

Abstract
Many studies of allosteric mechanisms use limited numbers of mutations to test whether residues play “key” roles. However, if a large percentage of the protein contributes to allosteric function, mutating any residue would have a high probability of modifying allostery. Thus, a predicted mechanism that is dependent on only a few residues could erroneously appear to be supported. We used whole-protein alanine-scanning mutagenesis to determine which amino acid side-chains of human liver pyruvate kinase (hL-PYK; approved symbol PKLR) contribute to regulation by fructose-1,6-bisphosphate (activator) and alanine (inhibitor). Each non-alanine/non-glycine residue hL-PYK was mutated to alanine to generate 431 mutant proteins. Allosteric functions in active proteins were quantified by following substrate affinity over a concentration range of effectors. Results show that different residues contribute to the two allosteric functions. Only a small fraction of mutated residues perturbed inhibition by alanine. In contrast, a large percentage of mutated residues influenced activation by Fru-1,6-BP; inhibition by alanine is not simply the reverse of activation by Fru-1,6-BP. Moreover, the results show that Fru-1,6-BP activation would be extremely difficult to elucidate using limited numbers of mutations. Additionally, this large mutational data set will be useful to train and test computational algorithms aiming to predict allosteric mechanisms.
Keywords: alanine-scanning mutagenesis, allostery, allosteric regulation, allosteric mechanism, liver pyruvate kinase, PKLR
Introduction
In recent years, there has been a renewed interest in understanding molecular details that give rise to allosteric regulation, the second secret of life [Monod, 1977]. This interest has generated a growing body of literature across a wide range of protein systems. Many studies either have used or are poised for a future use of mutations as experimental support for a proposed allosteric mechanism. Such studies are often initiated with a theoretical hypothesis being used to predict a potential allosteric mechanism. Next, the proposed mechanism is tested by mutating a limited number of residues that are “key” to the proposed mechanism. When mutations modify allosteric function, data are used as evidence to support the validity of the proposed mechanism and at times extrapolated as support for the original theoretical hypothesis. However for many examples in which mutagenesis has already been used, negative controls - mutations at protein residues that are predicted to have no role in allostery - are often lacking. The possibility that alternative mechanisms might be influenced by the same mutated “key” residues are often not considered. In fact, if a large fraction of the protein contributes to allosteric function, mutating any residue would have a high probability of modifying allosteric function. Thus, a predicted allosteric mechanism that is dependent on only a few residues could erroneously appear to be supported when only a limited number of residues are probed using mutagenesis.
The current study was initially designed to investigate the possibility that a very large percentage of a protein can contribute to allosteric function. If so, mutagenesis of many protein residues would likely alter allosteric function. An example of a system in which a large portion of a protein contributes to allosteric function would require improved strategies for future uses of mutational probes of allosteric mechanisms (e.g., increased mutational coverage of a protein including both areas predicted to contribute to allostery and those predicted to have no role in allostery). To test whether a large percentage of a protein contributes to allosteric regulation, we used a whole-protein alanine-scanning mutagenesis of an allosteric protein. The outcomes of this study are consistent with, within the caveats of what mutations can and cannot reveal about allostery[Carlson and Fenton, 2016], the potential that a large percentage of a protein can contribute to allosteric function.
The allosteric system that we have evaluated is the hyperglycemic target human liver pyruvate kinase (hL-PYK; approved symbol PKLR; MIM# 609712). This enzyme catalyzes the final reaction in glycolysis in the liver. Since the liver generates 90% of blood glucose derived from gluconeogenesis, glycolysis (and, in turn, hL-PYK) in the liver must be regulated in coordination with gluconeogeneic function. Allosteric regulation of hL-PYK’s affinity for its substrate, phosphoenolpyruvate (PEP), is one form of regulation that fills this need. Alanine allosterically inhibits hL-PYK. Alanine is generated in peripheral tissues during starvation when protein is being broken down for energy generation. Alanine acts both to carry nitrogen to the urea cycle in the liver and as a substrate for hepatic gluconeogenesis. Fructose-1,6-bisphosphate (Fru-1,6-BP) allosterically activates hL-PYK. Fru-1,6-BP is the product of the glycolytic phosphofructokinase reaction and acts as a feed-forward activator within the glycolytic pathway. The development of drugs that either mimic Fru-1,6-BP or that bind competitively with alanine without eliciting an allosteric response are both potential goals to increase hL-PYK activity in the treatment of hyperglycemia.
The definition of allostery applicable to studies of hL-PYK is the affinity of the enzyme for its substrate in the absence versus presence of an allosteric effector, recognizing that the effector binds to a site distinct from the active site [Fenton, 2008]. This definition provides a means to quantify allosteric function in the form of the allosteric coupling constant (Qax) [Fenton, 2008; Reinhart, 1983; Weber, 1972]:
| Equation 1 |
where Kia is the binding constant for substrate in the absence of effector, Kia/x is the binding constant for substrate when effector concentration is sufficiently high to saturate the effector binding site, Kix is the binding constant for effector in the absence of substrate, and Kix/a is the binding constant for effector when substrate concentration is sufficiently high to saturate the active site. When Qax ≠1, the binding of substrate (A) to a protein is allosterically coupled to the binding of the allosteric effector (X) to the same protein. We previously investigated the allosteric function of hL-PYK and other PYK isozymes using a number of techniques [Alontaga and Fenton, 2011; Carlson and Fenton, 2016; Fenton and Tang, 2009; Holyoak, et al., 2013; Ishwar, et al., 2015; Prasannan, et al., 2012; Prasannan, et al., 2013; Urness, et al., 2013; Williams, et al., 2006]. A key finding of our studies to-date has been the recognition that the residues that make the greatest contributions to ligand binding may be different from the residues that contribute the most to the allosteric coupling constant, Qax. In the current study, we will primarily consider whether Qax is or is not modified as a result of mutating individual residue of hL-PYK [Carlson and Fenton, 2016].
Knowledge of the structure of hL-PYK will be useful in presenting outcomes of mutations (Figure 1). hL-PYK is a homotetramer. Each subunit contains 4 domains. A single peptide chain in a subunit traces through the N-terminus, the first part of the A-domain, the B-domain, the second part of the A-domain, and the C-domain. There are two types of subunit interfaces in the tetramer. The first subunit interface is created primarily between two adjacent A-domains, but also includes contacts from C-domain and N-terminal domains (A-A interface). The second subunit interface is created exclusively by two C-domains (C-C interface). The B-domain projects away from other parts of the protein. Each subunit contains an active site between the A and B domains, an allosteric amino acid binding site between the A and C domains and an allosteric Fru-1,6-BP binding site in the C-domain.
Figure 1.
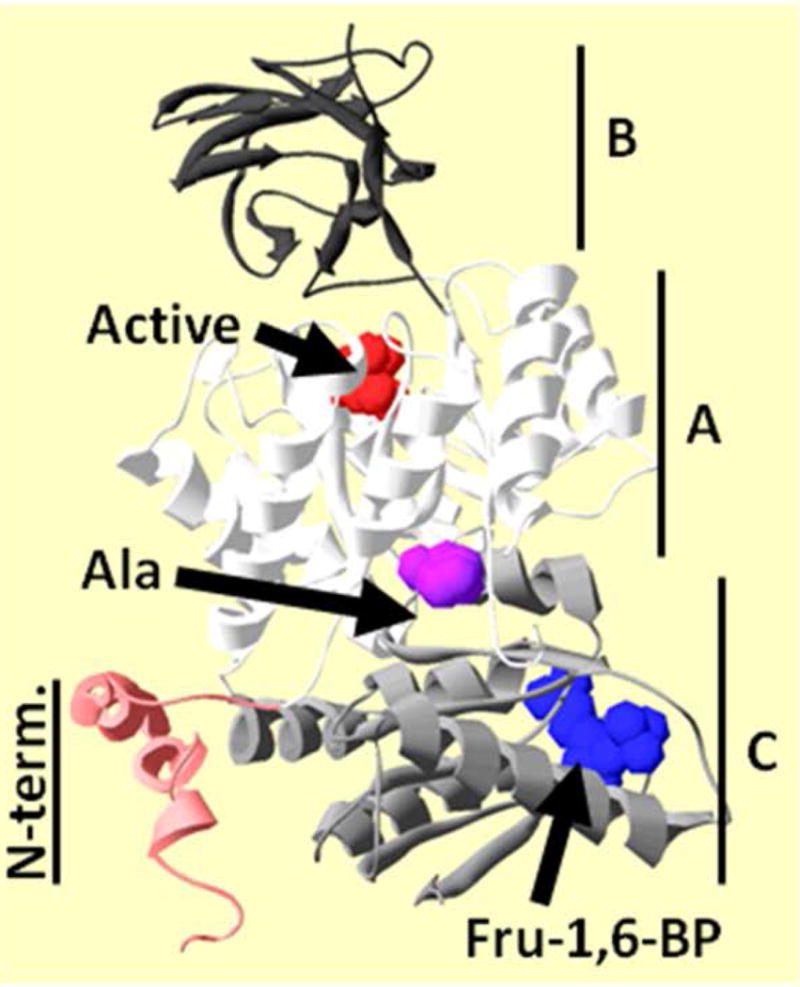
A single subunit from the homotetramer of hL-PYK as determined by X-ray crystallography (Protein Data Bank entry 4IMA [Holyoak, et al., 2013]). Each subunit contains four domains: N-terminus (red), A (white), B (dark gray), and C (light gray). The active site of this subunit contains citric acid (red spacefill). Fru-1,6-BP bound in the C-domain is in blue spacefill. This structure does not have the amino acid inhibitor bound. Therefore, H476, a residue that directly interacts with the inhibitor when it is bound, is in magenta spacefill to indicate the location of the alanine binding site. The subunit orientation used here is preserved in all other protein structure figures in this work.
