the point mutation rate was unchanged in ADP1-ISx. Deletion of an error-prone polymerase (dinP) and a DNA damage response regulator (umuDAb [the umuD gene of A. baylyi]) from the ADP1-ISx genome did not further reduce mutation rates. Surprisingly, ADP1-ISx exhibited increased transformability. This improvement may be due to less autolysis and aggregation of the engineered cells than of the wild type. Thus, deleting IS elements from the ADP1 genome led to a greater than expected increase in evolutionary reliability and unexpectedly enhanced other key strain properties, as has been observed for other clean-genome bacterial strains. ADP1-ISx is an improved chassis for metabolic engineering and other applications.
IMPORTANCE Acinetobacter baylyi ADP1 has been proposed as a next-generation bacterial host for synthetic biology and genome engineering due to its ability to efficiently take up DNA from its environment during normal growth. We deleted transposable elements that are capable of copying themselves, inserting into other genes, and thereby inactivating them from the ADP1 genome. The resulting “clean-genome” ADP1-ISx strain exhibited larger reductions in the rates of inactivating mutations than expected from spontaneous mutation rates measured via whole-genome sequencing of lineages evolved under relaxed selection. Surprisingly, we also found that IS element activity reduces transformability and is a major cause of cell aggregation and death in wild-type ADP1 grown under normal laboratory conditions. More generally, our results demonstrate that domesticating a bacterial genome by removing mobile DNA elements that have accumulated during evolution in the wild can have unanticipated benefits.
KEYWORDS: clean genome, insertion sequence, mutation accumulation, genome engineering, synthetic biology
INTRODUCTION
Acinetobacter baylyi ADP1 is a nonpathogenic Gram-negative soil bacterium that has been used as a platform for metabolic engineering and synthetic biology due to its high natural transformability (1–4). ADP1 naturally catabolizes aromatic compounds and can detoxify inhibitory molecules that are liberated during degradation of lignocellulosic feedstocks (5, 6), and it can be engineered for the production of biofuel components and high-value compounds, including wax esters and triacylglycerides (7, 8). ADP1 has also been used to construct cell-based biosensors for salicylate and components of crude oil (9, 10). Facile engineering of the ADP1 genome to edit and add to metabolic and sensing pathways is made possible by its constitutive expression of a competence apparatus that enables it to efficiently take up DNA from its environment in a non-sequence-specific manner (11). ADP1 can also transform fragmented and damaged DNA or mutagenized PCR products to introduce sequence variation at specific sites in its genome for optimizing engineered functions (12, 13).
Insertion sequence (IS) elements are minimal transposons that are widespread in bacteria (14). They contribute to mutagenesis by encoding transposases that move or copy their DNA sequences to new locations in a genome and by acting as long repeated sequences that can mediate homologous recombination events that lead to deletions and other large-scale genome rearrangements (15, 16). IS elements can be significant sources of genetic instability in engineered bacterial cells. For example, they are often the dominant source of spontaneous mutations that rapidly inactivate the heterologous expression of costly recombinant proteins from plasmids (17, 18).
The genome of A. baylyi ADP1 contains a total of six IS elements, all of the same IS1236 type (19, 20). IS1236 elements are members of the IS3 family (21). They operate by a copy-paste mechanism and insert randomly without any strong target site sequence bias. Five of the six IS1236 copies have identical sequences. Two of these are located in close proximity on the chromosome, forming a composite transposon (Tn5613) that can also mobilize two hypothetical genes located between the IS copies (22). The sixth copy (IS1236ψ) has evolved to be nonautonomous. It shares only 82% nucleotide identity with the other copies and has a frameshift mutation in its transposase gene (23).
We and others have observed that a considerable proportion of the mutations that occur in A. baylyi ADP1 are due to IS1236 activity. This transposable element family was initially characterized due to the prevalence of IS1236 insertions among mutations inactivating certain protocatechuate catabolism genes (20). IS1236 elements constitute 25 to 40% of all new mutations that accumulate during long-term adaptive laboratory evolution experiments with ADP1 (24, 25), including mutations in competence genes that often arise during laboratory culture and reduce or eliminate the transformability of the strain (25, 26). The DNA cleavage activity associated with IS1236 elements may also contribute to certain gene amplification events (23, 27).
In this study, we sequenced the genomes of A. baylyi ADP1 isolates that had been evolved under relaxed selection for 300 days and found that IS1236 elements were directly responsible for 26% of all spontaneous mutations. Therefore, to improve the genetic stability of ADP1, we engineered an IS-less version of the strain (ADP1-ISx) by deleting all six IS1236 copies. Characterization of the final strain revealed that ADP1-ISx exhibited a much larger reduction in mutation rates than expected and also significantly greater cell yields and transformation frequencies. These beneficial traits in the “clean-genome” ADP1-ISx strain make it an improved platform for bioengineering applications that take advantage of its metabolic versatility and high natural transformability.
RESULTS AND DISCUSSION
Spontaneous mutation rates in A. baylyi ADP1.
We measured genome-wide spontaneous mutation rates in ADP1 by sequencing 17 strains from a previous mutation accumulation (MA) experiment (25). Each of these clonal isolates represents the endpoint of a lineage that evolved for ∼7,500 generations (cell doublings) over 300 days of passaging colonies on LB agar. The filtering effect of natural selection is greatly reduced in MA experiments because every lineage experiences a single-cell bottleneck when a random colony is selected each day for propagation. This design enables genetic drift to dominate over selection and causes the rates at which mutations appear in MA lineages to be very close to the rates at which they spontaneously arise in a genome (28).
We found a total of 66 mutations across all 17 clones, with zero to nine mutations in each individual clone (Fig. 1A). Despite this wide range, the distribution of mutation counts across these independent lineages was still compatible with no difference in the underlying genomic mutation rate that each experienced (P = 0.45; Kruskal-Wallis test). The overall estimated mutation rate for ADP1 is 0.52 mutations per genome per 1,000 generations (Poisson 95% confidence interval, 0.40 to 0.66).
FIG 1.

Mutations observed in A. baylyi ADP1 by whole-genome sequencing of 17 strains that each evolved separately for 7,500 generations under relaxed selection in the MA experiment. (A) Mutations observed in each clone from the MA experiment divided into major categories. Overall, 26% of the mutations were directly related to IS1236 element activity. The precise location and nature of each mutation is reported in Table S1 in the supplemental material. (B) Summed observations of each base pair substitution across all sequenced clones from the MA experiment.
A majority of the mutations observed in the MA lines were base substitutions (n = 36). The estimated total base substitution mutation rate is 0.79 × 10−10 per base pair per generation (Poisson 95% confidence interval, 0.55 × 10−10 to 1.09 × 10−10). This value is similar to those found in other gammaproteobacteria, including Escherichia coli (29), Vibrio spp. (30), and Pseudomonas aeruginosa (31), for which estimates from MA experiments range from 0.7 × 10−10 to 2.1 × 10−10 substitutions per base pair per generation. Other mutational signatures in ADP1 were also similar to those observed in other bacterial species. Transitions were more common than transversions, and G·C→A·T base pair substitutions were the dominant type of point mutation (Fig. 1B).
Of the 29 base substitutions in protein-coding regions, 8 were synonymous. If these mutations were redistributed purely by chance in coding portions of the ADP1 genome while preserving the observed number of each type of base pair substitution (e.g., 20 G·C→A·T mutations), then 13.8% to 37.9% of the mutations would be expected to be synonymous (95% confidence interval). Thus, the observation of 27.6% synonymous mutations is compatible with greatly reduced selection for fitness during the experiment.
Of the 66 total mutations, 7 were insertions of IS1236 elements at new locations in the genome and 10 were deletions with at least one end adjacent to an IS1236 element (Fig. 1A). These 17 IS-mediated mutations were spread throughout 12 of the 17 sequenced clones with a single event per genome, except that one clone had five of these mutations and another had two. This proportion of IS-mediated mutations (17/66) was not significantly different from that observed for a set of largely beneficial mutations (30/75) found in a previous adaptive evolution experiment (25) (two-tailed Fisher's exact test; P = 0.073), implying that, when considered together, IS element-mediated mutations are not systematically much more or less beneficial than other types of mutations in A. baylyi. Four of the IS1236-mediated deletions were recombination events between the two IS elements flanking Tn5613 that eliminated its two cargo genes of unknown function. These deletions were also commonly observed in the adaptive evolution experiment (25). The remaining 13 nonpoint mutations included 6 indels of ≤50 bp, 5 larger deletions, and two larger duplications (Fig. 1A).
ADP1-ISx strain construction.
To eliminate IS elements as a source of mutations that could inactivate engineered DNA constructs, we deleted all six of the IS1236 elements in the ADP1 genome in five sequential cycles of genome editing to create strain ADP1-ISx (Fig. 2A). To avoid the unwanted spread of new IS1236 copies into unknown regions of the genome during strain construction, we used quantitative PCR (qPCR) to verify that the IS1236 copy number in the genome decreased after each deletion (Fig. 2B).
FIG 2.
