

ABSTRACT
Bacterioplankton are fundamental components of marine ecosystems and influence the entire biosphere by contributing to the global biogeochemical cycles of key elements. Yet, there is a significant gap in knowledge about their diversity and specific activities, as well as environmental factors that shape their community composition and function. Here, the distribution and diversity of surface bacterioplankton along the coastline of the Gulf of Naples (GON; Italy) were investigated using flow cytometry coupled with high-throughput sequencing of the 16S rRNA gene. Heterotrophic bacteria numerically dominated the bacterioplankton and comprised mainly Alphaproteobacteria, Gammaproteobacteria, and Bacteroidetes. Distinct communities occupied river-influenced, coastal, and offshore sites, as indicated by Bray-Curtis dissimilarity, distance metric (UniFrac), linear discriminant analysis effect size (LEfSe), and multivariate analyses. The heterogeneity in diversity and community composition was mainly due to salinity and changes in environmental conditions across sites, as defined by nutrient and chlorophyll a concentrations. Bacterioplankton communities were composed of a few dominant taxa and a large proportion (92%) of rare taxa (here defined as operational taxonomic units [OTUs] accounting for <0.1% of the total sequence abundance), the majority of which were unique to each site. The relationship between 16S rRNA and the 16S rRNA gene, i.e., between potential metabolic activity and abundance, was positive for the whole community. However, analysis of individual OTUs revealed high rRNA-to-rRNA gene ratios for most (71.6% ± 16.7%) of the rare taxa, suggesting that these low-abundance organisms were potentially active and hence might be playing an important role in ecosystem diversity and functioning in the GON.
IMPORTANCE The study of bacterioplankton in coastal zones is of critical importance, considering that these areas are highly productive and anthropogenically impacted. Their richness and evenness, as well as their potential activity, are very important to assess ecosystem health and functioning. Here, we investigated bacterial distribution, community composition, and potential metabolic activity in the GON, which is an ideal test site due to its heterogeneous environment characterized by a complex hydrodynamics and terrestrial inputs of varied quantities and quality. Our study demonstrates that bacterioplankton communities in this region are highly diverse and strongly regulated by a combination of different environmental factors leading to their heterogeneous distribution, with the rare taxa contributing to a major proportion of diversity and shifts in community composition and potentially holding a key role in ecosystem functioning.
KEYWORDS: bacterioplankton, 16S rRNA gene and rRNA, potential metabolic activity, Illumina sequencing, Gulf of Naples
INTRODUCTION
Microbes dominate the abundance, diversity, and activity of marine ecosystems, being key components of marine food webs and playing vital roles in major biogeochemical cycles, climate regulation, and the remineralization of organic matter (1, 2). Bacteria (and archaea) numerically dominate the microbial fraction, including both autotrophic primary producers that carry out photosynthesis and heterotrophic organisms that recycle the dissolved organic carbon and nutrients, processing nearly one-half of the global marine primary production (3). Bacterioplankton communities exhibit high phylogenetic and physiological diversity (4), with a remarkable capacity to transform and adapt to the environment around them (5). As a consequence, knowledge of microbial community composition and diversity patterns is critical to determining the health and functioning of marine ecosystems (1).
High-throughput sequencing of ribosomal genes has facilitated an increasing recognition of the vast diversity of marine microbes (6). Bacterioplankton diversity and distribution patterns have been extensively studied both at local (7, 8) and global scales (9–11) using this technique, yet some oceanic regions, particularly in highly populated coastal areas, remain underexplored (12). Determining the metabolic activity of bacterioplankton is crucial for estimating their potential contribution to ecosystem processes. Previous studies have examined bulk activity in terms of biomass production and growth rates, for instance, using bacterial production assays (13, 14) or by measuring incorporated labeled precursors (15, 16). However, much less is known of the activity of specific taxa within complex bacterioplankton communities, an aspect that can be analyzed using 16S rRNA-to-rRNA gene ratios as a proxy of potential growth rate relative to abundance (17, 18). Although this approach is not free from bias, for instance, due to more than one copy of the rRNA gene per cell (see references 17 and 19 and references therein), it can readily provide taxon-specific growth potential information, thus acting as a proxy for their contribution to ecosystem processes, as well as facilitating the identification of the key drivers of such processes (19). Previously, it was presumed that under steady-state conditions and in the absence of top-down regulation, most active bacteria are the ones that are most abundant, with higher growth rates leading to higher biomass (17). Consequently, the rare bacteria were generally considered to represent a “seed bank” encompassing slow-growing or dormant individuals ready to respond only when environmental conditions became favorable again (20). However, a few recent studies showed that rare operational taxonomic units (OTUs) can have higher levels of 16S rRNA (ribosomes) per rRNA gene (cell number) and that they may be disproportionately active relative to their abundances (21, 22). These findings have encouraged studies on the distribution patterns and significance of the rare biosphere in ecosystem functioning and prompted further analyses at finer taxonomic resolution.
The Gulf of Naples (GON) is a semienclosed deep embayment opening into the southern Tyrrhenian Sea, in the midwestern Mediterranean basin, with an average depth of 170 m and a surface area of 870 km2. Situated beside the urbanized and densely populated city of Naples, Italy, and its surroundings, the GON is subjected to severe anthropogenic pressures, such as land and industrial runoffs, improperly treated sewage discharge, and maritime trafficking (23). In its southern part, the GON receives several inputs from the Sarno River (one of the most polluted rivers in Europe), particularly following heavy rain events or from uncontrolled urban discharges. The GON is characterized by complex hydrodynamics, with a variable boundary between coastal and offshore waters, whose position depends upon the general circulation of the Tyrrhenian Sea, which intrudes eddies into the GON, mainly in the fall and winter (24). The extension of the boundary between mesotrophic coastal and oligotrophic offshore waters strongly depends on local physical geography and bottom topography (25) but also on seasonal variability (26, 27). At times when northerly currents dominate, freshwater inputs from the Volturno and Garigliano Rivers can be traced into the GON from the northern Gulf of Gaeta (28). The high morphological diversity and dynamic nature of the GON make it an ideal site to study microbial community structure and dynamics in response to environmental factors. Extensive investigations in this region have surveyed planktonic eukaryotes (23, 29), but bacterial communities have received much less attention, except for cyanobacteria (30, 31) and pathogens (e.g., see references 32 and 33). However, there has been a recent focus on using picophytoplankton to track physical processes, such as eddies, vertical mixing, and upwelling events (e.g., see references 30 and 31).
The aim of this study was to characterize GON bacterioplankton communities in terms of abundance and distribution and to relate them to the general environmental conditions present along the coastline of the GON. To facilitate this, we used flow cytometry and high-throughput sequencing of 16S rRNA gene amplicons to obtain high-resolution taxonomic identification. At three sites, potential metabolic activity relative to abundance was also investigated using 16S rRNA-to-16S rRNA gene ratios of individual operationally defined taxa.
RESULTS AND DISCUSSION
Temperature, salinity, inorganic nutrients, and chlorophyll a.
To gain comprehensive information about bacterial diversity and community composition in surface waters of the GON, a total of 16 stations spanning both coastal and offshore sites were sampled (Fig. 1). The values of all measured environmental parameters are reported in Table 1. Temperature and salinity values were quite homogeneous across the sampled region, ranging from 15.75°C (SA6) to 16.56°C (TA2) and from 36.13 practical salinity units (PSU) (MC) to 37.52 PSU (PO3), respectively, indicating active mixing in the area. As a consequence, stations located in the Sarno River catchment (SA1 to SA5) did not show significantly lower salinity values, as expected from freshwater discharge, although relatively higher nutrient concentrations (especially of N and P) detected at these stations (especially SA1) were interpreted as indicators of river influence (Table 1). The highest salinity (37.40 PSU and 37.52 PSU) and lowest chlorophyll a (0.93 and 0.62 μg · liter−1) values were recorded at stations PO3 and VE3, respectively, indicative of offshore oligotrophy.
FIG 1.

Map of the Gulf of Naples highlighting the location of the stations sampled. Samples for flow cytometry and 16S rRNA gene sequencing were collected at all stations, whereas samples for 16S rRNA sequencing were obtained only from stations PO1, TA1, and VE3 (indicated with red symbols). Red rectangle indicates Sarno River-influenced stations. The map was created with Ocean Data View software (version 4.7.3).
TABLE 1.
