

Abstract
The outcome of structure-guided mutational analyses are often used in support of postulated mechanisms of protein allostery. However, the limits of how informative mutations can be in understanding allosteric mechanisms are not completely clear. Here, we report an exercise to evaluate if mutational data can support a simplistic mechanistic model, developed with minimal data inputs. Due to the lack of a mechanism to explain how alanine allosterically modifies the affinity of human liver pyruvate kinase (hL-PYK; approved symbol PKLR) for its substrate, phosphoenolpyruvate (PEP), we proposed a speculative allosteric mechanism for this system. Within the allosteric amino acid binding site (something in the effector site must, of necessity, contribute to the allosteric mechanism), we implemented multiple mutational strategies: 1) site-directed random mutagenesis at positions that contact bound alanine and 2) mutations to probe specific questions. Despite acknowledged inadequacies used to formulate the speculative mechanism, many mutations modified the allosteric coupling constant (Qax) consistent with that mechanism. The observed support for this speculative mechanism leaves us to ponder the best use of mutational data in structure-function studies of allosteric mechanisms. The mutational databank derived from this exercise has an independent value for training and testing algorithms specific to allostery.
Keywords: Allosteric regulation, allostery, allosteric regulation, allosteric mechanism, liver pyruvate kinase
Introduction
In recent years, a common experimental design to study allosteric mechanisms has been to utilize structural data to propose a potential mechanism. These mechanisms can be based on a single protein structure or on a comparison of differentially ligated structures. Regardless of the structural data available, these mechanisms are often subsequently ‘tested’ through mutagenic analysis of “key” residues that have been assigned specific functions in the proposed allosteric mechanism. However, we have started to question if speculate-and-mutate strategies are an ideal approach to study allosteric mechanisms [Carlson and Fenton, 2016].
In an effort to better understand the limits of how informative mutational data can be in understanding allosteric mechanisms, we report here an exercise to evaluate if mutational data can support a simplistic mechanistic proposal. We include notes on why our simplistic model, although considerate of available structures, is developed with known gaps in structural information that is relevant to understanding the allosteric mechanism. Within the context of those acknowledged deficiencies, we then explore if mutational probes can generate data to support the proposed mechanism. To parallel mutational studies that are commonly represented in the literature, our mutational probings target the allosteric site. In fact, the effector binding site is one of a limited number of areas of a protein in which something must of necessity contribute to allosteric function, no matter what constitutes the true atomic-level allosteric mechanism.
For the goal of the exercise outlined here, our ideal allosteric system is one for which there is little or no previously speculated mechanisms to describe the allosteric mechanism. Allosteric inhibition of pyruvate kinase (PYK) isozymes appears to be limited to higher organisms. Even in the mammalian PYK enzymes, there are differences, with alanine acting as the inhibitor of liver PYK and phenylalanine playing that role for the muscle M1 isozyme. Despite extensive studies [Cheng, et al., 1996; Consler, et al., 1990; Consler, et al., 1992; Consler and Lee, 1988; Consler, et al., 1988; Consler, et al., 1989; Dombrauckas, et al., 2005; Friesen, et al., 1998a; Friesen, et al., 1998b; Friesen and Lee, 1998; Lee, 2008; Lee and Herman, 2011; Morgan, et al., 2013; Oberfelder, et al., 1984a; Oberfelder, et al., 1985; Oberfelder and Lee, 1985; Oberfelder, et al., 1984b; Wooll, et al., 2001; Yu, et al., 2003], there are few speculations for how amino acid effector binding modifies substrate, phosphoenolpyruvate (PEP), affinity in the active site. We are not overlooking previously proposed mechanisms for how amino acid binding might alter subunit interfaces [Morgan, et al., 2013], but are instead considering the lack of atomic level insights into how effector-related changes result in altered PEP affinity. That lack of previous speculations is ideal for our current exercise. Here we consider the allosteric inhibition of human Liver PYK (hL-PYK; approved symbol PKLR; MIM# 609712) by alanine.
The allosteric influence on hL-PYK affinity for its substrate, PEP, by the allosteric inhibitor alanine is best stated as the affinity of the enzyme for its substrate in the absence versus presence of an allosteric effector, recognizing that the effector binds to a site distinct from the active site (Figure 1) [Carlson and Fenton, 2016; Fenton, 2008]. Defining allosteric regulation in this way, allows for allosteric regulation to be quantified by the allosteric coupling constant (Qax) [Fenton, 2008; Reinhart, 1983; Weber, 1972]:
Figure 1.
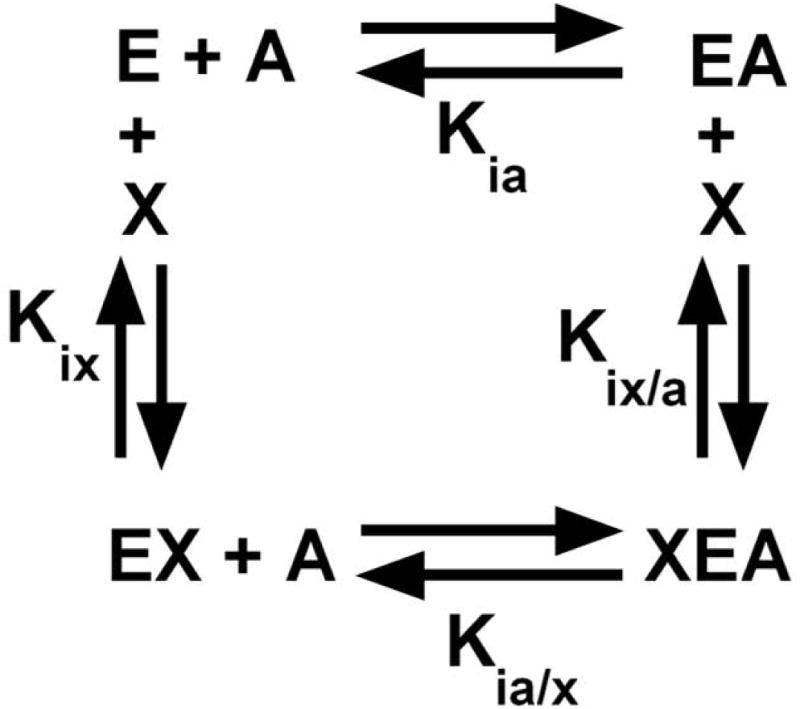
The allosteric thermodynamic cycle, in which enzyme (E) binds substrate (A) and effector (X).
| Equation 1 |
where Kia is the binding constant for substrate in the absence of effector, Kia/x is the binding constant for substrate when effector concentration is sufficiently high to saturate the effector binding site, Kix is the binding constant for effector in the absence of substrate, and Kix/a is the binding constant for effector when substrate concentration is sufficiently high to saturate the active site. When Qax ≠1, the binding of substrate (A) to a protein is allosterically coupled to the binding of the allosteric effector (X) to the same protein. In the exercise reported here, we primarily use the alterations in Qax to interpret if mutations made in the allosteric site of hL-PYK influence the allosteric mechanism [Carlson and Fenton, 2016].
