

Abstract
The relationship between inflammation and cancer has been recognized since the 17th century (6), and we now know much about the cells, cytokines and physiological processes that are central to both inflammation and cancer (39, 62, 66–68, 86, 92, 125). Chronic inflammation can induce certain cancers (11, 14, 27, 59, 61, 76, 79, 82), and solid tumors, in turn, can initiate and perpetuate local inflammatory processes that foster tumor growth and dissemination (7, 17, 67, 96). Consequently, inflammatory pathways have been targeted in attempts to control cancer (22, 46, 47). Inflammation is a central aspect of the innate immune system’s response to tissue damage or infection, and also facilitates the recruitment of circulating cells and antibodies of the adaptive immune response to the tissue. Components of the innate immune response carry out a robust, but sometimes overly-conservative response, sacrificing specificity for the sake of preservation. Thus, when innate immunity goes awry, it can have profound implications. How the innate and adaptive immune systems cooperate to neutralize pathogens and repair damaged tissues is still an area of intense investigation. Further, how these systems can respond to cancer, which arises from normal “self” cells that undergo an oncogenic transformation, has profound implications for cancer therapy. Recently, immunotherapies that activate adaptive immunity have shown unprecedented promise in the clinic, producing durable responses and dramatic increases in survival rate in patients with advanced stage melanoma (84, 119, 121). Consequently, the relationship between cancer and inflammation has now returned to the forefront of clinical oncology.
Graphical abstract
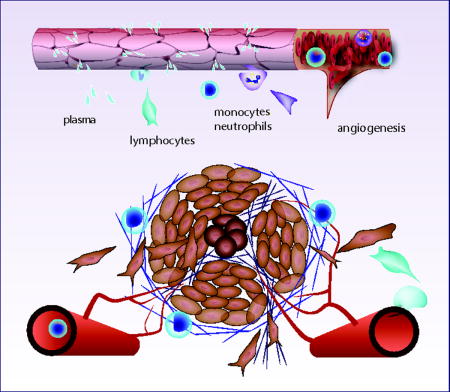
What is inflammation?
Inflammation is a primitive but robust response that serves as a rapid defense mechanism to contain potential pathogens, limit further tissue damage, and stimulate repair mechanisms. It is initiated by tissue-resident cells that detect pathogens or trauma and send alarm signals in the form of chemical messengers, which amplify the local response and recruit other cells to the area.
The four clinically-observable characteristics that indicate an inflammatory process are still often referred to by their latin terms (Figure 1): dolor (pain), calor (heat), rubor (redness), and tumor (swelling). Another symptom often associated with inflammation – loss of function – results from a neurological response that shuts down motor nerve function, presumably to limit motion, and thus minimize further damage.
Figure 1.

Differences in the progression of acute and cancer-induced inflammation. While many of the cells and processes are common, acute inflammation resolves after the pathogen is eliminated and/or tissue architecture has been stabilized. In cancer-induced inflammation, there is continuous mechanical disruption, cytokine and growth factor production and cell recruitment, so inflammation persists.
The process can be triggered by pathogens or trauma to the tissue, caused, for example, by mechanical damage, chemicals or extreme temperatures. The response to these diverse insults is surprisingly similar, and produces a number of biological and physiological changes (Figure 2a).
Figure 2.
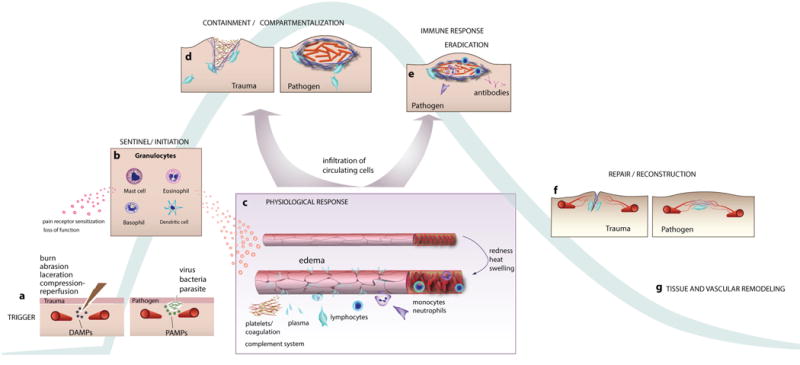
Inflammation induced by tissue insult or pathogens. a) Tissue damage produces cell and matrix debris (damage-associated molecular patterns, DAMPs) that binds to receptors on resident granuloctyes (mast cells, eosinophils, basophils). Similarly, pathogens release substances (pathogen -associated molecular patterns, PAMPs) that bind to receptors on these cells. b Activation of the granulocytes releases cytokines and chemokines that act on pain receptors and vascular cells. c) In response, blood vessels dilate and become leaky. The extravasating plasma carries proteins that modify the ECM, and the changes in blood flow and EC adhesion molecules facilitate the infiltration of circulating neutrophils, monocytes and other myeloid cells into the tissue. d) Macrophages and neutrophils clean up debris, attack any pathogens and fortify the extracellular matrix. e) In the case of pathogenic response, the cells and antibodies produced in the adaptive immune response arrive and help clear the infection. f) Tissue architecture is restored by macrophages and fibroblasts, which contract the wound and normalize the ECM. Angiogenic vessels re-perfuse the tissue. g) Remodeling of the tissue continues for weeks/months as blood vessels and matrix structures are refined.
Invading pathogens release foreign peptides (e.g., bacterial wall components, flagella), carbohydrates, and nucleic acids (bacteria or virus-specific DNA or RNA). Similarly, tissue damage releases substances such as uric acid, extracellular ATP and other debris from cells and the extracellular matrix. In either case, these substances can bind to pattern recognition receptors (PRRs) on the surface of resident macrophages, dendritic cells, histiocytes, mast cells and other granulocytes. Toll-like receptors (TLRs) are an important family of pattern recognition receptors involved in the initial response to many pathogens. In addition to recognizing foreign molecular patterns, TLRs respond to host molecules produced in response to stresses, including reactive oxygen and nitrogen species (RONS), HMGB1 (high-mobility group box protein 1), surfactant protein A, fibrinogen, degraded matrix components, heat shock proteins (hsp) and eosinophil-derived neurotoxin (EDN) (20).
Once engaged and activated, granulocytes respond by releasing the contents of their granules, which includes a potent cocktail of antimicrobial agents, enzymes, cytokines and reactive oxygen and nitrogen species (RONS; Figure 2b). These substances have dramatic effects on the blood vessels. For example, histamine, tumor necrosis factor-alpha (TNFα) and nitric oxide (NO) cause the blood vessels to dilate, increasing blood flow into the tissue. This is accompanied by a change in the endothelial cell junctions in the blood vessel wall, which allows plasma proteins to leak into the tissue (Figure 2c). These vascular effects are responsible for three of the classical hallmarks of inflammation: the increased blood perfusion causes the redness and heat, while the leakage of fluid into the tissue causes swelling (see Figure 1). The fourth – pain – is due to cytokines (including prostaglandins, TNFα, and interleukins) that sensitize neuro-receptors to amplify pain.
If there is sufficient damage to the blood vasculature, platelets and plasma proteins spill into the tissue, contacting matrix components and causing rapid clot formation. Platelet thrombosis is a robust mechanism for limiting blood loss, and is only transiently necessary (although can be an ongoing process if there is repeated injury). Platelets are versatile particles, simultaneously forming an interconnected polymeric fibrin clot and releasing multiple growth factors that attract inflammatory cells that help repair the damage (Figure 2c).