Bacterial abundances, geographic coordinates, and environmental parameters at each sampling station in the GONa
| Station IDb | Coordinates | Temp (°C) | Salinity (PSU) | Concnc |
Abundance (cells · ml−1) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chl a (μg · liter−1) | NO3 (μM) | NO2 (μM) | NH4 (μM) | PO4 (μM) | SiO4 (μM) | Prochlorococcus | Synechococcus | Heterotrophic bacteria | ||||
| SA1 | 40°43′58.08″N, 14°27′48.96″E | 15.87 | 36.99 | 1.15 | 33.67 | 1.30 | 8.22 | 0.82 | 46.50 | 0 | 1.33 × 104 | 2.50 × 106 |
| SA2 | 40°43′19.92″N, 14°27′52.92″E | 16.00 | 37.08 | 3.45 | 2.53 | 0.25 | 4.33 | 0.17 | 5.22 | 0 | 6.16 × 103 | 1.14 × 106 |
| SA3 | 40°43′28.92″N, 14°27′41.04″E | 15.99 | 36.98 | 3.11 | 4.23 | 0.31 | 3.86 | 0.30 | 8.00 | 0 | 8.9 × 103 | 5.65 × 105 |
| SA4 | 40°44′8.88″N, 14°27′21.96″E | 15.95 | 37.36 | 2.64 | 14.51 | 0.59 | 5.14 | 0.24 | 21.90 | 1.21 × 103 | 1.22 × 104 | 1.33 × 106 |
| SA5 | 40°43′36.84″N, 14°27′12.96″E | 16.13 | 37.06 | 3.10 | 11.34 | 0.49 | 2.55 | 0.17 | 17.10 | 2.52 × 103 | 1.46 × 104 | 1.64 × 106 |
| SA6 | 40°42′51.12″N, 14°28′12.72″E | 15.75 | 37.15 | 2.49 | 4.30 | 0.20 | 1.36 | 0.12 | 5.65 | 0 | 1.56 × 104 | 3.74 × 104 |
| SA7 | 40°44′5.64″N, 14°26′39.48″E | 16.20 | 36.88 | 2.59 | 5.52 | 0.27 | 1.85 | 0.15 | 8.67 | 0 | 1.42 × 104 | 1.80 × 106 |
| SA8 | 40°42′33.84″N, 14°27′34.92″E | 16.36 | 36.92 | 2.15 | 1.23 | 0.12 | 0.67 | 0.07 | 2.11 | 0 | 1.3 × 104 | 1.60 × 106 |
| VE3 | 40°40′59.88″N, 14°24′28.80″E | 16.13 | 37.4 | 0.62 | 2.12 | 0.12 | 0.80 | 0.07 | 1.93 | 1.80 × 103 | 1.71 × 104 | 1.44 × 106 |
| TA1 | 40°45′5.40″N, 14°23′54.60″E | 16.35 | 36.55 | 2.12 | 0.05 | 0.06 | 0.45 | 0.05 | 0.34 | 0 | 1.32 × 104 | 9.95 × 105 |
| TA2 | 40°44′39.48″N, 14°22′33.60″E | 16.56 | 36.87 | 1.80 | 0.00 | 0.03 | 0.64 | 0.01 | 0.14 | 1.32 × 103 | 1.6 × 104 | 1.44 × 106 |
| PO1 | 40°48′18.00″N, 14°19′.12″E | 16.23 | 36.61 | 3.09 | 2.43 | 0.17 | 0.87 | 0.06 | 2.99 | 0 | 7.6 × 103 | 9.77 × 105 |
| TG2 | 40°46′28.20″N, 14°20′12.12″E | 16.29 | 37.08 | 2.82 | 3.99 | 0.22 | 0.95 | 0.08 | 5.07 | 0 | 8.38 × 103 | 8.16 × 105 |
| TG1 | 40°46′44.40″N, 14°21′4.32″E | 16.21 | 36.72 | 3.60 | 0.15 | 0.05 | 0.63 | 0.04 | 0.18 | 0 | 2.05 × 103 | 8.17 × 105 |
| PO3 | 40°47′27.60″N, 14°16′33.60″E | 15.96 | 37.52 | 0.93 | 0.04 | 0.05 | 0.58 | 0.02 | 0.30 | 0 | 2.31 × 103 | 7.65 × 105 |
| MC | 40°48′42.84″N, 14°15′.00″E | 16.26 | 36.13 | 5.17 | 5.50 | 0.47 | 4.72 | 0.25 | 6.12 | 2.24 × 103 | 1.4 × 104 | 5.47 × 105 |
Bacterial abundance.
The abundances of phototrophic and heterotrophic bacteria enumerated by flow cytometry are reported in Table 1 and in Fig. S1 in the supplemental material. Heterotrophs dominated numerically, with average ± standard deviation (SD) abundances of 1.15 × 106 ± 0.57 × 106 cells · ml−1, while autotrophs were dominated by Synechococcus (average ± SD values, 1.11 × 104 ± 0.45 × 104 cells · ml−1) at all stations. Prochlorococcus was detected at only 5 stations, at concentrations between 1.11 × 103 and 2.52 × 103 cells · ml−1. No clear patterns in Synechococcus abundance were observed, while heterotrophic bacteria were more abundant at the MC station and near the Sarno River and less abundant at the offshore stations VE3 and PO3.
Alpha and beta diversity.
The rarefaction curves of the observed OTUs (Fig. S2a) approached saturation only for stations SA1, TA1, TA2, SA2, and SA5, indicating insufficient sampling depth for the others. Although the Chao1 curves (Fig. S2b) were steeper, they did not saturate for each library. However, the Shannon rarefaction curves (Fig. S2c), which consider both richness and evenness, showed saturation and a stable pattern in all samples, suggesting that sampling was sufficient to accurately describe the trends in alpha diversity. Furthermore, Good's coverage was >99% for all the samples (Table 2), indicating that, while possibly more rare taxa could be recovered by a deeper sampling, the rarefied sequencing depth we used satisfactorily represented the alpha diversity at the sampling sites.
TABLE 2.
Good's coverage and diversity indices at GON stationsa
| Station IDb | Good's coverage (avg ± SD) (%) | ACE*c | ACEd (avg ± SD) | Chao1 (avg ± SD) | Berger-Parker dominance index | Shannon index (avg ± SD) | Invsimpson (avg ± SD) |
|---|---|---|---|---|---|---|---|
| PO3 A | 99.7 ± 0.02 | 399 | 354 ± 42 | 331 ± 27 | 0.24 | 3.25 ± 0.008 | 11.98 ± 0.11 |
| VE3 A | 99.7 ± 0.02 | 459 | 350 ± 39 | 347 ± 29 | 0.31 | 3.05 ± 0.010 | 7.97 ± 0.08 |
| TG1 B | 99.6 ± 0.02 | 542 | 455 ± 69 | 373 ± 36 | 0.14 | 3.23 ± 0.007 | 14.64 ± 0.10 |
| PO1 B | 99.6 ± 0.02 | 507 | 479 ± 68 | 392 ± 40 | 0.13 | 3.17 ± 0.007 | 14.43 ± 0.10 |
| TG2 B | 99.6 ± 0.02 | 551 | 461 ± 56 | 401 ± 31 | 0.13 | 3.23 ± 0.005 | 15.00 ± 0.08 |
| TA1 B | 99.8 ± 0.02 | 614 | 333 ± 54 | 277 ± 36 | 0.13 | 2.66 ± 0.008 | 9.15 ± 0.05 |
| TA2 B | 99.8 ± 0.02 | 730 | 340 ± 60 | 279 ± 33 | 0.14 | 2.78 ± 0.008 | 10.01 ± 0.06 |
| SA5 B | 99.5 ± 0.03 | 714 | 594 ± 74 | 482 ± 48 | 0.16 | 3.26 ± 0.008 | 15.12 ± 0.11 |
| SA8 B | 99.6 ± 0.03 | 523 | 520 ± 55 | 428 ± 37 | 0.13 | 3.36 ± 0.007 | 16.66 ± 0.11 |
| SA7 C | 99.6 ± 0.03 | 724 | 609 ± 80 | 438 ± 49 | 0.32 | 2.73 ± 0.008 | 7.29 ± 0.06 |
| SA1 C | 99.5 ± 0.03 | 753 | 727 ± 108 | 498 ± 62 | 0.25 | 2.85 ± 0.009 | 8.93 ± 0.07 |
| SA2 C | 99.3 ± 0.04 | 935 | 962 ± 111 | 688 ± 64 | 0.26 | 2.96 ± 0.009 | 8.97 ± 0.08 |
| SA3 C | 99.4 ± 0.02 | 782 | 788 ± 74 | 561 ± 42 | 0.21 | 3.18 ± 0.006 | 11.95 ± 0.07 |
| SA4 C | 99.5 ± 0.03 | 829 | 706 ± 74 | 542 ± 50 | 0.16 | 3.26 ± 0.007 | 13.85 ± 0.10 |
| SA6 C | 99.7 ± 0.02 | 378 | 343 ± 42 | 293 ± 25 | 0.19 | 3.00 ± 0.005 | 11.18 ± 0.06 |
| MC | 99.6 | 418 | 418 | 400 | 0.22 | 3.17 | 11.74 |
Standard deviation values were generated by mothur after multiple resampling of the OTU abundance values in order to normalize the data in order to have equal number of sequences in each library.
Different uppercase letters next to the station names indicate significant differences between groups of stations (as in Fig. 2a) as obtained using the ANOSIM R statistic (Fig. S2).
ACE*, richness estimation of the nonnormalized 16S rRNA gene libraries.
ACE, richness estimation of 16S rRNA gene libraries normalized to the library with the lowest number of sequences.