In this exploration, 129 mutant proteins were characterized. In addition to our acknowledgment that the simple mechanism proposed is based on structural data with known knowledge gaps, there are a sufficient number of mutational outcomes that do not support our speculated mechanism to warrant us stating that our speculation is not valid. Nonetheless, a large percentage of the mutations can be interpreted to be in agreement with an oversimplified mechanism. Furthermore, those mutations that could be consistent with the invalid mechanism are the simpler designs (single point mutations) that are often used in studies of allostery. This outcome opens the conversation of how mutational studies can best be used in studies of allosteric mechanisms.
Beyond our exercise to evaluate the limits of mutations in studies of allosteric mechanisms, at the onset of this study, we had the benefit of knowing that any large mutational database would be useful to develop and test computational algorithms attempting to model allosteric mechanisms. Therefore, any and all outcomes from a truly exploratory use of mutational probings would be useful to the long term study of allostery. In particular, machine learning approaches are dependent on having available datasets to be used in learning phases: there are few if any such datasets available that specifically focused on quantifying allosteric coupling. Large mutational data sets are also useful to test algorithms, independent of how they were initially developed.
Materials and Methods
The GenBank RefSeq for the PKLR gene is NM_000298.5. Amino acid sequencing follows the HGVS rules, including using the methionine coded for by ATG translation initiation codon as residue 1.
Materials
The potassium salts of ADP and PEP were purchased from Chem-Impex International, Inc. Alanine and NADH was from Sigma. L-lactate dehydrogenase (Type III bovine heart) was purchased from Calzyme Laboratories, Inc. Other buffer components were from Fisher Scientific and Sigma.
Methods
Methods used in this work have been exhaustively detailed elsewhere [Fenton and Alontaga, 2009; Fenton and Hutchinson, 2009; Ishwar, et al., 2015]. Mutagenesis of the hL-PYK gene was performed with Quikchange (Stratagene), using both site-directed random mutagenesis via primers that were degenerate at the target codon in some cases, and single point mutations generated with specifically designed primers in other cases. All proteins were expressed in the FF50 strain of Escherichia coli [Fenton and Hutchinson, 2009]. Mutant proteins were partially purified using ammonium sulfate fractionation followed by dialysis [Fenton and Tang, 2009; Ishwar, et al., 2015]. Activity measurements were carried out at 30°C using a lactate dehydrogenase coupled assay in either HEPES or bicine buffer, pH 7.5 [Fenton and Alontaga, 2009].
Data fitting was with the nonlinear least-squares analysis of Kaleidagraph (Synergy) software. Fits of PEP titrations of initial rates (ν) used to obtain Kapp-PEP were as previously described [Fenton and Alontaga, 2009; Fenton and Hutchinson, 2009]. Kapp-PEP values as a function of effector concentration were fit to equation 1 [Reinhart, 2004]:
| Equation 2 |
where Ka = Kapp-PEP when [Effector] = 0; Kix = the dissociation constant for effector (X) binding to the protein in the absence of substrate (A); and Qax is the allosteric coupling constant discussed in detail elsewhere [Reinhart, 2004].
When effector affinity is reduced sufficiently that the upper plateau cannot be accessed within the 10mM maximal PEP concentration [Fenton and Hutchinson, 2009], then data were fit to [Williams, et al., 2006]:
| Equation 3 |
This latter fitting allows for an estimation of Kix. However, it does not provide information about the magnitude of the allosteric coupling.
In this study, there were several mutations that reduced activity below the detection limit of the assay. It seems very likely that some of these mutant proteins may still have some level of activity. However, due to the quantity of mutations included in this study no effort was made to further purify, concentrate and reanalyze mutant proteins that did not have detectible activity in our original assay design. Mutant proteins for which activity was not detected are noted by “No Data” in figures, below. Most notably, mutation of P483 was particularly sensitive to mutation and only P483G retained activity.
Results
Defining an allosteric mechanism
For the purpose of our exercise, we chose to speculate an allosteric mechanism that, while consistent with available structural information [Dombrauckas, et al., 2005; Fenton, et al., 2010a; Holyoak, et al., 2013; Larsen, et al., 1998; Larsen, et al., 1997; Larsen, et al., 1994; Morgan, et al., 2013; Valentini, et al., 2002; Williams, et al., 2006], lacks further validation by additional methods. Structural data for hL-PYK and other mammalian isozymes in complex with PEP and pyruvate analogues bound in the active site is available. In addition, the location of the allosteric amino acid binding site is known based upon co-crystallization of rabbit M1-PYK with alanine and M2-PYK with phenylalanine. Therefore, the only information we used to generate a model to describe the allosteric coupling in PYK were the locations of the PEP and alanine binding sites in the hL-PYK structure (Figure 2). Inspection of the structural data demonstrates that the PEP and alanine binding sites lie at opposite ends of the same secondary structure elements. We will consider this grouping of secondary structures elements (residues 57-61 and 340-397) as a singular, rigid structural unit and, for the purpose of discussion in this work, refer to it as the “2nd unit.” We therefore propose a mechanism in which in the absence of alanine, PEP binding causes the protein to adopt a conformation in which the 2nd unit is shifted into the alanine binding site. This shift allows the protein to sterically accommodate the binding of the PEP molecule in the active site. Next, recall that as an alternative to inhibition of substrate binding being caused by the removal of interactions that contribute positively to binding, inhibition can also result from the addition of interactions that contribute negatively to substrate binding. Therefore, when alanine binding precedes PEP binding, the same accommodating 2nd unit shift cannot occur when PEP binds. Therefore, in the presence of alanine, PEP binding incurs some level of steric hindrance due to the intrasubunit molecular crowding (i.e., modified plasticity). As a result, PEP binds to the active site with less favorable binding energy when alanine is present in the effector site than when alanine is absent.