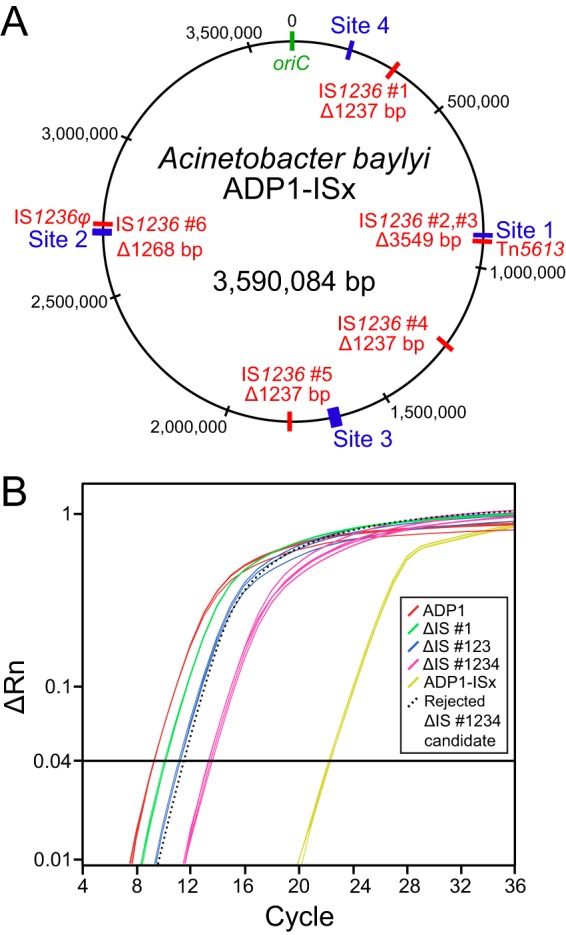
Construction of A. baylyi ADP1-ISx. (A) Five unmarked genomic deletions were made to remove all six IS1236 elements, designated by numbers 1 to 6, found in the wild-type ADP1 chromosome (shown in red) to create the ADP1-ISx strain. Sites 1 to 4 (shown in blue) were used for transformation and mutation assays. IS1236 elements number 2 and number 3 form a composite transposon (Tn5613), and element number 6 is inactive (IS1236φ). (B) Example of qPCR data used to monitor IS element deletion steps. The six IS copies per genome found in wild-type ADP1 register above a given fluorescence threshold (ΔRn value) during early PCR cycles when amplifying a 119-bp fragment located within the IS1236 transposase gene. The sequential removal of IS elements in the deletion strains progressively increases the number of cycles necessary to reach this threshold from the same input quantity of genomic DNA. One example of a rejected candidate strain that accumulated a new IS element insertion elsewhere in the genome such that its IS copy number did not decrease after the deletion of IS number 4 is shown (dotted line).
Whole-genome sequencing of candidate ADP1-ISx strains revealed that they were free of IS1236 elements but had all accumulated two secondary mutations (see Table S1 in the supplemental material). The altered genes were rpoD, which sustained an in-frame deletion of 3 bp in an (AAG)3 repeat, and cyoB, which acquired a nonsynonymous point mutation. RpoD is the ADP1 homolog of σ70, the major housekeeping sigma factor (32). CyoB is a subunit of the cytochrome bo terminal oxidase complex that is involved in respiration (33).
The ADP1 genome-editing procedure involves repeated transformations, growth of liquid cultures in LB broth, and growth of colonies on LB agar in the presence of kanamycin (Kan) or 3′-azido-2′,3′-dideoxythymidine (AZT) (1). Secondary mutations can accumulate by chance if they happen alongside the desired edits whenever a single colony is picked during this procedure (much like in the MA experiment). Alternatively, given the important roles of rpoD and cyoB in aerobic growth, it is possible that one or both of these mutations might have been favored under some or all of these culture conditions. In particular, the cyoB mutation may have been beneficial in response to repeated kanamycin exposure (34).
Under normal laboratory conditions, an ADP1-ISx candidate strain with these two mutations exhibited greatly compromised growth (Fig. 3). It had significantly longer lag times and doubling times in liquid cultures than wild-type ADP1 in both LB broth and minimal succinate (MS) defined medium (P values were <10−6 for two-tailed tests on each parameter in both media, as described in Materials and Methods). For example, the doubling time of the ADP1-ISx-rpoD-cyoB strain was 65 min in LB broth compared to 35 min for wild-type ADP1. Therefore, we reasoned that it might be possible to repair one or both of these mutations by transforming an ADP1 culture with PCR products containing the wild-type versions of the two genes and then passaging the cells. After <30 generations of serial transfer and regrowth in LB broth, we found that every large colony that we sequenced had reverted to wild-type versions of both genes. Whole-genome sequencing verified that one of the colonies now had no mutations in its genome aside from the five planned deletions of IS1236 elements. Growth curves of this final ADP1-ISx strain (Fig. 3) showed that its doubling time during exponential growth was now indistinguishable from that of wild-type ADP1 in both LB and MS media (P = 0.94 and P = 0.82, respectively). Interestingly, ADP1-ISx was able to grow to saturation more rapidly because it had a significantly shorter lag time than wild-type ADP1 in both LB and MS media (P = 0.011 and P < 10−6, respectively).
FIG 3.
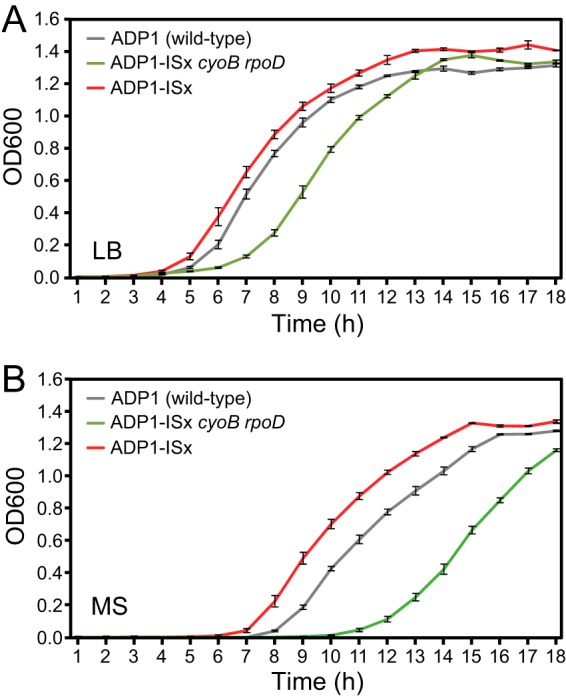
Repair of unplanned cyoB and rpoD mutations sustained during ADP1-ISx strain construction. Growth curves in LB broth (A) and MS defined medium (B) for wild-type ADP1, ADP1-ISx with the cyoB and rpoD mutations, and ADP1-ISx with these genes repaired to the wild-type sequence (as verified by whole-genome sequencing) were recorded by monitoring the OD600. The error bars represent standard deviations of three biological replicates.
Reduced rate of inactivating mutations in ADP1-ISx.
We next used a forward-mutation assay to determine if mutation rates were reduced in the engineered ADP1-ISx strain. Specifically, we measured the rates of loss-of-function mutations in a tdk reporter gene when it was integrated at several different chromosomal locations in each strain background (Fig. 2A). Loss of the function of this gene gives resistance to killing by azidothymidine (AZTr). We found ∼21-fold-reduced (site 1), ∼7-fold-reduced (site 2), and ∼13-fold-reduced (site 3) mutation rates in ADP1-ISx compared to wild-type ADP1 (Fig. 4A).
FIG 4.
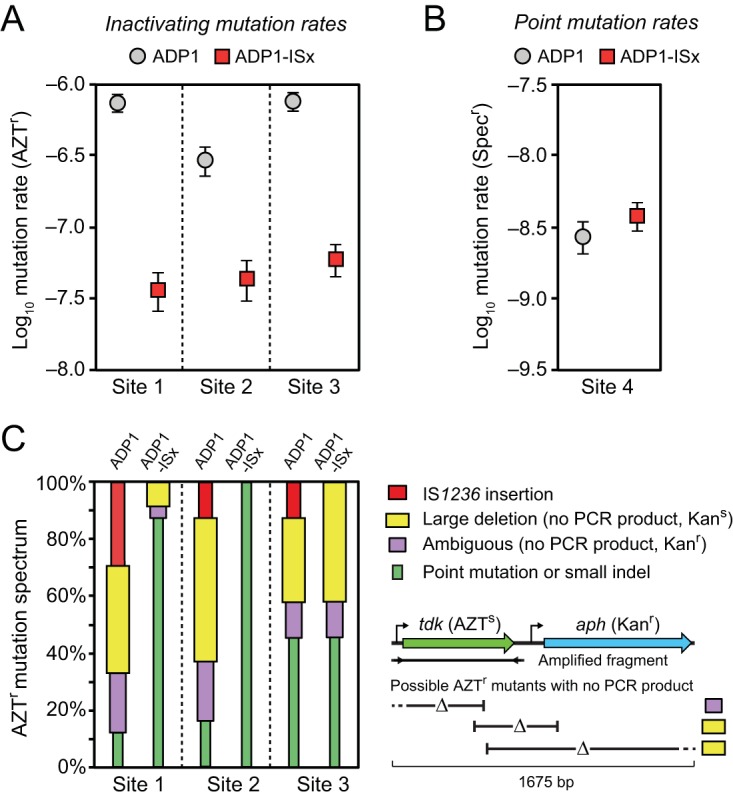
Reduced rates of inactivating mutations in ADP1-ISx. (A) Rates of mutations leading to loss of function of a counterselectable marker gene (tdk) were determined using fluctuation tests that selected for resistance to azidothymidine (AZTr). The error bars are 95% confidence intervals. The marker was placed at three different sites in the A. baylyi genome that differed in how near they were to IS1236 elements in the wild-type ADP1 genome (Fig. 2A). (B) Mutation rates were measured for reversion of a stop codon in the leader region of an antibiotic marker gene that restored spectinomycin resistance (Specr). This assay is expected to primarily reflect the point mutation rate. The error bars are 95% confidence intervals. (C) Spectrum of inactivating mutations determined by using PCR to amplify a fragment containing the tdk gene from the genome of one AZTr mutant from each of the 24 biological replicates of the fluctuation tests in panel A. IS1236 mutations and point mutations were inferred from an expected size change or no size change in this fragment, respectively. When no PCR product resulted and the function of the adjacent aph marker gene that was inserted alongside the tdk gene was also lost (yielding a Kans versus a Kanr phenotype), then the inactivating mutation was inferred to be a large deletion. If there was no PCR product and the mutant remained Kanr, then it was likely a large deletion that overlapped the tdk gene but not the aph gene. However, since the PCR could have failed to amplify a band for other reasons, we conservatively classified this as an “ambiguous” result. This logic for classifying mutations is illustrated in the schematic below the legend and further explained in the text (see “Altered mutational spectrum in ADP1-ISx”). As expected, no IS1236 insertions were found in ADP1-ISx. The proportion of large deletions among the inactivating mutations was also greatly reduced at site 1 and site 2.