The Chao1 diversity index was higher at most stations near the mouth of the Sarno River (Table 2) and was positively correlated with ammonium (Pearson's r = 0.66, P < 0.05). This pattern has been previously observed along coast to offshore transects, for instance, in the Southern Adriatic Sea (34), Blanes Bay in the northwestern Mediterranean Sea (35), the South China Sea (36), and Moreton Bay in Australia (37). The higher richness in coastal areas may be due to terrestrial inputs, to the availability of organic and inorganic matter at high concentrations, and to continuous mixing of local and external communities, resulting in numerous favorable niches for bacterioplankton communities. The higher rates of primary production in coastal areas provide a larger fraction of organic matter in the form of detritus, most of which is degraded by heterotrophic bacteria prior to entering higher trophic levels, supporting bacterial communities with higher richness in these areas (38).
The Bray-Curtis dissimilarity dendrogram clustered the samples into four different groups (Fig. 2a). Group I grouped the offshore PO3 and VE3 stations, confirming the similarity inferred from salinity and chlorophyll a data. Group II included stations TG1, TG2, and PO1, located in front of the heavily urbanized areas of the Neapolitan province. Group III included stations TA1, TA2, SA5, and SA8, north and south of the zone of influence of the Sarno River, while group IV included stations SA7, SA1, SA2, SA3, SA4, and SA6, which are closest to the Sarno River. Groups III and IV subclustered stations TA1 and TA2 (IIIa) and SA5 and SA8 (IIIb). Group IV subclustered stations SA1 and SA2 (IVa) and SA4 and SA6 (IVb). The MC station was not included in any of the clusters. Bray-Curtis clustering of samples was confirmed to be significant using the ANOSIM R statistic (Table S2), except for the lack of difference between groups II and III (R = 0.75, P > 0.05); hence, these two groups are considered one cluster (II+III). The OTUs responsible for Bray-Curtis similarity within groups were identified using similarity percentage (SIMPER) analysis (Table S3). Bacterial communities in groups I, II+III, and IV were 88.7%, 88.6%, and 88.8% similar, respectively, mainly due to similar proportions of the same OTUs (Table S2). The MC station showed relatively high proportions of some OTUs (including some rare ones) compared to all other stations, such as those attributed to Nereida spp., Flavobacteriaceae, Gammaproteobacteria, Oceanospirillaceae, the Roseobacter clade, Rickettsiales, Flavobacterium. and Cryomorphaceae, which are probably responsible for its outgrouping in the Bray-Curtis dendrogram.
FIG 2.

Station clustering based on Bray Curtis dissimilarity (a) and the distance matrix generated by calculating pairwise UniFrac metrics (b). The scale bar in panel b shows the distance between clusters in UniFrac units; if two or more environments have similar lineages, they have a distance of 0. The significance of the cluster nodes was determined using the jackknife analysis. Jackknife significance values are >99.9% = 1, 90 to 99% = 2, 70 to 90% = 3; 50 to 70% = 4, <50% = 5, and higher values indicate a higher adaptation of communities to the existing environmental conditions.
Bacterial community composition.
A total of 1,235 OTU, after excluding singletons, were used to assess bacterial community composition; 99% of these OTUs could be classified at the phylum level. These were affiliated with 17 major phyla and 3 candidate divisions (Fig. 3a). Overall, the surface bacterioplankton at all stations was dominated by Proteobacteria (∼69%), followed by Bacteriodetes (∼27%), as in other studies from the Mediterranean Sea (39). Proteobacteria was also the most diverse bacterial group, comprising the highest number of retrieved OTUs (47.2% of the whole data set). Of these, the most frequently occurring OTUs belonged to Gammaproteobacteria and Alphaproteobacteria (35% and 32% of Proteobacteria, respectively), mainly represented by Oceanospirillales and Alteromonadales (18% and 14% of the Gammaproteobacteria, respectively) and Rickettsiales (19% and 12% of the Alphaproteobacteria, respectively).
FIG 3.

(a) Bacterial community composition at the phylum level at each GON station sampled. The phylum Proteobacteria is split into several classes (Alphaproteobacteria, Betaproteobacteria, Deltaproteobacteria, Gammaproteobacteria, and Epsilonproteobacteria). Bacterial OTUs that could not be classified were labeled as “other bacteria.” (b) The relative abundances of the top 12 abundant phylotypes in the GON at a lower taxonomic level (order/family level).
Similar to studies conducted in surface waters on a global scale (9, 10), the dominant components of bacterial communities of the GON were present at all stations but in different proportions (Fig. 3b). Based on SIMPER analysis and following OTU grouping, Rhodobacterales resulted in the highest contributing clade at coastal sites (group II+III stations), while SAR11 and SAR86 were the most abundant offshore (group I stations). This is consistent with previous observations in the Southern Adriatic Sea (34) and the northwestern Mediterranean Sea (40) about SAR11 distribution. Members of SAR11 and SAR86 are well known for their ability to grow at low substrate concentrations, due to their streamlined genomes (41, 42) and light-harvesting proteorhodopsins for ATP production (42, 43), likely conferring them a selective advantage in oligotrophic waters (44). In contrast, the dominance of members of the Rhodobacterales (mainly of the genus Nereida and the Roseobacter clade) at coastal stations is likely due to the higher nutrient concentrations and chlorophyll a levels at these sites, given their known ability to thrive in more eutrophic environments with high primary productivity (45) and to use phytoplankton-derived dissolved organic carbon, especially that originating from diatoms (e.g., see reference 46), which abundantly bloom in the GON (47). At river-influenced stations (group IV), Alteromonadales (mainly Glaciecola and SAR92) dominated, related to their preference for high nutrient concentrations, a feature in good agreement with their copiotrophic nature (48), the exception being the SAR92 clade, which was more abundant at offshore stations, as already reported (49).
Cyanobacteria represented ∼1% of the total bacteria, with Synechococcus and Prochlorococcus present at all stations but with their contributions increased at both coastal and offshore (Synechococcus) or offshore stations alone (Prochlorococcus), only partially matching flow cytometry data (Table 1 and Fig. S1). This shows that sequencing was robust enough to retrieve Prochlorococcus even in samples where it was below the detection limits of flow cytometry. Contrary to other studies (e.g., see references 34, 50, and 51), Cyanobacteria did not dominate in the GON and did not show a clear distribution pattern, which may support their high genetic diversity with physiologically diverse clades in terms of light and nutrient adaptation (52–54). Thus, a more precise mapping of the microdiversity among closely related cyanobacterial lineages is critical to understanding their distribution pattern in this area.
In order to identify a significant differential distribution of bacterial groups at the OTU level in relation to environmental parameters, linear discriminant analysis effect size pipeline (LEfSe) analysis was applied. For this test, we considered stations within group I (offshore), II and III together (coastal), and IV (river influenced) to be the ones identified by the Bray-Curtis dendrogram (Fig. 2a) and confirmed by ANOSIM (Table S2). A total of 87 OTU, including both rare and abundant (<0.1% and >0.1% of total sequences, respectively) OTUs, were found to be differentially abundant in the three areas, with 14 OTU specialized for river-influenced stations, 18 OTU for coastal stations, and 55 OTU for offshore stations (Fig. 4a). However, many OTUs expected to be specialized at offshore stations (e.g., SAR11 and SAR92) were not highlighted by this test, due to the fact that they showed similar numerical distributions at both coastal and river-influenced stations, and LEfSe takes into account only the OTUs that are differentially abundant between the groups considered. Therefore, we performed a second LEfSe test considering river-influenced and coastal stations together (groups II, III, and IV), named “coastal+river-influenced,” and compared this merged group with the offshore group (group I). This second LEfSe test showed that a total of 113 OTU, including both rare and abundant OTUs, were differentially abundant, with 82 OTU specialized for offshore stations and 31 OTU for coastal+river-influenced stations (Fig. 4b). It is important to note that the offshore area consisted of a higher number of significantly differentially abundant OTUs than the coastal areas, contributed mainly by rare OTUs, suggesting a niche adaptation strategy adopted by these low-abundance and highly diverse taxa (55). Analysis at the OTU level is necessary to understand bacterial distribution patterns with the highest resolution. Our results highlight that the variability in bacterial community compositions across different sites in the GON is driven not only by abundant but also by rare species, whose spatial dynamics needs further thorough description and understanding.
FIG 4.

LEfSe analysis, indicating significantly differential distribution of taxa in the different groups of stations as identified by Bray-Curtis dendrogram (Fig. 2a) followed by ANOSIM R statistic (a), and after grouping coastal+river-influenced stations (b). Red asterisks indicate abundant OTUs that were present in all the libraries, while blue asterisks indicate OTUs that shifted between abundant and rare among stations. OTUs without an asterisk are rare (<0.1% of total sequences of the libraries).
OTU frequency.