Figure 2.
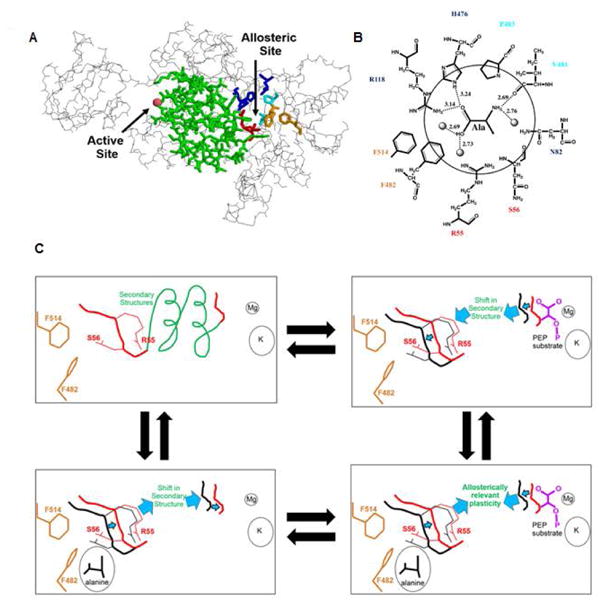
Development of a structure-based allosteric mechanistic model. A. A backbone trace of one subunit of hL-PYK. The active site and allosteric sites are known from various PYK structures with ligands co-crystalized (PDBID: 1A49, 1A5U, 1AQF, 2VGB, 1PKN, 4IM4, 4IP7, 1T5A, 4FXJ, 4FXF, 3N25, 2G50). The active site divalent cation is shown as a sphere. The secondary structures that reside between the active site and allosteric site are in green. Residues R118, H476, and N82 are shown as blue sticks. Residues R55 and S56 are in red sticks. Residues F482 and F514 are in orange stick. Residues V481 and P483 are in cyan stick. The backbone chain in green includes secondary structures that are considered as a single unit, labeled the 2nd unit. Residues 57-61 and 340-397 are included in this 2nd unit and collectively they define both β-sheets and α-helices that contribute to the α/β-barrel of the A-domain. B. A representation of the effector binding site derived from the 1.65 Å structure of M1-PYK in complex with alanine (PDBID:2G50) [Williams, et al., 2006], but with hL-PYK residue numbering and amino acid type. Potential hydrogen bonds formed between the bound alanine and protein residues are illustrated with dashed lines and the distances indicated. Three water molecules are also shown and rendered as spheres. C. Cartoon representation of potential shifts in secondary structure upon binding ligands: these representations for conformational change are speculative with no supporting structural information. The four corners of this panel are intended to represent the enzyme complexes represented in Figure 1. Note in lower right, the structural shifts suggested to occur when one ligand binds in isolation cannot occur when alanine and PEP simultaneously bind.
The identity represented in Equation 1 indicates that the magnitude of the influence that alanine has on PEP binding must be equal to the influence that PEP has on alanine binding. Any proposed allosteric mechanism should account for this reciprocity. Therefore based on our model, when alanine binds in the absence of PEP, the 2nd unit shifts into the PEP binding site. In a manner that is inverse to that stated above, that same shift cannot be accommodated when PEP precedes the binding of alanine. Therefore, in the ternary complex, neither PEP nor alanine bind as tightly to the enzyme as compared to when one ligand binds to the enzyme in the absence of the second ligand.
Reasons why our proposed mechanism is less than ideal
We think that the ideal information required to speculate an allosteric mechanism would be structural information obtained on each of the four enzyme forms that define the thermodynamic cycle in Figure 1. Any structural changes identified in the comparison of the E and EA forms would be compared to structural changes between the XE and XEA forms. Changes that are common whether A binds in the presence or in the absence of X are likely important to the binding of A, but those changes would cancel out in considerations of allostery (due to Equation 1) [Fenton, 2008]. By definition only, those structural changes to the protein that occur differently when A binds in the absence vs. in the presence of alanine would be the focus of an allosteric mechanism. Based on the relationship in Figure 1 it is obvious that the reciprocal process is also true: those structural changes that result from the binding of X in the absence of substrate (compare E and XE) that are equivalent to changes that result from the binding of X in the presence of substrate (compare EA to XEA) should contribute to X binding, but not to allostery. However, structural changes that occur differently when X binds in the presence vs. absence of substrate would be the focus of an allosteric mechanism.
Unfortunately, we do not have the required structural information on all four enzyme forms of hL-PYK that constitute the thermodynamic cycle in Figure 1. Even as more systems become available for which there is structural information for all four corners of the thermodynamic cycle, it will be important to consider if those structures reflect relevant solution structure: Are the structures allosterically competent? (Wording chose to parallel questions related to crystallized enzymes being enzymatically competent.) In the case of X-ray crystallographic studies, it should be relatively easy to test if the allosteric properties of a protein are retained in the solution conditions used for crystal growth. It will be more challenging to understand if lattice contacts in the protein crystal bias the structure away from allosterically-relevant solution structures. Clearly, there is a considerable lack of information that would offer the most ideal starting point to speculate an allosteric mechanism for hL-PYK. Therefore, we consider our proposed mechanism to be highly speculative and likely overly simplistic.
As a further acknowledgment of model simplicity, consider that each of the four subunits in the homotetrameric hL-PYK has one PEP binding site and one alanine binding site. Our simplistic speculation only considers how the alanine binding site in one subunit might result in reduced PEP affinity in the same subunit. Our mechanistic prediction makes no attempt to consider how the alanine binding in one subunit might modify PEP binding in the other three subunits. The types of evidence available in other systems [Fenton, et al., 2004; Fenton and Reinhart, 2002; Fenton and Reinhart, 2003; Fenton and Reinhart, 2009] that effector binding in one subunit alters substrate binding in other subunits is largely not available for hL-PYK [Hubbard and Cardenas, 1975].
Predicted mutational outcomes from our simplistic mechanistic model: alanine binding
From our model, we can make the assumption that some residues in the effector binding site must contribute to effector binding and it is reasonable to suggest that at least some of those effector-protein contacts occur through side-chains interactions. Therefore, mutating residues that establish side chain interactions with the effector are expected to reduce the ability of the protein to bind alanine. This prediction should be constant for all side-chain substitutions that do not preserve the original wild-type interactions.