Damage to the vasculature releases platelets and blood cells into the tissue, but even in the absence of frank physical trauma, granulocyte cytokines such as TNFα and histamine can increase the permeability of the endothelial junctions. This allows plasma fluid and proteins to filter across the vessel wall – without the loss of blood cells– and bathe the surrounding tissue. This serves multiple purposes. First, plasma contains provisional matrix components such as fibronectin and fibrinogen that condition the existing ECM to facilitate the imminent cell infiltration and repair process. Second, there are growth factors carried by the plasma that convect into the tissue and bind to matrix structures to aid in the recruitment and guidance of infiltrating immune cells. In addition to carrying plasma proteins and growth factors into the traumatized tissue, the fluid itself can serve as a signal as it flows through the interstitium, remodeling matrix structures (42) and activating mechanobiological pathways to guide the migration of leukocytes, fibroblasts and angiogenic blood vessels (94, 98, 106).
Vascular dilation and hyperpermeability – the early events of inflammation – also enhance the transport of circulating immune cells to the tissue, and this aspect of inflammation has been reviewed extensively (17, 104). As the plasma is filtered across the vessel, the blood gets more concentrated, increasing hematocrit. This, combined with the vessel dilation, slows blood velocity, enhances erythrocyte aggregation, and creates an erythrocyte-depleted zone near the vessel wall (102). Leukocytes are excluded from the central mass of erythrocytes and consequently travel near the vessel wall, where they can contact adhesion molecules that mediate leukocyte binding (70, 72, 75, 103). Endothelial adhesion molecules including ICAM-1, VCAM-1, PECAM-1, E-selectin and P-selectin are upregulated by granulocyte-derived growth factors such as histamine, interleukins, RONS and TNFα (25, 55, 71, 99). In this way, the leukocytes roll along the blood vessel wall, come to a stop and extravasate into the tissue.
Some of the cytokines produced in inflammation act as chemokines, which create chemical gradients to provide cues for cells as they extravasate and migrate into the tissue. Chemokines of the CXC family (112) are implicated in the recruitment of neutrophils and other leukocytes as well as angiogenic blood vessels (83, 89). In response to these chemokines, neutrophils and monocytes enter the tissue (57). The neutrophils begin phagocytosing microbes and cell debris and releasing additional cytokines to amplify the response. They also contain granules and can release anti-microbial molecules similar to the resident granulocytes and can create “neutrophil extracellular traps” (NETs). NETs are networks of DNA-rich fibers embedded with histones and granule proteins including neutrophil elastase, cathepsin G and myeloperoxidase that can trap and kill bacteria and fungi extracellularly, without phagocytosis (40, 45, 93, 107). The monocytes can differentiate into macrophages when they enter the tissue and also phagocytose cells and debris. Macrophages help orchestrate adaptive immunity by producing both pro- and anti-inflammatory factors (46, 56, 96). New blood vessels are also created in inflammatory responses. Damage to the tissue and blood vessels, the recruitment of cells, and the increased cell activity can all contribute to hypoxia, which initiates the recruitment of these new blood vessels.
In acute inflammation, neovascularization is self-limiting. The new blood vessels increase oxygen delivery, reducing hypoxia and the production of angiogenic growth factors such as VEGF. In a successful inflammatory response, these steps (immune cell recruitment and elimination of pathogens and repair of damage tissue) are sufficient to restore homeostasis (Figure 2e–g)). When there are no more pathogens or damage-associated activators to sustain the process, the inflammation resolves and the tissue continues remodeling over time to optimize the microanatomy and vasculature. The resolution of acute inflammation is aided by short half-lives of the biochemical mediators of the innate immune response. Because they are rapidly cleared, inflammation resolves soon after the triggering stimulus is removed. Unfortunately, this fail-safe mechanism is circumvented in pathological, chronic inflammation. This can happen, for example, when the pathogen cannot be completely eradicated by the adaptive and innate immune responses, resulting in a “stand-off” (eg., in tuberculosis in the lung, hepatitis infection in the liver, and H. pylori infection in the gut). It is also the case in auto-immune inflammation, where there is a constant source of self-antigen to perpetuate the response. And, as we will see later, resolution of inflammation can be thwarted by a growing tumor, which continuously disrupts the tissue structure, produces inflammatory cytokines, and recruits inflammatory cells (Figure 1, bottom).
The role of inflammation in cancer initiation
There is a wealth of evidence showing that chronic inflammation can promote cancer development and that tumor-induced inflammation creates a “snowball” effect, perpetuating tumor progression (64). It is estimated that as many as 20% of cancers are instigated by chronic inflammation or persistent infections (115). Inflammatory cytokines and cells are implicated in tumorigenesis and tumor progression in most sites, and have been extensively documented in cancers of the stomach (116, 118), colon (16, 35, 63), skin (3, 65, 78, 126, 128), liver (4, 8, 41), breast (2, 21, 48), lung (32, 83, 100), and head/neck (12, 14, 15).
Some of the important pathways linking inflammation to tumor initiation or progression have been identified. The inflammatory microenvironment has elevated levels of nuclear factor kappa B, RONS, cytokines, prostaglandins and microRNAs that affect cell proliferation, cell death, cellular senescence, DNA mutation rates, DNA methylation and angiogenesis (90). Cytokines such as IL-6 are present in many cancers, and have been implicated in tumorigenesis and metastasis (108) by regulating NF-kappaB and STAT3 (29). Inflammation after partial liver resection can instigate hepatocellular carcinoma (8), through Akt and ARRB1 pathways (123). And in mouse models, it has been shown that TNFα increases spontaneous mammary cancer (88). This list is not exhaustive, and it is likely that many, if not all, of the cells and biomolecules produced in inflammation play some role in the initiation and maintenance of tumor growth.
The association between inflammation and cancer is perhaps best documented in the digestive system, where inflammation caused by diet, gut bacteria, infection or auto-immune disorders is relatively common. For example, chronic ulcerative colitis and Crohn’s disease increase the risk for colon cancer, acid reflux is associated with esophageal cancer, Hepatitis C infection and alcohol consumption increase the risk of hepatocellular carcinoma, and Helicobacter pylori infection increases the risk of gastric and colon cancer. The chronic inflammation that precedes tumorigenesis exhibits many of the same features as the acute, physiological process. For example, animal models have shown that inflammation causes activation of mucosal mast cells, which recruit CD11b(+)Gr1(+) myeloid cells from the circulation that help promote colitis-related colon cancer (122).
Foods can contribute to chronic inflammation that enables gastrointestinal cancers. Although the mechanistic links between diet and inflammation are not completely understood, the relationship is well-established in some cases. Chronic consumption of alcohol activates mast cells, causes polyp formation, and enhances tumor formation and invasion in a mouse model of colon cancer (120). In some cases, specific antigens have been identified that can initiate and sustain inflammation. For example, it has been shown that red meat contains high levels of a nonhuman form of the sialic acid N-glycolylneuraminic acid (Neu5Gc). This foreign antigen can get incorporated into tissue and attract inflammatory cells (87).
Whether the source is from auto-immunity, infection or food-related, inflammation has important implications for the gut microenvironment. In the colon, inflammation induces DNA damage through RONS (69). The RONS are produced, in part, by Gr-1+cells: Gr-1+ neutrophils produce nitric oxide via inducible nitric oxide synthase (iNOS) to exacerbate inflammation, hyperplasia, dysplasia, and tumor progression in the colon. Blocking iNOS-mediated RONS production inhibits carcinogenesis (33).