Forty-eight OTU (95.8% of the total sequences) were abundant (here defined as OTUs accounting for >0.1% of the total sequences) at either all or ≥50% of the total stations. Forty-eight OTU (2.6% of the total sequences) were observed to be abundant at <50% of all stations, while 1,139 OTU (1.6% of the total sequences) were considered rare (here defined as OTUs accounting for <0.1% of the total sequences). These numbers suggest that a tremendous bacterial diversity in the GON is accounted for by the so-called “rare biosphere,” as described in reference 56, which is in agreement with the findings of other studies (34, 35, 57). Among the rare OTUs, Arcobacter, Chlamydiae, Firmicutes (mainly Ruminococcaceae, Veillonellaceae, Lachnospiraceae, and Staphylococcaceae), Fusobacteria, Lentisphaeraceae (Lentisphaera and Victivallis), Spirochaetes, Synergistetes, and Vibrio spp. were observed (≤0.01% of the total sequences) and were attributed to sewage-associated-bacteria or pathogens (e.g., see references 58–60), indicating the dispersal of fecal bacteria from the anthropogenically affected coastal area and suggesting that they can survive in marine waters and therefore represent a reservoir of pathogenicity potentially harmful to human health. Similarly, the presence of Rhodocyclales, which are known to degrade several pollutants (61), is suggestive of pollutant biodegradation occurring in the GON, which is related to the many anthropogenic activities along the GON coastline.
Bacterial communities and environmental factors.
In order to identify the specific environmental factors that might explain bacterial community composition in the GON, canonical correspondence analysis (CCA) was performed using OTU abundances and the measured environmental parameters (Fig. 5). The first two axes explained 78% and 18% of the cumulative variance, respectively. Monte Carlo permutations (999 permutations) indicated that only salinity (P = 0.004), chlorophyll a (P = 0.001), and ammonium concentrations (P = 0.012) were significant determinants of bacterial community structure in terms of abundance of OTUs, based on the envfit parameter. Ammonium was a determinant at the Sarno River-influenced stations (Fig. 2a, group IV), which were dominated by Alteromonadales. Likewise, the bacterial community structure at the MC station, which is largely explained by chlorophyll a, was dominated by members of the Rhodobacterales. Salinity was the dominant variable at the oligotrophic sites VE3 and PO3 that were mainly dominated by SAR11. The effects of other environmental parameters, such as temperature, nitrate, nitrite, silicate, and phosphate, were probably masked by the complex physical features of the GON at the time of sampling, including mixing. These results indicate that, similar to other marine areas (34, 62), it is a combination of several environmental factors that influence the local distribution of bacterial populations in the GON. Indeed, the unexplained variation in the CCA is likely due to unmeasured environmental variables and processes, with the processes including top-down controls, like grazing and viral lysis, which are also known to shape bacterial community composition.
FIG 5.
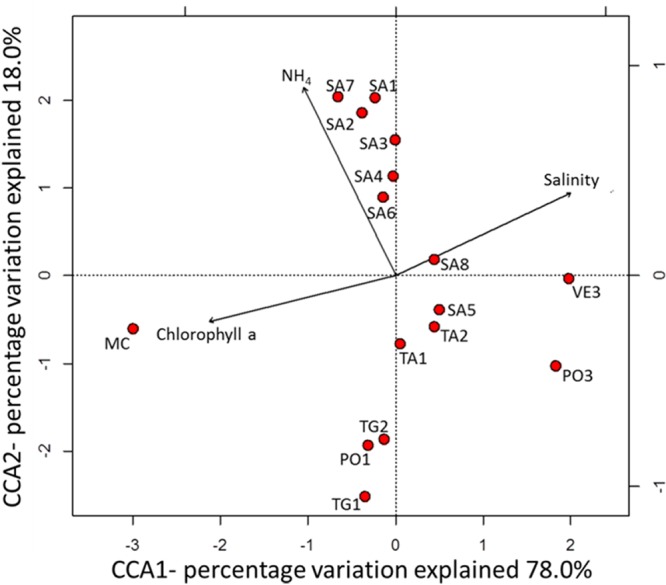
CCA ordination plot depicting the relationship between environmental parameters and bacterial community structure, as represented by 16S rRNA gene sequence data.
In order to further compare the phylogenetic diversity of the bacterial communities between stations, UniFrac distance metrics were calculated based on a maximum likelihood tree constructed using one representative sequence from each OTU (Fig. 2b). Weighted hierarchical clustering showed that community composition was more similar at those sites located in close vicinity to each other or with similar features. Although similar clustering was also mirrored by a Bray-Curtis dendrogram (Fig. 2a) and CCA (Fig. 5), UniFrac analysis revealed that environmental heterogeneity not only regulates the spatial distribution of abundance of different taxonomic groups at the local scale in the GON but may also have resulted in distinct phylogenetic diversity (see Fig. 2b).
Bacterial potential metabolic activity.
The potential activity of each operationally defined taxon and the correlation between potential activity (16S rRNA frequency) and abundance (16S rRNA gene frequency) were assessed at the three stations where good-quality RNA could be obtained: VE3, PO1, and TA1. These stations are also representative of the coastal (TA1 and PO1) and offshore areas (VE3). The 16S rRNA-to-rRNA gene ratios may have been affected by the extraction of DNA and RNA from different lysates, but libraries were normalized by an equal number of sequences before performing calculations or statistical tests, in order to avoid any bias (as also indicated in Materials and Methods).
Overall, rRNA and rRNA gene libraries were positively correlated at all the three sites (Fig. 6) (Spearman's and Kendall's P < 0.0001; Kendall's τ, VE3 = 0.22, PO1 = 0.52, TA1 = 0.53). The average 16S rRNA-to-rRNA gene ratios for rare (<0.1% of total sequence abundances) and abundant bacteria were 2.3 (from 0.2 to 8.47, n = 209) and 0.71 (from 0.09 to 1.51, n = 79), respectively, at station VE3; 1.63 (from 0.27 to 6.67, n = 103) and 0.95 (from 0.18 to 2.2, n = 57), respectively, at station PO1; and 1.2 (from 0.22 to 4.90, n = 87) and 0.8 (from 0.19 to 1.92, n = 57), respectively, at station TA1, with the ratio for rare bacteria being significantly higher than the ratio for abundant bacteria (P < 0.05, t test). At all three stations, most (79.80% at VE3, 54.40% at PO1, and 70.20% at TA1) of the abundant taxa had a ratio of <1, whereas most (84.60% at VE3, 77.66% at PO1, and 52.84% at TA1) of the rare taxa had a ratio of ≥1. For instance, an abundant SAR11 OTU (ID-10) showed ratios of <1 at all the three sites (Fig. 7), similar to what was observed in other studies for SAR11 (22). Likewise, OTUs attributed to Rhodobacteraceae and Flavobacteriaceae showed a 16S rRNA-to-rRNA gene ratio of <1. However, cultivated members of these groups have been previously reported to possess more than one ribosomal operon (22), which may explain this ratio. As for the rare OTUs, Blastopirellula (Planctomycetaceae) showed a very high 16S rRNA-to-rRNA gene ratio (8.5) at the VE3 site. The possibility of a sequencing artifact for this high ratio was excluded, since the same behavior was observed for other Blastopirellula OTUs identified at the same station. These results suggest that although potential activity and abundance showed a significant positive correlation at the whole-community level, this may not hold true at the individual taxon level; therefore, abundance may not reflect the degree of their potential activity, as was also observed in previous studies in other geographical areas (17, 18). While the role of the abundant phylotypes in ecosystem functioning is well recognized, the contribution of the rare ones remained largely underestimated until recently, due to the previous assumption of them being merely the slow-growing and dormant members of the community (20). In general, the abundant bacterial phylotypes altogether contribute a higher proportion of biomass production or activity, but not always on a per-cell basis, while rare bacteria can have 16S rRNA-to-rRNA gene ratios of >1, therefore exhibiting higher potential activity than the abundant ones, as reported by us and others (17, 18, 22). Unfortunately, due to our small sample size, correlations between 16S rRNA and rRNA genes for individual OTUs could not be statistically tested.
FIG 6.
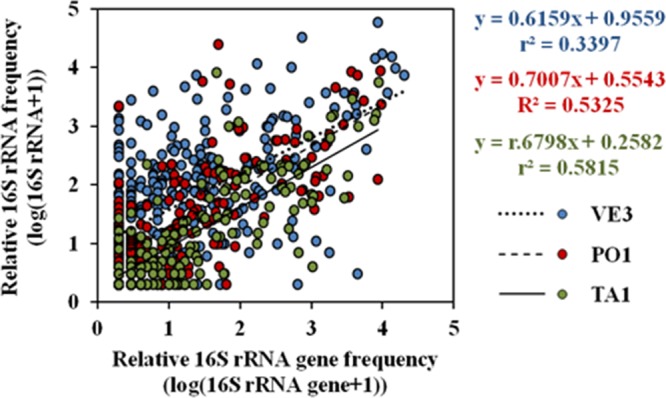
Correlation between 16S rRNA and 16S rRNA gene frequencies at stations VE3, PO1, and TA1 with slopes for each station (slope for the total data is y = 0.6191x + 0.7343; r2 = 0.3845). Individual data points correspond to paired log(rRNA + 1) and log(rRNA gene + 1) for each individual OTU. Data were log transformed in order to eliminate bias. Therefore, correlations are limited to OTUs where both rRNA and rRNA gene sequences were present.
FIG 7.