Mutational insights into side chain contributions to alanine binding
To look for evidence that is consistent with our proposed model, we chose to create a series of mutations at several positions that comprise the the alanine binding site (Figure 3). Consistent with earlier suggestions [Carlson and Fenton, 2016], rather than dictating the nature of the substituted side chain, site-directed random mutagenesis (using primers degenerate at the codon of interest) was used to generate a substitution series at each position. The nature of the substitution was determined by DNA sequencing. Each mutated gene was isolated for protein expression. For those mutant proteins that retained activity, the response of activity to PEP concentration was used to determine Kapp-PEP and the response of Kapp-PEP to effector concentrations was fit to Equation 2 to evaluate Ka-PEP, Kix-Ala, and Qax (or Equation 3 for the estimation of Ka-PEP and Kix-Ala, when simultaneous saturation with both PEP and alanine was not possible).
Figure 3.
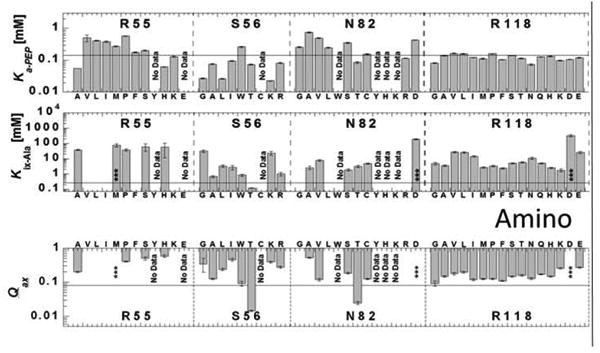
Ka-PEP, Kix-Ala, and Qax determined for mutant proteins designed in the site-directed random study. Mutated residues are labeled in the respective sections of the graph; sections dedicated to each residue position are divided by vertical dashed lines, and replacement residues at the respective positions are listed on the x-axis. For each position, replacement residues are roughly ordered from smallest to largest hydrophobic, smallest to largest hydrophilic, positive charge and negative charge from left to right. In each panel, a horizontal line is included to indicate the respective value for wild-type protein. Several mutations were not active and are noted with “No Data.” Examples that show some response to alanine, but with sufficiently decreased affinity to prevent saturation with the inhibitor are indicated by an asterisk (***). The absence of any data/symbol in graphs of Kix-Ala and Qax indicates no allosteric response.
Inspection of Figure 3, with a specific consideration of location of the residues in the alanine binding site (Figure 2), indicates that H476 and R118 collectively play a primary role in alanine binding. With exception of H476Q, substitutions at this position reduce the enzyme's affinity for alanine sufficiently to prevent a full evaluation of allosteric coupling. The nature of the replacement (hydrophobic or hydrophilic) does not seem to be a determining factor in the outcome, allowing us to assign the loss in affinity to a loss of the His side-chain, rather than assigning that outcome to the addition of replacement side-chains [Carlson and Fenton, 2016]. When evaluation is possible, the severity of the loss of alanine binding leads to the speculation that those mutations of the 476 position that completely lack a response to alanine may be due to a complete failure of alanine to bind (as opposed to alanine binding without eliciting an allosteric response). The single exception that demonstrates a milder influence on alanine affinity is H476Q. Interestingly the ε-amino group of glutamine can be position equivalent to the ε-amino on the histidine ring. Taken together, the ε-amino group of H476 plays a dominant role in binding of the amino acid effector.
At the R118 position, the increase in Kix-Ala appears independent of the nature of the substitution. Therefore, it seems likely that the reduced alanine binding is a result of removal of the native arginine at 118. The exception to this conclusion is that the addition of negative charge at position 118 (R118D, R118E) causes a more drastic effect compared to the other mutations. Therefore, it is proposed that the more significant loss in alanine binding in the R118D and R118E substitutions is the combination of the loss of positive interactions between the carboxyl group of alanine and the native R118 sidechain, as well as the addition of new negative interactions between the negatively charged aspartate and glutamate side chains and the carboxyl group of the alanine effector.
The structural data suggest that N82 positions a water molecule that, in turn, interacts with the amino group of the bound effector. The mutagenesis data suggest that the enzyme loses activity upon substitution of N82 with the majority of large side chain substitutions. For the smaller side chains substitutions, the data trend is similar to that of R118. That is, most responses indicated similar increases in Kix-Ala, with the exception of introducing a negative charge (N82D). Using the same logic applied to the interpretation of data for mutations at R118, we conclude that the loss of the native N82 side chain increases Kix-Ala, but introduction of a negative charge at that residue position causes an additional negative influence on alanine binding.
The lack of response of proteins with mutations at position 482 should be considered cautiously. In our assay, the absence of a response to alanine could indicate either that alanine does not bind, or that alanine binds, but fails to elicit an allosteric response on PEP affinity. The mutations that retain an allosteric response include either a ring structure and/or the ability to form a pi bond. (In Figure 3, consider that in addition to ring structures, arginine can contribute pi interactions.) A ring structure/pi bond potential at position 482 may be required for either alanine binding, allosteric coupling, or both. Due to the proximity with R55 and the greatly reduced binding caused by mutations at the 55 position, F482 and R55 may function jointly to define one side of the alanine binding site. A joint role of these two residues might also help explain the indicated role that R55 has in alanine binding, since there is no structural evidence that R55 makes direct interactions with bound effector
The effect of mutations at the S56 position are more dependent on the chemical nature of the replacement side chain, including S56T, one of only two mutant proteins with increased alanine binding. Not surprisingly, mutations at V481 and F514 have modest influences on alanine binding, as the structural data demonstrate that it is the backbone of the 481 position that interacts with the bound alanine molecule. Lastly, with the exception of P483G, mutations at the 483 position cause a loss of enzyme activity.