Variation among the mutation rates measured at each of these three sites is consistent with greater stabilization of the ADP1-ISx chromosome at locations that are nearer to active IS1236 elements in wild-type ADP1. The largest mutation rate reduction was found at site 1, which is only 1.3 kb away from Tn5613. Site 3 is located 76.9 kb from the nearest IS element and has an intermediate level of reduction in mutation rates. Site 2, which exhibited the smallest mutation rate reduction, is also located very near an IS element, but it is the mutated IS1236φ copy. Therefore, this result provides further evidence that the element is inactive. Taking this into account, site 2 is actually the furthest of the three sites from the nearest active IS element, which is 910.4 kb away.
Overall, the rates of inactivating mutations in wild-type ADP1 at different sites in the chromosome varied by around 3-fold, and these mutation rates were highest at the two sites that were within 100 kb of active IS elements. This range of variation is broadly consistent with those in previous studies that have looked at the proportions of inactivating mutations caused by insertions of IS1236 and Tn5613 at various genetic loci in ADP1 (20, 22). However, all of the measurements comparing ADP1-ISx to wild-type ADP1 found much larger decreases in mutation rates than the ∼1.3-fold expected from the overall contribution of IS elements to mutagenesis found in the MA experiment, where they accounted for just 26% of all spontaneous mutations. Even considering that IS insertions are more likely to disrupt a gene than point mutations, inactivating mutations were much rarer than we expected in ADP1-ISx.
Unchanged point mutation rate in ADP1-ISx.
In order to understand if lower rates of point mutations in ADP1-ISx contributed to the greater than expected improvement in its genetic stability compared to wild-type ADP1, we next performed a reverse-mutation assay (Fig. 4B). Specifically, we inserted a spectinomycin resistance (Specr) gene with a stop codon introduced near the beginning of its reading frame into the chromosome of each strain at site 4 (Fig. 2A). Selecting for spectinomycin-resistant mutants was expected to be specific for measuring the rates of base substitutions that restore gene expression by changing the stop codon to a sense codon. The mutation rate estimated for ADP1-ISx (3.8 × 10−9 per cell per generation) for this assay was slightly higher than that found for wild-type ADP1 (2.7 × 10−9 per cell per generation), but this difference in mutation rates was not statistically significant (P = 0.054; likelihood ratio test) (35). Thus, point mutation rates appear to be unchanged or, if anything, slightly higher in ADP1-ISx.
Altered mutational spectrum in ADP1-ISx.
Next, we compared the types of inactivating mutations that occurred in ADP1-ISx versus wild-type ADP1 (Fig. 4C). For each of the six test strains, we analyzed 24 independent AZTr mutants isolated during the fluctuation assays by attempting to amplify a 767-bp PCR product containing the tdk gene from their genomes. Insertion of a new IS1236 copy within the tdk gene would increase the size of this PCR product to ∼2,000 bp. Point mutations and small insertions or deletions in the tdk gene would lead to no visible change in PCR product size. Larger deletions that removed a portion of the tdk gene and adjacent genome regions extending past the primer binding sites would lead to no amplification product. To corroborate these PCR results, we further tested for whether function of the Kanr gene located adjacent to tdk in the reporter cassette was maintained in each mutant. If a strain with no PCR product was also kanamycin sensitive (Kans), then it was further evidence that a larger deletion that overlapped both the tdk gene and the adjacent Kanr marker gene had occurred. If, on the other hand, a strain with no PCR product remained kanamycin resistant, then we classified the mutation as an “ambiguous” diagnosis. This situation could indicate that there was a deletion overlapping the tdk gene but not the Kanr marker gene, or it could be the result of a PCR that failed for some other reason.
As expected, we found no evidence of IS insertions in the ADP-ISx strains, though they were responsible for 12.5% to 29.2% of the mutations in wild-type ADP1, depending on where the reporter gene was integrated into the chromosome. For site 1 and site 2, a large fraction of the inactivating mutations in wild-type ADP1 resulted in no PCR band: 58.3% for site 1 and 70.8% for site 2. A majority of these mutants were also kanamycin sensitive (10/14 for site 1 and 12/17 for site 2), which is consistent with the mutants having large deletions that overlap both the tdk and Kanr genes. The spectrum of mutations shifted markedly in ADP1-ISx. Only point mutations were found at site 2 in ADP1-ISx, and 87.5% of mutations at site 1 were point mutations. Thus, deletion of IS elements from ADP1 resulted in a lower rate of IS insertions, as expected, but also fewer large deletions, which are apparently mediated by the action of IS elements. This result could potentially explain the greater than expected reduction in mutation rates in ADP1-ISx.
Site 3 exhibited a different change in mutational signature. Here, there were fewer mutants with no PCR band in wild-type ADP1 (41.7%), and the proportion of these mutants was similar in ADP1-ISx (54.2%). Again, these mutants were mostly kanamycin sensitive, 7/10 for wild-type ADP1 and 10/13 for ADP1-ISx. These results are consistent with large deletions occurring at this site via a mechanism that is not directly dependent on IS activity. We found a 14/15-bp match for a sequence within the aph (Kanr) gene to a region 3,108 bp upstream of the tdk-Kanr gene cassette insertion site that is a candidate for homology that might mediate a deletion hot spot there (36). However, it remains unclear why there is an overall decrease in the mutation rate at site 3 without a commensurate shift in the mutational spectrum away from IS-related mutations.
Increased transformability with reduced autolysis in ADP1-ISx.
As we constructed strains for testing mutation rates by transforming reporter cassettes, we noticed that this process appeared to be more efficient in ADP1-ISx. Directly testing for a change showed that ADP1-ISx exhibited significantly increased transformation frequencies for PCR products containing the tdk-Kanr gene cassette (into site 2) or a Specr gene (into site 4) (Fig. 5A). ADP1-ISx was 7.6-fold and 3.3-fold more transformable than wild-type ADP1 in the site 2 and site 4 assays, respectively.
FIG 5.

ADP1-ISx exhibits increased transformability and reduced autolysis. (A) Transformation frequencies measured as transformants per CFU under normal growth conditions in LB medium for PCR products containing the tdk-Kanr cassette (into site 2) or a spectinomycin resistance marker (into site 4). (B) CFU measured by plating dilutions of cultures on LB agar after saturating growth. (C) Reduced settling behavior of ADP1-ISx compared to ADP1 in saturated cultures left at room temperature for 48 h after growth. (D) Accumulation of extracellular DNA over time in the supernatants of MS cultures grown under standard conditions. (E) Fraction of cells staining as dead in LB cultures after 24 h of growth. The error bars are 95% confidence intervals.
We also observed a slight but significant increase in the final optical density (OD) of cultures of ADP1-ISx (Fig. 3) in both LB and MS media (P = 0.00098 and P = 0.0028; two-tailed Welch's t tests comparing ODs after 18 h in LB and MS media, respectively). In further exploring this difference, we found that there were ∼4 times as many CFU in saturated LB cultures of ADP1-ISx as there were for wild-type ADP1 (Fig. 5B). Additionally, when these cultures were left unshaken on a bench, wild-type ADP1 settled to a pellet in the bottom while ADP1-ISx cells remained in suspension for days (Fig. 5C). We have seen differences in cellular aggregation in evolved ADP1 strains previously, though it was increased aggregation in this case (25). Taken together, these observations seemed to indicate that there were either more viable cells, less aggregation of those cells (leading to more CFU per cell), or both in ADP1-ISx cultures.
One possible explanation for the reduced viable-cell counts and transformability of wild-type ADP1 compared to ADP1-ISx could be that deleting IS1236 elements decreases autolysis. Genomic DNA released from dead cells would be expected to compete with the exogenous DNA added in transformation assays for uptake into cells. In line with this hypothesis, we found that the concentrations of extracellular DNA that accumulated in cultures grown in MS medium were higher for wild-type ADP1 (154 ng/ml) than they were in ADP1-ISx (63.2 ng/ml) at 24 h, and this difference became even more pronounced after 48 h (Fig. 5D). LIVE/DEAD staining found large amounts of autolysis in normal cultures of wild-type ADP1 grown in LB broth, while ADP1-ISx cultures exhibited much less cell death (Fig. 5E). The same trend was found in MS medium, though the fraction of cells staining as dead was lower for both strains under these conditions: 6.4% ± 4.6% for wild-type ADP1 and 1.8% ± 1.4% for ADP-ISx (95% CI). Thus, reduced autolysis of ADP1-ISx may contribute to its improved transformability.
It is also possible that the improved transformability of ADP1-ISx is due to other factors. For example, the activity of IS1236 elements could inhibit successful transformation after exogenous DNA is inside a cell. This effect could be direct, if the IS1236 transposase interacts with or inactivates DNA structures that are intermediates involved in successful integration into the chromosome. Alternatively, IS1236 activity could indirectly modulate DNA repair pathways that are also required for transformation (37), by drawing the necessary factors away to sites of transposition, for example. Increased aggregation of cells in a wild-type ADP1 culture compared to an ADP1-ISx culture related to the settling phenotype might also reduce transformability by restricting the surface area of ADP1 cells that is accessible to added DNA for uptake.
Importantly, increased aggregation might complicate our measurements of transformation frequencies and mutation rates if 1 CFU contains multiple (and more) cells for wild-type ADP1 compared to ADP1-ISx. Only one cell in an aggregate needs to be transformed or mutated for it to form a colony on a selective plate, yet it is counted as a single CFU on a nonselective plate regardless of the number of cells in the aggregate. The consequence is that what are normally assumed to be “per-cell” transformation and mutation rates will be overestimated for a strain exhibiting more aggregation because they actually reflect “per-aggregate” rates. Essentially, there are multiple chances (one for each cell) for the aggregate as a whole to give a signal of transformation or mutation.
Considering the potential effect of aggregation on our measurements, it is clear that it could not explain our finding that ADP1-ISx is more transformable than wild-type ADP1. The opposite relationship, with ADP1 appearing to be more transformable, would be expected if aggregation were the only factor. In contrast, an aggregation artifact could potentially explain why there was a larger decrease in the apparent per-cell inactivating mutation rates estimated for ADP1-ISx relative to wild-type ADP1 than was expected from the overall contribution of IS1236 to mutagenesis (Fig. 4A), including the large decrease at site 3 without much change in the mutational spectrum (Fig. 4C). However, we would also expect to find a commensurate difference in our measurements of point mutation rates between wild-type ADP1 and ADP1-IS, and we did not (Fig. 4B). Therefore, we conclude that any change in aggregation between wild-type ADP1 and ADP1-ISx does not explain the dominant trends we see in these measurements.
