

Abstract
Computational tools are widely used for ranking and prioritizing variants for characterizing their disease relevance. Since numerous tools have been developed, they have to be properly assessed before being applied. Critical Assessment of Genome Interpretation (CAGI) experiments have significantly contributed towards the assessment of prediction methods for various tasks. Within and outside the CAGI, we have addressed several questions that facilitate development and assessment of variation interpretation tools. These areas include collection and distribution of benchmark datasets, their use for systematic large scale method assessment, and the development of guidelines for reporting methods and their performance. For us, CAGI has provided a chance to experiment with new ideas, test the application areas of our methods, and network with other prediction method developers. In this article, we discuss our experiences and lessons learned from the various CAGI challenges. We describe our approaches, their performance, and impact of CAGI on our research. Finally, we discuss some of the possibilities that CAGI experiments have opened up and make some suggestions for future experiments.
Keywords: CAGI, variation interpretation, PON-P, PON-P2, performance assessment measures, variation benchmarks, mutation prediction
INTRODUCTION
Numerous genetic variants are known to be involved in diseases. The disease relevance of the majority of variants identified in exomes and complete genomes remains elusive. Each exome is estimated to code for about 11,000 amino acid substitutions (AASs) in comparison to a reference sequence (Abecasis, et al., 2010). Prediction tools are required for sorting and interpreting novel variants because all of them cannot be investigated experimentally. Systematic assessment of performance of prediction tools is important for identifying the best methods. Although some reports of systematic assessment of variant effect prediction methods have been published (Khan and Vihinen, 2010; Thusberg, et al., 2011; Grimm, et al., 2015), the Critical Assessment of Genome Interpretation (CAGI) experiments (Adebali, et al., in preparation) have significantly contributed towards method validation, proof of concept testing, and general attitude for experimentally tested methods.
Our group has a long experience and interest in investigating variants and their effects and includes protein engineering experiments to improve enzyme properties (Vihinen and Mäntsälä, 1990; Vihinen, et al., 1990; Vihinen, et al., 1991; Vihinen, et al., 1994a; Nera, et al., 2000; Rasila, et al., 2012), variant collection and distribution on locus specific variation databases (LSDBs) (Vihinen, et al., 1995; Piirilä, et al., 2006; Väliaho, et al., 2006), interpretation of variants and their effects (Vihinen, et al., 1994b; Lee, et al., 2014; Väliaho, et al., 2015), and the development of recommendations and standards for variation data (Celli, et al., 2012; Vihinen, et al., 2012; Vihinen, et al., 2016) as well as the development of various prediction tools to filter and interpret harmful variants (Ali, et al., 2012; Olatubosun, et al., 2012; Niroula, et al., 2015; Niroula and Vihinen, 2015a; Niroula and Vihinen, 2016a; Yang, et al., 2016). In addition, we have promoted the importance of systematic performance assessments (Khan and Vihinen, 2010; Thusberg, et al., 2011), systematic measures and reporting of prediction methods (Vihinen, 2012; Vihinen, 2013), and need for benchmark datasets (Nair and Vihinen, 2013; Schaafsma and Vihinen, 2015b) and for systematics and nomenclature for describing variants (Byrne, et al., 2012; Vihinen, 2014b; Vihinen, 2015d; Vihinen, 2015b). Currently, we curate about 130 LSDBs, mainly for primary immunodeficiencies (PIDs) (Piirilä, et al., 2006; Schaafsma and Vihinen, 2015a) and also for protein kinase and Src homology 2 (SH2) domain variants (Ortutay, et al., 2005; Lappalainen, et al., 2008). These datasets have allowed studies for understanding disease mechanisms, especially in PIDs (Vihinen, et al., 1995; Thusberg and Vihinen, 2006; Väliaho, et al., 2006; Lanzi, et al., 2010; Lee, et al., 2014; Schaafsma and Vihinen, 2015a) and also in cancers (Niroula and Vihinen, 2015b).
Challenge-based experiments enable testing novel ideas and approaches to address the challenges. Several experiments or competitions have been organized in different fields of science (Bender, 2016; Saez-Rodriguez, et al., 2016). These competitions enable participants from different backgrounds to solve common problems and promote collaborations. The concept of blind predictions is a good way of testing the current status of methods. We are familiar with such experiments ever since the first Critical Assessment of protein Structure Prediction (CASP) where we participated with some protein structure predictions (Mosimann, et al., 1995). Thus, we welcomed the CAGI and have been involved in all the four experiments. Here, we reflect and recollect on our experiences of the CAGI experiments. Since the results of individual challenges are published elsewhere (Adebali, et al., in preparation; Carraro, et al., submitted; Daneshjou, et al., submitted; Pejaver, et al., submitted), we concentrate on describing how the CAGI experiments have impacted our method development and assessment as well as other aspects of variant effect prediction.