Predicted mutational outcomes from our simplistic mechanistic model: signatures of modified allostery
Due to the speculation that allostery results from intrasubunit crowding/modified plasticity due to simultaneous binding of both alanine and PEP, a mutation that increases bulk in the effector site is expected to accentuate the allosteric inhibition (decrease in the Qax value). We can envision that a mutation in the alanine binding site that increases the total effector site bulk/intrasubunit crowding would reduce the ability of the 2nd unit to shift upon PEP binding. In this scenario, a mutation that increases bulk in the effector site could “mimic” the proposed allosteric role for bound alanine. However, extra bulk from an introduced side-chain might be accommodated in the structure with no influence on PEP affinity, until alanine is also present. Therefore, mutations that introduce bulk into the alanine binding site and decrease Qax, either with or without an associated increase in Ka-PEP, would be consistent with our simplistic model. Likewise, mutations that reduce overall bulk in the effector site and that cause Qax to approach 1, either with or without an associated decrease in Ka-PEP, would be consistent with our simplistic model [Carlson and Fenton, 2016].
Mutational insights into side-chain contributions to allosteric regulation by alanine
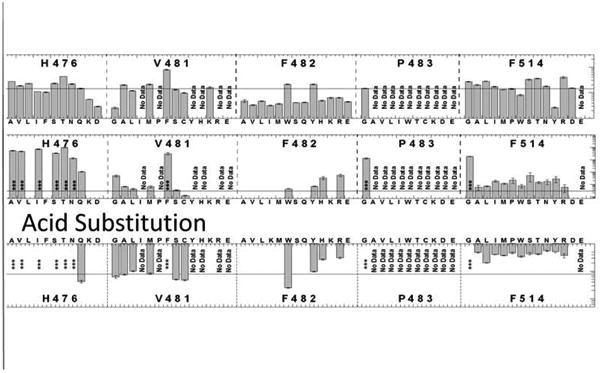
The most dramatic set of mutations with altered allosteric coupling are those mutations made at positons F514 and R55. These mutations all cause Qax to increase, approaching 1. Although a few of these changes in Qax are accompanied with decreased Ka-PEP values, most are not. Of those, most of the substitutions should cause reduced overall bulk. However, F514W and R115H do not fit neatly into this consideration.
As noted above, the absence of an allosteric signal when F482 is replaced with many side-chain types does not indicate if alanine fails to bind to the mutant proteins, or if alanine binds, but fails to elicit an effect on PEP binding (also applicable to several mutations at the R55 and other positions). We lean towards the idea that alanine might bind without eliciting an allosteric response in those mutants. Our reason for leaning towards that interpretation is that F482H and F482R both show reduced allosteric inhibition (increased Qax), rather than significant decreases in alanine binding. In addition, the removal of side-chain bulk at the 482 position causes PEP to bind tighter (reduced Ka-PEP), the opposite of the mimic proposed if additional side chain bulk is added to cause reduced PEP binding. Based on that interpretation, if we group F482 with R55 and F514, then an area of the alanine binding site has been identified to play a role in the allosteric mechanism.
Qax values for mutations at the S56 and N82 positions are more dependent on the chemical nature of the side chain replacement. Interestingly, two of the three mutations (S56T, N82T, and F482W) that cause a respectable decrease in Qax occur at those two positions (none of the three demonstrate an appreciable increase in Ka-PEP).
Any additional attempt to interpret alanine binding site residues for roles in allostery based on mutational studies is far from convincing. We could consider that the R55 and S56 positions reside sequentially on a loop that constitutes the end of 2nd unit (Figure 2). In turn, the R55/S56 loop neighbors the side-chains of F482 and F514. One speculation could be that the additional hydrophobic bulk at positions 482 and/or 514 might cause R55/S56, and in turn the 2nd unit, to shift towards the active site and result in the partial steric hindrance of PEP binding. However, that structure based consideration is not supported from our mutational data set (e.g., added bulk caused by the S56W does not appreciably alter Qax and it causes only a modest reduction in Ka-PEP). Nonetheless, the location of R55 may respond to additional bulk added at the R55, F482, or F514 positions, and that response may influence PEP affinity. Results from our site-directed random mutational study provided many outcomes that could be explained by our simplistic mechanism. Importantly, there are also data that do not appear to be consistent with that mechanism.
Specific questions: Does altered rigidity near the alanine binding site influence Qax?
Rather than start a study with a large mutational data set and attempt to interpret results within a proposed mechanism, many studies design mutants to specifically test aspects predicted by the speculated mechanism. Therefore, we next used that same approach with no consideration of the outcome or evaluation of the site-directed random mutagenesis study presented above.
If, as our model suggests, extra bulk in the effector binding site (either via the binding of alanine, or through mutations that increase steric bulk of amino acid side chains lining the binding site) inhibits PEP binding because of a shift in the 2nd unit, we reasoned that introducing amino acid substitutions that increase the flexibility of the 2nd unit would increase the ability of the allosteric site to accommodate additional bulk without eliciting an allosteric response. More specifically, reduced steric hindrance when PEP and alanine are simultaneously bound would modify the influence alanine binding has on PEP affinity. To keep our focus in the allosteric binding site itself, we considered that R55 and S56 reside on a loop at the end of the 2nd unit and within the alanine binding site. To increase flexibility, we characterized both the A54G and the T57G mutations. The T57G mutation caused loss of activity. The alanine inhibition by the A54G mutant displays reduced allosteric coupling by alanine (Figure 4). This modified allosteric coupling can be viewed as a reduced distance between the lower and upper plateaus in the absence of and at very high levels of alanine, respectively. This reduced allosteric coupling caused by the A54G mutation could be interpreted as having some level of consistency with the outcomes predicted by our model.
Figure 4.