Comparison of the relative abundances and potential activities of different phylogenetic groups within and between different sites in the GON. (A) Maximum likelihood phylogenetic tree of the 50 most abundant OTUs present in all the equally subsampled 16S rRNA and 16S rRNA gene libraries; Methanococcoides burtonii, an archaeal species, was included as an outgroup. (B) Heatmap showing the log abundance [log (observations + 1)] for each 16S rRNA and 16S rRNA gene OTU present in the tree. (C) Specific potential activity relative to abundance of each OTU present at the VE3, PO1, and TA1 stations calculated using the rRNA-to-rRNA gene ratio.
OTUs were ranked on the basis of their abundance in each library. Differences in the relative rank of the same OTU among different sites, and between rRNA gene and rRNA libraries of the same site, were observed. For instance, the majority of the top 50 most abundant OTUs were shared by all six libraries, but the relative abundances of these OTUs varied between sites. In addition, variations in the sequence abundance of the same OTU between rRNA gene and rRNA libraries resulted in substantial variation in the 16S rRNA-to-rRNA gene ratio among OTUs, both within and between stations (Fig. 7). These differences can be attributed to different biotic and abiotic environmental factors (as in references 18 and 63) or to differences in evenness of diversity between rRNA genes and rRNA at different stations (as also found by Hunt et al. [17]). Both hypotheses are possible in our case, since the average ± SD Shannon index values of the rRNA gene and rRNA libraries were 3.05 ± 0.24 and 2.44 ± 0.80, respectively, and the values of the measured environmental factors were also different. Differences in the 16S rRNA-to-rRNA gene ratio can also be due to life strategies adopted by some bacteria, for instance, the increase in ribosomal concentration as they enter dormancy to achieve higher protein synthesis potential to be used after recovery to a vegetative stage in response to favorable cues (64).
Conclusions.
The observed strong spatial structuring of bacterial communities in the GON is tightly regulated by environmental factors and local hydrological features, suggesting a niche-based community assembly, in accordance with the famous dictum “everything is everywhere, but the environment selects” (65). Further evaluation of the bacterial association and the cooccurrence network is needed to gain additional insights into the potential relationships within these communities, as well as to reveal ecological processes, such as top-down and bottom-up regulation and community succession, as suggested by other researchers (e.g., see reference 66). Regular monitoring of bacterioplankton community structure and activity, both at the seasonal and annual scale, will facilitate a better understanding and predictions of their responses to the changing environment. In particular, in our data, salinity appears to play a relevant role, and in this context, the GON may represent an interesting test site to predict the responses of the microbial components to the changing climate-induced salinity increase in the oceans (67). Our study also highlights the relevance of the rare phylotypes in the heterogeneous bacterial diversity and their potential role in ecosystem functioning, suggesting that they harbor a persistent functional pool of ecological potential rather than acting merely as a “seed bank” (68). Therefore, an assessment of their spatiotemporal dynamics and mechanisms controlling their population size is crucial.
MATERIALS AND METHODS
Sample collection, chlorophyll, and nutrient analysis.
Sixteen surface seawater samples (Fig. 1) were collected from 10 to 29 April 2013 on board the R/V Minerva Uno and the R/V Vettoria, using a rosette sampler equipped with 10-liter Niskin bottles and a conductivity-temperature-depth (CTD) probe (SBE 911plus; SeaBird Electronics, USA). At all stations, seawater samples for chlorophyll a, inorganic nutrient (NO2, NO3, NH4, PO4, and SiO4) analyses, flow cytometry counts, and 16S rRNA gene and 16S rRNA library preparation were collected from the same Niskin bottle. Duplicate samples for flow cytometry (1 ml each) were placed in 1.5-ml cryovials, fixed with a mixture of 0.05% (vol/vol) glutaraldehyde and 1% (wt/vol) paraformaldehyde (final concentrations) for 10 min in the dark, frozen in liquid nitrogen, and stored at −80°C until analysis, as performed in reference 31. For DNA collection, 2 liters of each sample was prefiltered through a 10-μm-pore-size mesh net to remove large eukaryotes and debris and successively filtered through 47-mm glass filter/A filters (Whatman, UK), whose nominal pore size (1.67 μm) is larger than most free-living marine bacteria (55). Cells were then collected onto a 0.22-μm-pore-size Sterivex filter cartridge (Millipore, USA) using a peristaltic pump at 24 rpm. Sterivex filters were then sealed, frozen in liquid nitrogen, and stored at −80°C until DNA extraction. For RNA collection, the procedure was exactly the same as for the DNA extraction, except for the last filter, which was a 0.22-μm-pore-size 47-mm Durapore filter (Millipore), using a vacuum pump at low vacuum (2.9 lb/in2). After filtration, each filter was stored in a cryovial, frozen in liquid nitrogen, and stored at −80°C until RNA extraction.
Samples for the measurement of total chlorophyll a were filtered onto 25-mm Whatman GFF filters, frozen, and stored in liquid nitrogen until analysis. Concentrations were determined using a Shimadzu (Japan) RF-5301PC spectrofluorometer, as previously described (69). Discrete samples for inorganic nutrient concentrations (NO3, NO2, NH4, PO4, and SiO4) were frozen and stored at −20°C until analysis. Concentrations were determined using a FlowSys AutoAnalyzer (Systea S.p.A., Italy) (70). The detection limits were 0.01 μM for nitrates, nitrites, and phosphates, 0.05 μM for ammonia, and 0.1 μM for silicates.
Flow cytometry.
Bacterioplankton cell concentrations were estimated using a FACSCalibur (Becton Dickinson, USA), according to standard procedures (71). For an assessment of heterotrophic bacteria, frozen samples were thawed and stained with SYBR green I (Molecular Probes, Inc., USA). Cell abundances were determined by extraction using CellQuest software (Becton Dickinson).
Nucleic acid extraction and sequencing.
DNA extraction was performed using the DNeasy blood and tissue kit (Qiagen, USA), according to the manufacturer's instructions, with the only minor modification being overnight incubation at 56°C following addition of the Qiagen lysis buffer and proteinase K to the Sterivex filters. RNA extraction was performed using the PowerWater RNA isolation kit (Mo Bio Laboratories, USA), according to the manufacturer's protocol. DNA contamination was checked by PCR of the 16S rRNA gene using standard primers and conditions (72). The minimum amount (20 ng) of extracted RNA was reverse transcribed with random primers using the iScript cDNA synthesis kit (Bio-Rad, USA), according to the manufacturer's instructions. DNA and cDNA samples were shipped to the GeneCore Genomics Core Facility at the European Molecular Biology Laboratory, Heidelberg, Germany, for paired-end multiplexed sequencing using the Illumina MiSeq platform. The PCR protocol used for amplifying DNA and cDNA was standardized to use the minimum amount of template. The primers used for library preparation covered the V4 and V5 hypervariable regions (515F-926R) of the 16S rRNA gene, as in reference 73, with only one base difference (515F, 5′-GTGCCAGCMGCCGCGGTAA-3′; 906R, 5′-CCGYCAATTYMTTTRAGTTT-3′).
Sequence analyses.
Low-quality raw reads with a contaminating adapter sequence or with a Phred value less than 25 were excluded using Trimmomatic (74) The resulting sequences were further processed and analyzed using the mothur software package (version 1.33.1 [75]), according to the MiSeq standard operating procedure (SOP) (http://www.mothur.org/wiki/MiSeq_SOP [76]). Reads were joined into contigs using the sequence and quality score data from the Fastq files. Primers were trimmed, and sequences were further screened according to the following parameters: minimum length, 370 bp; maximum length, 376 bp; maximum number of Ns, 0; and maximum homopolymers, 8. Unique sequences, i.e., randomly screened sequences from groups of identical sequences present in two or more copies, were screened out in order to speed up the process and to avoid computational memory problems. The resulting sequences were aligned against the SILVA reference database (version 102) and preclustered, allowing a maximum of three differences between sequences. Chimeric sequences were detected with the UCHIME algorithm (77) and removed. After this filtration, taxonomic assignment was realized using the SILVA taxonomy string with the k-nearest-neighbor algorithm at a bootstrap confidence score of 100 and a minimum similarity threshold of 80%. Across all 16S rRNA gene libraries, a total of 8,811,540 raw reads were obtained, from which 1,216,213 high-quality sequences with an average read length of 370 to 376 bp were used for taxonomic assignment. Only a very small number (10 sequences) of archaeal sequences assigned to Euryarchaeota and Crenarchaeota appeared in the whole data set, and these were not included in further analysis. Sequences flagged as chloroplasts, mitochondria, or eukaryotes were also excluded. The remaining 1,210,304 sequences were clustered de novo using the average neighbor algorithm at a distance cutoff 97% similarity, resulting in 2,681 OTU. OTUs identified as singletons were excluded from the data set to limit the inflation caused by spurious OTUs (PCR artifacts), and a consensus taxonomy for each of the remaining 1,235 OTU was obtained. The full lists of OTUs for the 16S rRNA gene and the 16S rRNA libraries are provided in Data Sets S1 and S2 in the supplemental material.
Diversity estimates and statistical analyses.