PERFORMANCE ASSESSMENT AND MEASURES
Since numerous variant impact predictors have been developed, it is essential to compare their performances. The developer-reported performance scores are not always reliable and representative (Vihinen, 2012). The predictor performance may vary depending on the dataset used for assessment. Therefore, for a fair comparison, an independent assessment using a gold standard dataset is required (Fig. 1). Until recently, such benchmark datasets were missing. We have collected benchmark variation datasets for various prediction tasks to VariBench (Nair and Vihinen, 2013). VariSNP is another benchmark database containing neutral variants from dbSNP after filtering out disease-causing and cancer variants (Schaafsma and Vihinen, 2015b). When using benchmark datasets for testing the performance of machine learning (ML) tools, the training and test datasets have to be disjoint (Vihinen, 2013; Walsh, et al., 2016). One of the goals when developing variation ontology (VariO) for describing the effects, consequences and mechanisms of variants was to assist searchers for reliable, properly annotated, experimentally verified cases for training and testing of prediction methods (Vihinen, 2014b).
Figure 1.
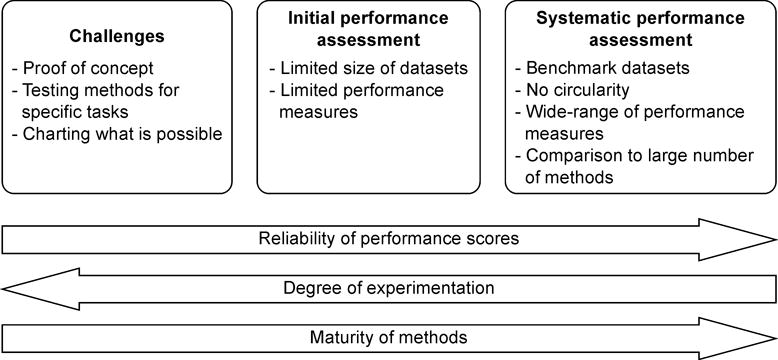
Different schemes used for method performance assessment. CAGI belongs to challenges, which are important for testing ideas and finding what is possible with the current approaches. Initial performance assessments typically represent those included in the original publications describing prediction methods. They often suffer from limitations of small dataset and may also be selective in regards to reporting performance measures. Sometimes they approach the thoroughness of systematic performance assessment. Especially the availability of benchmark datasets has improved the quality of method comparisons. It is essential that the cases used for training the methods are not used for testing the performance and that all the necessary performance measures and details are provided. For a meaningful comparison, the assessment should be extensive and include related methods, especially those with good performance. Challenges provide estimates of the method performance while the systematic comparisons facilitate their ranking. Challenges allow more freedom for experimenting with prediction methods, while mature methods require systematic optimization.
We and others have assessed the performance of methods for predicting impact of variants on tolerance/pathogenicity (Thusberg, et al., 2011; Bendl, et al., 2014; Grimm, et al., 2015; Niroula, et al., 2015; König, et al., 2016), protein localization (Laurila and Vihinen, 2009), protein stability (Potapov, et al., 2009; Khan and Vihinen, 2010), RNA splicing (Jian, et al., 2014), protein disorder (Ali, et al., 2014), and solubility (Yang, et al., 2016). The performance assessments showed vast differences between methods and indicated a need for improved tools. One of the problems in performance assessments has been circularity i.e. overlap between the training and the test datasets at variant, protein, or protein family level. Performance of many methods decreased when they were tested on circularity-free test datasets (Grimm, et al., 2015; Vihinen, 2015a). Circularity-free test datasets should be used when assessing performance of methods.
Method performance assessment requires appropriate measures to describe the performance systematically. A single score is not sufficient to capture all aspects of performance; thus, several performance metrics should be used (Vihinen, 2012; Lever, et al., 2016). In several of the CAGI challenges, various performance measures have been used to assess the methods’ performances. Selective use of performance measures may give biased and misleading impression of the capabilities of tools. Therefore, details of algorithms, their application area, and their performance scores should be documented and reported (Vihinen, 2013; Vihinen, 2015c). These guidelines are now a requirement for submission of articles describing prediction methods to Human Mutation and are applied e.g. on the articles in this special issue.
CAGI AND IMPROVEMENT OF OUR PREDICTORS
The CAGI experiments allow assessment of methods through diverse challenges. The challenges can be specific for genes, proteins, mechanisms, or diseases. Some of them require predicting the impacts of variants on specific genes, proteins, or diseases (e.g. CBS, CHEK2, BRCA, RAD50, etc.) and others require mapping genomes to the individual phenotype or predicting the diseased and healthy exomes (PGP, Sick Kids, Crohn’s disease, bipolar disorder, etc.).