Inflammation can also modify the population of microbes in the gut, favoring the growth of harmful bacteria such as E. coli. Indeed, E. coli is found in higher concentrations in patients with inflammatory bowel disease and colorectal cancer, suggesting that colitis can enable tumorigenesis by shifting the microbiome toward a population more capable of inducing gene damage and mutagenesis (5). Another cancer-implicated shift in the microbiome is infection with Heliobacter pylori, which can induce gastric inflammation through the action of specific proteins such as CagA (105), and by activating Toll-like receptor (TLR), NFKB and Cyclooxygenase-2 (COX-2) -prostaglandin E2 (PGE2) pathways (31). In an inflammatory environment, which contains many proteases and RONS, DNA damage becomes more likely. This is especially important in the gut, where ongoing epithelial proliferation renders the genetic material more vulnerable. In addition to direct damage to DNA, epigenetic alterations such as hypermethylation of DNA methylation valleys have also been documented in the context of inflammation (1).
Inflammation is also implicated in obesity-related cancers. With the increasing prevalence of obesity in the world, there is much interest in understanding the elevated risk of cancer in overweight and obese patients. Much of the effort is focused on obesity-induced inflammation, which has now been linked to cancer progression at a number of sites (10, 36, 44, 53, 111). As new adipose tissues form, angiogenesis creates additional blood vessels to supply the tissue. The angiogenesis associated with adipose expansion plays a role in the chronic inflammation observed in obesity (36). In models of pancreatic ductal adenocarcinoma development in obese mice, the obesity promotes desmoplasia by recruiting macrophages and neutrophils, and by activating pancreatic stellate cells; this desmoplasia facilitates tumor growth (49, 51). Blocking PlGF or its receptor VEGFR1 signaling prevents the obesity-induced tumor growth (49, 51). Thus, there appears to be collusion amongst resident cells, recruited cells and adipocytes that promotes tumor progression in obesity. Breast cancer progression can also be affected by obesity. In tissues of obese patients, macrophages can be seen surrounding dead or dying adipocytes, creating “crown-like” structures in a process known as white adipose tissue (WAT) inflammation. The presence of WAT inflammation is accompanied by elevated levels of insulin, glucose, leptin, C-reactive protein and IL6, potentially contributing to the poorer prognosis of cancer patients who are overweight or obese (53).
A similar relationship between inflammation and carcinogenesis is found in prostate cancer (26, 109), although the association is not always consistent (74). In cases when inflammation predisposes prostate cancer, macrophage inhibitory cytokine-1 (MIC-1) expression, driven by IL-1beta and TNF-alpha pathways, is involved in the carcinogenesis (30, 54).
When chronic inflammation exists, cytokines and other related species are found in the blood circulation. There are now efforts aimed at identifying and measuring these species to inform treatment strategy or identify patients likely to develop cancer from at-risk populations. The hope is that these inflammatory biomarkers will allow oncologists to determine the risk of tumorigenesis or tumor recurrence after surgery. Supporting this approach, inflammation-related biomarkers in plasma have been found to be associated with a higher risk of developing lung cancer: C-reactive protein, serum amyloid A, soluble tumor necrosis factor receptor-2 and monokine induced by gamma interferon (CXCL9/MIG) were more prevalent in the blood of patients who went on to develop cancer (95). Other biomarker approaches are simpler and easily implemented. By calculating serum albumin levels and the ratio of neutrophils to lymphocytes in the blood, it is possible to calculate a “prognostic inflammation score”. This metric is a significant prognostic marker in ovarian cancer (117). Another metric has been proposed to assess inflammation in cervical cancer (cervical cancer systemic inflammation score, CCSIS). Based on a retrospective study, this score used the platelet/lymphocyte ratio in conjunction with serum albumin level. CCSIS correlated with cancer stage, and high scores were associated with poor tumor differentiation and higher numbers of inflammatory cells (129). Monitoring inflammation is not only limited to blood-borne biomarkers. To assess inflammation in the gut, it is possible to detect biomarkers in the feces that predict the occurrence of mucinous colon adenocarcinoma, as has been demonstrated in a mouse model of Helicobacter bilis-infection (34).
Cancer initiates and perpetuates inflammation
Of course, inflammation is not necessary for mutagenesis, and many cancers form in non-inflamed tissues. But even in these cases, the tumor itself creates inflammation (Figure 3a) (28). The extent and nature of this inflammation varies, but in general, tends to facilitate tumor progression (67). Tumors can induce inflammatory reactions through a number of mechanisms. First, the cancer cells can express cytokines that recruit neutrophils and macrophages (13). These recruited cells then release additional inflammatory molecules, amplifying the response. Second, the growing tumor can physically damage the normal tissue, releasing damage-associated molecular patterns (DAMPs) that activate receptors on resident granulocytes, which initiate inflammatory repair mechanisms. Similarly, growing tumors can compress blood and lymphatic vessels, shutting off the supply of oxygen and nutrients (19, 101). The resulting hypoxia leads to the production of cytokines and angiogenic growth factors, which recruit new blood vessels and macrophages (Figure 3b–d). In contrast to acute inflammation, which eventually resolves, these inflammatory processes persist as long as the tumor continues to damage normal structures and produce relevant cytokines (Figure 3f).
Figure 3.
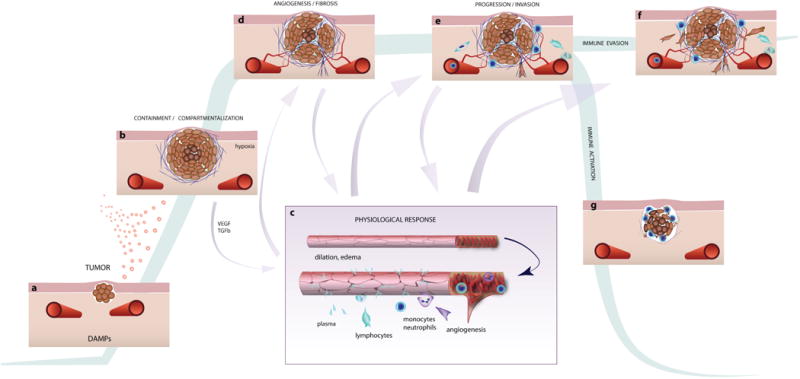
Involvement of inflammatory processes during tumor development and progression. a) After mutagenesis, cancer cells proliferate, disrupting the surrounding tissue, drawing the attention of resident macrophages and fibroblasts. Cancer cell-produced cytokines and growth factors may also play a role in this early stage. Resident granulocytes may also respond to DAMPs released by damaged cells and matrix as the tumor disrupts normal tissue. b) As the tumor expands, macrophages and fibroblasts are recruited to the area, and matrix production is increased. Oxygen diffusion limitations cause hypoxia in the tumor core, and angiogenic growth factors such as VEGF are produced by the tumor. c) The growth factors act on nearby blood vessels, causing dilation and leakiness. d) The growth factor environment, modified ECM and fluid flow through the tissue all encourage formation of new vessels that enter and feed the tumor. e) This environment, and the new vasculature, accelerates the infiltration of innate immune cells, which produce additional inflammatory cytokines that further affect the blood vessels and recruit more cells. f) Without specific anti-tumor immunity, these events amplify out of control, and the abundant/modified ECM provides pathways for cancer invasion. g) Immunotherapies can activate the adaptive immune response, allowing cytotoxic T-cells to kill the cancer cells.
As part of the damage repair mechanism, fibroblasts modify the extracellular matrix structure (Figure 3d,e). When cancer grows in breast tissue, this process causes a dense collagen matrix that is mechanically stiff, which can serve as an early indicator of a tumor by self-examination. Such fibrosis can activate the migration and invasion of cancer cells through the MAPK and PI3K pathways (60, 85), and in breast cancer patients, dense, aligned patterns of collagen are associated with poorer prognosis (23). One of the enzymes implicated in modifying collagen structure in breast cancer is lysil oxidase (LOX). LOX is expressed by activated fibroblasts in tumors, and its production is enhanced in response to high levels of TGF-beta. LOX causes collagen crosslinking, which strengthens focal adhesions, upregulates PI3K, and facilitates cell migration. Consequently, blocking the ability of LOX to induce collagen crosslinking decreases metastasis (81).