The 16S rRNA gene libraries prepared using samples collected from all the 16 stations produced 26,177 to 151,291 reads, 487 to 1,362 unique sequences, and 281 to 715 OTU after quality filtration (details for each station are in Table S1). Rarefaction curves for the observed OTUs, Chao1, and Shannon diversity index values were generated using the “rarefaction.single” command built in mothur (Fig. S2) and compared as in reference 78 to evaluate the sampling effort and the alpha diversity of the different communities. Good's coverage was >99% for all the samples, indicating that the majority of the species present were sufficiently sampled. Good's coverage, alpha diversity indices (Shannon-Wiener [79, 80] and invsimpson [81]), richness estimators (Chao1 [82]), and abundance-based richness estimation (ACE) (83) were computed based on multiple random resamplings to make the sample size equal to the sample with the lowest number of sequences (26,177, MC station). The Berger-Parker dominance index (84) was also calculated but on nonnormalized data. Beta diversity measures were performed on normalized sample sizes in R using the vegan package. Sample clustering using the Bray-Curtis dissimilarity index was carried out using the functions “vegdist” and “hclust” in the R software. A statistical comparison of Bray-Curtis dissimilarity values between community clusters and subclusters was performed using pairwise analysis of similarity (ANOSIM) using the software Primer 6 (85). The main operationally defined taxa contributing to >90% similarity within Bray-Curtis clusters of bacterial communities were identified using similarity percentage (SIMPER) analysis (86) in Primer 6 (85). Based on SIMPER analysis, we were able to identify which taxa are driving the differences observed in the composition between sites in the GON, and then we grouped all the OTUs with the same affiliation at the order/family level.
The relationships between bacterial communities and environmental parameters were explored using canonical correspondence analysis (CCA) using the vegan package in the R software, using the envfit parameter. Data were log(x + 1) transformed prior to any statistical analysis. The distribution patterns of all the abundant taxa in each station were visualized by generating heatmaps in the MeV program (http://mev.tm4.org/#/welcome).
A linear discriminant analysis (LDA) effect size pipeline (LEfSe [87]), available at http://huttenhower.sph.harvard.edu/galaxy/, was used to determine significantly differentially distributed OTUs. A threshold of 2.0 was chosen as threshold for the logarithmic LDA scores.
A sequence from each OTU was randomly selected using the “get.oturep” command in mothur and used for alignment in ClustalW, incorporated as an accessory application in the BioEdit software package (version 7.2.5) (88). Maximum likelihood phylogenetic distances were calculated using the aligned representative sequences in FastTree (89). The corresponding tree was visualized in MEGA (version 6) (90) and was used for UniFrac analysis (91) in order to compare the phylogenetic distances between microbial communities as a function of different sites. In order to assess which stations in the tree had similar bacterial assemblages, a hierarchical clustering with abundance weight was performed, based on the distance matrix generated by computing pairwise UniFrac distance. The robustness of the clustering was determined by weighted Jackknife analysis, with random resampling of the sequences and clustering with 1,000 permutations while calculating support for each node.
Potential metabolic activity.
The 16S rRNA-to-rRNA gene ratio was used to assess the potential specific activity relative to the abundance of each operationally defined taxon at the three stations marked in Fig. 1 (VE3, PO1, and TA1) from where we could obtain good-quality RNA. All samples were extracted, amplified, and analyzed using the same procedure to avoid any bias. Singletons were removed, and data were natural log(x + 1) transformed prior to statistical testing. All tests were performed on normalized data sets with an equal number of sequences for the 16S rRNA gene and 16S rRNA libraries of each sample. Since assumptions of parametric regressions were not fulfilled even after normalization of the data, nonparametric Spearman and Kendall's tau tests were performed in the R software. Heatmaps were generated to visualize differences in the 16S rRNA gene and 16S rRNA abundances of the same OTU between sites and of different OTUs within each site. Differences in 16S rRNA-to-rRNA gene ratios between OTUs were also assessed within and between sites. Sequences were weighted to bacterial abundances.
Accession number(s).
The sequences obtained in this study have been deposited in the European Nucleotide Archive (ENA) with accession numbers ERS1162689 to ERS1162707 (study BioProject accession no. PRJEB14040).
Supplementary Material
ACKNOWLEDGMENTS
We thank Jed A. Fuhrman for his advice on the 16S rRNA gene primers used in this study, Patrick Schloss and his team for their suggestions regarding mothur, Adriana Zingone for advice, Francesco Musacchia for Trimmomatic analysis, the Captains and crew of the R/V Minerva Uno and R/V Vettoria, and Fabio Conversano, Gianluca Zazo, Violante Stefanino, and Marco Cannavacciuolo for valuable help during sampling and for chlorophyll analyses. We also thank two anonymous referees who provided useful comments to improve the manuscript.
This study was supported by the RITMARE Flag Project from the Italian Ministry of the University and Research (MIUR). K.R. was supported by a fellowship from Stazione Zoologica “Anton Dohrn” in the framework of the SZN-Open University Ph.D. Program. K.R. prepared the DNA, analyzed the flow cytometry (FCM) samples, performed the bioinformatics analyses, elaborated all data, and wrote the first draft of the manuscript. C.B. collected the samples and ran part of the flow cytometry samples, R.P. helped with the statistical analyses, V.B. performed the sequencing, E.B. and M.B. performed a previous library preparation, F.M. coordinated the nutrient and chlorophyll analyses, A.P. coordinated the sampling and performed nutrient analyses, M.S. participated in sampling, R.S. supported and supervised K.R. on bioinformatics, D.J.S. supervised K.R.'s work and reviewed the manuscript, and R.C. conceived the study, coordinated the different contributions, supervised K.R., and elaborated, prepared, and reviewed the manuscript together with K.R.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00494-17.
REFERENCES
- 1.Fuhrman JA. 2009. Microbial community structure and its functional implications. Nature 459:1–17. doi: 10.1038/nature08058. [DOI] [PubMed] [Google Scholar]
- 2.Amaral-Zettler L, Artigas LF, Baross J, Bharathi L, Boetius A, Chandramohan D, Herndl G, Kogure K, Neal P, Pedrós-Alió C, Ramette A, Schouten S, Stal L, Thessen A, Leeuw JD, Sogin M. 2010. A global census of marine microbes, p 223–245. In McIntyre AD. (ed), Life in the world's oceans. Blackwell Publishing Ltd., Oxford, United Kingdom. [Google Scholar]
- 3.Benner R, Herndl G. 2011. Bacterially derived dissolved organic matter in the microbial carbon pump, p 46–48. In Jiao N, Azam F, Sanders S (ed), Microbial carbon pump in the ocean. American Association for the Advancement of Science, Washington, DC. [Google Scholar]
- 4.Pace NR. 1997. A molecular view of microbial diversity and the biosphere. Science 276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- 5.Follows MJ, Dutkiewicz S, Grant S, Chisholm SW. 2007. Emergent biogeography of microbial communities in a model ocean. Science 315:1843–1846. doi: 10.1126/science.1138544. [DOI] [PubMed] [Google Scholar]
- 6.DeLong EF, Karl DM. 2005. Genomic perspectives in microbial oceanography. Nature 439:1014. doi: 10.1038/nature04573. [DOI] [PubMed] [Google Scholar]
- 7.Herlemann DP, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ, Andersson AF. 2011. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5:1571–1579. doi: 10.1038/ismej.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng X, Dai X, Huang L. 2016. Spatial variations of prokaryotic communities in surface water from India Ocean to Chinese marginal seas and their underlining environmental determinants. Front Mar Sci 3:17–26. doi: 10.3389/fmars.2016.00017. [DOI] [Google Scholar]
- 9.Pommier T, Canbäck B, Riemann L, Boström K, Simu K, Lundberg P, Tunlid A, Hagström Å. 2007. Global patterns of diversity and community structure in marine bacterioplankton. Mol Ecol 16:867–880. doi: 10.1111/j.1365-294X.2006.03189.x. [DOI] [PubMed] [Google Scholar]