Deleting error-prone polymerases does not further reduce the mutation rate.
Deletion of the three stress-induced error-prone polymerases Pol II, Pol IV, and Pol V from IS-less E. coli strain MDS42 has been shown to reduce its inactivating mutation rate by approximately 50% (38). We investigated whether a similar improvement was possible in ADP1-ISx. ADP1 encodes homologs of two of these E. coli polymerases: Pol IV (dinP) and Pol V (umuDC). However, the polymerase function of the umuDC complex is no longer intact. ADP1 umuC is a pseudogene interrupted by a transposase fragment derived from an ISEhe3-like element (39). The size and regulation of the ADP1 umuD gene differs from that of E. coli, and this distinct protein subfamily (encoded by a gene designated umuDAb [the umuD gene of A. baylyi]) acts as a regulator of the DNA damage response in Acinetobacter species (39–41).
We created ADP1-ISx strains with dinP and umuDAb deleted, singly and in combination. No significant difference in the rate of inactivating mutations in the tdk gene integrated into the chromosome at site 2 was found among these three deletion strains (Fig. 6). To test whether there was a difference in mutagenesis under conditions in which error-prone polymerase expression might be induced to a higher level, we repeated the assay in the presence of subinhibitory concentrations of nalidixic acid. Nalidixic acid causes double-strand breaks and thereby induces the expression of error-prone polymerases via the SOS response in E. coli and other bacteria (42). We found that nalidixic acid increased the rate of mutations in the ΔumuDAb mutant and ADP1-ISx to the same extent. Thus, deleting dinP and umuDAb appears to have no benefit with respect to further reducing the mutation rate of ADP1-ISx.
FIG 6.
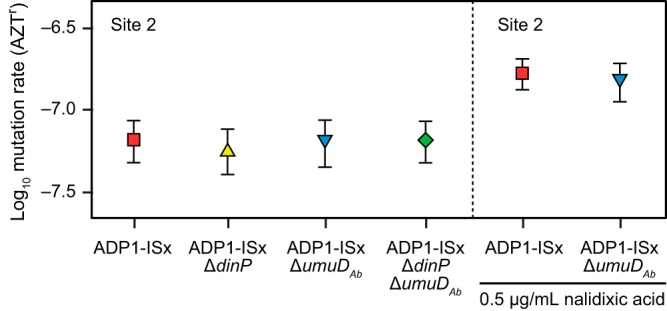
Inactivating mutation rates are unchanged in ADP1-ISx after deletion of error-prone polymerase DinP and DNA damage response regulator UmuDAb. The effects of deleting each gene on the rates of mutations inactivating the tdk counterselectable marker gene inserted into the genome at site 2 were measured for each strain. Mutation rates in ADP1-ISx and the umuDAb deletion strain were also determined in the presence of a subinhibitory concentration of nalidixic acid, which induces double-strand breaks in DNA. The error bars are 95% confidence intervals.
Comparison to other clean-genome bacterial strains.
Several other studies have deleted selfish DNA elements and nonessential genes from bacterial genomes to enhance the biotechnology performance characteristics of the bacteria (43–52). One comprehensive effort has been the development of the streamlined E. coli strain MDS42 (43). MDS42 was derived from E. coli K-12 by deleting all active IS elements, all prophage remnants, and many nonessential genes from its chromosome. The rates of mutations inactivating a chromosomal reporter gene are lower in MDS42 than in its progenitor strain (43), and these mutation rates were further reduced in subsequent work by deleting all three error-prone polymerases from its genome (38). Genetic stability is improved in MDS42 to the point that plasmids encoding highly toxic proteins that are rapidly inactivated in wild-type E. coli can be propagated in the strain (17). MDS42 also exhibited an unanticipated increase in electroporation efficiency. The exact reason for this improvement is unknown, as many genes were deleted in the strain, but it is thought to result from the deletion of fimbriae that might inhibit access of DNA to the cell surface.
Pseudomonas putida EM383, a streamlined chassis strain derived from KT2440, also showed enhanced properties after the deletion of flagellar genes, four prophages, and both of its active transposons (Tn7 and Tn4652) (52). EM383 exhibits increased retention of functional plasmids during prolonged culture and has improved growth characteristics. Similarly, deleting either of two families of active IS elements from the Corynebacterium glutamicum ATCC 13032 genome improved its properties (44). The resulting WJ004 (with all four ISCg1 elements deleted) and WJ008 (with all four ISCg2 elements deleted) strains exhibited up to 6-fold-increased electroporation efficiency and greater recombinant protein yields while maintaining the same growth rates as the parental strain. Reduced-genome strains of Bacillus subtilis 168 can exhibit improved cell yield and enzyme or chemical production (45). In this species, there is typically a gradual decrease in transformability as large regions of the chromosome are deleted (45, 53). However, this loss can be compensated for, and transformation can even be improved over wild-type levels, by overexpressing competence regulators (54). Overall, our results with ADP1-ISx are broadly similar to the results of these studies of other clean-genome bacterial strains. We have explicitly shown that fewer inactivating mutations occur in ADP1-ISx, and it exhibits an unanticipated improvement in the efficiency of natural transformation.
Conclusions.
Directed evolution is a powerful tool for creating novel biomolecules that do not exist in nature and for optimizing complex cellular systems, but when inadvertent and unchecked, unplanned evolution can impede progress in bioengineering by introducing mutations that lead to unpredictable failures of DNA-encoded functions. Our work constructing ADP1-ISx further highlights the effectiveness of a rational genome “cleanup” strategy to improve a cellular chassis and the collateral benefits that often accompany domesticating an organism in this way. In microbial species that harbor a manageable number of IS elements, deleting these agents of instability may often be the simplest fail-safe approach for a high payoff in terms of improving genome stability and cell productivity. A. baylyi ADP1-ISx is an upgraded chassis for synthetic biology and genetic studies that wish to take advantage of its natural transformability.
MATERIALS AND METHODS
Strains and culture conditions.
A. baylyi ADP1 cultures were grown in LB or MS medium at 30°C with orbital shaking, as previously described (25). The media were supplemented with 50 μg/ml Kan, 60 μg/ml Spec, or 200 μg/ml AZT, as specified. Engineered ADP1 strains were archived at −80°C in 15 to 20% (vol/vol) glycerol. Prior to all experiments, cell stocks were thawed on ice, diluted 1:1,000 in the appropriate media, and grown overnight. Then, these revived cultures were diluted 1:1,000 and grown for an additional 24 ± 1 h for preconditioning to the culture conditions of an experiment. Cell dilutions for plating on agar were made in sterile saline (0.85% [wt/vol] NaCl).
MA experiment.
The MA lines were generated in prior work (25). Briefly, 18 ADP1 lineages were passaged through 300 daily single-cell bottlenecks by picking an arbitrary colony from LB agar, streaking it out to single colonies, and allowing regrowth for 24 h before selecting the next colony to transfer. This procedure gives ∼25 cell divisions (generations of regrowth) each day for an estimated total of ∼7,500 generations of evolution over the course of the entire MA experiment.
Genome sequencing.
DNA was isolated from evolved clones from the MA experiment and genome-edited clones from the construction of ADP1-ISx and then prepared for sequencing as described previously (25). These samples were sequenced to >100× coverage using the Illumina HiSeq 2500 platform at the University of Texas at Austin Genome Analysis and Sequencing Facility (GSAF). Read files were analyzed by using the breseq computational pipeline (v0.28.0) (55, 56) to predict mutations, including IS element insertions and other types of structural variation, relative to the ADP1 reference genome (GenBank accession no. NC_005966.1) (19). When counting mutations in the MA experiment, we removed genome differences that we previously identified as being in our original laboratory strain (25), and we also did not include an insertion of a single G that was present in 15 of the 17 sequenced genomes at a site 74 bp upstream of the atpI gene because it was most likely already present in the original culture that was used to found these lineages. Statistical tests were performed in R (v3.3.2) (57). The final mutation predictions are provided in Table S1 in the supplemental material.
Genome modification.
Enzymes were purchased from New England BioLabs (Ipswich, MA). Phusion polymerase was used for all PCRs. Primer sequences are provided in Table 1. The tdk-Kanr gene dual-selection cassette was PCR amplified from the per gene knockout strain from the ADP1 single-gene deletion collection (58) using primers P1-F and P2-R. For each IS or gene deletion, ∼1,000 bp of flanking sequence on each side of the deleted region was amplified in two additional PCRs. Primers P3-F and P1′P4-R were used for the upstream region, and primers P2′P5-F and P6-R were used for the downstream region. The PCR products were cleaned up using the GeneJet purification kit (Thermo Scientific). The two flanking-region products and the tdk-Kanr gene cassette were joined by overlap PCR to create the “knockout cassette.” Two PCR products for the flanking regions were joined in a separate overlap PCR to produce the “rescue cassette” used for scarless tdk-Kanr gene cassette removal. To add overlap in this case, the upstream region was amplified with primers P3-F and P5′P4-R, and the downstream region was amplified with primers P4′P5-F and P6-R. Each type of overlap PCR amplification began with 15 temperature cycles with only the templates present (no primer addition), followed by 20 further cycles with the external primers P3-F and P6-R. The final amplified cassettes were digested with 40 U DpnI by adding 2 μl of enzyme to each 50-μl PCR mixture and incubating for 1 h at 37°C. Then, they were purified again using the GeneJet PCR purification kit before transformation.
TABLE 1.