In this study, changes in the magnitude of Qax (Equation 1), caused by side chain mutation to alanine, were mapped as a color gradient onto the structure of hL-PYK. This simple presentation quickly conveys our overall conclusion that a large percentage of a protein can contribute to allosteric function. We also present several observations that are relevant to subgroups of mutations; in several cases, these observations resulted in limitations of which mutated positions provided quantitative data for inclusion in the whole protein study.
Materials and Methods
The GenBank RefSeq for the PKLR gene is NM_000298.5. Amin acid sequencing follows the HGVS rules, including using the methionine coded for by ATG translation initiation codon as residue 1.
The overall methods and the assay conditions used in this study have been described in detail elsewhere [Ishwar, et al., 2015]. Of special note, proteins used in this study are only partially purified using ammonium sulfate fractionations and extensive dialysis. We have previously demonstrated that the allosteric responses of protein prepared in this way are equivalent to those of the purified protein [Ishwar, et al., 2015]. Importantly, one description of allostery that is consistent with outcomes from the current work is that a sum of many small changes (conformational or dynamic) distributed across a large part of the protein constitute an allosteric mechanism. It is common knowledge that changes in system conditions (pH, pressure, temperature) uniquely influence different types of interactions within a protein (e.g., charge-charge interactions between two amino acid side chains, or hydrogen bonds between a residue backbone and an amino acid side chain). Therefore, change in allosteric coupling (Qax) that result from change in solution conditions (e.g., pH [Fenton and Hutchinson, 2009]) are likely a result of many small solution condition-dependent modifications distributed throughout the protein. This image implies sensitivity to the solution conditions used to evaluate allosteric functions and therefore, we refer the reader to our very detailed description of our assay design [Ishwar, et al., 2015].
The general work flow for data collection is presented in Figure 2. As previously noted, the dependency of enzymatic activity on PEP concentration is biphasic [Fenton and Hutchinson, 2009]. Kapp-PEP values were obtained by fitting initial rates obtained from kinetic assay to:
| Equation 2 |
where Vmax is the maximum velocity associated with the high-affinity PEP phase, Kapp-PEP is the concentration of substrate that yields a rate equal to one-half the Vmax, and nH is the Hill coefficient associated with the high-affinity PEP phase. nH values were allowed to float from 1 to 4, but not beyond those limits. In many cases, c was evaluated at high Fru-1,6-BP concentration and held constant across all other concentrations of Fru-1,6-BP and alanine. Kapp-PEP values (obtained from Equation 2) were graphed as a function of effector concentration and this response was fit to [Reinhart, 2004]:
| Equation 3 |
where Ka = Kapp-PEP when [Effector] = 0; Kix = the dissociation constant for effector (X) binding to the protein in the absence of substrate (A); and Qax is the allosteric coupling constant [Reinhart, 2004]. In examples of inhibition when there is no evidence for the upper plateau even at very high concentrations of inhibitor, Equation 3 reduces to Equation 4 [Johnson and Reinhart, 1997; Williams, et al., 2006]:
| Equation 4 |
Figure 2.
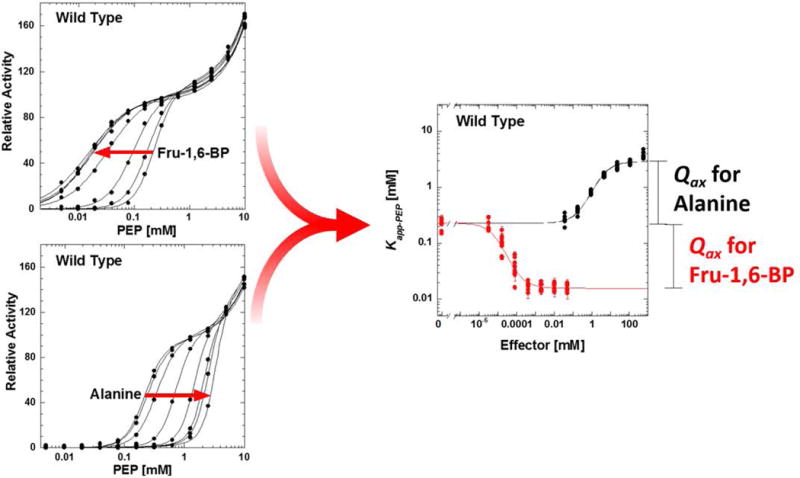
The work flow for data collection. Left: Activity as a function of PEP concentration was fit to Equation 2 to generate Kapp-PEP at a known concentration of effector. Each panel on the left includes 96 data points collected on a 96-well plate. Right: Kapp-PEP as a function of effector concentration was fit to Equation 3 to derive Qax. Error bars in the right hand panel are parameter errors estimated during data fitting to Equation 2; these same errors were used to weight Kapp-PEP values when fitting to Equation 3. Ten replicates (i.e., 10 data points at each effector concentration) from different preparations of wild type enzyme are included on the right.
Results
Modified allosteric coupling
The overall goal of this study was to evaluate how mutating individual residues of hL-PYK to alanine influences Qax associated with Fru-1,6-BP activation and Qax associated with alanine inhibition. The outcome of that goal is shown in Figures 3 and 4. To present which mutations modify Qax-Ala and Qax-Fru-1,6-BP values were converted to the free energies associated with allostery and compared to that of the wild type protein. Differences in free energies of allostery were scaled to one of the greatest changes and relative changes were color coded onto the protein structure (Figures 3 and 4). Please carefully note the definitions for color changes in these two figures. As exemplified by the Fru-1,6-BP regulation in Figure 3, a large percentage of a protein can contribute to allosteric function. However, the inhibiting regulation by alanine (Figure 4) offers a contrast in which a much lower percentage of the protein contributes to allostery.
Figure 3.