When the first CAGI was announced, we were developing an ML-based method, PON-P, for predicting pathogenicity of variants (Olatubosun, et al., 2012). Although several tools were available for that purpose, independent performance assessment showed that they were suboptimal (Thusberg, et al., 2011). We aimed at exploiting the benefits of existing prediction methods by developing a meta-predictor. Predictions of four tolerance predictors: PhD-SNP (Capriotti, et al., 2006), SIFT (Ng and Henikoff, 2003), PolyPhen-2 (Adzhubei, et al., 2010), and SNAP (Bromberg and Rost, 2007) and a stability effect predictor, I-Mutant (Capriotti, et al., 2008) were used to train PON-P after performing a feature selection. PON-P was trained and tested on VariBench datasets (Nair and Vihinen, 2013). By bootstrapping, totally 200 random forest (RF) predictors were trained. Reliability score was computed based on the results of the RF predictors and the variants with a high reliability score were classified as pathogenic or neutral and those with a lower reliability as unknown. PON-P was benchmarked against state-of-the-art methods and it showed superior performance (Olatubosun, et al., 2012). We used PON-P predictions in the first two CAGIs.
Although PON-P had a good performance, it was slow and it became difficult to maintain due to the third party components. NGS data analysis demands for fast and reliable tools. Therefore, we developed a novel tool PON-P2 which does not rely on any other predictors, instead utilizes features describing evolutionary conservation, biochemical properties of amino acids, Gene Ontology (GO) annotations, and functional and structural annotations of variant sites (Niroula, et al., 2015). We used VariBench dataset for training and testing. To avoid circularity in testing, we separated the training and the test datasets so that all variants in proteins from the same protein family were kept either in the training or in the test dataset. We trained 200 RF predictors using bootstrap training data and the method classifies the variants into pathogenic, neutral, and unknown, similar to PON-P. Using a probabilistic approach, PON-P2 integrates information for functional and structural annotations of the variant site together with the RF predictions. PON-P2 was benchmarked using several datasets, and it consistently obtained the best performance scores (Niroula, et al., 2015; Riera, et al., 2016). PON-P2 predictions were used in the CAGIs 3 and 4.
CAGI CHALLENGES AND LESSONS LEARNED
We have participated in several challenges in the CAGI experiments. They fall into two categories, those that require predicting the impacts of genetic variants and those that require predicting the cases and controls from exome data. Both PON-P and PON-P2 have been developed for predicting variant tolerance (pathogenicity). Although many of the cases used for training the methods have functional effects, our tools are not originally intended for such predictions. During the four CAGI experiments, there were two challenges to predict pathogenicity of variants for which our methods, PON-P and PON-P2, are intended. We applied the tools also in some other challenges to test the applicability of the methods for new tasks. When using prediction methods, it is essential to know their strengths and limitations (Vihinen, 2014a). When applied outside the primary application area, problems may occur. We have shown that generic protein disorder predictors are not suitable for analysis of the effects of AASs on protein disorder (Ali, et al., 2014) or generic protein solubility methods for AASs affecting protein solubility (Yang, et al., 2016). The CAGI challenges allowed us to test our methods even outside the intended application area to learn about possible applications of methods and to find the best approaches for optimal application. We have learnt a number of lessons from the challenges. Here, we briefly discuss the challenges we have participated and our approaches for prediction.
Challenges requiring prediction of variant impact
The variant impact prediction challenges required predicting effects of variants on pathogenicity and protein function. Although PON-P and PON-P2 are not intended for predicting the functional effect of variants, we used their predictions in some of these challenges by transforming the predicted pathogenicity scores. In the CHEK2 and RAD50 challenges, the task was to predict whether the given variants are present in diseased cases or healthy individuals. In these challenges, we hypothesized the pathogenic variants to be in the diseased cases and the benign variants to have equal chances to be in both diseased and healthy individuals. So, we transformed the predicted pathogenicity scores in the range 0.5 to 1 and used them as the probabilities for occurring in the diseased individual.
The CBS challenge required predicting the effect of AASs to the relative growth in yeast complementation assay in high and low pyridoxine concentrations. If x is the predicted probability of pathogenicity and se is the standard error obtained from PON-P, we transformed the predicted pathogenicity scores to relative growth rates as follows
The growth rates were modeled based on an assumption that there was a threshold for CBS enzyme activity below which the strains could not grow at low pyridoxine concentration. Above the threshold, the growth was assumed to be directly proportional to the PON-P predicted score. Among several models derived using different thresholds, this model showed the best performance.
We achieved average to top performance, depending on the challenge. Our submissions were rated among the best methods in the CBS, RAD50, and CHEK2 challenges, and average in the BRCA and p16 challenges. The numbers of AASs in these challenges were small, 10 for BRCA and for p16, 34 for CHEK2, 35 for RAD50, and 51 and 84 for CBS in CAGI 1 and CAGI 2, respectively. In the CBS challenge in CAGI 1, PON-P predictions were consistently ranked among the top five methods based on various scores e.g. Spearman’s correlation coefficient, Receiver Operating Characteristic (ROC) curve, area under the ROC curve (AUC), Z-score, and root mean square deviation (RMSD). The method obtained the top ranks for both high and low pyridoxine concentrations. In CAGI 2, our method obtained average to high ranks based on different performance measures. In the CHEK2 challenge (CAGI 1) and the RAD50 challenge (CAGI 2), our predictions with PON-P were ranked among the best five tools. In the RAD50 challenge, PON-Pall included all variants that PON-P could predict and PON-P0.95 included variants that were predicted reliably at probability level 0.95 (Olatubosun, et al., 2012). PON-P0.95 showed the best performance for all AASs as well as for AASs in the domains (P-loop hydrolase and Zinc hook).