Another ECM component prevalent in solid tumors is hyaluronan (HA). It can create compressive forces in the tumor leading to vessel collapse (18, 101) and activate cancer cells by binding to cell-surface receptors (73). Upon fragmentation by RONS, HA becomes pro-inflammatory as it binds to CD44 and RHAMM receptors on tumor and host cells (91).
Interestingly, it is not only tumor growth that can induce inflammation, but also the attempts to eradicate cancer. This is especially evident with radiation therapy, which is known to induce inflammatory responses in the tumor and surrounding tissue. This results in a number of classical processes involved in damage-induced inflammation, including increased vascular hyperpermeability mediated by mast cell degranulation (77). The inflammation and RONS production can affect the regrowth of residual tumor cells, and this is mediated, in part, by bone marrow-derived CD11b+ cells. The myeloid BMDCs, primarily monocytes, rapidly appear in tumors after local irradiation, attracted by tumor-produced chemokines including stromal-derived factor 1alpha (SDF-1α). Blocking the association of SDF-1α with its receptor CXCR4 inhibits tumor regrowth (58).
Targeting inflammation to prevent or treat cancer
Because the link between inflammation and cancer has been known for decades, there have been a host of treatments developed or prevent or treat cancer by blocking inflammation. One approach is to inhibit cyclooxygenase (COX) activity using non-steroidal anti-inflammatory drugs (NSAIDs) or selective blockers. COX-2 fuels cancer-related inflammation by converting arachidonic acid into prostaglandin E2, which is pro-inflammatory. NSAIDs such as aspirin and ibuprofen inhibit COX enzymes, and their long-term use can decrease systemic inflammation and the incidences of colon (24, 113, 114) and lung (38) cancers. Celecoxib, a COX-2 specific NSAID, reduced duodenal polyposis (an early event in bowel carcinogenesis) after six months of treatment (80), and when combined with paclitaxel, it can improve lung cancer therapy (37). Other studies have shown less encouraging results (52, 130), and additional clinical studies are needed to determine the potential benefits and risks of long-term NSAIDs for cancer prevention and treatment.
Another strategy is to normalize the tumor-induced desmoplasia in an attempt to remove the associated signals that sustain the inflammation as tumors expand. Angiotensin II type 1 (AT1) receptors are regulated by oxidative stress and hypoxia, and are therefore important in cancer-related inflammation (97). AT1 receptor signaling activates fibroblasts, contributing to fibrosis. Blocking AT1 receptor signaling reduces the production of stromal collagen and hyaluronan (19). This has the effect of normalizing the extracellular matrix and decompressing blood vessels, allowing better delivery of anti-tumor drugs. Attempts to normalize the inflammatory environment in this way in pancreatic cancer have shown promise. Pancreatic ductal adenocarcinoma is a highly desmoplastic tumor, characterized by infiltration of macrophages and fibroblasts that create fibrosis and perpetuate the inflammation. By blocking AT1 signaling in a mouse model of the disease, it is possible to partially resolve the inflammatory process and inhibit tumor growth (50). HA is another potential therapeutic target over-produced in desmoplastic tissue. Agents that digest HA can reduce these inflammatory effects and have shown promising responses in pancreatic ductal adenocarcinoma (19, 43, 101).
Many other pathways and processes involved in inflammation have been targeted, alone or in combination with anti-cancer drugs, with varied results. However, recent efforts to manipulate the immunosuppression that is prevalent in cancer-related inflammation have eclipsed these previous strategies, and are promising to change the field of immuno-oncology. Inflammation plays a role in the immunosuppression that helps tumors evade immune attack. As mentioned previously, one of the goals of inflammation and the innate immune response is to enable the transit of cells from the blood stream into the tissue by changing blood flow, vascular permeability, endothelial adhesion molecules and interstitial matrix composition. The cells that arrive include not only myeloid derived cells involved in innate immunity, but also cells of the adaptive immune response such as helper T cells and cytotoxic T cells. Once in the tissue, these cells, which have the ability to recognize specific antigens and eradicate the cells that evade innate immunity, are subject to multiple control mechanisms.
The adaptive immune response is highly regulated, and even if specific cytotoxic cells are available, they can be prevented from mobilizing, activating, or proliferating. Many regulatory mechanism can suppress these processes, and two of the systems currently receiving much attention involve cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed death-ligand -1 (PD-L1). CTLA4 expressed by regulatory T cells causes suppression of T cell activation. Similarly, PD-L1 expressed by cells in the inflammatory environment can bind to its receptor, PD-1 on T cells, preventing their proliferation and activation. Antibodies against PD-1 block the engagement by PD-L1 and remove the suppression, allowing for activation of the cytotoxic response. This results in tumor regression and greatly improves patient outcomes (Figure 3g) (110, 121). Importantly, myeloid derived cells recruited by the inflammatory process express PD-L1 and are involved in the immunosuppression (9, 124, 127). Current research in many labs and clinics is focused on further understanding and improving upon this strategy to “wake up” the immune system in other indications, and in patients who are refractory to anti-PD-1 treatment.
Conclusions
Here, I have given a broad overview of the current understanding of inflammation, and how it is co-opted by solid tumors, focusing on the physiological aspects and the prospects for synergistic therapies involving inflammation and immunity. The topic is extremely broad, and there is much more known about each inflammatory cell and the biochemical pathways than could be included here. Nevertheless, there is still much work needed to understand inflammation from a systems biology perspective: while many key components are known, we still lack a fundamental understanding of the control mechanisms. It contains many positive and negative feedback systems that, under normal circumstances, result in a well-behaved and robust system. As we learn more about how it is controlled, systems biology analyses will allow unification of concepts and construction of a more rationale framework to explain how inflammation works, in general, and how it contributes to cancer, specifically.
Acknowledgments
I thank Drs. Tim Padera and Dai Fukumura for their helpful suggestions on the manuscript.