Primers used in this study
| Primer | Description | Sequence |
|---|---|---|
| tdk-Kanr gene cassette | ||
| P1-F | tdk-Kanr gene amplification for deletion and rescue cassettes | CCCAGCCTCCAATTCAAATCA |
| P2-R | tdk-Kanr gene amplification for deletion and rescue cassettes | CCAGCTCCGCATGCTTAGAA |
| tdk-R | Used with P1-F to amplify just the tdk gene from the tdk-KanR gene cassette | ACATCAGAGATTTTGAGACACAACGTG |
| Deletion of IS1236 elements to create ISx-ADP1 | ||
| IS#1-P3-F | Amplifies 5′ flank for IS1236 no. 1 deletion and rescue cassettes | GCTTGAACCTCACTCCTGA |
| IS#1-P1′P4-R | Amplifies 5′ flank with IS#1-P3-F for IS1236 no. 1 deletion cassette | TGATTTGAATTGGAGGCTGGGACAATTGCATAAAAATGAAAGTAACTC |
| IS#1-P2′P5-F | Amplifies 3′ flank with IS#1-P6-R for IS1236 no. 1 deletion cassette | TTCTAAGCATGCGGAGCTGGTGTCCAGTCAATTGCTTTAATG |
| IS#1-P6-R | Amplifies 3′ flank for IS1236 no. 1 deletion and rescue cassettes | TCGATAGACTAAAATGACTAGACAGG |
| IS#1-P5′P4-R | Amplifies 5′ flank with IS#1-P3-F for IS1236 no. 1 deletion cassette | CATTAAAGCAATTGACTGGACAACAATTGCATAAAAATGAAAGTAACTC |
| IS#1-P4′P5-F | Amplifies 3′ flank with IS#1-P6-R for IS1236 no. 1 deletion cassette | GAGTTACTTTCATTTTTATGCAATTGTTGTCCAGTCAATTGCTTTAATG |
| IS#23-P3-F | Amplifies 5′ flank for Tn5613 deletion and rescue cassettes | TAAAGGACCTGATAAGGCGAT |
| IS#23-P1′P4-R | Amplifies 5′ flank with IS#23-P3-F for Tn5613 deletion cassette | TGATTTGAATTGGAGGCTGGGCTTTAATGATTAAATAATCAATTTTCATCT |
| IS#23-P2′P5-F | Amplifies 3′ flank with IS#23-P6-R for Tn5613 deletion cassette | TTCTAAGCATGCGGAGCTGGTCAATTGACCGCTTGATCA |
| IS#23-P6-R | Amplifies 3′ flank for Tn5613 deletion and rescue cassettes | GATCTATTTGGCAATATCATAAACATC |
| IS#23-P5′P4-R | Amplifies 5′ flank with IS#23-P3-F for Tn5613 deletion cassette | TGATCAAGCGGTCAATTGACTTTAATGATTAAATAATCAATTTTCATCT |
| IS#23-P4′P5-F | Amplifies 3′ flank with IS#23-P6-R for Tn5613 deletion cassette | AGATGAAAATTGATTATTTAATCATTAAAGTCAATTGACCGCTTGATCA |
| IS#4-P3-F | Amplifies 5′ flank for IS1236 no. 4 deletion and rescue cassettes | GGCGATTAAGAGAGTAATTCCA |
| IS#4-P1′P4-R | Amplifies 5′ flank with IS#4-P3-F for IS1236 no. 4 deletion cassette | TGATTTGAATTGGAGGCTGGGATGAACATCTTCAACATTTAGATCAAG |
| IS#4-P2′P5-F | Amplifies 3′ flank with IS#4-P6-R for IS1236 no. 4 deletion cassette | TTCTAAGCATGCGGAGCTGGCATGCATTGCACGTTCG |
| IS#4-P6-R | Amplifies 3′ flank for IS1236 no. 4 deletion and rescue cassettes | AATGCCAGATCTACACTGACG |
| IS#4-P5′P4-R | Amplifies 5′ flank with IS#4-P3-F for IS1236 no. 4 deletion cassette | CGAACGTGCAATGCATGATGAACATCTTCAACATTTAGATCAAG |
| IS#4-P4′P5-F | Amplifies 3′ flank with IS#4-P6-R for IS1236 no. 4 deletion cassette | CTTGATCTAAATGTTGAAGATGTTCATCATGCATTGCACGTTCG |
| IS#5-P3-F | Amplifies 5′ flank for IS1236 no. 5 deletion and rescue cassettes | GAATTTAAAGTTAAAGATATTTTTGCG |
| IS#5-P1′P4-R | Amplifies 5′ flank with IS#5-P3-F for IS1236 no. 5 deletion cassette | TGATTTGAATTGGAGGCTGGGGTTTGAGTTTATCGGAAAAACTATC |
| IS#5-P2′P5-F | Amplifies 3′ flank with IS#5-P6-R for IS1236 no. 5 deletion cassette | TTCTAAGCATGCGGAGCTGGAACAAAAGGCCATGGAG |
| IS#5-P6-R | Amplifies 3′ flank for IS1236 no. 5 deletion and rescue cassettes | GTACCTTGCCGAGCAAA |
| IS#5-P5′P4-R | Amplifies 5′ flank with IS#5-P3-F for IS1236 no. 5 deletion cassette | CTCCATGGCCTTTTGTTGTTTGAGTTTATCGGAAAAACTATC |
| IS#5-P4′P5-F | Amplifies 3′ flank with IS#5-P6-R for IS1236 no. 5 deletion cassette | GATAGTTTTTCCGATAAACTCAAACAACAAAAGGCCATGGAG |
| IS#6-P3-F | Amplifies 5′ flank for IS1236 no. 6 deletion and rescue cassettes | GGAGTATCGTGATCCCAAAG |
| IS#6-P1′P4-R | Amplifies 5′ flank with IS#6-P3-F for IS1236 no. 6 deletion cassette | TGATTTGAATTGGAGGCTGGGCTACTCAGTTAGTCGAAAGTATCATCC |
| IS#6-P2′P5-F | Amplifies 3′ flank with IS#6-P6-R for IS1236 no. 6 deletion cassette | TTCTAAGCATGCGGAGCTGGGTTCAAGTGGGGTTTACTGG |
| IS#6-P6-R | Amplifies 3′ flank for IS1236 no. 6 deletion and rescue cassettes | CAGTGATATGCATGCTGAGG |
| IS#6-P5′P4-R | Amplifies 5′ flank with IS#6-P3-F for IS1236 no. 6 deletion cassette | CCAGTAAACCCCACTTGAACCTACTCAGTTAGTCGAAAGTATCATCC |
| IS#6-P4′P5-F | Amplifies 3′ flank with IS#6-P6-R for IS1236 no. 6 deletion cassette | GGATGATACTTTCGACTAACTGAGTAGGTTCAAGTGGGGTTTACTGG |
| qPCR to monitor IS1236 deletion | ||
| qIS-F | Amplifies 119-bp fragment common to all IS1236 elements with qIS-R | GTTTTCGCCAGGCATAATA |
| qIS-R | Amplifies 119-bp fragment common to all IS1236 elements with qIS-F | CAGATCATGCCAAGAAAAGTAC |
| Repair of rpoD and cyoB mutations | ||
| rpoD-F | Amplifies fragment containing rpoD mutation with rpoD-R | GCCTTCAAACAATTCACTTAACC |
| rpoD-R | Amplifies fragment containing rpoD mutation with rpoD-F | GCAATCGAAATAACGAGACG |
| rpoD-S | Sequence mutation in rpoD gene | GCTGAGGTTAACGATCATCT |
| cyoB-F | Amplifies fragment containing cyoB mutation with cyoB-R | GGTACGCTTTTATCTGGTGTAA |
| cyoB-R | Amplifies fragment containing cyoB mutation with cyoB-F | CAGCAGAATTATGATCATGACTC |
| cyoB-S | Sequence mutation in cyoB gene | GACACGTCGTTTGAATACAT |
| Integration of tdk-Kanr gene cassette for measuring inactivating mutation rates and transformation frequency | ||
| Site1-P3-F | Amplifies 5′ flank for integration of tdk-Kanr gene cassette | ATAAAAAATTTTTTATTATAATTAATAATTAAGTTGTG |
| Site1-P1′P4-R | Amplifies 5′ flank with Site1-P3-F for integration of tdk-Kanr gene cassette | TGATTTGAATTGGAGGCTGGGATCTAAACCTTGTGATATCAACTGTT |
| Site1-P2′P5-F | Amplifies 3′ flank with Site1-P6-R for integration of tdk-Kanr gene cassette | TTCTAAGCATGCGGAGCTGGGTGCAAAGAATATTCATCTGAATG |
| Site1-P6-R | Amplifies 3′ flank for integration of tdk-Kanr gene cassette | TACTTCAGATTCTGGGAATTTCT |
| Site2-P3-F | Amplifies 5′ flank for integration of tdk-Kanr gene cassette | CAAATCCATTGAAAAGAAATGC |
| Site2-P1′P4-R | Amplifies 5′ flank with Site2-P3-F for integration of tdk-Kanr gene cassette | TGATTTGAATTGGAGGCTGGGCTGATCATCATGGACAAGCT |
| Site2-P2′P5-F | Amplifies 3′ flank with Site2-P6-R for integration of tdk-Kanr gene cassette | TTCTAAGCATGCGGAGCTGGTGTGATCTTTTTTTCAAATTCATT |
| Site2-P6-R | Amplifies 3′ flank for integration of tdk-Kanr gene cassette | GGTAATGTTCTCTCCTACGAAAAG |
| Site3-P3-F | Amplifies 5′ flank for integration of tdk-Kanr gene cassette | CCTGATCCAGAAAGATATATGACTT |
| Site3-P1′P4-R | Amplifies 5′ flank with Site3-P3-F for integration of tdk-Kanr gene cassette | TGATTTGAATTGGAGGCTGGGTGAAAAGTTATCAAATATTGACGTATG |
| Site3-P2′P5-F | Amplifies 3′ flank with Site3-P6-R for integration of tdk-Kanr gene cassette | TTCTAAGCATGCGGAGCTGGTAAAGTGATCGAGCAGCCA |
| Site3-P6-R | Amplifies 3′ flank for integration of tdk-Kanr gene cassette | AACTATACATATTGAATTTTTAAATAATAATTTAATTC |
| Integration of Specr genes in region 1 for measuring point mutation rates and transformation frequency | ||
| R1-P3-F | Amplifies R1 5′ flank for integration of tdk-Kanr gene cassette | ACGCCGAGTCCTCTTGAGTACAGG |
| R1-P1′P4-R | Amplifies R1 5′ flank with Site1-P3-F for integration of tdk-Kanr gene cassette | TGATTTGAATTGGAGGCTGGGGATTTTCCGCCCATCTCAC |
| R1-P2′P5-F | Amplifies R1 3′ flank with Site1-P6-R for integration of tdk-Kanr gene cassette | TTCTAAGCATGCGGAGCTGGCAGAAATTATAAAACGCACATCA |
| R1-P6-R | Amplifies R1 3′ flank for integration of tdk-Kanr gene cassette | ACTCGCTGCAATAGTGGCAAAAGC |
| Specr-F | Amplifies Specr gene to construct rescue cassette | TTCAAATATGTATCCGCTCATGAGACAATAAC |
| Specr-R | Amplifies Specr gene to construct rescue cassette | TTATTTGCCGACTACCTTGGTGATC |
| Specr-E8*-R | Amplifies 5′ end of Specr gene with Specr-F to introduce stop codon | CTGATAGTTGAGTCGATACTTAGGCGA |
| Specr-E8*-F | Amplifies 3′ end of Specr gene with Specr-R to introduce stop codon | GCCTAAGTATCGACTCAACTATCAGAGGTA |
| Specr-R1-R | Amplifies R1 5′ flank with R1-P3-F to create overlap with Specr gene | ATGAGCGGATACATATTTGAAGATTTTCCGCCCATCTCAC |
| Specr-R1-F | Amplifies R1 3′ flank with R1-P6-R to create overlap with Specr gene | ACCAAGGTAGTCGGCAAATAATCTTTACGAGTCATGCCAGCAC |
| Deletion of dinP and umuDAb | ||
| dinP-P3-F | Amplifies 5′ flank for dinP deletion and rescue cassettes | CAAAGCGATAATGTAGAAAAAAC |