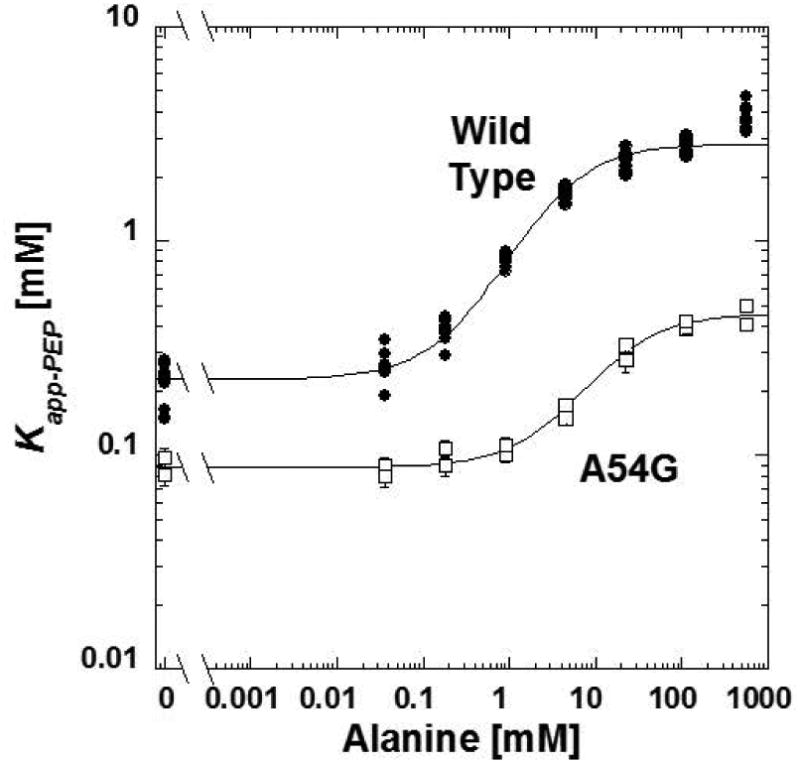
The response of the Kapp-PEP for A54G to concentrations of alanine. Compared to the response of the wild type protein (solid circles), the A54G mutant protein (open squares) has a reduced allosteric coupling. In this presentation, allosteric coupling is the difference between the horizontal plateaus low effector concentration, compared to the horizontal plateau at very high alanine plateaus. Lines represent the best fits to Equation 2. When error bars are not apparent, they are smaller than data point symbols.
Specific questions: Does S56 determine inhibitor specificity in PYK isozymes?
A direct comparison of residues in the amino acid binding sites of hL-PYK and rabbit M1-PYK indicates that the only differences is Ser at residue 56 in hL-PYK vs Asn in M1-PYK. However, unlike hL-PYK, M1-PYK is allosterically inhibited by phenylalanine. Alanine binds to M1-PYK competitively with phenylalanine, but the smaller ligand has a negligible influence on PEP affinity [Fenton, et al., 2010b; Prasannan, et al., 2011; Prasannan, et al., 2012; Williams, et al., 2006]. With respect to our mechanistic speculations, our simplistic mechanism might be even more justified in studies of PYK isozymes that are inhibited by phenylalanine, as compared to the hL-PYK isozyme that is inhibited by alanine. Primarily, a structure of human M2-PYK co-crystalized with phenylalanine bound indicates that the phenyl ring from the phenylalanine inhibitor wedges between the side chains of the residues equivalent to F482 and R55 in hL-PYK [Morgan, et al., 2013]. That binding of the phenylalanine effector has been speculated to cause the side chain of F482 to rotate [Morgan, et al., 2013] (Figure 5), even though the same flip occurs upon alanine binding [Williams, et al., 2006]. F514 is also immediately adjacent to where the effector phenyl ring is located in the M2-PYK structure. Given the lack of other changes in the effector site, we ask if the S56N would modify the specificity of hL-PYK for the identity of the amino acid that acts an inhibitor. This question is relevant to our speculated mechanism, given that a phenyl ring of M1-PYK-bound phenylalanine likely interacts with F482 and/or F514. Therefore, the different side chains at the 56 position could modulate different allosteric responses in the two enzymes. Neither the S56N nor the S56Q mutations were generated in our earlier random mutagenesis design. Therefore, we created the S56N and S56Q mutations to evaluate their influence on alanine and phenylalanine inhibition of hL-PYK.
Figure 5.
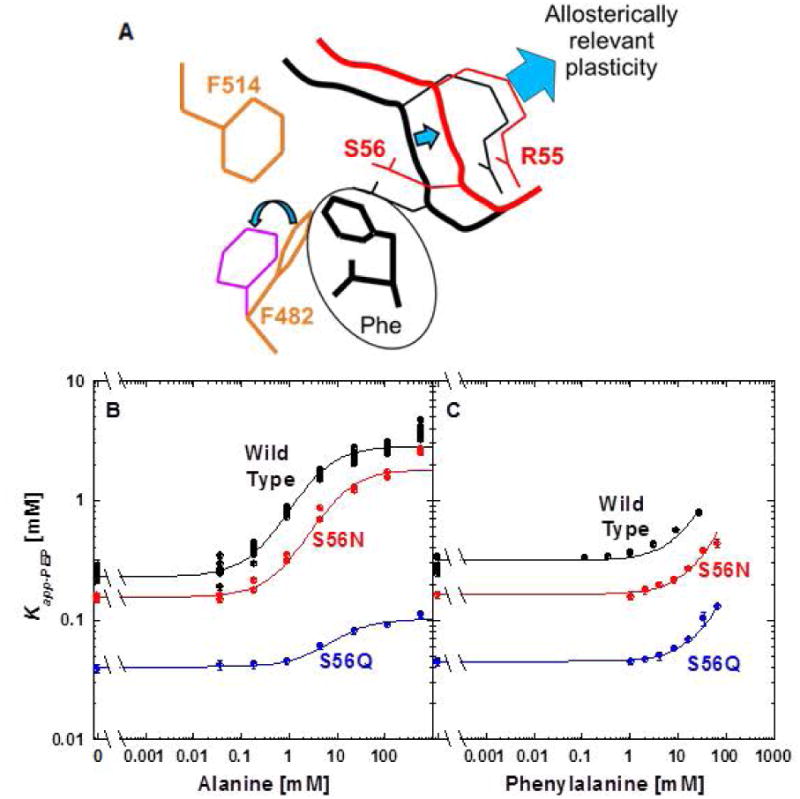
A. A cartoon of the ring flipping of F482 that appears to result from binding of phenylalanine to M1-PYK and M2-PYK isozymes [Morgan, et al., 2013]. B. The response of S56N and S56Q to alanine and phenylalanine. The responses to alanine were fit to Equation 2 and the responses to phenylalanine were fit to Equation 3. When error bars are not apparent, they are smaller than data point symbols.
The response of hL-PYK to inhibition by phenylalanine showed little change in response to creating the S56N or S56Q mutations. Interestingly, although not the original intent of this design, the S56Q mutation clearly reduces the allosteric response to alanine. However, the reduced PEP affinity of the S56Q mutation is an outcome that does not neatly support our simplistic mechanism.
Specific questions: Do the potential interactions between R55 and N82 contribute to allostery?