NIH grant R01HL128168
References
- 1.Abu-Remaileh M, Bender S, Raddatz G, Ansari I, Cohen D, Gutekunst J, Musch T, Linhart H, Breiling A, Pikarsky E, Bergman Y, Lyko F. Chronic inflammation induces a novel epigenetic program that is conserved in intestinal adenomas and in colorectal cancer. Cancer Res. 2015;75:2120–30. doi: 10.1158/0008-5472.CAN-14-3295. [DOI] [PubMed] [Google Scholar]
- 2.Al Murri AM, Bartlett JM, Canney PA, Doughty JC, Wilson C, McMillan DC. Evaluation of an inflammation-based prognostic score (GPS) in patients with metastatic breast cancer. Br J Cancer. 2006;94:227–30. doi: 10.1038/sj.bjc.6602922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alam M, Khan M, Veledar E, Pongprutthipan M, Flores A, Dubina M, Nodzenski M, Yoo SS. Correlation of Inflammation in Frozen Sections With Site of Nonmelanoma Skin Cancer. JAMA Dermatol. 2016;152:173–6. doi: 10.1001/jamadermatol.2015.3649. [DOI] [PubMed] [Google Scholar]
- 4.Alderton GK. Inflammation: the gut takes a toll on liver cancer. Nat Rev Cancer. 2012;12:379. doi: 10.1038/nrc3283. [DOI] [PubMed] [Google Scholar]
- 5.Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, Rhodes JM, Stintzi A, Simpson KW, Hansen JJ, Keku TO, Fodor AA, Jobin C. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–3. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 7.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22:33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Barash H, E RG, Edrei Y, Ella E, Israel A, Cohen I, Corchia N, Ben-Moshe T, Pappo O, Pikarsky E, Goldenberg D, Shiloh Y, Galun E, Abramovitch R. Accelerated carcinogenesis following liver regeneration is associated with chronic inflammation-induced double-strand DNA breaks. Proc Natl Acad Sci U S A. 2010;107:2207–12. doi: 10.1073/pnas.0908867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–35. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12:465–77. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhatelia K, Singh K, Singh R. TLRs: linking inflammation and breast cancer. Cell Signal. 2014;26:2350–7. doi: 10.1016/j.cellsig.2014.07.035. [DOI] [PubMed] [Google Scholar]
- 12.Bian Y, Hall B, Sun ZJ, Molinolo A, Chen W, Gutkind JS, Waes CV, Kulkarni AB. Loss of TGF-beta signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene. 2012;31:3322–32. doi: 10.1038/onc.2011.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonavita E, Galdiero MR, Jaillon S, Mantovani A. Phagocytes as Corrupted Policemen in Cancer-Related Inflammation. Adv Cancer Res. 2015;128:141–71. doi: 10.1016/bs.acr.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 14.Bonomi M, Patsias A, Posner M, Sikora A. The role of inflammation in head and neck cancer. Adv Exp Med Biol. 2014;816:107–27. doi: 10.1007/978-3-0348-0837-8_5. [DOI] [PubMed] [Google Scholar]
- 15.Bornstein S, White R, Malkoski S, Oka M, Han G, Cleaver T, Reh D, Andersen P, Gross N, Olson S, Deng C, Lu SL, Wang XJ. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest. 2009;119:3408–19. doi: 10.1172/JCI38854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bromberg J, Wang TC. Inflammation and cancer: IL-6 and STAT3 complete the link. Cancer Cell. 2009;15:79–80. doi: 10.1016/j.ccr.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caronni N, Savino B, Bonecchi R. Myeloid cells in cancer-related inflammation. Immunobiology. 2015;220:249–53. doi: 10.1016/j.imbio.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 18.Chauhan VP, Lanning RM, Diop-Frimpong B, Mok W, Brown EB, Padera TP, Boucher Y, Jain RK. Multiscale measurements distinguish cellular and interstitial hindrances to diffusion in vivo. Biophys J. 2009;97:330–6. doi: 10.1016/j.bpj.2009.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P, Popovic Z, Huang P, Bawendi MG, Boucher Y, Jain RK. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4:2516. doi: 10.1038/ncomms3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen K, Huang J, Gong W, Iribarren P, Dunlop NM, Wang JM. Toll-like receptors in inflammation, infection and cancer. Int Immunopharmacol. 2007;7:1271–85. doi: 10.1016/j.intimp.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 21.Cohen EN, Gao H, Anfossi S, Mego M, Reddy NG, Debeb B, Giordano A, Tin S, Wu Q, Garza RJ, Cristofanilli M, Mani SA, Croix DA, Ueno NT, Woodward WA, Luthra R, Krishnamurthy S, Reuben JM. Inflammation Mediated Metastasis: Immune Induced Epithelial-To-Mesenchymal Transition in Inflammatory Breast Cancer Cells. PLoS One. 2015;10:e0132710. doi: 10.1371/journal.pone.0132710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colombo MP, Mantovani A. Targeting myelomonocytic cells to revert inflammation-dependent cancer promotion. Cancer Res. 2005;65:9113–6. doi: 10.1158/0008-5472.CAN-05-2714. [DOI] [PubMed] [Google Scholar]
- 23.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–32. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol. 2015;12:584–96. doi: 10.1038/nrclinonc.2015.105. [DOI] [PubMed] [Google Scholar]
- 25.De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA, Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–8. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Marzo AM, Platz EA, Sutcliffe S, Xu J, Gronberg H, Drake CG, Nakai Y, Isaacs WB, Nelson WG. Inflammation in prostate carcinogenesis. Nat Rev Cancer. 2007;7:256–69. doi: 10.1038/nrc2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deng T, Lyon CJ, Bergin S, Caligiuri MA, Hsueh WA. Obesity, Inflammation, and Cancer. Annu Rev Pathol. 2016;11:421–49. doi: 10.1146/annurev-pathol-012615-044359. [DOI] [PubMed] [Google Scholar]
- 28.Diakos CI, Charles KA, McMillan DC, Clarke SJ. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014;15:e493–503. doi: 10.1016/S1470-2045(14)70263-3. [DOI] [PubMed] [Google Scholar]
- 29.Dmitrieva OS, Shilovskiy IP, Khaitov MR, Grivennikov SI. Interleukins 1 and 6 as Main Mediators of Inflammation and Cancer. Biochemistry (Mosc) 2016;81:80–90. doi: 10.1134/S0006297916020024. [DOI] [PubMed] [Google Scholar]
- 30.Dubey S, Vanveldhuizen P, Holzbeierlein J, Tawfik O, Thrasher JB, Karan D. Inflammation-associated regulation of the macrophage inhibitory cytokine (MIC-1) gene in prostate cancer. Oncol Lett. 2012;3:1166–70. doi: 10.3892/ol.2012.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Echizen K, Hirose O, Maeda Y, Oshima M. Inflammation in gastric cancer: Interplay of the COX-2/prostaglandin E2 and Toll-like receptor/MyD88 pathways. Cancer Sci. 2016;107:391–7. doi: 10.1111/cas.12901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engels EA, Wu X, Gu J, Dong Q, Liu J, Spitz MR. Systematic evaluation of genetic variants in the inflammation pathway and risk of lung cancer. Cancer Res. 2007;67:6520–7. doi: 10.1158/0008-5472.CAN-07-0370. [DOI] [PubMed] [Google Scholar]