Our methods have learned to generalize based on variants from a large number of different proteins, approximately 29,000 variants in 7,600 proteins in the case of PON-P2. The performance of predictors varies protein-wise (Riera, et al., 2016). The CAGI challenges taught us to be careful with the application areas of methods. On the other hand, the results indicated that generic tolerance prediction can be transformed quite reliably to functional effects, such as the growth rates in the CBS challenge, as discussed also in (Pejaver, et al., submitted). However, if large enough datasets are available, it might be better to develop a dedicated tool. When such data are lacking, reliable tolerance scores can provide a basis for estimates. The datasets in the challenges were too small for obtaining reliable estimation of method performance and for their ranking. We have previously used 40,000 cases for tolerance method performance assessment (Thusberg, et al., 2011).
Challenges requiring identification of patients and healthy individuals using exome data
We participated in Crohn’s disease and bipolar disorder exome challenges, which required distinguishing diseased and healthy individuals based on exome sequencing data. In both challenges, our strategy was to use PON-P or PON-P2 to identify harmful variants in candidate genes and then to stratify the diseased cases and controls based on these predictions. As the exomes contain large numbers of variants including AASs in numerous proteins, the candidate gene approach allows reducing the data dimensionality. In the CAGI 2, we collected a list of candidate genes previously implicated in Crohn’s disease in genome-wide association studies (GWAS). Then, we computed a statistical score based on the odds ratio for locations identified in GWAS and the predicted pathogenicity of variants. The statistical score was used to distinguish Crohn’s disease cases and controls.
In the CAGI 4, we used ML-approach for both Crohn’s disease and bipolar disorder exome challenges. The training datasets provided by the CAGI were split into training and validation sets. The training dataset was used for generating and selecting features and for training models. Then, the validation dataset was used to evaluate the models. We collected candidate genes for both diseases from literature. Then, the pathogenicity of variants in the datasets was predicted using PON-P2. Based on the predicted pathogenicity of variants in the candidate genes, we derived several features for training ML-methods. We performed extensive feature selection approach to identify useful features and used them for predictor training.
Our approaches failed to distinguish the diseased exomes from the healthy ones. Although, the ML-approaches for bipolar disorder exome challenge showed approximately 70% accuracy in our validation, they performed close to random on the challenge datasets. Among other limitations, the size of the training data set was too small considering the feature space. In addition, we limited our analysis to variants in the candidate genes. Several genes have been suggested to be related to Crohn’s disease and bipolar disorder, but have not been verified. We might have missed relevant genes while concentrating on the candidate genes. Another limitation of our approach was that, we left out truncating variants; and the splicing effect of variants was not considered either.
We tested various approaches to improve the predictions on these challenges. The co-occurrence of harmful variants in pairs of genes was often identified to be useful during the feature selection. The predictors trained on different but overlapping training datasets showed similar performance which was slightly improved when the predictions were combined. Although the utilized approach did not work well, the knowledge and experience gained from these challenges will be useful when addressing the challenges in the future CAGI experiments.
LOOKING FORWARD
The CAGI experiments have enabled method developers to test their ideas and learn about the benefits and limitations of the tools. In our opinion, the strongest contribution of the CAGI has been in facilitating proof of concept tests for novel ideas (Fig. 1). CAGI experiments have included various types of challenges. We have been able to test applicability of our methods for different purposes. We have submitted predictions from alternative models for the same tasks and compared them.
New tools for variant interpretation are developed constantly and others updated. Although most journals require the developers to report performance of the methods compared to the state-of-the-art methods, it is not uncommon to find mistakes, biased and incomplete reporting, and overblown statements. The quantity and quality of the training and test datasets, the scope of feature space utilized, algorithm implementation, and performance assessment, all affect the performance of predictors (Niroula and Vihinen, 2016b). Independent large scale performance assessment studies provide reliable performance scores for methods. Community-wide efforts are required to develop, maintain, and use benchmark datasets and assessment systems. The CAGI challenges are useful for assessing method performance for specific tasks, but it is not a suitable approach for extensive ranking of the methods due to limited sizes of datasets (Fig. 1). Large validated datasets have higher power and enable reliable ranking of predictors; however, it is difficult to obtain such datasets.
Although the methods for variant interpretation are not perfect, in some areas especially in pathogenicity prediction, they are approaching a plateau. With the increasing numbers of validated variants, it has become possible to develop some gene/protein or disease specific predictors. Such specific predictors benefit from gene or disease-specific information and can have better performance than generic prediction methods (Jordan, et al., 2011; Ali, et al., 2012; Masica, et al., 2012; Thompson, et al., 2013; Thompson, et al., 2014; Niroula and Vihinen, 2015a; Väliaho, et al., 2015; Niroula and Vihinen, 2016a; Vazquez, et al., 2016). However, a recent study indicated that generic and specific predictors have complementary roles and our PON-P2 tool was better than protein specific predictors in 70 out of 82 cases (85%) (Riera, et al., 2016). The availability of increased numbers of validated cases will facilitate development of novel tools for specific tasks.