The side chain of R55 and N82 are pointed towards each other in the hL-PYK structure. This proximity is hard to visualize when viewing the protein from the exterior, but can be appreciated from a view point within the subunit (Figure 6). It remains possible, that alanine binds via interactions with H476, R118 and N82, and this binding is relayed to R55 due to the binding event altering interactions between R55 and N82. That change in the R55 side chain interaction could then be what controls the 2nd unit mobility. Please note that this idea is a slight variation from our original mechanism in that the focus in the alanine site shifts from total bulk to the R55/N82 interaction.
Figure 6.
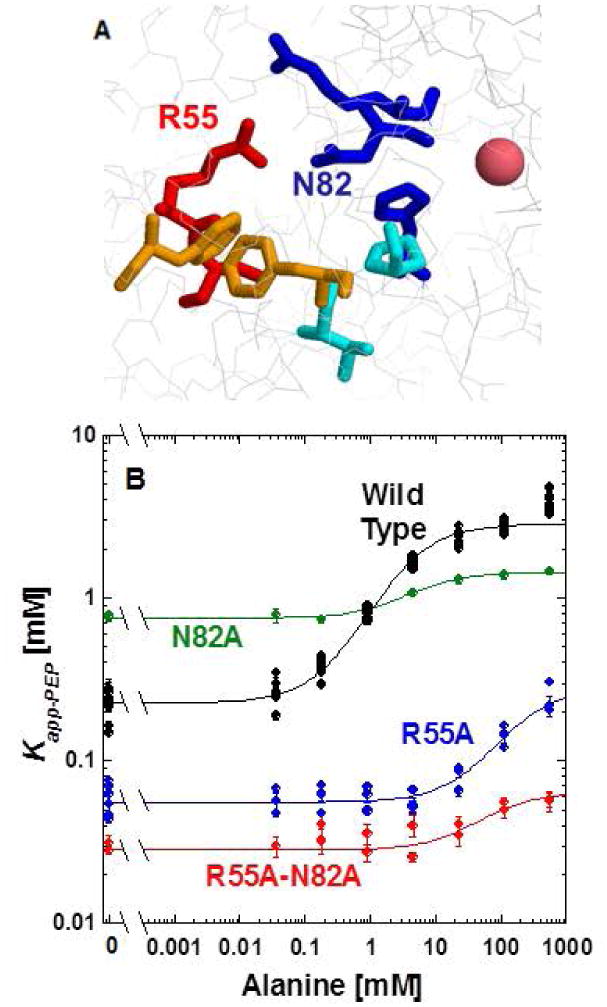
A. An image of the potential side chain interactions between R55 and N82 of hL-PYK structure [Holyoak, et al., 2013], with a view point from the interior of the protein. B. The response of the wild type protein (black), R55A (blue), N82A (green) and the R55A-N82A (red) double mutant proteins to alanine. In this figure, the responses to alanine were fit to Equation 2. However, the data are not convincing that the upper plateau has been reached for R55A and the line for the R55A data should only be considered as an indication of data trend. The responses to phenylalanine were fit to Equation 3. When error bars are not apparent, they are smaller than data point symbols.
We reasoned that if alanine and/or PEP binding disrupts the R55 and N82 interactions that exist in the absence of ligand, then we should be able to detect synergistic effects between mutations at these two positions. In other words, a mutation at R55 would result in modified alanine and/or PEP binding and a mutation at N82 would result in modified alanine and/or PEP binding. However, if the two mutations are added simultaneously, the second mutation would not contribute any additional influence on ligand binding (compared to either of the individual mutations alone). To test this idea of synergistic function, we allowed our selection of replacement amino acids to be biased by the outcomes of the site directed random mutagenesis study reported above. However, even with that bias, the R55K-N82T mutation was not active. In our second attempt, we used the alanine substitutions at both positions (Figure 6). However, the synergy we were evaluating is not supported in comparison of Ka-PEP values (left y-intercept comparison). We could also evaluate synergy in Kix-Ala and Qax. However, the data for R55 do not convincingly reach a plateau and, therefore, we chose not to continue this line of comparison.
Specific questions: Can mutations be combined to maximize protein properties?
Up to this point in presentation of results, we have focused on outcomes that could show consistency with the speculated simplistic mechanism. However, in this section, we will now present evidence that is strongly inconsistent with that mechanism. To introduce bulk at multiple locations in the effector site, we again selected mutations based on outcomes from the site directed random mutagenesis study. Combinations of mutations that increased bulk in the effector site showed propensities to increase PEP affinity (reduced Ka-PEP) and to increase the allosteric inhibition caused by alanine (reduced Qax). Both of these outcomes are inconsistent with expectations from our proposed mechanism.
Of the four mutant proteins represented in Figure 7, S56T-F482W had the largest allosteric response and, coincidentally, the highest affinity for PEP. Due to the absence of obtaining the upper horizontal plateau at very high concentrations of alanine, we could not quantify Qax for these mutants. However in Figure 7, the increased allosteric influence in the S56T-F482W protein can be appreciated by considering the difference from the plateau in the absence of alanine and the highest Kapp-PEP value. In contrast to our simplistic speculated mechanism, both of the mutations in the S56T-F482W protein add to, not decrease, the total bulk in the effector site. Of course we simply used an empirical approach in our attempt to maximize PEP affinity and to maximize the allosteric response to alanine. Other empirical attempts that did not maximize targeted properties as extensively as the S56T-F482W were S56T-H476Q-F482W, S56T-N82T-F482W, and S56T-N82T-H475Q-F482W (Figure 7).
Figure 7.