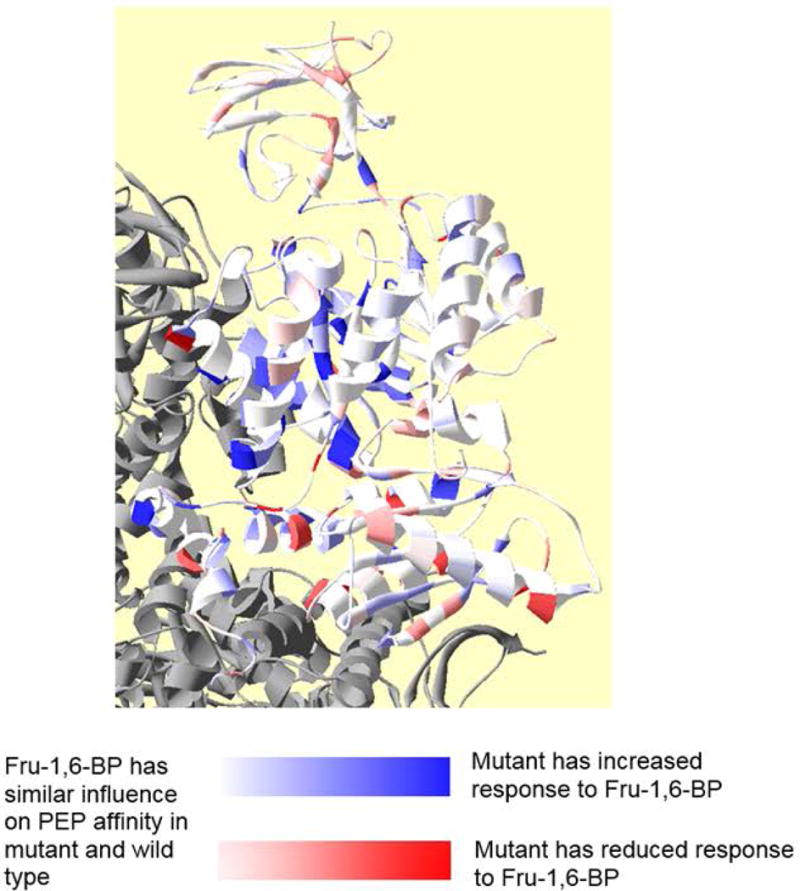
Residue positions at which side-chain removal alters allosteric coupling for Fru-1,6-BP activation (Qax-Fru-1,6-BP). The active site and the Fru-1,6-BP allosteric site within one subunit of the homotetramer are marked; other subunits in gray. In this figure, the white to red gradient indicates decreased allosteric activation (deeper red, reduced Qax, and less activated than wild type). The white to blue gradient indicates increased allosteric activation (more blue, increased Qax, and more activated than wild type). To further help relate color gradients in this figure and in Figure 4 to quantitative changes caused by a mutation, the deepest blue and deepest red indicate ≥1 kcal/mol change in the ΔGax associated with Qax. The lightest discernable shades of color indicate ~0.35 kcal/mol change.
Figure 4.
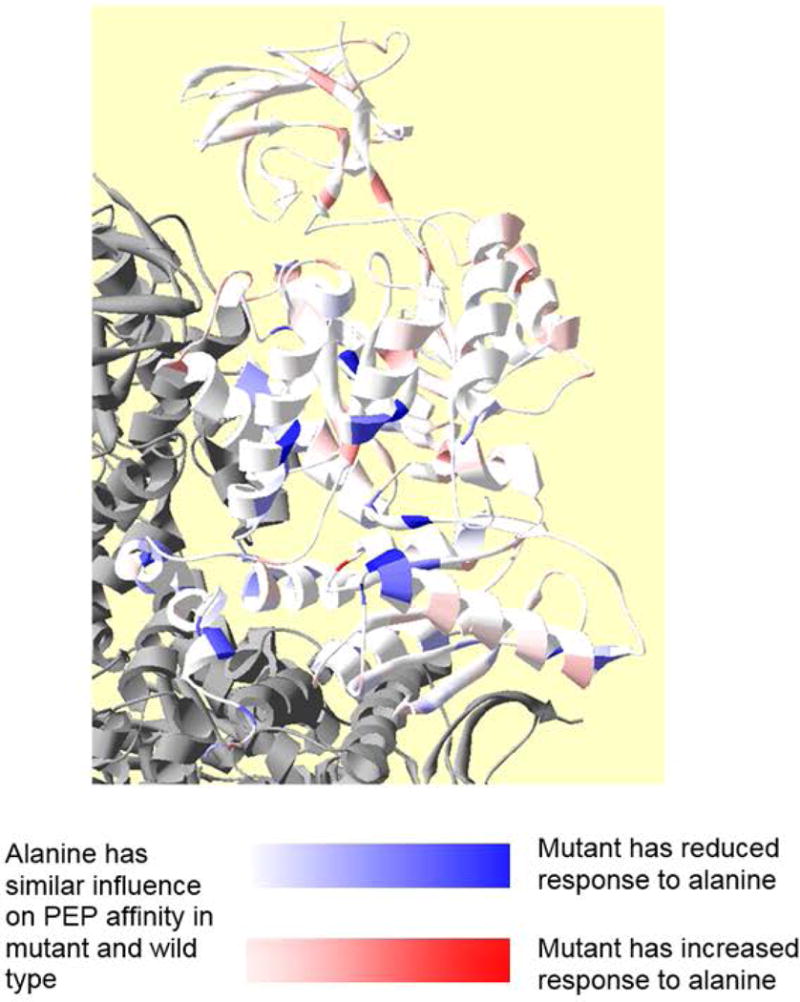
Residue positions at which side-chain removal alters allosteric coupling for alanine inhibition (Qax-Ala). In this figure, the white to red gradient indicates increased allosteric inhibition (deeper red, greater Qax, and more inhibited than wild type). The white to blue gradient indicates decreased allosteric inhibition (more blue, reduced Qax, and less inhibited than wild type). Please note that because the increase in Qax has the exact opposite meaning for an inhibitor and an activator (increased Qax for an activator indicates more allosteric activation, whereas increased Qax approaching 1 for an inhibitor indicates reduced allosteric inhibition), color coding in this figure has exact opposite meaning compared to that in Figure 3.
As a more detailed consideration of the results, there were several mutations that increased the allosteric activation by Fru-1,6-BP (deeper blue in Figure 3). The locations of those mutations that reduce allosteric activation by Fru-1,6-BP are largely in the A-domain, with several near the A-A subunit interface. There are also a number of mutations that reduced the allosteric activation by Fru-1,6-BP (deeper red in Figure 3). Interestingly, mutations that reduce activation by Fru-1,6-BP largely reside in the C-domain, with several in the C-C subunit interface.
Most mutations that increased allosteric inhibition by alanine (deeper red in Figure 4) only did so marginally. Mutations that reduced inhibition by alanine (deeper blue in Figure 4) are in the alanine binding site and in the A-A subunit interface, including residues in the N-terminus.
Retention of activity
The overall summary of the current work is that a large percentage of a protein can contribute to allosteric regulation (Figures 3 and 4), however, this study also identified a range of outcomes that are specific to much smaller subsets of mutations. Because those outcomes influence which positions are and are not represented in Figures 3 and 4, they will be presented here. However, throughout the discussion of details, the reader should not lose sight of the summary that a large percentage of a protein can contribute to allosteric regulation.
Supp. Figure S1 highlights residue positions of the protein for which mutational probing did not provide information. We did not mutate protein positions that are naturally alanine or glycine in the wild type protein. Alanine and glycine residues are evenly distributed throughout the protein (in red in Supp. Figure S1). In addition, not all mutated proteins retained activity, which was used to progressively evaluate PEP affinity and then Qax. Of the 431 non-alanine/non-glycine residues in hL-PYK, the side-chains of 384 positions (89%) could be truncated without complete loss of activity. The positions of mutations that abolished activity (or reduce activity below a detectable limit) are in blue in Supp. Figure S1. As expected, there is a higher representation of mutants that cause loss of activity in the hydrophobic core (primarily in the A-domain). The mutant proteins that retained activity represent 70.7% coverage of the total (543) residues in hL-PYK. The positions that retained activity were advanced for further analysis. However, positions that are naturally alanine or glycine in the wild type protein and those that result in loss of activity upon mutagenesis did not contribute to our overall conclusion that a large percentage of a protein can contribute to allosteric function.
Modified activity response to PEP concentration: Fru-1,6-BP dependent triphasic response
For those mutant proteins that retained activity, initial velocity was determined at varying concentrations of PEP (Figures 2 and 5). These data sets were fit to Equation 2 to determine Kapp-PEP. Importantly, Equation 2 includes both the standard form of the Hill equation and an additional term that is necessary to account for a gain of activity at very high PEP concentrations (i.e., “low-affinity”). More information on the low-affinity phase of the PEP response curve is provided in the next section. The high-affinity phase of the response curve, fit to a Hill equation, is the one affected by allosteric effectors.
Figure 5.