- 33.Erdman SE, Rao VP, Poutahidis T, Rogers AB, Taylor CL, Jackson EA, Ge Z, Lee CW, Schauer DB, Wogan GN, Tannenbaum SR, Fox JG. Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc Natl Acad Sci U S A. 2009;106:1027–32. doi: 10.1073/pnas.0812347106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ericsson AC, Myles M, Davis W, Ma L, Lewis M, Maggio-Price L, Franklin C. Noninvasive detection of inflammation-associated colon cancer in a mouse model. Neoplasia. 2010;12:1054–65. doi: 10.1593/neo.10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fazio C, Piazzi G, Vitaglione P, Fogliano V, Munarini A, Prossomariti A, Milazzo M, D’Angelo L, Napolitano M, Chieco P, Belluzzi A, Bazzoli F, Ricciardiello L. Inflammation increases NOTCH1 activity via MMP9 and is counteracted by Eicosapentaenoic Acid-free fatty acid in colon cancer cells. Sci Rep. 2016;6:20670. doi: 10.1038/srep20670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukumura D, Incio J, Shankaraiah RC, Jain RK. Obesity and Cancer: An Angiogenic and Inflammatory Link. Microcirculation. 2016;23:191–206. doi: 10.1111/micc.12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gasparini G, Meo S, Comella G, Stani SC, Mariani L, Gamucci T, Avallone A, Lo Vullo S, Mansueto G, Bonginelli P, Gattuso D, Gion M. The combination of the selective cyclooxygenase-2 inhibitor celecoxib with weekly paclitaxel is a safe and active second-line therapy for non-small cell lung cancer: a phase II study with biological correlates. Cancer J. 2005;11:209–16. doi: 10.1097/00130404-200505000-00007. [DOI] [PubMed] [Google Scholar]
- 38.Gomes M, Teixeira AL, Coelho A, Araujo A, Medeiros R. The role of inflammation in lung cancer. Adv Exp Med Biol. 2014;816:1–23. doi: 10.1007/978-3-0348-0837-8_1. [DOI] [PubMed] [Google Scholar]
- 39.Hagemann T, Balkwill F, Lawrence T. Inflammation and cancer: a double-edged sword. Cancer Cell. 2007;12:300–1. doi: 10.1016/j.ccr.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol. 2011;7:75–7. doi: 10.1038/nchembio.496. [DOI] [PubMed] [Google Scholar]
- 41.He G, Karin M. NF-kappaB and STAT3 – key players in liver inflammation and cancer. Cell Res. 2011;21:159–68. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helm CL, Fleury ME, Zisch AH, Boschetti F, Swartz MA. Synergy between interstitial flow and VEGF directs capillary morphogenesis in vitro through a gradient amplification mechanism. Proc Natl Acad Sci U S A. 2005;102:15779–84. doi: 10.1073/pnas.0503681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM, Tjulandin SA, Gladkov OA, Holcombe RF, Korn R, Raghunand N, Dychter S, Jiang P, Shepard HM, Devoe CE. Phase Ib Study of PEGylated Recombinant Human Hyaluronidase and Gemcitabine in Patients with Advanced Pancreatic Cancer. Clin Cancer Res. 2016;22:2848–54. doi: 10.1158/1078-0432.CCR-15-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howe LR, Subbaramaiah K, Hudis CA, Dannenberg AJ. Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin Cancer Res. 2013;19:6074–83. doi: 10.1158/1078-0432.CCR-12-2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu SC, Yu HS, Yen FL, Lin CL, Chen GS, Lan CC. Neutrophil extracellular trap formation is increased in psoriasis and induces human beta-defensin-2 production in epidermal keratinocytes. Sci Rep. 2016;6:31119. doi: 10.1038/srep31119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Y, Snuderl M, Jain RK. Polarization of tumor-associated macrophages: a novel strategy for vascular normalization and antitumor immunity. Cancer Cell. 2011;19:1–2. doi: 10.1016/j.ccr.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, Martin JD, Martin MR, Vianello F, Leblanc P, Munn LL, Huang P, Duda DG, Fukumura D, Jain RK, Poznansky MC. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109:17561–6. doi: 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hung RJ, Ulrich CM, Goode EL, Brhane Y, Muir K, Chan AT, Marchand LL, Schildkraut J, Witte JS, Eeles R, Boffetta P, Spitz MR, Poirier JG, Rider DN, Fridley BL, Chen Z, Haiman C, Schumacher F, Easton DF, Landi MT, Brennan P, Houlston R, Christiani DC, Field JK, Bickeboller H, Risch A, Kote-Jarai Z, Wiklund F, Gronberg H, Chanock S, Berndt SI, Kraft P, Lindstrom S, Al Olama AA, Song H, Phelan C, Wentzensen N, Peters U, Slattery ML, Gecco, Sellers TA, Foci, Casey G, Gruber SB, Corect, Hunter DJ, Drive, Amos CI, Henderson B, G-O Network Cross Cancer Genomic Investigation of Inflammation Pathway for Five Common Cancers: Lung, Ovary, Prostate, Breast, and Colorectal Cancer. J Natl Cancer Inst. 2015;107 doi: 10.1093/jnci/djv246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, Ng MR, Nia HT, Grahovac J, Kao S, Babykutty S, Huang Y, Jung K, Rahbari NN, Han X, Chauhan VP, Martin JD, Kahn J, Huang P, Desphande V, Michaelson J, Michelakos TP, Ferrone CR, Soares R, Boucher Y, Fukumura D, Jain RK. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discov. 2016 doi: 10.1158/2159-8290.CD-15-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Incio J, Suboj P, Chin SM, Vardam-Kaur T, Liu H, Hato T, Babykutty S, Chen I, Deshpande V, Jain RK, Fukumura D. Metformin Reduces Desmoplasia in Pancreatic Cancer by Reprogramming Stellate Cells and Tumor-Associated Macrophages. PLoS One. 2015;10:e0141392. doi: 10.1371/journal.pone.0141392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Incio J, Tam J, Rahbari NN, Suboj P, McManus DT, Chin SM, Vardam TD, Batista A, Babykutty S, Jung K, Khachatryan A, Hato T, Ligibel JA, Krop IE, Puchner SB, Schlett CL, Hoffmman U, Ancukiewicz M, Shibuya M, Carmeliet P, Soares R, Duda DG, Jain RK, Fukumura D. PlGF/VEGFR-1 Signaling Promotes Macrophage Polarization and Accelerated Tumor Progression in Obesity. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Iwama T, Akasu T, Utsunomiya J, Muto T. Does a selective cyclooxygenase-2 inhibitor (tiracoxib) induce clinically sufficient suppression of adenomas in patients with familial adenomatous polyposis? A randomized double-blind placebo-controlled clinical trial. Int J Clin Oncol. 2006;11:133–9. doi: 10.1007/s10147-005-0548-z. [DOI] [PubMed] [Google Scholar]
- 53.Iyengar NM, Zhou XK, Gucalp A, Morris PG, Howe LR, Giri DD, Morrow M, Wang H, Pollak M, Jones LW, Hudis CA, Dannenberg AJ. Systemic Correlates of White Adipose Tissue Inflammation in Early-Stage Breast Cancer. Clin Cancer Res. 2016;22:2283–9. doi: 10.1158/1078-0432.CCR-15-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karan D, Holzbeierlein J, Thrasher JB. Macrophage inhibitory cytokine-1: possible bridge molecule of inflammation and prostate cancer. Cancer Res. 2009;69:2–5. doi: 10.1158/0008-5472.CAN-08-1230. [DOI] [PubMed] [Google Scholar]
- 55.Khan BV, Harrison DG, Olbrych MT, Alexander RW, Medford RM. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc Natl Acad Sci U S A. 1996;93:9114–9. doi: 10.1073/pnas.93.17.9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Khatami M. Chronic Inflammation: Synergistic Interactions of Recruiting Macrophages (TAMs) and Eosinophils (Eos) with Host Mast Cells (MCs) and Tumorigenesis in CALTs. M-CSF, Suitable Biomarker for Cancer Diagnosis! Cancers (Basel) 2014;6:297–322. doi: 10.3390/cancers6010297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Klintrup K, Makinen JM, Kauppila S, Vare PO, Melkko J, Tuominen H, Tuppurainen K, Makela J, Karttunen TJ, Makinen MJ. Inflammation and prognosis in colorectal cancer. Eur J Cancer. 2005;41:2645–54. doi: 10.1016/j.ejca.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 58.Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010;70:5679–85. doi: 10.1158/0008-5472.CAN-09-4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kwon OJ, Zhang L, Ittmann MM, Xin L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc Natl Acad Sci U S A. 2014;111:E592–600. doi: 10.1073/pnas.1318157111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li G, Wang Z, Ye J, Zhang X, Wu H, Peng J, Song W, Chen C, Cai S, He Y, Xu J. Uncontrolled inflammation induced by AEG-1 promotes gastric cancer and poor prognosis. Cancer Res. 2014;74:5541–52. doi: 10.1158/0008-5472.CAN-14-0968. [DOI] [PubMed] [Google Scholar]