The existing prediction methods are mainly targeted for the coding part of the genome. Despite the limited amount of validated non-coding variation data, some tools are available in this domain (Kircher, et al., 2014; Ritchie, et al., 2014; Zhou and Troyanskaya, 2015). The numbers of non-coding variants associated with diseases are increasing and eventually will allow the development of novel tools. Most methods predict impacts of single variants as independent events, but many disease phenotypes are due to the simultaneous contributions of multiple variants and/or factors. Predictors capable of exploring variant interactions are of great need to improve our abilities to understand the mechanisms of complex diseases. There have been several CAGI challenges for such diseases and we believe there will be more challenges of similar kind in the future.
Variant interpretation has largely focused on predicting the pathogenicity or impacts of variants and classifying them into binary groups, harmful and benign. Some methods predict continuous scores; but their predictions are based on the binary data. While binary predictions provide useful information for variant screening, they do not capture the complete range of pathogenicity. Most diseases have a continuum of phenotypes ranging from benign to severe (Vihinen, 2017). Variants in several genes or diseases have been classified into groups of disease severity. These classifications are often based on clinical characteristics of patients and molecular analysis (Weinreb, et al., 2010; McCormick, et al., 2013). The classification of disease severity is useful for studying disease mechanisms, disease prognosis, genotype-phenotype correlations, and for designing interventions for personalized/precision medicine. Although many variants in several genes/proteins and diseases have been associated with certain phenotypic severity, extensive classification is available only for a few of them. Recently, we collected variants causing mild, moderate, or severe disease phenotypes from 91 proteins and developed a tool, PON-PS, for predicting the severity of disease-causing AASs (Niroula and Vihinen, 2017). As the disease severity is influenced by several factors, gene/protein or disease specific predictors could provide useful information. In the CAGI 4, Crohn’s disease exome challenge included a sub-challenge for predicting those individuals who developed the disease before the age of 10. Such a challenge is very interesting and has a high clinical impact. Challenges for predicting the severity of diseases could be of high interest and impact, but it may be difficult to obtain data for such challenges.
In conclusion, the CAGI experiments have witnessed improvements in the prediction methods, novel tools and applications, as well as indicated defects in our ability to interpret variants, especially in relation to diseases. The increasing size of data available for some of the CAGI challenges enables the participants to develop more reliable methods. Novel challenges are needed to encourage method developers to cover novel application areas. One problem preventing this is that the prediction seasons are relatively short, therefore do not allow extensive method development. The impact of CAGI experiments could increase if they promote collaboration between the data providers, assessors, and predictors/developers especially after the prediction and assessment season. This could lead to the development of more accurate methods by combining the expertise of method developers, lessons learned from the challenges, and the know-how of the data producers about the application area.
Acknowledgments
We want to thank the late Ayodeji Olatubosun and Jouni Väliaho for their contributions in CAGIs 1 and 2 and for the development of PON-P methods. We acknowledge Swedish Research Council, Barncancerfonden, and Faculty of Medicine, Lund University for funding this work. We also thank the CAGI organizers for their support during the preparation of this manuscript. The CAGI experiment coordination is supported by NIH U41 HG007446 and the CAGI conference by NIH R13 HG00 6650.
Footnotes
We have no conflict of interest to declare.
References
- Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali H, Olatubosun A, Vihinen M. Classification of mismatch repair gene missense variants with PON-MMR. Hum Mutat. 2012;33(4):642–650. doi: 10.1002/humu.22038. [DOI] [PubMed] [Google Scholar]
- Ali H, Urolagin S, Gurarslan O, Vihinen M. Performance of protein disorder prediction programs on amino acid substitutions. Hum Mutat. 2014;35(7):794–804. doi: 10.1002/humu.22564. [DOI] [PubMed] [Google Scholar]
- Bender E. Challenges: Crowdsourced solutions. Nature. 2016;533(7602):S62–64. doi: 10.1038/533S62a. [DOI] [PubMed] [Google Scholar]
- Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, Brezovsky J, Damborsky J. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol. 2014;10(1):e1003440. doi: 10.1371/journal.pcbi.1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007;35(11):3823–3835. doi: 10.1093/nar/gkm238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne M, Fokkema IF, Lancaster O, Adamusiak T, Ahonen-Bishopp A, Atlan D, Béroud C, Cornell M, Dalgleish R, Devereau A, Patrinos GP, Swertz MA, et al. VarioML framework for comprehensive variation data representation and exchange. BMC Bioinformatics. 2012;13:254. doi: 10.1186/1471-2105-13-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics. 2006;22(22):2729–2734. doi: 10.1093/bioinformatics/btl423. [DOI] [PubMed] [Google Scholar]
- Capriotti E, Fariselli P, Rossi I, Casadio R. A three-state prediction of single point mutations on protein stability changes. BMC Bioinformatics. 2008;9(Suppl 2):S6. doi: 10.1186/1471-2105-9-S2-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli J, Dalgleish R, Vihinen M, Taschner PE, den Dunnen JT. Curating gene variant databases (LSDBs): toward a universal standard. Hum Mutat. 2012;33(2):291–297. doi: 10.1002/humu.21626. [DOI] [PubMed] [Google Scholar]
- Grimm DG, Azencott CA, Aicheler F, Gieraths U, MacArthur DG, Samocha KE, Cooper DN, Stenson PD, Daly MJ, Smoller JW, Duncan LE, Borgwardt KM. The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Hum Mutat. 2015;36(5):513–523. doi: 10.1002/humu.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014;42(22):13534–13544. doi: 10.1093/nar/gku1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan DM, Kiezun A, Baxter SM, Agarwala V, Green RC, Murray MF, Pugh T, Lebo MS, Rehm HL, Funke BH, Sunyaev SR. Development and validation of a computational method for assessment of missense variants in hypertrophic cardiomyopathy. Am J Hum Genet. 2011;88(2):183–192. doi: 10.1016/j.ajhg.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Vihinen M. Performance of protein stability predictors. Hum Mutat. 2010;31(6):675–684. doi: 10.1002/humu.21242. [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König E, Rainer J, Domingues FS. Computational assessment of feature combinations for pathogenic variant prediction. Mol Genet Genomic Med. 2016;4(4):431–446. doi: 10.1002/mgg3.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzi G, Ferrari S, Vihinen M, Caraffi S, Kutukculer N, Schiaffonati L, Plebani A, Notarangelo LD, Fra AM, Giliani S. Different molecular behavior of CD40 mutants causing hyper-IgM syndrome. Blood. 2010;116(26):5867–5874. doi: 10.1182/blood-2010-03-274241. [DOI] [PubMed] [Google Scholar]
- Lappalainen I, Thusberg J, Shen B, Vihinen M. Genome wide analysis of pathogenic SH2 domain mutations. Proteins. 2008;72(2):779–792. doi: 10.1002/prot.21970. [DOI] [PubMed] [Google Scholar]
- Laurila K, Vihinen M. Prediction of disease-related mutations affecting protein localization. BMC Genomics. 2009;10:122. doi: 10.1186/1471-2164-10-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YN, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, Al-Herz W, Haddad E, LeDeist F, Bleesing JH, Henderson LA, Pai SY, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. 2014;133(4):1099–1108. doi: 10.1016/j.jaci.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever J, Krzywinski M, Altman N. Points of significance: Classification evaluation. Nat Methods. 2016;13(8):603–604. [Google Scholar]
- Masica DL, Sosnay PR, Cutting GR, Karchin R. Phenotype-optimized sequence ensembles substantially improve prediction of disease-causing mutation in cystic fibrosis. Hum Mutat. 2012;33(8):1267–1274. doi: 10.1002/humu.22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick EM, Hopkins E, Conway L, Catalano S, Hossain J, Sol-Church K, Stabley DL, Gripp KW. Assessing genotype-phenotype correlation in Costello syndrome using a severity score. Genet Med. 2013;15(7):554–557. doi: 10.1038/gim.2013.6. [DOI] [PubMed] [Google Scholar]
- Mosimann S, Meleshko R, James MN. A critical assessment of comparative molecular modeling of tertiary structures of proteins. Proteins. 1995;23(3):301–317. doi: 10.1002/prot.340230305. [DOI] [PubMed] [Google Scholar]
- Nair PS, Vihinen M. VariBench: a benchmark database for variations. Hum Mutat. 2013;34(1):42–49. doi: 10.1002/humu.22204. [DOI] [PubMed] [Google Scholar]
- Nera KP, Brockmann E, Vihinen M, Smith CI, Mattsson PT. Rational design and purification of human Bruton’s tyrosine kinase SH3-SH2 protein for structure-function studies. Protein Expr Purif. 2000;20(3):365–371. doi: 10.1006/prep.2000.1316. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niroula A, Urolagin S, Vihinen M. PON-P2: prediction method for fast and reliable identification of harmful variants. PLoS One. 2015;10(2):e0117380. doi: 10.1371/journal.pone.0117380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niroula A, Vihinen M. Classification of amino acid substitutions in mismatch repair proteins using PON-MMR2. Hum Mutat. 2015a;36(12):1128–1134. doi: 10.1002/humu.22900. [DOI] [PubMed] [Google Scholar]