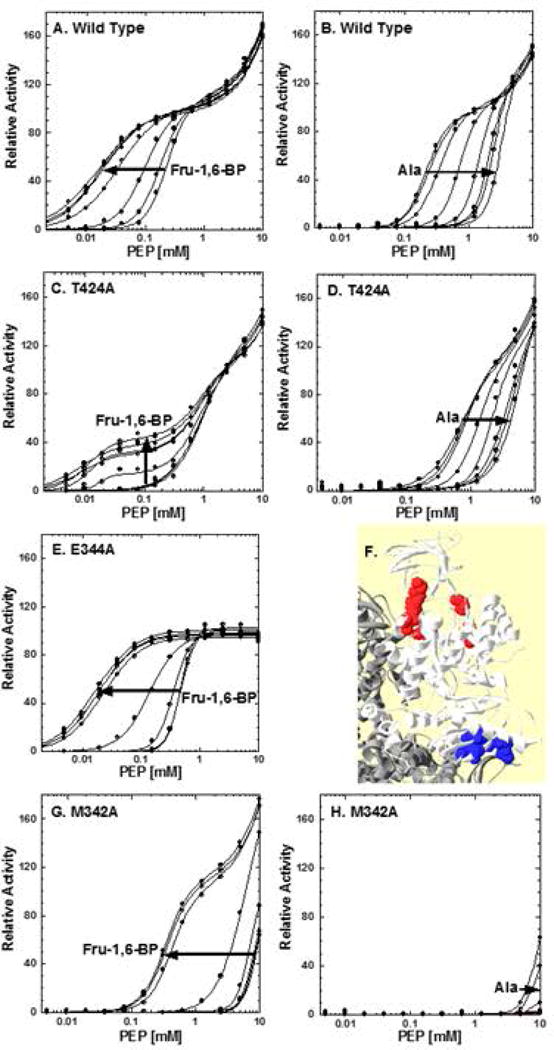
Representatives of mutations that alter the activity response profile to a PEP titration. The activity response of the wild type protein is included (panels A and B) for comparison. Arrows indicate increasing concentration of the indicated effector. T424A (panels C and D) exemplifies the data pattern obtained for a group of mutations that respond to Fru-1,6-BP additions by a V-type activation that creates a third phase in the activity response to PEP. E344A (panel E) represents a set of mutations that removal of the “low affinity” phase of the activity response to PEP concentration. Alanine inhibition of E344A shows no additional features and is not included here. Panel F indicates the locations of mutations that alter the shape of the response curve to PEP concentrations. Mutations that introduce a Fru-1,6-BP dependent V-type response group in the C-domain near the C-C domain interface (blue spacefill). Mutations that remove the “low affinity” response group around the active site (red spacefill). M342A (panels G and H) exemplifies mutants that retain activity but with greatly reduced PEP affinity, such that Kapp-PEP could only be evaluated at high Fru-1,6-BP. When was Kapp-PEP reduced even further (exemplified by M346A; data not shown), Vmax, and therefore Kapp-PEP, could not be evaluated, even at very high Fru-1,6-BP and despite a qualitatively observation that PEP affinity continues to respond to effectors. Lines represent data fits to Equation 2.
Several mutations altered the shape of the activity vs. [PEP] response (high-affinity phase): R418A, T424A, D499A, V524A, and M537A (Figure 5C and 5D). The mutations in this group cluster on the protein near the C-C subunit interface (Figure 5F). These mutations all cause the PEP affinity associated with the high-affinity phase to decrease in the absence of allosteric effectors. However, the biphasic response is still very distinct in the absence of allosteric effectors. PEP affinity is further diminished by the addition of alanine, very similar to the response of the wild type protein. The most unique feature of this group of mutants is that the addition of Fru-1,6-BP causes a V-type activation; i.e., a change in the maximal activity. Moreover, the V-type activation induces yet another transition in the response curve, leading to a triphasic response; the newly identified Fru-1,6-BP-dependent phase has a Kapp-PEP that is lower than those associated with either of the phases present in the absence of Fru-1,6-BP. The two phases that are present even in the absence of Fru-1,6-BP were not responsive to Fru-1,6-BP additions. The complexity of the Fru-1,6-BP-dependent response excluded their use in further evaluations of allosteric regulation. The same mutations are, however, included in evaluations of allosteric inhibition by alanine. Even though the triphasic response discussed here precluded 5 positions from being included in the whole protein study, it is the allosteric regulation by Fru-1,6-BP that provides our example that a large percentage of a protein can contribute to allosteric function.
Modified activity response to PEP concentration: loss of biphasic response
Several mutations remove the “low affinity” response that occurs at very high PEP: P129, D189, D190, K259, T340 and E344 (Figure 5E). Despite the fairly large distribution in sequence space, all of these mutations cluster around the active site (Figure 5F). The low affinity phase occurs only at high PEP concentration and is not likely to be physiologically relevant. This “low affinity” response can be fit to a Hill equation (fitting the biphasic response over the full range of PEP concentrations to a Hill+Hill equation with a fixed Vmax value for the low affinity phase [Fenton and Hutchinson, 2009]) and, as used here, to a background linear response (fitting the full biphasic response to a Hill+linear equation [Fenton and Alontaga, 2009]). We have also suggested three potential explanations for the “low affinity” phase of the biphasice response: 1) Phosphate contamination in PEP may be causing activation over a concentration range of PEP to generate the “low affinity” phase; phosphate ions act as a V-type activator of the high affinity PEP phase and high concentrations of phosphate cause the biphasic response to approach a monophasic response [Ishwar, et al., 2015]. 2) Based on crystal structures, there may be an altered-sites mechanism, in which two of the four active sites in a homotetramer have high affinity for PEP and the second two have low PEP affinity [Fenton and Hutchinson, 2009]; 3) Because anion concentration is known to modify the allosteric response, the decrease in Cl− ions associated with increasing PEP concentration while maintaining constant K+ may result in an artifact; this latter suggestion is less likely to explain the observation due to the relatively small percent change in Cl− ion concentration [Fenton and Alontaga, 2009]. Mutations discussed in this section may be useful in future studies that attempt to delineate between the various potential sources of the low affinity phase of the PEP response curve. However, these observations did not exclude any positions from being represented in the whole protein study.
Greatly reduced PEP affinity: a potential cause of loss of activity
A complete absence of activity could be the outcome of protein misfolding and/or disruption of the catalytic mechanism. However, a third potential cause of complete loss of activity is that the protein fails to bind substrate over the concentration range of substrate that is experimentally accessible. In the absence of data, we cannot identify mutations that cause such extreme reduction in PEP affinity. However, if such mutations are possible, then we also expect to find mutations that substantially reduce PEP affinity to the edge of the detectible range.
M342A and L365A reduce PEP sufficiently that the only condition under which saturation by PEP was observed was at very high concentrations of Fru-1,6-BP (Figure 5G and 5H). As a result, Kapp-PEP could only be evaluated under these high Fru-1,6-BP concentrations. We did not test L365A for inhibition by Ala. As yet another level of reduced PEP affinity, M346A and D366A clearly show activity at very high PEP, but there is no indication that Vmax is approached within the working PEP concentration, even in the presence of Fru-1,6-BP (Supp. Figure S4). Therefore, Kapp-PEP cannot be evaluated for these mutations even at very high concentrations of Fru-1,6-BP. However empirically, the activity at very high PEP concentrations indicates a response to the allosteric effector alanine.
M342A, L365A, M346A, and D366A, serve as examples of the greatly reduced PEP affinity that we anticipated in at least some mutant proteins. Based on the identification of these mutations, it seems likely that some mutate proteins lack observable activity due to even greater reductions in PEP affinity. For the immediate goal of determining if a large percentage of a protein can contribute to allostery, the lack of quantifiable values for Qax values for the four mutant proteins discussed in this section resulted in those positions not being represented in the whole protein study.
Absence of simultaneous saturation by PEP and alanine
The next group of mutations includes those for which Kapp-PEP does not reach a plateau at very high alanine concentration (Figure 6A). This outcome can be a result of reduced PEP affinity and/or reduced alanine affinity. As previously demonstrated [Johnson and Reinhart, 1997; Williams, et al., 2006], when saturating concentrations of inhibitor cannot be obtained (i.e., only a partial inhibition response is obtainable within the solubility limits of the effector), the response of Kapp-PEP to inhibitor concentrations can be fit to Equation 4 to evaluate Ka and Kix (Figure 6A). This fitting approach was used for the group of 28 mutations for which simultaneous saturation with PEP and alanine was not possible: F38A, R55A, I60A, T62A, S89A, T126A, N222A, P224A, D237A, S255A, F256A, M303A, I313A, E316A, V318A, N330A, P352A, V360A, N362A, I371A, M372A, E376A, M389A, Y402A, L410A, D419A, E422A, and H476A. Importantly, the inability to simultaneously saturate mutant enzymes with both PEP and alanine has no influence on the ability to evaluate allosteric activation by Fru-1,6-BP. Therefore, positions represented in this section were included in the whole protein study of Fru-1,6-BP activation, but not the study of alanine inhibition.