| dinP-P1′P4-R | Amplifies 5′ flank with dinP-P3-F for dinP deletion cassette | TGATTTGAATTGGAGGCTGGGTCGTATTTTTTTTACACAAAAA |
| dinP-P2′P5-F | Amplifies 3′ flank with dinP-P6-R for dinP deletion cassette | TTCTAAGCATGCGGAGCTGGTCATTTTACTGTATTTGTGATTT |
| dinP-P6-R | Amplifies 3′ flank for IS1236 no. 1 deletion and rescue cassettes | ATGCTCTCGATGAGTATTGG |
| dinP-P5′P4-R | Amplifies 5′ flank with dinP-P3-F for dinP deletion cassette | CTCAAATCACAAATACAGTAAAATGATCGTATTTTTTTTACACAAAAATTTAA |
| dinP-P4′P5-F | Amplifies 3′ flank with dinP-P6-R for dinP deletion cassette | TTAAATTTTTGTGTAAAAAAAATACGATCATTTTACTGTATTTGTGATTTGAG |
| umuD-P3-F | Amplifies 5′ flank for umuD deletion and rescue cassettes | TTGGCTCCACTACTCACAGA |
| umuD-P1′P4-R | Amplifies 5′ flank with umuD-P3-F for umuD deletion cassette | TGATTTGAATTGGAGGCTGGGTCATGAGTCAGAGAATCTTTGC |
| umuD-P2′P5-F | Amplifies 3′ flank with umuD-P6-R for umuD deletion cassette | TTCTAAGCATGCGGAGCTGGAATGTTTTCTCAAGTTAAAATAATCTAA |
| umuD-P6-R | Amplifies 3′ flank for umuD deletion and rescue cassettes | ATCTATACTAGTAGATTATACGGACGATG |
| umuD-P5′P4-R | Amplifies 5′ flank with umuD-P3-F for umuD deletion cassette | TTAGATTATTTTAACTTGAGAAAACATTTCATGAGTCAGAGAATCTTTGC |
| umuD-P4′P5-F | Amplifies 3′ flank with umuD-P6-R for umuD deletion cassette | GCAAAGATTCTCTGACTCATGAAATGTTTTCTCAAGTTAAAATAATCTAA |
Each genome modification involved two transformation steps. First, 70 μl of ADP1 cells from a preconditioned LB culture of the strain being edited was added to 1 ml of LB broth containing 100 ng of knockout cassette DNA. After overnight growth (16 to 24 h), 100 μl of the transformation culture and 100 μl of a 1:10 dilution were plated on LB-Kan plates, and 100 μl of a 106 dilution were plated on LB medium to monitor transformation frequencies. In most cases, plating 100 μl from a 1:10 dilution of a transformation culture resulted in ∼30 colonies on LB-Kan. In the second step, the integrated tdk-Kanr gene cassette was replaced using the corresponding rescue cassette PCR product with the same transformation procedure, except plating on LB-AZT plates to select for removal of the tdk-Kanr gene cassette. Each step was typically carried out with three or more replicates so that independently derived clones could be screened for success. Whole-cell PCR was used to confirm successful integration and loss of the tdk-Kanr gene cassette at each step.
Each IS element was deleted one at a time using this procedure, except for the two IS1236 elements in Tn5613 (number 2 and number 3), which were deleted simultaneously. This double IS deletion also removed two putative genes of unknown function (ACIAD0955 and ACIAD0956) located between the IS elements and was designed to preserve a stop codon in an adjacent gene of unknown function (ACIAD0959) that overlaps IS1236 number 3.
The dinP and umuDAb deletions were constructed in the repaired ADP1-ISx strain background. The double mutant was made by adding the umuDAb deletion to the ΔdinP strain. The ΔumuDAb single mutant produced small colonies on LB agar and did not reach saturation in LB liquid after 24 h of growth. Its growth defect appears to be due to a secondary mutation that occurred during our strain construction, as ΔumuDAb strains constructed by others have been reported to exhibit normal growth (39). The ΔdinP and ΔdinP ΔumuDAb strains that we constructed grew normally.
qPCR monitoring of IS deletion.
We used qPCR to monitor the relative copy numbers of IS1236 elements during ADP1-ISx strain construction. Genomic DNA (gDNA) was purified from at least three of the clones confirmed by PCR to have a successful deletion of each targeted IS copy. The Qubit dsDNA BR assay kit (Invitrogen) was used to determine DNA concentrations in these samples.
qPCRs were set up with gDNA (1.1 ng/μl), IS-F, and IS-R primers (0.5 μM each) and SYBR green dye real-time PCR master mix (Applied Biosystems). These primers amplify a 119-bp product common to all six IS1236 elements (Table 1). More cycles of PCR amplification (Rn) were required to surpass an arbitrary signal threshold in each successive strain that removed additional IS1236 elements from the ADP1 genome. Clones with a higher Rn value were used to continue the deletion procedure.
Growth curves.
Growth curves for wild-type ADP1 and ADP1-ISx were initiated by making 1:1,000 dilutions of cultures preconditioned in LB or MS medium in 50 ml of the same medium in 250-ml Erlenmeyer flasks. The optical density of samples removed from these cultures was measured at 600 nm (OD600) to monitor growth. These assays were carried out in triplicate. Time points at which the mean of the OD600 values across replicates was less than 0.25 were used for nonlinear least-squares fitting to a model with exponential growth rate and lag time parameters using R (v3.3.2) (57). Differences in growth rates or lag times between two strains were evaluated by simultaneously fitting the OD600 data for both strains to a model that allowed just one global value for this parameter for both strains and then examining the significance of an added offset that allowed for per-strain variation in that parameter.
Reversion of cyoB and rpoD mutations.
Regions of the wild-type ADP1 genome consisting of ∼1,000 bp upstream and ∼1,000 bp downstream of the cyoB or rpoD mutation were amplified by PCR. These PCR products were DpnI digested and gel purified. Then, 250 ng of each PCR product was transformed into the same culture of ADP1-ISx under standard conditions. After 12 h of growth, 100 μl of this culture was transferred into 10 ml fresh LB broth. After 6 h of growth, 100 μl was transferred again into 10 ml LB broth. After a final 6 h of growth, 100 μl of a 106 dilution was plated on LB agar. PCR and Sanger sequencing showed that all 12 of the large colonies picked from this LB plate had reverted to wild-type sequences for both genes. After whole-genome sequencing to confirm that it had not accumulated any new secondary mutations, one of these clones was designated ADP1-ISx and used in all further experiments.
Gene inactivation mutation rate assays.
Three pairs of strains derived from ADP1-ISx and wild-type ADP1 were constructed, each with the tdk-Kanr gene cassette inserted at a different genomic site using the methods described above. For each of these strains, 30 independent 40-μl LB broth cultures, each containing ∼500 CFU from a dilution of a preconditioned culture, were started in 96-well microplates. These plates were sealed with adhesive foil and incubated at 30°C with orbital shaking at 250 rpm. Following overnight growth, the entire volume of 24 of the cultures for each of the six strains was independently plated on LB-AZT plates. A 106 dilution of the remaining six 40-μl cultures of each strain was plated on nonselective LB plates. After incubation at 30°C for 30 h, colonies on the selective and nonselective plates were counted. Mutation rates were calculated from these counts using FALCOR (59).
To compare the types of mutations leading to AZT resistance in each strain, one colony was picked from each selective plate in the fluctuation tests and grown overnight in liquid LB medium. We then used PCR with primers P1-F and tdk-R to amplify the tdk portion of the tdk-Kanr gene cassette from the genome of each of these mutants.
Point mutation rate assays.
We utilized a spectinomycin resistance gene in which the eighth codon was mutated from GAA to the TAA stop codon to measure point mutation rates. To create this Specr-E8* mutational reporter, we amplified the spectinomycin resistance gene from pIM1463 (60) in two halves with primers that introduced the G-to-T mutation in a region common to each PCR product, and then we stitched the two pieces together in a round of overlap PCR. We inserted this marker into the A. baylyi genome in region 1, a site where we had inserted a Kanr gene cassette in a previous study (25). Specifically, strains ADP1-Specr-E8* and ADP1-ISx-Specr-E8* were created by first integrating a tdk-Kanr gene cassette in region 1 and then using the mutant Specr-E8* DNA assembled with regions of flanking homology as a rescue cassette.