- Niroula A, Vihinen M. Harmful somatic amino acid substitutions affect key pathways in cancers. BMC Med Genomics. 2015b;8(1):53. doi: 10.1186/s12920-015-0125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niroula A, Vihinen M. PON-mt-tRNA: a multifactorial probability-based method for classification of mitochondrial tRNA variations. Nucleic Acids Res. 2016a;44(5):2020–2027. doi: 10.1093/nar/gkw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niroula A, Vihinen M. Variation interpretation predictors: principles, types, performance and choice. Hum Mutat. 2016b;37(6):579–597. doi: 10.1002/humu.22987. [DOI] [PubMed] [Google Scholar]
- Niroula A, Vihinen M. Predicting severity of disease-causing variants. Hum Mutat in press. 2017 doi: 10.1002/humu.23173. [DOI] [PubMed] [Google Scholar]
- Olatubosun A, Väliaho J, Härkönen J, Thusberg J, Vihinen M. PON-P: integrated predictor for pathogenicity of missense variants. Hum Mutat. 2012;33(8):1166–1174. doi: 10.1002/humu.22102. [DOI] [PubMed] [Google Scholar]
- Ortutay C, Väliaho J, Stenberg K, Vihinen M. KinMutBase: a registry of disease-causing mutations in protein kinase domains. Hum Mutat. 2005;25(5):435–442. doi: 10.1002/humu.20166. [DOI] [PubMed] [Google Scholar]
- Piirilä H, Väliaho J, Vihinen M. Immunodeficiency mutation databases (IDbases) Hum Mutat. 2006;27(12):1200–1208. doi: 10.1002/humu.20405. [DOI] [PubMed] [Google Scholar]
- Potapov V, Cohen M, Schreiber G. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Eng Des Sel. 2009;22(9):553–560. doi: 10.1093/protein/gzp030. [DOI] [PubMed] [Google Scholar]
- Rasila TS, Vihinen M, Paulin L, Haapa-Paananen S, Savilahti H. Flexibility in MuA transposase family protein structures: functional mapping with scanning mutagenesis and sequence alignment of protein homologues. PLoS One. 2012;7(5):e37922. doi: 10.1371/journal.pone.0037922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera C, Padilla N, de la Cruz X. The complementarity between protein-specific and general pathogenicity predictors for amino acid substitutions. Hum Mutat. 2016;37(10):1013–1024. doi: 10.1002/humu.23048. [DOI] [PubMed] [Google Scholar]
- Ritchie GR, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nat Methods. 2014;11(3):294–296. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Rodriguez J, Costello JC, Friend SH, Kellen MR, Mangravite L, Meyer P, Norman T, Stolovitzky G. Crowdsourcing biomedical research: leveraging communities as innovation engines. Nat Rev Genet. 2016;17(8):470–486. doi: 10.1038/nrg.2016.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaafsma GC, Vihinen M. Genetic variation in Bruton tyrosine kinase. In: Plebani A, Lougaris V, editors. Agammaglobulinemia. Switzerland: Springer International Publishing; 2015a. pp. 75–85. [Google Scholar]
- Schaafsma GC, Vihinen M. VariSNP, a benchmark database for variations from dbSNP. Hum Mutat. 2015b;36(2):161–166. doi: 10.1002/humu.22727. [DOI] [PubMed] [Google Scholar]
- Thompson BA, Goldgar DE, Paterson C, Clendenning M, Walters R, Arnold S, Parsons MT, Michael DW, Gallinger S, Haile RW, Hopper JL, Jenkins MA, et al. A multifactorial likelihood model for MMR gene variant classification incorporating probabilities based on sequence bioinformatics and tumor characteristics: a report from the Colon Cancer Family Registry. Hum Mutat. 2013;34(1):200–209. doi: 10.1002/humu.22213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, Bapat B, Bernstein I, Capella G, den Dunnen JT, du Sart D, Fabre A, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014;46(2):107–115. doi: 10.1038/ng.2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg J, Olatubosun A, Vihinen M. Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat. 2011;32(4):358–368. doi: 10.1002/humu.21445. [DOI] [PubMed] [Google Scholar]
- Thusberg J, Vihinen M. Bioinformatic analysis of protein structure-function relationships: case study of leukocyte elastase (ELA2) missense mutations. Hum Mutat. 2006;27(12):1230–1243. doi: 10.1002/humu.20407. [DOI] [PubMed] [Google Scholar]
- Walsh I, Pollastri G, Tosatto SC. Correct machine learning on protein sequences: a peer-reviewing perspective. Brief Bioinform. 2016;17(5):831–840. doi: 10.1093/bib/bbv082. [DOI] [PubMed] [Google Scholar]
- Vazquez M, Pons T, Brunak S, Valencia A, Izarzugaza JM. wKinMut-2: Identification and interpretation of pathogenic variants in human protein kinases. Hum Mutat. 2016;37(1):36–42. doi: 10.1002/humu.22914. [DOI] [PubMed] [Google Scholar]