Figure 6.
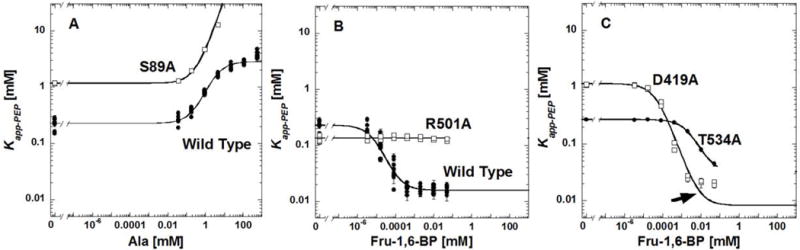
Examples of altered responses to allosteric effectors. Panel A includes an example of the use of Equation 4 to fit the response of Kapp-PEP to alanine when either alanine or PEP affinity is reduced sufficiently to prevent detection of the upper plateau. Example data are for S89A (open squares). The response of the wild type protein (filled circles) is included for comparison (fit to Equation 3). In panel B, the response of the Wild Type (filled circles) protein to Fru-1,6-BP concentration is compared to R501A (open squares) as an example of mutations that completely remove the response to Fru-1,6-BP. In panel C, D419A (open squares) represents the mutations for which the response to Fru-1,6-BP does not fit well to Equation 3 (note poor fit indicated by an arrow). Data from mutations that cause this type of response were not included in the analysis. Also in panel C, T534A (filled circles) exemplifies a response for a mutation that greatly reduces affinity for Fru-1,6-BP, but does not abolish the allosteric response; we were unable to fit Qax in these examples.
Modified PEP affinity
Given the caveats above, fits of the response of Kapp-PEP over a concentration range of effector resulted in values for Ka-PEP. As a way of presenting modified PEP affinities, Ka-PEP values from fits to Equation 3 or 4 were treated like true dissociation constants (pyruvate kinase isozymes are generally considered to be rapid equilibrium enzymes [Boyer, 1962]) and converted to ΔGa. These ΔGa values for mutations were compared to that of the wild type protein. Once differences were calculated, one of the largest changes in the set of mutations was used as the maximum change and all others were normalized as a percentage of that largest change. The percentage value was then used as a color code to map these relative differences onto the protein structure (Figure 7A). Mutations that increase PEP affinity are color coded with a white to red gradient, the deeper red being the mutation that causes the greatest increase in PEP affinity. A white to blue gradient indicates mutations that reduce PEP affinity. Residues left in white include those for which no data is available, as well as those that can accommodate an alanine substitution with no impact on PEP affinity. As seen in Figure 7A, very few mutations increase PEP affinity and those that do are located primarily in the C-domain, with a few in the B-domain. Mutations that reduce PEP affinity cluster in the A-domain along the A-A subunit interface, with a few in the N-terminus as well.
Figure 7.
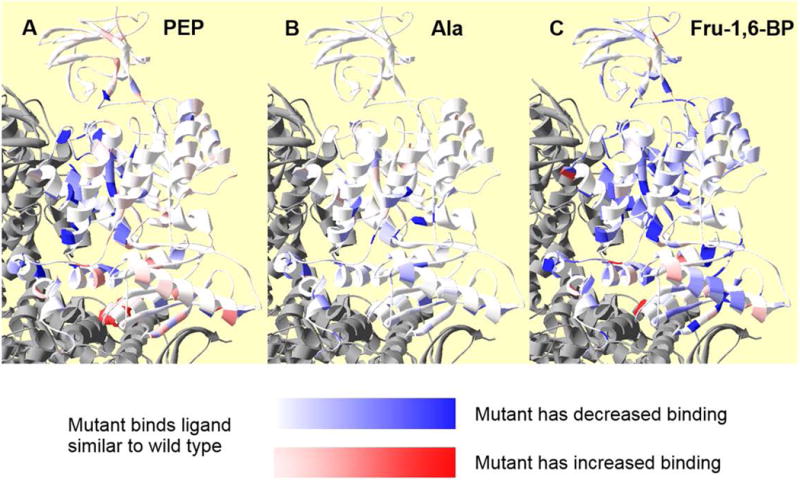
Residue positions within a subunit at which side-chain removal alters ligand affinity/binding. In each panel, coloring represents relative change in apparent affinity (PEP) or binding (Fru-1,6-BP and Ala) when comparing values from mutant and wild type proteins. Neighboring subunits are in gray. The white to red gradient indicates increased affinity/binding (i.e., the deeper the red, the higher the affinity for the respective ligand to the protein containing a mutation at that residue position). The white to blue gradient indicates decreased affinity/binding (i.e., the deeper the blue, the lower the affinity for the respective ligand to the protein containing a mutation at that residue position). Panel A, indicates residue positions at which side-chain removal alters Ka-PEP. Positions 342, 346, 365, and 366 are in deep blue to reflect a large shift in PEP affinity, despite the lack of a quantitative assessment of this affinity. Unfortunately, the T340A mutation, which has the largest increase in PEP affinity, is not visible in this orientation of this ribbon structure; to improve visibility of this residue, T340 is in spacefill red and can be seen at the C-C subunit interface. Panel B indicates residue positions at which side-chain removal alters Kix-Ala. Panel C indicates residue positions at which side-chain removal alters Kix-Fru-1,6-BP.
Although we treated Ka-PEP like a dissociation constant for the purpose of data presentation, this parameter is the concentration of substrate that result in half the Vmax activity in the absence of allosteric effector. Since Ka-PEP is obtained from initial velocity data, it is a complex rate constant. Therefore, changes in many properties of the protein can alter this parameter. Nonetheless, most of the areas of the protein at which a point mutation caused the largest changes in Ka-PEP are the same areas of the protein that have been identified for allosteric roles by other methods [Ishwar, et al., 2015; Prasannan, et al., 2013]. The whole protein presentation of positions that influence Ka-PEP (Figure 7A) provides a visual contrast with the whole protein presentation of positions that influence Qax-Fru-1,6-BP and Qax-Ala (Figures 3&4).
Modified alanine binding
When alanine binding could be determined, the treatment of Kix-Ala values parallels that for Ka-PEP values above. Mainly, these dissociation constants were converted to ΔGix and these ΔGix were compared to that of the wild type protein; differences were used to color the respective residues on the protein (Figure 7B). However, K323A, D369A, E396A, F482A, and R512A completely remove the response to alanine. In the absence of a response to an allosteric effector and without additional binding information for the effector, we cannot distinguish if 1) alanine binds, but fails to elicit an allosteric response or 2) if alanine fails to bind. This group of mutations is not represented in Figure 7B.
Again, color gradient mapping of results onto the hL-PYK structure provides a visual comparison of which positions influence Kix-Ala (Figure 7B) vs. those that influence Ka-PEP (Figure 7A) Qax-Fru-1,6-BP and Qax-Ala (Figures 3&4). In contrast to the previous consideration of PEP affinity (Figure 7A), we did not identify mutations that increased alanine binding to any appreciable level (Figure 7B). Mutations that reduce alanine binding are localized near the A-C domain interface, which is the location of the amino acid binding site. However, there are also mutations on the N-terminus domain that influence alanine binding.
Modified Fru-1,6-BP binding
A few mutations remove all traces of activation by Fru-1,6-BP: T340A, W494A, R501A, and L540A (Figure 6B). In the absence of a response and without additional binding information for the effector, we cannot distinguish whether Fru-1,6-BP binds, but fails to elicit an allosteric response or whether Fru-1,6-BP fails to bind.
T534A was the single mutation that reduced binding of Fru-1,6-BP, such that the response of Kapp-PEP to this effector is not fully defined (i.e., the lower plateau is not obtainable within the working concentration of Fru-1,6-BP). Unfortunately, an equation analogous to Equation 4 for inhibition is not available to analyze activation. Therefore, Fru-1,6-BP binding could not be assessed for T534A (Figure 6C).