- 10.Zinger L, Amaral-Zettler LA, Fuhrman JA, Horner-Devine MC, Huse SM, Welch DBM, Martiny JB, Sogin M, Boetius A, Ramette A. 2011. Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS One 6:e24570. doi: 10.1371/journal.pone.0024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Armbrust EV, Palumbi SR. 2015. Uncovering hidden worlds of ocean biodiversity. Science 348:865–867. doi: 10.1126/science.aaa7378. [DOI] [PubMed] [Google Scholar]
- 12.Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, Djahanschiri B, Zeller G, Mende DR, Alberti A, Cornejo-Castillo FM, Costea PI, Cruaud C, d'Ovidio F, Engelen S, Ferrera I, Gasol JM, Guidi L, Hildebrand F, Kokoszka F, Lepoivre C, Lima-Mendez G, Poulain J, Poulos BT, Royo-Llonch M, Sarmento H, Vieira-Silva S, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Tara Oceans Coordinators, Bowler C, de Vargas C, Gorsky G, Grimsley N, Hingamp P, Iudicone D, Jaillon O, Not F, Ogata H, Pesant S, Speich S, Stemmann L, Sullivan MB, Weissenbach J, Wincker P, Karsenti E, Raes J, Acinas SG, et al. 2015. Structure and function of the global ocean microbiome. Science 348:1261359. doi: 10.1126/science.1261359. [DOI] [PubMed] [Google Scholar]
- 13.Ducklow H. 2000. Bacterial production and biomass in the oceans, p 85–120. In Kirchman DL. (ed), Microbial ecology of the oceans. John Wiley and Sons, New York, NY. [Google Scholar]
- 14.Fouilland E, Mostajir B. 2010. Revisited phytoplanktonic carbon dependency of heterotrophic bacteria in freshwaters, transitional, coastal and oceanic waters. FEMS Microbiol Ecol 73:419–429. doi: 10.1111/j.1574-6941.2010.00896.x. [DOI] [PubMed] [Google Scholar]
- 15.Fuhrman J, Azam F. 1982. Thymidine incorporation as a measure of heterotrophic bacterioplankton production in marine surface waters: evaluation and field results. Mar Biol 66:109–120. doi: 10.1007/BF00397184. [DOI] [Google Scholar]
- 16.Kirchman D, K'nees E, Hodson R. 1985. Leucine incorporation and its potential as a measure of protein synthesis by bacteria in natural aquatic systems. Appl Environ Microbiol 49:599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunt DE, Lin Y, Church MJ, Karl DM, Tringe SG, Izzo LK, Johnson ZI. 2013. Relationship between abundance and specific activity of bacterioplankton in open ocean surface waters. Appl Environ Microbiol 79:177–184. doi: 10.1128/AEM.02155-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campbell BJ, Kirchman DL. 2013. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J 7:210–220. doi: 10.1038/ismej.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blazewicz SJ, Barnard RL, Daly RA, Firestone MK. 2013. Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J 7:2061–2068. doi: 10.1038/ismej.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lennon JT, Jones SE. 2011. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol 9:119–130. doi: 10.1038/nrmicro2504. [DOI] [PubMed] [Google Scholar]
- 21.Jones SE, Lennon JT. 2010. Dormancy contributes to the maintenance of microbial diversity. Proc Natl Acad Sci U S A 107:5881–5886. doi: 10.1073/pnas.0912765107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell BJ, Yu L, Heidelberg JF, Kirchman DL. 2011. Activity of abundant and rare bacteria in a coastal ocean. Proc Natl Acad Sci U S A 108:12776–12781. doi: 10.1073/pnas.1101405108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zingone A, Dubroca L, Iudicone D, Margiotta F, Corato F, d' Alcalà MR, Saggiomo V, Sarno D. 2010. Coastal phytoplankton do not rest in winter. Estuar Coasts 33:342–361. doi: 10.1007/s12237-009-9157-9. [DOI] [Google Scholar]
- 24.Cianelli D, Uttieri M, Buonocore B, Falco P, Zambardino G, Zambianchi E. 2011. Dynamics of a very special Mediterranean coastal area: the Gulf of Naples, p 129–150. In Williams GS. (ed), Mediterranean ecosystems: dynamics, management and conservation, Nova Science Publishers, Hauppauge, NY. [Google Scholar]
- 25.Grieco L, Tremblay LB, Zambianchi E. 2005. A hybrid approach to transport processes in the Gulf of Naples: an application to phytoplankton and zooplankton population dynamics. Cont Shelf Res 25:711–728. doi: 10.1016/j.csr.2004.10.014. [DOI] [Google Scholar]
- 26.Carrada GC, Fresi E, Marino D, Modigh M, Ribera d'Alcalà M. 1981. Structural analysis of winter phytoplankton in the Gulf of Naples. J Plankton Res 3:291–314. doi: 10.1093/plankt/3.2.291. [DOI] [Google Scholar]
- 27.Marino D, Modigh M, Zingone A. 1984. General features of phytoplankton communities and primary production in the Gulf of Naples and adjacent waters, p 89–100. In Holm-Hansen O, Bolis L, Gilles L (ed), Marine phytoplankton and productivity. Springer, Berlin, Germany. [Google Scholar]
- 28.Carrada G, Hopkins T, Bonaduce G, Ianora A, Marino D, Modigh M, d'Alcalà MR, di Scotto CB. 1980. Variability in the hydrographic and biological features of the Gulf of Naples. Mar Ecol 1:105–120. doi: 10.1111/j.1439-0485.1980.tb00213.x. [DOI] [Google Scholar]
- 29.Mazzocchi MG, Dubroca L, García-Comas C, Di Capua I, d'Alcalà MR. 2012. Stability and resilience in coastal copepod assemblages: the case of the Mediterranean long-term ecological research at Station MC (LTER-MC). Prog Oceanogr 97:135–151. doi: 10.1016/j.pocean.2011.11.003. [DOI] [Google Scholar]
- 30.Modigh M, Saggiomo V, d'Alcala MR. 1996. Conservative features of picoplankton in a Mediterranean eutrophic area, the Bay of Naples. J Plankton Res 18:87–95. doi: 10.1093/plankt/18.1.87. [DOI] [Google Scholar]
- 31.Casotti R, Brunet C, Aronne B, d'Alcala MR. 2000. Mesoscale features of phytoplankton and planktonic bacteria in a coastal area as induced by external water masses. Mar Ecol Prog Ser 195:15–27. doi: 10.3354/meps195015. [DOI] [Google Scholar]
- 32.Izzo G, Tosti E, Volterra L. 1983. Fecal contamination of marine sediments in a stretch of the Gulf of Naples. Water Air Soil Pollut 20:191–198. doi: 10.1007/BF00279629. [DOI] [Google Scholar]
- 33.Volterra L, Tosti E, Vero A, Izzo G. 1985. Microbiological pollution of marine sediments in the southern stretch of the Gulf of Naples. Water Air Soil Pollut 26:175–184. doi: 10.1007/BF00292067. [DOI] [Google Scholar]
- 34.Quero GM, Luna GM. 2014. Diversity of rare and abundant bacteria in surface waters of the Southern Adriatic Sea. Mar Genomics 17:9–15. doi: 10.1016/j.margen.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Pommier T, Neal PR, Gasol JM, Coll M, Acinas SG, Pedrós-Alió C. 2010. Spatial patterns of bacterial richness and evenness in the NW Mediterranean Sea explored by pyrosequencing of the 16S rRNA. Aquat Microb Ecol 61:221–233. doi: 10.3354/ame01484. [DOI] [Google Scholar]
- 36.Du J, Xiao K, Li L, Ding X, Liu H, Lu Y, Zhou S. 2013. Temporal and spatial diversity of bacterial communities in coastal waters of the South China Sea. PLoS One 8:e66968. doi: 10.1371/journal.pone.0066968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hewson I, Fuhrman JA. 2004. Richness and diversity of bacterioplankton species along an estuarine gradient in Moreton Bay, Australia. Appl Environ Microbiol 70:3425–3433. doi: 10.1128/AEM.70.6.3425-3433.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mou X, Sun S, Edwards RA, Hodson RE, Moran MA. 2008. Bacterial carbon processing by generalist species in the coastal ocean. Nature 451:708–711. doi: 10.1038/nature06513. [DOI] [PubMed] [Google Scholar]
- 39.Luna GM. 2015. Diversity of marine microbes in a changing Mediterranean Sea. Rend Lincei 26:49–58. doi: 10.1007/s12210-014-0333-x. [DOI] [Google Scholar]
- 40.Salter I, Galand PE, Fagervold SK, Lebaron P, Obernosterer I, Oliver MJ, Suzuki MT, Tricoire C. 2015. Seasonal dynamics of active SAR11 ecotypes in the oligotrophic Northwest Mediterranean Sea. ISME J 9:347–360. doi: 10.1038/ismej.2014.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grote J, Thrash JC, Huggett MJ, Landry ZC, Carini P, Giovannoni SJ, Rappé MS. 2012. Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio 3(5):e00252-12. doi: 10.1128/mBio.00252-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R, Novotny M, Yee-Greenbaum J, Selengut JD, Haft DH, Halpern AL, Lasken RS, Nealson K, Friedman R, Venter JC. 2012. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6:1186–1199. doi: 10.1038/ismej.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giovannoni SJ, Bibbs L, Cho JC, Stapels MD, Desiderio R, Vergin KL, Rappé MS, Laney S, Wilhelm LJ, Tripp HJ, Mathur EJ, Barofsky DF. 2005. Proteorhodopsin in the ubiquitous marine bacterium SAR11. Nature 438:82–85. doi: 10.1038/nature04032. [DOI] [PubMed] [Google Scholar]
- 44.Gómez-Pereira PR, Hartmann M, Grob C, Tarran GA, Martin AP, Fuchs BM, Scanlan DJ, Zubkov MV. 2013. Comparable light stimulation of organic nutrient uptake by SAR11 and Prochlorococcus in the North Atlantic subtropical gyre. ISME J 7:603–614. doi: 10.1038/ismej.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brinkhoff T, Giebel HA, Simon M. 2008. Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch Microbiol 189:531–539. doi: 10.1007/s00203-008-0353-y. [DOI] [PubMed] [Google Scholar]