- 62.Li N, Grivennikov SI, Karin M. The unholy trinity: inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell. 2011;19:429–31. doi: 10.1016/j.ccr.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, Takabe K, Kordula T, Milstien S, Spiegel S. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23:107–20. doi: 10.1016/j.ccr.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–83. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lund AW, Medler TR, Leachman SA, Coussens LM. Lymphatic Vessels, Inflammation, and Immunity in Skin Cancer. Cancer Discov. 2016;6:22–35. doi: 10.1158/2159-8290.CD-15-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mantovani A. Cancer: inflammation by remote control. Nature. 2005;435:752–3. doi: 10.1038/435752a. [DOI] [PubMed] [Google Scholar]
- 67.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 68.Marx J. Cancer research. Inflammation and cancer: the link grows stronger. Science. 2004;306:966–8. doi: 10.1126/science.306.5698.966. [DOI] [PubMed] [Google Scholar]
- 69.Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, McFaline JL, Schauer DB, Dedon PC, Fox JG, Samson LD. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–25. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Melder RJ, Koenig GC, Witwer BP, Safabakhsh N, Munn LL, Jain RK. During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nat Med. 1996;2:992–7. doi: 10.1038/nm0996-992. [DOI] [PubMed] [Google Scholar]
- 71.Melder RJ, Munn LL, Yamada S, Ohkubo C, Jain RK. Selectin- and integrin-mediated T-lymphocyte rolling and arrest on TNF-alpha-activated endothelium: augmentation by erythrocytes. Biophys J. 1995;69:2131–8. doi: 10.1016/S0006-3495(95)80087-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Migliorini C, Qian Y, Chen H, Brown EB, Jain RK, Munn LL. Red blood cells augment leukocyte rolling in a virtual blood vessel. Biophys J. 2002;83:1834–41. doi: 10.1016/S0006-3495(02)73948-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front Immunol. 2015;6:201. doi: 10.3389/fimmu.2015.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moreira DM, Nickel JC, Andriole GL, Castro-Santamaria R, Freedland SJ. Chronic baseline prostate inflammation is associated with lower tumor volume in men with prostate cancer on repeat biopsy: Results from the REDUCE study. Prostate. 2015;75:1492–8. doi: 10.1002/pros.23041. [DOI] [PubMed] [Google Scholar]
- 75.Munn LL, Dupin MM. Blood cell interactions and segregation in flow. Ann Biomed Eng. 2008;36:534–44. doi: 10.1007/s10439-007-9429-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ness RB, Cottreau C. Possible role of ovarian epithelial inflammation in ovarian cancer. J Natl Cancer Inst. 1999;91:1459–67. doi: 10.1093/jnci/91.17.1459. [DOI] [PubMed] [Google Scholar]
- 77.Park KR, Monsky WL, Lee CG, Song CH, Kim DH, Jain RK, Fukumura D. Mast Cells Contribute to Radiation-Induced Vascular Hyperpermeability. Radiat Res. 2016;185:182–9. doi: 10.1667/RR14190.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perez-Moreno M, Song W, Pasolli HA, Williams SE, Fuchs E. Loss of p120 catenin and links to mitotic alterations, inflammation, and skin cancer. Proc Natl Acad Sci U S A. 2008;105:15399–404. doi: 10.1073/pnas.0807301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol. 2004;14:433–9. doi: 10.1016/j.semcancer.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 80.Phillips RK, Wallace MH, Lynch PM, Hawk E, Gordon GB, Saunders BP, Wakabayashi N, Shen Y, Zimmerman S, Godio L, Rodrigues-Bigas M, Su LK, Sherman J, Kelloff G, Levin B, Steinbach G, FAPS Group A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut. 2002;50:857–60. doi: 10.1136/gut.50.6.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pickup MW, Laklai H, Acerbi I, Owens P, Gorska AE, Chytil A, Aakre M, Weaver VM, Moses HL. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-beta-deficient mouse mammary carcinomas. Cancer Res. 2013;73:5336–46. doi: 10.1158/0008-5472.CAN-13-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 83.Pold M, Zhu LX, Sharma S, Burdick MD, Lin Y, Lee PP, Pold A, Luo J, Krysan K, Dohadwala M, Mao JT, Batra RK, Strieter RM, Dubinett SM. Cyclooxygenase-2-dependent expression of angiogenic CXC chemokines ENA-78/CXC Ligand (CXCL) 5 and interleukin-8/CXCL8 in human non-small cell lung cancer. Cancer Res. 2004;64:1853–60. doi: 10.1158/0008-5472.can-03-3262. [DOI] [PubMed] [Google Scholar]
- 84.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, Salama AK, Taylor M, Ott PA, Rollin LM, Horak C, Gagnier P, Wolchok JD, Hodi FS. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–17. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–43. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pyne NJ, Pyne S. Sphingosine 1-phosphate is a missing link between chronic inflammation and colon cancer. Cancer Cell. 2013;23:5–7. doi: 10.1016/j.ccr.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 87.Samraj AN, Pearce OM, Laubli H, Crittenden AN, Bergfeld AK, Banda K, Gregg CJ, Bingman AE, Secrest P, Diaz SL, Varki NM, Varki A. A red meat-derived glycan promotes inflammation and cancer progression. Proc Natl Acad Sci U S A. 2015;112:542–7. doi: 10.1073/pnas.1417508112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sangaletti S, Tripodo C, Ratti C, Piconese S, Porcasi R, Salcedo R, Trinchieri G, Colombo MP, Chiodoni C. Oncogene-driven intrinsic inflammation induces leukocyte production of tumor necrosis factor that critically contributes to mammary carcinogenesis. Cancer Res. 2010;70:7764–75. doi: 10.1158/0008-5472.CAN-10-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schenk BI, Petersen F, Flad HD, Brandt E. Platelet-derived chemokines CXC chemokine ligand (CXCL)7, connective tissue-activating peptide III, and CXCL4 differentially affect and cross-regulate neutrophil adhesion and transendothelial migration. J Immunol. 2002;169:2602–10. doi: 10.4049/jimmunol.169.5.2602. [DOI] [PubMed] [Google Scholar]
- 90.Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010;31:37–49. doi: 10.1093/carcin/bgp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schwertfeger KL, Cowman MK, Telmer PG, Turley EA, McCarthy JB. Hyaluronan, Inflammation, and Breast Cancer Progression. Front Immunol. 2015;6:236. doi: 10.3389/fimmu.2015.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sethi G, Sung B, Aggarwal BB. TNF: a master switch for inflammation to cancer. Front Biosci. 2008;13:5094–107. doi: 10.2741/3066. [DOI] [PubMed] [Google Scholar]
- 93.Shan Q, Dwyer M, Rahman S, Gadjeva M. Distinct susceptibilities of corneal Pseudomonas aeruginosa clinical isolates to neutrophil extracellular trap-mediated immunity. Infect Immun. 2014;82:4135–43. doi: 10.1128/IAI.02169-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shi ZD, Tarbell JM. Fluid flow mechanotransduction in vascular smooth muscle cells and fibroblasts. Ann Biomed Eng. 2011;39:1608–19. doi: 10.1007/s10439-011-0309-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shiels MS, Katki HA, Hildesheim A, Pfeiffer RM, Engels EA, Williams M, Kemp TJ, Caporaso NE, Pinto LA, Chaturvedi AK. Circulating Inflammation Markers, Risk of Lung Cancer, and Utility for Risk Stratification. J Natl Cancer Inst. 2015;107 doi: 10.1093/jnci/djv199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. 2008;267:204–15. doi: 10.1016/j.canlet.2008.03.028. [DOI] [PubMed] [Google Scholar]
- 97.Smith GR, Missailidis S. Cancer, inflammation and the AT1 and AT2 receptors. J Inflamm (Lond) 2004;1:3. doi: 10.1186/1476-9255-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Song JW, Munn LL. Fluid forces control endothelial sprouting. Proc Natl Acad Sci U S A. 2011;108:15342–7. doi: 10.1073/pnas.1105316108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Spiecker M, Darius H, Kaboth K, Hubner F, Liao JK. Differential regulation of endothelial cell adhesion molecule expression by nitric oxide donors and antioxidants. J Leukoc Biol. 1998;63:732–9. [PubMed] [Google Scholar]