The point mutation rate was assessed for ADP1-Specr-E8* and ADP1-ISx-Specr-E8* using Luria-Delbrück fluctuation tests in LB medium. Preconditioned cultures were diluted and used to inoculate 1-ml test tube cultures with ∼30 to 50 cells. After growth for ∼18 to 24 h, cells from 24 of the tubes for each strain were pelleted via centrifugation, and the entire amount was plated on LB-Spec agar. These selective plates were incubated at 30°C for 4 days before counting colonies. The other six replicates were diluted 106 in sterile saline, plated on nonselective LB agar, and incubated overnight before counting. Point mutation rates were calculated and compared using the rSalvador R package (version 1.7) (35, 61).
Transformation frequency.
Six replicate 0.5-ml transformation reaction mixtures were prepared with 250 ng/ml of DNA consisting of the tdk-Kanr gene cassette PCR amplified with ∼1,000-bp flanking homologies targeting site 2 or the unmutated Specr gene cassette amplified with ∼1,000-bp flanking homologies targeting site 4. At the same time, dilutions from every 0.5-ml transformation mixture were plated onto LB agar to acquire CFU counts corresponding to each. Negative controls with DNA but no cells were included to rule out possible contamination issues. All samples and transformation mixtures were incubated for 16 h. Dilutions of each transformation were plated on LB-Kan and LB agar, and transformation frequencies were calculated by taking the ratios of CFU after overnight incubation at 30°C on selective versus nonselective plates.
eDNA concentration.
Eight 5-ml cultures in MS medium were inoculated with a 1:1,000 dilution of preconditioned cells and grown for 48 h. Extracellular-DNA (eDNA) measurements were made using the Qubit high-sensitivity (HS) kit on the supernatant from 1-ml samples of these cultures after centrifuging for 10 min at 21,130 relative centrifugal force (rcf).
LIVE/DEAD staining.
Cells from preconditioned cultures grown for 24 h in MS medium were analyzed using the LIVE/DEAD BacLight bacterial viability and counting kit (Molecular Probes). For each biological replicate, 10,000 events were captured on a BD LSR Fortessa flow cytometer and analyzed with FlowJo (v10.1). We analyzed five biological replicates in LB broth and three biological replicates in MS medium for each strain. LIVE/DEAD gating was determined using dead cell controls prepared by incubating cultures with 70% (vol/vol) isopropanol for 1 h at room temperature.
Accession number(s).
FASTQ files for each sequenced strain have been deposited in the NCBI Sequence Read Archive (accession no. SRP074541).
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health (R00-GM087550), the Defense Advanced Research Projects Agency (HR0011-15-C0095), the National Science Foundation BEACON Center for the Study of Evolution in Action (DBI-0939454), and a National Science Foundation CAREER award (CBET-1554179).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01025-17.
REFERENCES
- 1.Metzgar D, Bacher JM, Pezo V, Reader J, Döring V, Schimmel P, Marlière P, de Crécy-Lagard V. 2004. Acinetobacter sp. ADP1: an ideal model organism for genetic analysis and genome engineering. Nucleic Acids Res 32:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaneechoutte M, Young DM, Ornston LN, De Baere T, Nemec A, Van Der Reijden T, Carr E, Tjernberg I, Dijkshoorn L. 2006. Naturally transformable Acinetobacter sp. strain ADP1 belongs to the newly described species Acinetobacter baylyi. Appl Environ Microbiol 72:932–936. doi: 10.1128/AEM.72.1.932-936.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elliott KT, Neidle EL. 2011. Acinetobacter baylyi ADP1: transforming the choice of model organism. IUBMB Life 63:1075–1080. doi: 10.1002/iub.530. [DOI] [PubMed] [Google Scholar]
- 4.de Berardinis V, Durot M, Weissenbach J, Salanoubat M. 2009. Acinetobacter baylyi ADP1 as a model for metabolic system biology. Curr Opin Microbiol 12:568–576. doi: 10.1016/j.mib.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Young DM, Parke D, Ornston LN. 2005. Opportunities for genetic investigation afforded by Acinetobacter baylyi, a nutritionally versatile bacterial species that is highly competent for natural transformation. Annu Rev Microbiol 59:519–551. doi: 10.1146/annurev.micro.59.051905.105823. [DOI] [PubMed] [Google Scholar]
- 6.Kannisto MS, Mangayil RK, Shrivastava-Bhattacharya A, Pletschke BI, Karp MT, Santala VP. 2015. Metabolic engineering of Acinetobacter baylyi ADP1 for removal of Clostridium butyricum growth inhibitors produced from lignocellulosic hydrolysates. Biotechnol Biofuels 8:198. doi: 10.1186/s13068-015-0389-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Santala S, Efimova E, Kivinen V, Larjo A, Aho T, Karp M, Santala V. 2011. Improved triacylglycerol production in Acinetobacter baylyi ADP1 by metabolic engineering. Microb Cell Fact 10:36. doi: 10.1186/1475-2859-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santala S, Efimova E, Koskinen P, Karp MT, Santala V. 2014. Rewiring the wax ester production pathway of Acinetobacter baylyi ADP1. ACS Synth Biol 3:145–151. doi: 10.1021/sb4000788. [DOI] [PubMed] [Google Scholar]
- 9.Huang WE, Wang H, Zheng H, Huang L, Singer AC, Thompson I, Whiteley AS. 2005. Chromosomally located gene fusions constructed in Acinetobacter sp. ADP1 for the detection of salicylate. Environ Microbiol 7:1339–1348. [DOI] [PubMed] [Google Scholar]
- 10.Zhang D, He Y, Wang Y, Wang H, Wu L, Aries E, Huang WE. 2012. Whole-cell bacterial bioreporter for actively searching and sensing of alkanes and oil spills. Microb Biotechnol 5:87–97. doi: 10.1111/j.1751-7915.2011.00301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Averhoff B, Graf I. 2008. The natural transformation system of Acinetobacter baylyi ADP1: a unique DNA transport machinery, p 119–139. In Gerischer U. (ed), Acinetobacter: molecular biology. Caister AP, Norfolk, United Kingdom. [Google Scholar]
- 12.Overballe-Petersen S, Harms K, Orlando LAA, Mayar JVM, Rasmussen S, Dahl TW, Rosing MT, Poole AM, Sicheritz-Ponten T, Brunak S, Inselmann S, de Vries J, Wackernagel W, Pybus OG, Nielsen R, Johnsen PJ, Nielsen KM, Willerslev E. 2013. Bacterial natural transformation by highly fragmented and damaged DNA. Proc Natl Acad Sci U S A 110:19860–19865. doi: 10.1073/pnas.1315278110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buchan A, Ornston LN. 2005. When coupled to natural transformation in Acinetobacter sp. strain ADP1, PCR mutagenesis is made less random by mismatch repair. Appl Environ Microbiol 71:7610–7612. doi: 10.1128/AEM.71.11.7610-7612.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vandecraen J, Chandler M, Aertsen A, Van Houdt R. 13 April 2017. The impact of insertion sequences on bacterial genome plasticity and adaptability. Crit Rev Microbiol. doi: 10.1080/1040841X.2017.1303661. [DOI] [PubMed] [Google Scholar]
- 15.Raeside C, Gaffé J, Deatherage DE, Tenaillon O, Briska AM, Ptashkin RN, Cruveiller S, Médigue C, Lenski RE, Barrick JE, Schneider D. 2014. Large chromosomal rearrangements during a long-term evolution experiment with Escherichia coli. mBio 5:e01377-14. doi: 10.1128/mBio.01377-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee H, Doak TG, Popodi E, Foster PL, Tang H. 2016. Insertion sequence-caused large-scale rearrangements in the genome of Escherichia coli. Nucleic Acids Res 44:7109–7119. doi: 10.1093/nar/gkw647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umenhoffer K, Fehér T, Balikó G, Ayaydin F, Pósfai J, Blattner FR, Pósfai G. 2010. Reduced evolvability of Escherichia coli MDS42, an IS-less cellular chassis for molecular and synthetic biology applications. Microb Cell Fact 9:38. doi: 10.1186/1475-2859-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sleight SC, Bartley BA, Lieviant JA, Sauro HM. 2010. Designing and engineering evolutionary robust genetic circuits. J Biol Eng 4:12. doi: 10.1186/1754-1611-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbe V, Vallenet D, Fonknechten N, Kreimeyer A, Oztas S, Labarre L, Cruveiller S, Robert C, Duprat S, Wincker P, Ornston LN, Weissenbach J, Marlière P, Cohen GN, Médigue C. 2004. Unique features revealed by the genome sequence of Acinetobacter sp. ADP1, a versatile and naturally transformation competent bacterium. Nucleic Acids Res 32:5766–5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerischer U, D'Argenio DA, Ornston LN. 1996. IS1236, a newly discovered member of the IS3 family, exhibits varied patterns of insertion into the Acinetobacter calcoaceticus chromosome. Microbiology 142:1825–1831. doi: 10.1099/13500872-142-7-1825. [DOI] [PubMed] [Google Scholar]
- 21.Mahillon J, Chandler M. 1998. Insertion sequences. Microbiol Mol Biol Rev 62:725–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Segura A, Bünz PV, D'Argenio DA, Ornston LN. 1999. Genetic analysis of a chromosomal region containing vanA and vanB, genes required for conversion of either ferulate or vanillate to protocatechuate in Acinetobacter. J Bacteriol 181:3494–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cuff LE, Elliott KT, Seaton SC, Ishaq MK, Laniohan NS, Karls AC, Neidle EL. 2012. Analysis of IS1236-mediated gene amplification events in Acinetobacter baylyi ADP1. J Bacteriol 194:4395–4405. doi: 10.1128/JB.00783-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jezequel N, Lagomarsino MC, Heslot F, Thomen P. 2013. Long-term diversity and genome adaptation of Acinetobacter baylyi in a minimal-medium chemostat. Genome Biol Evol 5:87–97. doi: 10.1093/gbe/evs120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Renda BA, Dasgupta A, Leon D, Barrick JE. 2015. Genome instability mediates the loss of key traits by Acinetobacter baylyi ADP1 during laboratory evolution. J Bacteriol 197:872–881. doi: 10.1128/JB.02263-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bacher JM, Metzgar D, de Crécy-Lagard V. 2006. Rapid evolution of diminished transformability in Acinetobacter baylyi. J Bacteriol 188:8534–8542. doi: 10.1128/JB.00846-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seaton SC, Elliott KT, Cuff LE, Laniohan NS, Patel PR, Neidle EL. 2012. Genome-wide selection for increased copy number in Acinetobacter baylyi ADP1: locus and context-dependent variation in gene amplification. Mol Microbiol 83:520–535. doi: 10.1111/j.1365-2958.2011.07945.x. [DOI] [PubMed] [Google Scholar]