- 46.Taylor JD, Cottingham SD, Billinge J, Cunliffe M. 2014. Seasonal microbial community dynamics correlate with phytoplankton-derived polysaccharides in surface coastal waters. ISME J 8:245–248. doi: 10.1038/ismej.2013.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.d'Alcalà MR, Conversano F, Corato F, Licandro P, Mangoni O, Marino D, Mazzocchi M, Modigh M, Montresor M, Nardella M, Saggiomo V, Sarno D, Zingone A. 2004. Seasonal patterns in plankton communities in a pluriannual time series at a coastal Mediterranean site (Gulf of Naples): an attempt to discern recurrences and trends. Sci Mar 68:65–83. doi: 10.3989/scimar.2004.68s165. [DOI] [Google Scholar]
- 48.Liu M, Dong Y, Zhao Y, Zhang G, Zhang W, Xiao T. 2011. Structures of bacterial communities on the surface of Ulva prolifera and in seawaters in an Ulva blooming region in Jiaozhou Bay, China. World J Microb Biotechnol 27:1703–1712. doi: 10.1007/s11274-010-0627-9. [DOI] [Google Scholar]
- 49.Cho JC, Giovannoni SJ. 2004. Cultivation and growth characteristics of a diverse group of oligotrophic marine Gammaproteobacteria. Appl Environ Microbiol 70:432–440. doi: 10.1128/AEM.70.1.432-440.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crespo BG, Pommier T, Fernández-Gómez B, Pedrós-Alió C. 2013. Taxonomic composition of the particle-attached and free-living bacterial assemblages in the Northwest Mediterranean Sea analyzed by pyrosequencing of the 16S rRNA. Microbiologyopen 2:541–552. doi: 10.1002/mbo3.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li J, Li N, Li F, Zou T, Yu S, Wang Y, Qin S, Wang G. 2014. Spatial diversity of bacterioplankton communities in surface water of northern South China Sea. PLoS One 9:e113014. doi: 10.1371/journal.pone.0113014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zwirglmaier K, Jardillier L, Ostrowski M, Mazard S, Garczarek L, Vaulot D, Not F, Massana R, Ulloa O, Scanlan DJ. 2008. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol 10:147–161. [DOI] [PubMed] [Google Scholar]
- 53.Scanlan DJ, Ostrowski M, Mazard S, Dufresne A, Garczarek L, Hess WR, Post AF, Hagemann M, Paulsen I, Partensky F. 2009. Ecological genomics of marine picocyanobacteria. Microbiol Mol Biol Rev 73:249–299. doi: 10.1128/MMBR.00035-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kent AG, Dupont CL, Yooseph S, Martiny AC. 2016. Global biogeography of Prochlorococcus genome diversity in the surface ocean. ISME J 10:1856–1865. doi: 10.1038/ismej.2015.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alonso-Sáez L, Díaz-Pérez L, Morán XA. 2015. The hidden seasonality of the rare biosphere in coastal marine bacterioplankton. Environ Microbiol 17:3766–3780. doi: 10.1111/1462-2920.12801. [DOI] [PubMed] [Google Scholar]
- 56.Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci U S A 103:12115–12120. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Galand PE, Casamayor EO, Kirchman DL, Lovejoy C. 2009. Ecology of the rare microbial biosphere of the Arctic Ocean. Proc Natl Acad Sci U S A 106:22427–22432. doi: 10.1073/pnas.0908284106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vandamme P, Vancanneyt M, Pot B, Mels L, Hoste B, Dewettinck D, Vlaes L, van den Borre C, Higgins R, Hommez J. 1992. Polyphasic taxonomic study of the emended genus Arcobacter with Arcobacter butzleri comb. nov. and Arcobacter skirrowii sp. nov., an aerotolerant bacterium isolated from veterinary specimens. Int J Syst Bacteriol 42:344–356. doi: 10.1099/00207713-42-3-344. [DOI] [PubMed] [Google Scholar]
- 59.Campbell AM, Fleisher J, Sinigalliano C, White JR, Lopez JV. 2015. Dynamics of marine bacterial community diversity of the coastal waters of the reefs, inlets, and wastewater outfalls of southeast Florida. Microbiologyopen 4:390–408. doi: 10.1002/mbo3.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luna GM, Quero GM, Perini L. 2016. Next generation sequencing reveals distinct fecal pollution signatures in aquatic sediments across gradients of anthropogenic influence. Adv Oceanogr Limnol 25:157. [Google Scholar]
- 61.Loy A, Schulz C, Lücker S, Schöpfer-Wendels A, Stoecker K, Baranyi C, Lehner A, Wagner M. 2005. 16S rRNA gene-based oligonucleotide microarray for environmental monitoring of the betaproteobacterial order “Rhodocyclales.” Appl Environ Microbiol 71:1373–1386. doi: 10.1128/AEM.71.3.1373-1386.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu J, Fu B, Yang H, Zhao M, He B, Zhang XH. 2015. Phylogenetic shifts of bacterioplankton community composition along the Pearl Estuary: the potential impact of hypoxia and nutrients. Front Microbiol 6:64. 10.3389/fmicb.2015.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aanderud ZT, Vert JC, Lennon JT, Magnusson TW, Breakwell DP, Harker AR. 2016. Bacterial dormancy is more prevalent in freshwater than hypersaline lakes. Front Microbiol 7:853. doi: 10.3389/fmicb.2016.00853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sukenik A, Kaplan-Levy RN, Welch JM, Post AF. 2012. Massive multiplication of genome and ribosomes in dormant cells (akinetes) of Aphanizomenon ovalisporum (Cyanobacteria). ISME J 6:670–679. doi: 10.1038/ismej.2011.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baas-Becking LGM. 1934. Geobiologie of Inleiding Tot de Milieukunde. In W.P. Van Stockum & Zoon, The Hague, The Netherlands. [Google Scholar]
- 66.Vergin KL, Jhirad N, Dodge J, Carlson CA, Giovannoni SJ. 2017. Marine bacterioplankton consortia follow deterministic, non-neutral community assembly rules. Aquat Microb Ecol 79:165–175. doi: 10.3354/ame01824. [DOI] [Google Scholar]
- 67.Curry R, Dickson B, Yashayaev I. 2003. A change in the freshwater balance of the Atlantic Ocean over the past four decades. Nature 426:826–829. doi: 10.1038/nature02206. [DOI] [PubMed] [Google Scholar]
- 68.Lynch MD, Neufeld JD. 2015. Ecology and exploration of the rare biosphere. Nat Rev Microbiol 13:217–229. doi: 10.1038/nrmicro3400. [DOI] [PubMed] [Google Scholar]
- 69.Holm-Hansen O, Lorenzen CJ, Holmes RW, Strickland JD. 1965. Fluorometric determination of chlorophyll. ICES J Mar Sci 30:3–15. doi: 10.1093/icesjms/30.1.3. [DOI] [Google Scholar]
- 70.Hansen HP, Grasshoff K. 1983. Automated chemical analysis. In Grasshoff K, Ehrhardt M, Kremlin K (ed), Methods of seawater analysis. Verlag Chemie, Weinheim, Germany. [Google Scholar]
- 71.Balestra C, Alonso-Sáez L, Gasol JM, Casotti R. 2011. Group-specific effects on coastal bacterioplankton of polyunsaturated aldehydes produced by diatoms. Aquat Microb Ecol 63:123–131. doi: 10.3354/ame01486. [DOI] [Google Scholar]
- 72.Lane DJ. 1991. 16S/23S rRNA sequencing, p 115–147. In Stackebrandt E, Goodfellow M (ed), Nucleic acid techniques in bacterial systematics. John Wiley & Sons, New York, NY. [Google Scholar]
- 73.Parada A, Needham DM, Fuhrman JA. 2015. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time-series and global field samples. Environ Microbiol 18:1403–1414. doi: 10.1111/1462-2920.13023. [DOI] [PubMed] [Google Scholar]
- 74.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lundin D, Severin I, Logue JB, Östman Ö, Andersson AF, Lindström ES. 2012. Which sequencing depth is sufficient to describe patterns in bacterial α-and β-diversity? Environ Microbiol Rep 4:367–372. doi: 10.1111/j.1758-2229.2012.00345.x. [DOI] [PubMed] [Google Scholar]
- 79.Shannon CE, Weaver W. 1949. The mathematical theory of information. University of Illinois Press, Urbana, IL. [Google Scholar]
- 80.Krebs CJ. 1989. Ecological methodology. Harper & Row, New York, NY. [Google Scholar]
- 81.Simpson EH. 1949. Measurement of diversity. Nature 163:688. doi: 10.1038/163688a0. [DOI] [Google Scholar]
- 82.Chao A. 1984. Non-parametric estimation of the number of classes in a population. Scand J Stat 11:265–270. [Google Scholar]
- 83.Chao A, Lee SM. 1992. Estimating the number of classes via sample coverage. J Am Stat Assoc 87:210–217. doi: 10.1080/01621459.1992.10475194. [DOI] [Google Scholar]
- 84.Magurran M. 1988. Ecological diversity and its measurement. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 85.Clarke KR, Gorley RN. 2006. Primer v6: user manual/tutorial. PRIMER-E, Plymouth, United Kingdom. [Google Scholar]
- 86.Clarke KR. 1993. Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 18:117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x. [DOI] [Google Scholar]
- 87.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biol 12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser (Oxf) 41:95–98. [Google Scholar]
- 89.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.