Another change that limited assessment of Kix-Fru-1,6-BP involved the shape of the response of Kapp-PEP to Fru-1,6-BP. The best description of the problematic curve shape (Figure 6C) is a sharper transition between the upper and lower plateaus in the data trends. The best fit to Equation 3 generates a curve that extends below the lowest plateau in the data (Figure 6C). This outcome often reflects a change in the homotropic cooperativity associated with effector binding [Reinhart, 1988] and the data can be more accurately fit using an equation initially developed for dimers [Reinhart, 1988; Williams, et al., 2006]. However, due to the large number of variables in that dimer equation, it is best to restrict the nH values for effector binding based on a direct assessment of that information. No such evaluation of the homotropic cooperativity of Fru-1,6-BP binding has been attempted here. The mutations that cause this unique shape change include F38A, N222A, D237A, D419A, and V539A. Due to poor fits of these data, Fru-1,6-BP binding for these mutations is not available. These outcomes limited the representation of the respective positions in our whole protein study and our overall conclusion that a large percentage of a protein can contribute to allosteric function.
When Kix-Fru-1,6-BP values could be determined, our presentation parallels that for Ka-PEP and Kix-Ala values above. However, the mutations just discussed did not provide Kix-Fru-1,6-BP estimates and are not included in Figure 7C. In addition to these unique considerations, mutations that introduced the “triphasic” response as discussed above (R418A, T424A, D499A, V524A, and M537A) were not included in our evaluation of Fru-1,6-BP binding. Also note that we previously found that in our analysis the binding constants Kix-Fru-1,6-BP has highest error estimates compared to other Ka-PEP and Kix-Ala [Fenton and Alontaga, 2009]. Nonetheless, several data trends are apparent in Figure 7C. Mutations throughout the A and C domains modify Fru-1,6-BP binding. Mutations in the C-domain were much more likely to modify Fru-1,6-BP binding than modify either PEP affinity or alanine binding. Mutations that cause more severe reductions in Fru-1,6-BP binding (i.e., deeper blue color) occur in the C-C subunit interface and near the A-C domain interface in the subunit. The few mutations that cause increases in Fru-1,6-BP binding are scattered throughout the C-domain with a unique outlier (R354A) in the A-A subunit interface. With the addition of this final structural figure and the comparison with other structural figures (Figures 3, 4, and 7), it becomes clear that different regions of the protein contribute to Qax-Fru-1,6-BP, Qax-Ala, Ka-PEP, Kix-Ala, and Kix-Fru-1,6-BP. This conclusion is consistent with a running theme from other findings in our laboratory: residues that make the greatest contributions to ligand binding may be different from the residues that contribute the most to the allosteric coupling constant, Qax [Alontaga and Fenton, 2011; Carlson and Fenton, 2016; Fenton and Tang, 2009; Holyoak, et al., 2013; Ishwar, et al., 2015; Prasannan, et al., 2012; Prasannan, et al., 2013; Urness, et al., 2013; Williams, et al., 2006].
Data trends in positions that contribute to allostery
We returned to the evaluation of Qax values to determine if the outcomes from this alanine-scanning mutagenesis study highlights any individual structural feature for primary importance in allosteric functions. Using the 4IMA [Holyoak, et al., 2013] structure of hL-PYK, we obtained structural assignments from PDBsum [de Beer, et al., 2014], salt bridges from ESBRI [Costantini, et al., 2008], average depth of amino acid atoms from the protein surface from DEPTH [Tan, et al., 2011], and subunit and domain interfaces from visual inspection. For the division of inside vs. outside of the protein, we used the arbitrary cutoff of 5.5Å depth for the average of all atoms in a given residue. For positions included in each structural classification, mutation-dependent differences in Qax were course-grain binned, with no consideration of whether differences caused increased or decreased allosteric function (i.e., the absolute value of the difference). Three bins were created for the differences in Qax. Here ΔGax values (used for color coding above) were used to determine bin limits so that changes in Fru-1,6-BP activation could be comparable to changes in alanine inhibition. Bins are: minimal response (ΔGax= 0 to 0.4 kcal/mol), moderate response (ΔGax= 0.4 to 0.75 kcal/mol), and strong response (ΔGax > 0.75 kcal/mol). Within each structural classification in which there was data for 7 or more positions, the numbers of positions in each data bin were converted into a % of the total positions that contribute to the structural classification and it is this % of positions that is included in Figure 8. The alanine-scanning mutagenesis design resulted in data for only two positions that are tryptophan in the wild type protein and only four positions that are methionine. Therefore, these amino acid types are not included in Figure 8. Likewise, our design only obtained data for 5 positions in gamma turns and therefore, that structural classification is absent from Figure 8.
Figure 8.
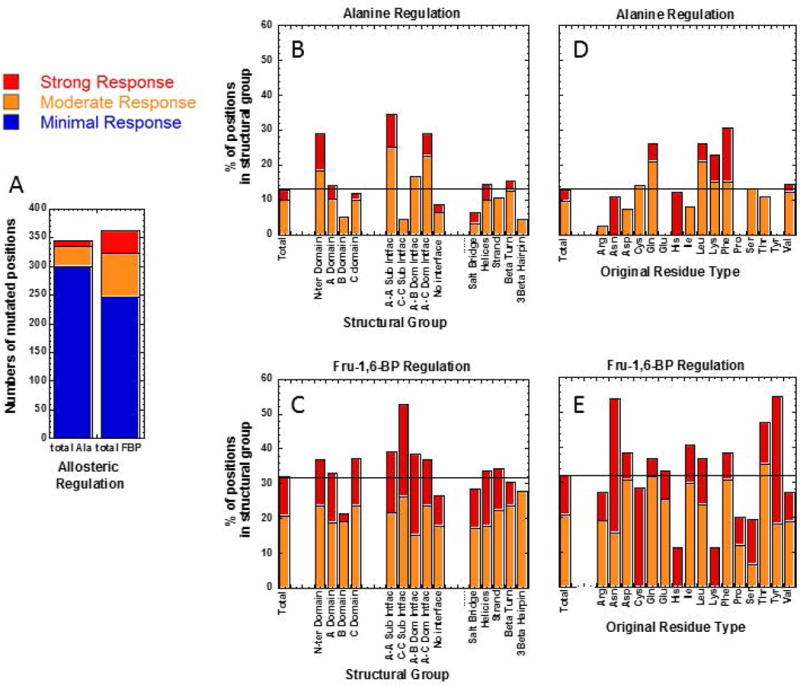
Mutated positions in various structural classes that result in altered allosteric coupling. Panel A presents numbers of positions that fall within our course-grain bins of minimal, moderate, and strong responses. Importantly, these bins consider only absolute value changes, with no consideration if allosteric responses are reduced or increased in the mutant protein. Panel B (regulation by alanine) and C (regulation by Fru-1,6-BP) show the percentage of positions in each structural group that respond to mutation moderately and strongly. Similarly Panel D (regulation by alanine) and E (regulation by Fru-1,6-BP) use original amino acid type as structural groups and, within these groups, show the percentage of positions that, upon mutation, result in moderate and strong responses. In panels B through E, only structural groups with 7 or more positions for which data were obtained are included: Methionine, Tryptophan and gamma-turns did not meet this qualification and those structural groups are not included.
Panel A of Figure 8 reinforces the earlier observation that mutational probing of many fewer positions result in moderate or strong changes in allosteric inhibition by alanine, as compared to the number of probed positions that influence allosteric regulation by Fru-1,6-BP. Only moderate and strong responses resulting from mutational probing are included in panels B through E of Figure 8 and those outcomes are represented as % of the total positions included in the respective structural group. In each panel, we include the % of strong and moderate responses in the total positions that yielded Qax values. Structural features of interest to allostery can then be identified by higher percentages of moderate and strong responders compared to this total. The N-terminus, the A-A subunit interface, the A-C subunit interface, Glutamine residues, leucine residues, lysine residues, and phenylalanine residues all play important roles in allosteric inhibition by alanine. The B-domain, the C-C subunit interface, residues that are not part of either subunit or domain interfaces, salt bridges, 3β-turns, arginine residues, aspartic acid residues, and isoleucine residues all have reduced roles in alanine inhibition. In contrast, the C-C subunit interface, asparagine residues, threonine residues and tyrosine residues have above average contributions to allosteric activation by Fru-1,6-BP. The B-domain, residues that are not part of either subunit or domain interfaces, arginine residues, histidine residues, lysine residues, proline residues, and serine residues all play fewer roles in Fru-1,6-BP activation. The overall patterns of structural groups that include higher and lower percentages of residues that influence Qax values is again consistent with Fru-1,6-BP activation and alanine inhibition functioning through very different mechanisms.