- 28.Barrick JE, Lenski RE. 2013. Genome dynamics during experimental evolution. Nat Rev Genet 14:827–839. doi: 10.1038/nrg3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee H, Popodi E, Tang H, Foster PL. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A 109:E2774–E2783. doi: 10.1073/pnas.1210309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dillon MM, Sung W, Lynch M, Cooper VS. 2017. Genome-wide biases in the rate and molecular spectrum of spontaneous mutations in Vibrio cholerae and Vibrio fischeri. Mol Biol Evol 34:93–109. doi: 10.1093/molbev/msw224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dettman JR, Sztepanacz JL, Kassen R. 2016. The properties of spontaneous mutations in the opportunistic pathogen Pseudomonas aeruginosa. BMC Genomics 17:27. doi: 10.1186/s12864-015-2244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wösten MMSM. 1998. Eubacterial sigma-factors. FEMS Microbiol Rev 22:127–150. [DOI] [PubMed] [Google Scholar]
- 33.Bekker M, De Vries S, Ter Beek A, Hellingwerf KJ, Teixeira De Mattos MJ. 2009. Respiration of Escherichia coli can be fully uncoupled via the nonelectrogenic terminal cytochrome bd-II oxidase. J Bacteriol 191:5510–5517. doi: 10.1128/JB.00562-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lázár V, Pal Singh G, Spohn R, Nagy I, Horváth B, Hrtyan M, Busa-Fekete R, Bogos B, Méhi O, Csörgő B, Pósfai G, Fekete G, Szappanos B, Kégl B, Papp B, Pál C. 2013. Bacterial evolution of antibiotic hypersensitivity. Mol Syst Biol 9:700. doi: 10.1038/msb.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng Q. 2016. Comparing mutation rates under the Luria-Delbrück protocol. Genetica 144:351–359. doi: 10.1007/s10709-016-9904-3. [DOI] [PubMed] [Google Scholar]
- 36.Jack BR, Leonard SP, Mishler DM, Renda BA, Leon D, Suárez GA, Barrick JE. 2015. Predicting the genetic stability of engineered DNA sequences with the EFM Calculator. ACS Synth Biol 4:939–943. doi: 10.1021/acssynbio.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kickstein E, Harms K, Wackernagel W. 2007. Deletions of recBCD or recD influence genetic transformation differently and are lethal together with a recJ deletion in Acinetobacter baylyi. Microbiology 153:2259–2270. doi: 10.1099/mic.0.2007/005256-0. [DOI] [PubMed] [Google Scholar]
- 38.Csörgo B, Fehér T, Tímár E, Blattner FR, Pósfai G. 2012. Low-mutation-rate, reduced-genome Escherichia coli: an improved host for faithful maintenance of engineered genetic constructs. Microb Cell Fact 11:11. doi: 10.1186/1475-2859-11-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hare JM, Perkins SN, Gregg-Jolly LA. 2006. A constitutively expressed, truncated umuDC operon regulates the recA-dependent DNA damage induction of a gene in Acinetobacter baylyi strain ADP1. Appl Environ Microbiol 72:4036–4043. doi: 10.1128/AEM.02774-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hare JM, Adhikari S, Lambert KV, Hare AE, Grice AN. 2012. The Acinetobacter regulatory UmuDAb protein cleaves in response to DNA damage with chimeric LexA/UmuD characteristics. FEMS Microbiol Lett 334:57–65. doi: 10.1111/j.1574-6968.2012.02618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hare JM, Ferrell JC, Witkowski TA, Grice AN. 2014. Prophage induction and differential RecA and UmuDAb transcriptome regulation in the DNA damage responses of Acinetobacter baumannii and Acinetobacter baylyi. PLoS One 9:e93861. doi: 10.1371/journal.pone.0093861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Newmark KG, O'Reilly EK, Pohlhaus JR, Kreuzer KN. 2005. Genetic analysis of the requirements for SOS induction by nalidixic acid in Escherichia coli. Gene 356:69–76. doi: 10.1016/j.gene.2005.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pósfai G, Plunkett G, Fehér T, Frisch D, Keil GM, Umenhoffer K, Kolisnychenko V, Stahl B, Sharma SS, de Arruda M, Burland V, Harcum SW, Blattner FR. 2006. Emergent properties of reduced-genome Escherichia coli. Science 312:1044–1046. doi: 10.1126/science.1126439. [DOI] [PubMed] [Google Scholar]
- 44.Choi JW, Yim SS, Kim MJ, Jeong KJ. 2015. Enhanced production of recombinant proteins with Corynebacterium glutamicum by deletion of insertion sequences (IS elements). Microb Cell Fact 14:207. doi: 10.1186/s12934-015-0401-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Zhu X, Zhang X, Fu J, Wang Z, Chen T, Zhao X. 2016. Characterization of genome-reduced Bacillus subtilis strains and their application for the production of guanosine and thymidine. Microb Cell Fact 15:94. doi: 10.1186/s12934-016-0494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirokawa Y, Kawano H, Tanaka-Masuda K, Nakamura N, Nakagawa A, Ito M, Mori H, Oshima T, Ogasawara N. 2013. Genetic manipulations restored the growth fitness of reduced-genome Escherichia coli. J Biosci Bioeng 116:52–58. doi: 10.1016/j.jbiosc.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 47.Martínez-García E, Nikel PI, Aparicio T, de Lorenzo V. 2014. Pseudomonas 2.0: genetic upgrading of P. putida KT2440 as an enhanced host for heterologous gene expression. Microb Cell Fact 13:159. doi: 10.1186/s12934-014-0159-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Unthan S, Baumgart M, Radek A, Herbst M, Siebert D, Brühl N, Bartsch A, Bott M, Wiechert W, Marin K, Hans S, Krümer R, Seibold G, Frunzke J, Kalinowski J, Rückert C, Wendisch VF, Noack S. 2015. Chassis organism from Corynebacterium glutamicum; a top-down approach to identify and delete irrelevant gene clusters. Biotechnol J 10:290–301. doi: 10.1002/biot.201400041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morimoto T, Kadoya R, Endo K, Tohata M, Sawada K, Liu S, Ozawa T, Kodama T, Kakeshita H, Kageyama Y, Manabe K, Kanaya S, Ara K, Ozaki K, Ogasawara N. 2008. Enhanced recombinant protein productivity by genome reduction in Bacillus subtilis. DNA Res 15:73–81. doi: 10.1093/dnares/dsn002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu D, Fu Y, Liu F, Xu H, Saris PEJ, Qiao M. 2017. Enhanced heterologous protein productivity by genome reduction in Lactococcus lactis NZ9000. Microb Cell Fact 16:1. doi: 10.1186/s12934-016-0616-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Komatsu M, Uchiyama T, Omura S, Cane DE, Ikeda H. 2010. Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc Natl Acad Sci U S A 107:2646–2651. doi: 10.1073/pnas.0914833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lieder S, Nikel PI, de Lorenzo V, Takors R. 2015. Genome reduction boosts heterologous gene expression in Pseudomonas putida. Microb Cell Fact 14:23. doi: 10.1186/s12934-015-0207-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reuß DR, Altenbuchner J, Mäder U, Rath H, Ischebeck T, Sappa PK, Thürmer A, Guérin C, Nicolas P, Steil L, Zhu B, Feussner I, Klumpp S, Daniel R, Commichau FM, Völker U, Stülke J. 2017. Large-scale reduction of the Bacillus subtilis genome: consequences for the transcriptional network, resource allocation, and metabolism. Genome Res 27:289–299. doi: 10.1101/gr.215293.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rahmer R, Heravi KM, Altenbuchner J. 2015. Construction of a super-competent Bacillus subtilis 168 using the PmtlA-comKS inducible cassette. Front Microbiol 6:1431. doi: 10.3389/fmicb.2015.01431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deatherage DE, Barrick JE. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol 1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barrick JE, Colburn G, Deatherage DE, Traverse CC, Strand MD, Borges JJ, Knoester DB, Reba A, Meyer AG. 2014. Identifying structural variation in haploid microbial genomes from short-read resequencing data using breseq. BMC Genomics 15:1039. doi: 10.1186/1471-2164-15-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 58.de Berardinis V, Vallenet D, Castelli V, Besnard M, Pinet A, Cruaud C, Samair S, Lechaplais C, Gyapay G, Richez C, Durot M, Kreimeyer A, Le Fèvre F, Schächter V, Pezo V, Döring V, Scarpelli C, Médigue C, Cohen GN, Marlière P, Salanoubat M, Weissenbach J. 2008. A complete collection of single-gene deletion mutants of Acinetobacter baylyi ADP1. Mol Syst Biol 4:174. doi: 10.1038/msb.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hall BM, Ma C-X, Liang P, Singh KK. 2009. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria-Delbrück fluctuation analysis. Bioinformatics 25:1564–1565. doi: 10.1093/bioinformatics/btp253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murin CD, Segal K, Bryksin A, Matsumura I. 2012. Expression vectors for Acinetobacter baylyi ADP1. Appl Environ Microbiol 78:280–283. doi: 10.1128/AEM.05597-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng Q. 2017. rSalvador 1.7: an R tool for the Luria-Delbrück fluctuation assay. http://eeeeeric.com/rSalvador/. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.