- 100.Spitz MR, Gorlov IP, Amos CI, Dong Q, Chen W, Etzel CJ, Gorlova OY, Chang DW, Pu X, Zhang D, Wang L, Cunningham JM, Yang P, Wu X. Variants in inflammation genes are implicated in risk of lung cancer in never smokers exposed to second-hand smoke. Cancer Discov. 2011;1:420–9. doi: 10.1158/2159-8290.CD-11-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, Smith BL, Ferrone CR, Hornicek FJ, Boucher Y, Munn LL, Jain RK. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci U S A. 2012;109:15101–8. doi: 10.1073/pnas.1213353109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sun C, Jain RK, Munn LL. Non-uniform plasma leakage affects local hematocrit and blood flow: implications for inflammation and tumor perfusion. Ann Biomed Eng. 2007;35:2121–9. doi: 10.1007/s10439-007-9377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun C, Migliorini C, Munn LL. Red blood cells initiate leukocyte rolling in postcapillary expansions: a lattice Boltzmann analysis. Biophys J. 2003;85:208–22. doi: 10.1016/S0006-3495(03)74467-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sundd P, Pospieszalska MK, Cheung LS, Konstantopoulos K, Ley K. Biomechanics of leukocyte rolling. Biorheology. 2011;48:1–35. doi: 10.3233/BIR-2011-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Suzuki N, Murata-Kamiya N, Yanagiya K, Suda W, Hattori M, Kanda H, Bingo A, Fujii Y, Maeda S, Koike K, Hatakeyama M. Mutual reinforcement of inflammation and carcinogenesis by the Helicobacter pylori CagA oncoprotein. Sci Rep. 2015;5:10024. doi: 10.1038/srep10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Swartz MA, Fleury ME. Interstitial flow and its effects in soft tissues. Annu Rev Biomed Eng. 2007;9:229–56. doi: 10.1146/annurev.bioeng.9.060906.151850. [DOI] [PubMed] [Google Scholar]
- 107.Tamarozzi F, Turner JD, Pionnier N, Midgley A, Guimaraes AF, Johnston KL, Edwards SW, Taylor MJ. Wolbachia endosymbionts induce neutrophil extracellular trap formation in human onchocerciasis. Sci Rep. 2016;6:35559. doi: 10.1038/srep35559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol. 2014;26:54–74. doi: 10.1016/j.smim.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 109.Taverna G, Pedretti E, Di Caro G, Borroni EM, Marchesi F, Grizzi F. Inflammation and prostate cancer: friends or foe? Inflamm Res. 2015;64:275–86. doi: 10.1007/s00011-015-0812-2. [DOI] [PubMed] [Google Scholar]
- 110.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Trinchieri G. Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol. 2012;30:677–706. doi: 10.1146/annurev-immunol-020711-075008. [DOI] [PubMed] [Google Scholar]
- 112.Verbeke H, Geboes K, Van Damme J, Struyf S. The role of CXC chemokines in the transition of chronic inflammation to esophageal and gastric cancer. Biochim Biophys Acta. 2012;1825:117–29. doi: 10.1016/j.bbcan.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 113.Wang D, Dubois RN. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene. 2010;29:781–8. doi: 10.1038/onc.2009.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang D, Dubois RN, Richmond A. The role of chemokines in intestinal inflammation and cancer. Curr Opin Pharmacol. 2009;9:688–96. doi: 10.1016/j.coph.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang K, Karin M. Tumor-Elicited Inflammation and Colorectal Cancer. Adv Cancer Res. 2015;128:173–96. doi: 10.1016/bs.acr.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 116.Wang TC, Goldenring JR. Inflammation intersection: gp130 balances gut irritation and stomach cancer. Nat Med. 2002;8:1080–2. doi: 10.1038/nm1002-1080. [DOI] [PubMed] [Google Scholar]
- 117.Wang YQ, Jin C, Zheng HM, Zhou K, Shi BB, Zhang Q, Zheng FY, Lin F. A novel prognostic inflammation score predicts outcomes in patients with ovarian cancer. Clin Chim Acta. 2016;456:163–9. doi: 10.1016/j.cca.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 118.Watanabe M, Kato J, Inoue I, Yoshimura N, Yoshida T, Mukoubayashi C, Deguchi H, Enomoto S, Ueda K, Maekita T, Iguchi M, Tamai H, Utsunomiya H, Yamamichi N, Fujishiro M, Iwane M, Tekeshita T, Mohara O, Ushijima T, Ichinose M. Development of gastric cancer in nonatrophic stomach with highly active inflammation identified by serum levels of pepsinogen and Helicobacter pylori antibody together with endoscopic rugal hyperplastic gastritis. Int J Cancer. 2012;131:2632–42. doi: 10.1002/ijc.27514. [DOI] [PubMed] [Google Scholar]
- 119.Weber JS, Gibney G, Sullivan RJ, Sosman JA, Slingluff CL, Jr, Lawrence DP, Logan TF, Schuchter LM, Nair S, Fecher L, Buchbinder EI, Berghorn E, Ruisi M, Kong G, Jiang J, Horak C, Hodi FS. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): an open-label, randomised, phase 2 trial. Lancet Oncol. 2016;17:943–55. doi: 10.1016/S1470-2045(16)30126-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wimberly AL, Forsyth CB, Khan MW, Pemberton A, Khazaie K, Keshavarzian A. Ethanol-induced mast cell-mediated inflammation leads to increased susceptibility of intestinal tumorigenesis in the APC Delta468 min mouse model of colon cancer. Alcohol Clin Exp Res. 2013;37(Suppl 1):E199–208. doi: 10.1111/j.1530-0277.2012.01894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xu L, Yi HG, Wu Z, Han W, Chen K, Zang M, Wang D, Zhao X, Wang H, Qu C. Activation of mucosal mast cells promotes inflammation-related colon cancer development through recruiting and modulating inflammatory CD11b(+)Gr1(+) cells. Cancer Lett. 2015;364:173–80. doi: 10.1016/j.canlet.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 123.Yang Y, Guo Y, Tan S, Ke B, Tao J, Liu H, Jiang J, Chen J, Chen G, Wu B. beta-Arrestin1 enhances hepatocellular carcinogenesis through inflammation-mediated Akt signalling. Nat Commun. 2015;6:7369. doi: 10.1038/ncomms8369. [DOI] [PubMed] [Google Scholar]
- 124.Yu GT, Bu LL, Huang CF, Zhang WF, Chen WJ, Gutkind JS, Kulkarni AB, Sun ZJ. PD-1 blockade attenuates immunosuppressive myeloid cells due to inhibition of CD47/SIRPalpha axis in HPV negative head and neck squamous cell carcinoma. Oncotarget. 2015;6:42067–80. doi: 10.18632/oncotarget.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang J, Chen L, Xiao M, Wang C, Qin Z. FSP1+ fibroblasts promote skin carcinogenesis by maintaining MCP-1-mediated macrophage infiltration and chronic inflammation. Am J Pathol. 2011;178:382–90. doi: 10.1016/j.ajpath.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, Rhim AD, Simeone DM, Beatty GL, Pasca di Magliano M. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut. 2016 doi: 10.1136/gutjnl-2016-312078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zheng D, Bode AM, Zhao Q, Cho YY, Zhu F, Ma WY, Dong Z. The cannabinoid receptors are required for ultraviolet-induced inflammation and skin cancer development. Cancer Res. 2008;68:3992–8. doi: 10.1158/0008-5472.CAN-07-6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zheng RR, Huang M, Jin C, Wang HC, Yu JT, Zeng LC, Zheng FY, Lin F. Cervical cancer systemic inflammation score: a novel predictor of prognosis. Oncotarget. 2016;7:15230–42. doi: 10.18632/oncotarget.7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhou YY, Hu ZG, Zeng FJ, Han J. Clinical Profile of Cyclooxygenase-2 Inhibitors in Treating Non-Small Cell Lung Cancer: A Meta-Analysis of Nine Randomized Clinical Trials. PLoS One. 2016;11:e0151939. doi: 10.1371/journal.pone.0151939. [DOI] [PMC free article] [PubMed] [Google Scholar]