A broad range of responses to point mutations
In addition to evaluating the usefulness of whole-protein alanine-scanning mutagenesis as a technique to evaluate allosteric mechanisms, the current mutational data set also offers a warning against structural comparison of two different members of a protein family. Such comparisons among various members of a protein family continue to be used in an attempt to predict allosteric mechanism. Those comparisons often overlook the drastic binding and allosteric differences that can result from a single point mutation and downplay the fact that sequence differences between any two family members in a protein family likely include multiple residues. An overlay of all best data fit lines for single point-mutant proteins evaluated in this study (Figure 9) leaves a very strong visual image of the wide range of altered binding and allosteric consequences that can result from point mutations. Among these changes, we note that the distribution of apparent PEP affinities in the absence of effectors (left hand y-intercept of both panels in Figure 9) and the apparent PEP affinities in the presence of alanine (right hand y-intercept in panel A) are much broader than the apparent PEP affinities in the presence of Fru-1,6-BP (right hand y-intercept in panel B). Due to this wide range of responses that result from point mutations, it follows that structural comparisons among different members in a protein family should be used with a high level of caution when studying allosteric mechanisms and all proposed mechanisms from those comparisons should be thoroughly tested with functional studies.
Figure 9.
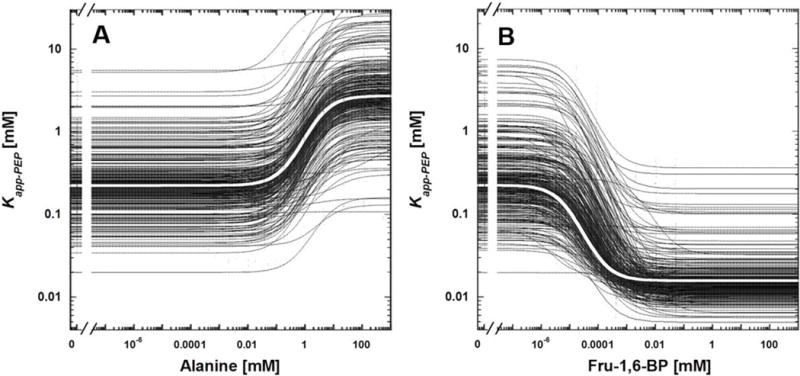
Data for all mutant proteins. Panel A includes inhibition by alanine. Panel B includes activation by Fru-1,6-BP. Black lines are fits for data from mutant proteins. In each panel, ther is a white line overlaid on top of black lines and those white lines are fits for data from wild type protein. Lines are the best fit of data to Equation 3. Data points have been minimized so that only the best fit lines are apparent. The distribution of Kapp-PEP values at high Fru-1,6-BP (i.e., the density of the right-hand y-intercepts in graph B) is more narrow than the distribution of Kapp-PEP values in the absence of effector (i.e., the density of the left-hand y-intercepts in graphs A and B) or in the presence of alanine (i.e., the density of the right-hand y-intercepts in graph A). However, this data presentation indicates the very wide range of altered binding and allosteric consequences that can result from single point mutations.
Discussion
This evaluation of hL-PYK allostery was useful to address both general questions about allostery and very specific questions about the allosteric regulation of hL-PYK. The first global question addressed herein is whether a large percentage of a protein can contribute to an allosteric mechanism. Indeed, activation of hL-PYK by Fru-1,6-BP appears to involve a large fraction of the protein (Figure 3). Based on the arbitrary cutoffs used to generate Figure 8, ~30% of the mutated positions for which data were obtained moderately or strongly influenced allosteric regulation by Fru-1,6-BP. In contrast, a large percentage of residue side-chains in hL-PYK can be truncated with minimal influence on allosteric inhibition by alanine (Figure 4). Collectively, these two allosteric responses indicate that an allosteric mechanism can, but does not always, involve large fractions of a protein. Although this overall conclusion can be briefly summarized, it has a substantial ramification for experimental designs/interpretations targeting allosteric mechanisms: 1) Attempts to illuminate (i.e., develop putative allosteric mechanisms) allosteric mechanisms should consider the potential that a large percentage of a protein might contribute to allostery. 2) The use of mutations to probe allosteric mechanisms should include large numbers of positions predicted to be both key to the mechanism and nonessential to the mechanism. Furthermore, although mutational outcomes do not directly provide insight into changes in the protein (conformational or dynamic [Carlson and Fenton, 2016]), the fact that a large number of positions can contribute to allostery seems consistent with an image in which lots of small changes (potentially in protein dynamics) constitute the allosteric mechanism.
The second global question addressed herein pertains to the relationship between inhibition and activation. Based on historic two-state mechanisms, there is a persistent idea that all allosteric mechanisms in a protein are related (e.g., allosteric inhibition involves changes that are “opposite” to those caused by activation). However, in our data, the set of residue positions highlighted for potential roles in allosteric activation by Fru-1,6-BP differ from those highlighted for potential roles in allosteric inhibition by alanine. These differences are apparent in Figures 3 and 4, as well as in Figure 8. This is consistent with other types of data indicating that the two regulations of hL-PYK function by independent mechanisms [Fenton and Hutchinson, 2009].
Third, there continues to be considerable discussion of pathways and networks that might connect two allosterically coupled binding sites within a protein. Here we will use “pathways” to refer to Rube Goldberg-type mechanisms (binding causes change A; change A causes change B; change B causes change C, etc.) and use “network” to denote a series of juxtapose residues with no implied Rube Goldberg-type mechanism [Carlson and Fenton, 2016]. Networks might be expected if side-chain repacking and/or side-chain dynamics were primary change associated with allostery[Carlson and Fenton, 2016]. Another option is that secondary structures and or domain rotations are the primary conformational and/or dynamic changes that contribute to allostery. The expected outcome from this latter possibility is that only residues involved in stabilizing interactions between two structural elements have roles in allostery [Carlson and Fenton, 2016]. In this study, we did not observe the existence of continuous networks of juxtapose residues that extend uninterrupted between an effector site and the active site (Figures 3 and 4). Instead, the scattered distribution of residues identified for roles in allostery is more consistent with allosteric regulation arising from conformational and/or dynamic changes in larger structural elements: secondary structure and/or domains. In that regard, hL-PYK appears to be similar to the Light-Activated Protein[Zayner, et al., 2013]. However, given the locations in hL-PYK of residues at which an alanine mutation did not result in new information (Supp. Figure S1), we cannot rule out that a localized area of hL-PYK in which a small network of residues contribute to allostery. Nonetheless, hL-PYK appears to serve as an example of an allosteric systems that does not use one continuous network of residues that link the respective ligand binding sites.
Although our study did not identify a network of residues, several general areas of the protein appear to contribute allosteric functions. In particular, regions near the two types of domain interfaces are highlighted to have allosteric functions. Interestingly, those general areas of hL-PYK highlighted here for roles in allosteric regulation by Fru-1,6-BP and alanine are the same general areas highlighted by hydrogen/deuterium exchange in rabbit muscle pyruvate kinase (M1-PYK) for roles in allosteric inhibition by phenylalanine [Prasannan, et al., 2013]. Given that there are sequence differences between hL-PYK and M1-PYK, that these two proteins are regulated by different effectors, and that there is evidence that the two isozymes use different protein/effector contacts in their respective allosteric mechanisms[Alontaga and Fenton, 2011], a new question can be asked in future studies: Are there only certain regions of a protein scaffold that can accommodate an allosteric mechanism? Within a protein family, it may be possible that the same regions of each family member contributes to allosteric function, even if the mechanistic details vary among family members.
At the completion of this study, we conclude that whole-protein alanine-scanning mutagenesis is a useful tool to identify protein residues that potentially contribute to allosteric mechanisms. In particular, this probing approach is likely to be complementary to the protein backbone monitoring offered by hydrogen/deuterium exchange [Beckett, 2012; Frantom, et al., 2009; Landgraf, et al., 2013; Prasannan, et al., 2013; Song, et al., 2014; Underbakke, et al., 2014]. This independent method will also be beneficial to guide structural comparisons to focus on which regions of a protein might experience allosterically relevant conformational and/or dynamic changes. Most importantly, the Fru-1,6-BP example where a large percentage of residues in the protein contribute to allostery suggests that mutational evaluations of allostery should include sufficient numbers of mutations to provide high coverage across the entire protein.