- Weinreb NJ, Cappellini MD, Cox TM, Giannini EH, Grabowski GA, Hwu WL, Mankin H, Martins AM, Sawyer C, vom Dahl S, Yeh MS, Zimran A. A validated disease severity scoring system for adults with type 1 Gaucher disease. Genet Med. 2010;12(1):44–51. doi: 10.1097/GIM.0b013e3181c39194. [DOI] [PubMed] [Google Scholar]
- Vihinen M. How to evaluate performance of prediction methods? Measures and their interpretation in variation effect analysis. BMC Genomics. 2012;13(Suppl 4):S2. doi: 10.1186/1471-2164-13-S4-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen M. Guidelines for reporting and using prediction tools for genetic variation analysis. Hum Mutat. 2013;34(2):275–282. doi: 10.1002/humu.22253. [DOI] [PubMed] [Google Scholar]
- Vihinen M. Majority vote and other problems when using computational tools. Hum Mutat. 2014a;35(8):912–914. doi: 10.1002/humu.22600. [DOI] [PubMed] [Google Scholar]
- Vihinen M. Variation Ontology for annotation of variation effects and mechanisms. Genome Res. 2014b;24(2):356–364. doi: 10.1101/gr.157495.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen M. The importance of proper testing of predictor performance. Hum Mutat. 2015a;36(5):iii–iv. [Google Scholar]
- Vihinen M. Muddled genetic terms miss and mess the message. Trends Genet. 2015b;31(8):423–425. doi: 10.1016/j.tig.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Vihinen M. No more hidden solutions in bioinformatics. Nature. 2015c;521(7552):261. doi: 10.1038/521261a. [DOI] [PubMed] [Google Scholar]
- Vihinen M. Types and effects of protein variations. Hum Genet. 2015d;134(4):405–421. doi: 10.1007/s00439-015-1529-6. [DOI] [PubMed] [Google Scholar]
- Vihinen M. How to Define Pathogenicity, Health and Disease? Hum Mutat. 2017;38(2):129–136. doi: 10.1002/humu.23144. [DOI] [PubMed] [Google Scholar]
- Vihinen M, Cooper MD, de Saint Basile G, Fischer A, Good RA, Hendriks RW, Kinnon C, Kwan SP, Litman GW, Notarangelo LD, et al. BTKbase: a database of XLA-causing mutations. International Study Group. Immunol Today. 1995;16(10):460–465. doi: 10.1016/0167-5699(95)80027-1. [DOI] [PubMed] [Google Scholar]
- Vihinen M, den Dunnen JT, Dalgleish R, Cotton RG. Guidelines for establishing locus specific databases. Hum Mutat. 2012;33(2):298–305. doi: 10.1002/humu.21646. [DOI] [PubMed] [Google Scholar]
- Vihinen M, Hancock JM, Maglott DR, Landrum MJ, Schaafsma GC, Taschner P. Human Variome Project quality assessment criteria for variation databases. Hum Mutat. 2016;37(6):549–558. doi: 10.1002/humu.22976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihinen M, Helin S, Mäntsälä P. Site-directed mutagenesis of putative active-site residues of Bacillus stearothermophilus α-amylase. Mol Eng. 1991;1(3):267–273. [Google Scholar]
- Vihinen M, Mäntsälä P. Conserved residues of liquefying alpha-amylases are concentrated in the vicinity of active site. Biochem Biophys Res Commun. 1990;166(1):61–65. doi: 10.1016/0006-291x(90)91911-b. [DOI] [PubMed] [Google Scholar]
- Vihinen M, Ollikka P, Niskanen J, Meyer P, Suominen I, Karp M, Holm L, Knowles J, Mäntsälä P. Site-directed mutagenesis of a thermostable alpha-amylase from Bacillus stearothermophilus: putative role of three conserved residues. J Biochem. 1990;107(2):267–272. doi: 10.1093/oxfordjournals.jbchem.a123037. [DOI] [PubMed] [Google Scholar]
- Vihinen M, Peltonen T, Iitia A, Suominen I, Mantsala P. C-terminal truncations of a thermostable Bacillus stearothermophilus alpha-amylase. Protein Eng. 1994a;7(10):1255–1259. doi: 10.1093/protein/7.10.1255. [DOI] [PubMed] [Google Scholar]
- Vihinen M, Vetrie D, Maniar HS, Ochs HD, Zhu Q, Vorechovsky I, Webster AD, Notarangelo LD, Nilsson L, Sowadski JM, et al. Structural basis for chromosome X-linked agammaglobulinemia: a tyrosine kinase disease. Proc Natl Acad Sci U S A. 1994b;91(26):12803–12807. doi: 10.1073/pnas.91.26.12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väliaho J, Faisal I, Ortutay C, Smith CI, Vihinen M. Characterization of all possible single-nucleotide change caused amino acid substitutions in the kinase domain of bruton tyrosine kinase. Hum Mutat. 2015;36(6):638–647. doi: 10.1002/humu.22791. [DOI] [PubMed] [Google Scholar]
- Väliaho J, Smith CI, Vihinen M. BTKbase: the mutation database for X-linked agammaglobulinemia. Hum Mutat. 2006;27(12):1209–1217. doi: 10.1002/humu.20410. [DOI] [PubMed] [Google Scholar]
- Yang Y, Niroula A, Shen B, Vihinen M. PON-Sol: prediction of effects of amino acid substitutions on protein solubility. Bioinformatics. 2016;32(13):2032–2034. doi: 10.1093/bioinformatics/btw066. [DOI] [PubMed] [Google Scholar]
- Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods. 2015;12(10):931–934. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]