CAGI competition: At the completion of our study and before submitting for publication, data generated in this study were used as a challenge in a Critical Assessment of Genome Interpretation “CAGI” competition. CAGI is a global community experiment to objectively assess computational methods for predicting phenotypic impacts of genomic variation. The outcome of the predictive competition using the hL-PYK mutational data set are presented elsewhere. However, the original data are made available here in hopes that they will be useful to any research groups who wish to train and/or test their own algorithms aiming to predict allosteric mechanisms and/or the outcome of mutations on those mechanisms.
Supplementary Material
Acknowledgments
This work was supported by NIH grant GM115340. We are greatly appreciative to Drs. Owen Nadeau and Alexey Ladokhin for their willingness to carefully read this manuscript and offer editorial comments. Data reported in this work were coordinated by Dr. Liskin Swint-Kruse for use in a CAGI-4 challenge in the Fourth Critical Assessment of Genome Interpretation (CAGI 4) organized by R.A. Hoskins, chaired by S.E. Brenner and J. Moult, and titled “Liver pyruvate kinase (L-PYK): predict the effects of missense mutations on kinase activity and allosteric regulation.”
References
- Alontaga AY, Fenton AW. Effector analogues detect varied allosteric roles for conserved protein-effector interactions in pyruvate kinase isozymes. Biochemistry. 2011;50:1934–9. doi: 10.1021/bi200052e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett D. Hydrogen-deuterium exchange study of an allosteric energy cycle. Methods Mol Biol. 2012;796:261–78. doi: 10.1007/978-1-61779-334-9_14. [DOI] [PubMed] [Google Scholar]
- Boyer PD. Pyruvate Kinase. In: Boyer PD, Lardy HA, Myrback K, editors. The Enzymes. New York: Academic Press; 1962. pp. 95–113. [Google Scholar]
- Carlson GM, Fenton AW. What Mutagenesis Can and Cannot Reveal About Allostery. Biophys J. 2016;110:1912–23. doi: 10.1016/j.bpj.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini S, Colonna G, Facchiano AM. ESBRI: a web server for evaluating salt bridges in proteins. Bioinformation. 2008;3:137–8. doi: 10.6026/97320630003137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beer TA, Berka K, Thornton JM, Laskowski RA. PDBsum additions. Nucleic Acids Res. 2014;42:D292–6. doi: 10.1093/nar/gkt940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW. Allostery: an illustrated definition for the ‘second secret of life’. Trends Biochem Sci. 2008;33:420–5. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW, Alontaga AY. The impact of ions on allosteric functions in human liver pyruvate kinase. Methods Enzymol. 2009;466:83–107. doi: 10.1016/S0076-6879(09)66005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW, Hutchinson M. The pH dependence of the allosteric response of human liver pyruvate kinase to fructose-1,6-bisphosphate, ATP, and alanine. Arch Biochem Biophys. 2009;484:16–23. doi: 10.1016/j.abb.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW, Reinhart GD. Isolation of a single activating allosteric interaction in phosphofructokinase from Escherichia coli. Biochemistry. 2002;41:13410–6. doi: 10.1021/bi026450g. [DOI] [PubMed] [Google Scholar]
- Fenton AW, Tang Q. An activating interaction between the unphosphorylated n-terminus of human liver pyruvate kinase and the main body of the protein is interrupted by phosphorylation. Biochemistry. 2009;48:3816–8. doi: 10.1021/bi900421f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantom PA, Zhang HM, Emmett MR, Marshall AG, Blanchard JS. Mapping of the allosteric network in the regulation of alpha-isopropylmalate synthase from Mycobacterium tuberculosis by the feedback inhibitor L-leucine: solution-phase H/D exchange monitored by FT-ICR mass spectrometry. Biochemistry. 2009;48:7457–64. doi: 10.1021/bi900851q. [DOI] [PubMed] [Google Scholar]
- Holyoak T, Zhang B, Deng J, Tang Q, Prasannan CB, Fenton AW. Energetic coupling between an oxidizable cysteine and the phosphorylatable N-terminus of human liver pyruvate kinase. Biochemistry. 2013;52:466–76. doi: 10.1021/bi301341r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishwar A, Tang Q, Fenton AW. Distinguishing the interactions in the fructose 1,6-bisphosphate binding site of human liver pyruvate kinase that contribute to allostery. Biochemistry. 2015;54:1516–24. doi: 10.1021/bi501426w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Reinhart GD. Failure of a two-state model to describe the influence of phospho(enol)pyruvate on phosphofructokinase from Escherichia coli. Biochemistry. 1997;36:12814–22. doi: 10.1021/bi970942p. [DOI] [PubMed] [Google Scholar]
- Landgraf RR, Goswami D, Rajamohan F, Harris MS, Calabrese MF, Hoth LR, Magyar R, Pascal BD, Chalmers MJ, Busby SA, et al. Activation of AMP-activated protein kinase revealed by hydrogen/deuterium exchange mass spectrometry. Structure. 2013;21:1942–53. doi: 10.1016/j.str.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J. Chance and Necessity: Essay on the Natural Philosophy of Modern Biology. Penguin Books, Ltd; 1977. [Google Scholar]
- Prasannan CB, Tang Q, Fenton AW. Allosteric Regulation of Human Liver Pyruvate Kinase by Peptides that Mimic the Phosphorylated/Dephosphorylated N-Terminus. Methods Mol Biol. 2012;796:335–49. doi: 10.1007/978-1-61779-334-9_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasannan CB, Villar MT, Artigues A, Fenton AW. Identification of regions of rabbit muscle pyruvate kinase important for allosteric regulation by phenylalanine, detected by H/D exchange mass spectrometry. Biochemistry. 2013;52:1998–2006. doi: 10.1021/bi400117q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart GD. The determination of thermodynamic allosteric parameters of an enzyme undergoing steady-state turnover. Arch Biochem Biophys. 1983;224:389–401. doi: 10.1016/0003-9861(83)90225-4. [DOI] [PubMed] [Google Scholar]
- Reinhart GD. Linked-function origins of cooperativity in a symmetrical dimer. Biophys Chem. 1988;30:159–72. doi: 10.1016/0301-4622(88)85013-0. [DOI] [PubMed] [Google Scholar]
- Reinhart GD. Quantitative analysis and interpretation of allosteric behavior. Methods Enzymol. 2004;380:187–203. doi: 10.1016/S0076-6879(04)80009-0. [DOI] [PubMed] [Google Scholar]
- Song H, Olsen OH, Persson E, Rand KD. Sites involved in intra- and interdomain allostery associated with the activation of factor viia pinpointed by hydrogen-deuterium exchange and electron transfer dissociation mass spectrometry. J Biol Chem. 2014;289:35388–96. doi: 10.1074/jbc.M114.614297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan KP, Varadarajan R, Madhusudhan MS. DEPTH: a web server to compute depth and predict small-molecule binding cavities in proteins. Nucleic Acids Res. 2011;39:W242–8. doi: 10.1093/nar/gkr356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underbakke ES, Iavarone AT, Chalmers MJ, Pascal BD, Novick S, Griffin PR, Marletta MA. Nitric oxide-induced conformational changes in soluble guanylate cyclase. Structure. 2014;22:602–11. doi: 10.1016/j.str.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urness JM, Clapp KM, Timmons JC, Bai X, Chandrasoma N, Buszek KR, Fenton AW. Distinguishing the chemical moiety of phosphoenolpyruvate that contributes to allostery in muscle pyruvate kinase. Biochemistry. 2013;52:1–3. doi: 10.1021/bi301628k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber G. Ligand binding and internal equilibria in proteins. Biochemistry. 1972;11:864–78. doi: 10.1021/bi00755a028. [DOI] [PubMed] [Google Scholar]
- Williams R, Holyoak T, McDonald G, Gui C, Fenton AW. Differentiating a Ligand’s Chemical Requirements for Allosteric Interactions from Those for Protein Binding. Phenylalanine Inhibition of Pyruvate Kinase. Biochemistry. 2006;45:5421–9. doi: 10.1021/bi0524262. [DOI] [PubMed] [Google Scholar]
- Zayner JP, Antoniou C, French AR, Hause RJ, Jr, Sosnick TR. Investigating models of protein function and allostery with a widespread mutational analysis of a light-activated protein. Biophys J. 2013;105:1027–36. doi: 10.1016/j.bpj.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.