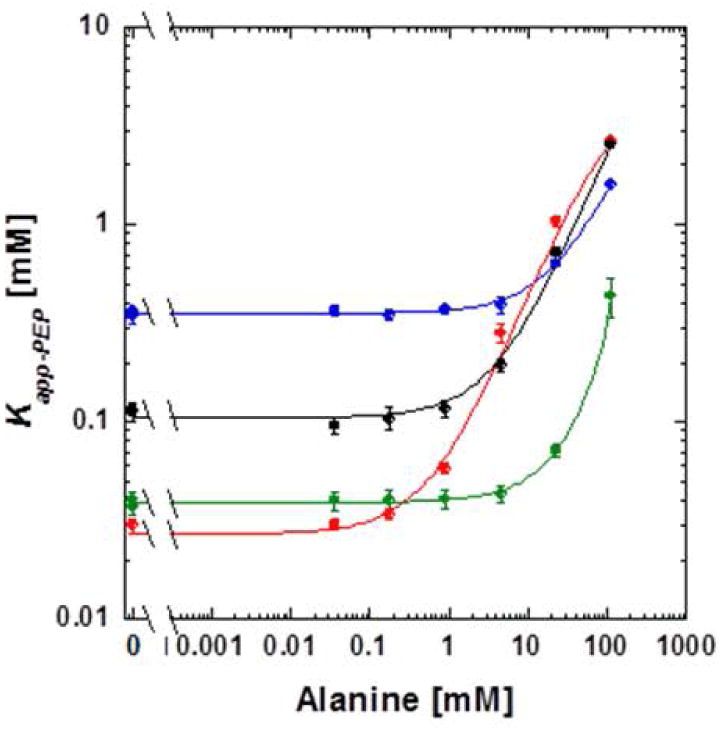
The response of Kapp-PEP to alaine for proteins with combinations of mutations, all of which are located in the alanine binding site. S56T-F482W (red), S56T-H476Q-F482W (green), S56T-N82T-F482W (black), and S56T-N82T-H475Q-F482W (blue). Lines represent data trends.
We also used combinations of mutations to demonstrate that the affinity for PEP could be maximized without a response to alanine. S56L-V481G, V481G-F482H, V481G-F514Y, and S56L-V481G-F482H-F514Y all had Ka-PEP values in the range of 0.02 to 0.03 mM, but no response to alanine. R55H-S56L-H476D-V481G-F514Y lacked activity.
Discussion
In this study, a range of mutations were introduced into the alanine binding site of hL-PYK and outcomes were compare to those expected from a speculated allosteric mechanism. Throughout most of the presentation of results, we purposefully focused on which outcomes could be consistent with predictions from a simplistic mechanism. We have also been careful to outline why the original considerations to derive that hypothesis were based on insufficient structural insights. We reserved our stronger arguments against the simplistic mechanism for the final result: proteins with multiple mutations added to the effector site (Figure 7). However in contrast to data that are not consistent with the speculated mechanism, we were surprised at the number of individual mutational outcomes that could be interpreted to show consistency with our poorly conceived mechanistic hypothesis.
Given this outcome and considering that mutagenesis is an “easy” tool to use in mechanistic studies, we are then left to ask how mutations can best be interpreted in the context of understanding allosteric mechanisms. To answer that, we contrast the overall outcome of this study (129 mutant proteins characterized) with our previous work using mutations to evaluate the Fru-1,6-BP allosteric site of hL-PYK [Ishwar, et al., 2015]. In that previous study, both an effector analogue series and the outcomes from 88 mutant proteins collectively supported a mechanism that was originally conceived with additional structural insights. Importantly, it was the large number of outcomes that were collectively consistent with a single mechanism that indicates strong support for that mechanism. Therefore, we suggest mutational studies of allostery should 1) probe a broad range of positions including both those predicted to be important to and those predicted to have no role in a proposed allosteric mechanism, 2) to use a range of substitutions at each probed position [Carlson and Fenton, 2016], and 3) be interpreted with a focus on inconsistent outcomes, rather than focusing on results that might be consistent with a speculated mechanism. It remains possible that the same mutagenesis strategy would be the most ideal to study any protein function.
In truth, there are other types of data that we overlooked in developing our simplistic model. In particular, an effector analogue series was already used to identify that the L-2-aminopropanaldehyde substructure of the amino acid effector is the primary determinant of amino acid effector binding affinity in both M1-PYK and hL-PYK [Alontaga and Fenton, 2011; Williams, et al., 2006]. That substructure interacts with the equivalent positions of R55, N82, and H476 (hL-PYK residue identification) in M1-PYK [Williams, et al., 2006]. Therefore, the assignment of these same residues for primary functions in amino acid binding is confirmatory. However, like the mutagenesis study here, the analogue study of hL-PYK did not provide many clues into which parts of the inhibitor or the binding site contribute to allostery.
Despite the mechanistic insights that remain unknown, the large mutagenesis data set created by this activity fills a needed gap. Therefore, the mutations presented in this work, along with those previously published [Ishwar, et al., 2015], will be useful in training and testing computational algorithms that focus on understanding allosteric mechanisms.
CAGI competition: The outcomes of mutagenesis studies were used as a prediction challenge in a Critical Assessment of Genome Interpretation “CAGI” competition. CAGI is a global community experiment to objectively assess computational methods for predicting phenotypic impacts of genomic variation. The results of that CAGI competition are presented elsewhere, but the data for mutants in this study are provided here.
Supplementary Material
Acknowledgments
This work was supported by NIH grant GM115340. Data generated in this study were coordinated by Dr. Liskin Swint-Kruse for use in a CAGI-4 challenge in the Fourth Critical Assessment of Genome Interpretation (CAGI 4) organized by R.A. Hoskins, chaired by S.E. Brenner and J. Moult, and titled “Liver pyruvate kinase (L-PYK): predict the effects of missense mutations on kinase activity and allosteric regulation.”
Footnotes
This work was supported by NIH grant GM115340.
References
- Alontaga AY, Fenton AW. Effector analogues detect varied allosteric roles for conserved protein-effector interactions in pyruvate kinase isozymes. Biochemistry. 2011;50:1934–9. doi: 10.1021/bi200052e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GM, Fenton AW. What Mutagenesis Can and Cannot Reveal About Allostery. Biophys J. 2016;110:1912–23. doi: 10.1016/j.bpj.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Friesen RH, Lee JC. Effects of conserved residues on the regulation of rabbit muscle pyruvate kinase. J Biol Chem. 1996;271:6313–21. doi: 10.1074/jbc.271.11.6313. [DOI] [PubMed] [Google Scholar]
- Consler TG, Jennewein MJ, Cai GZ, Lee JC. Synergistic effects of proton and phenylalanine on the regulation of muscle pyruvate kinase. Biochemistry. 1990;29:10765–71. doi: 10.1021/bi00500a007. [DOI] [PubMed] [Google Scholar]
- Consler TG, Jennewein MJ, Cai GZ, Lee JC. Energetics of allosteric regulation in muscle pyruvate kinase. Biochemistry. 1992;31:7870–8. doi: 10.1021/bi00149a018. [DOI] [PubMed] [Google Scholar]
- Consler TG, Lee JC. Domain interaction in rabbit muscle pyruvate kinase. I. Effects of ligands on protein denaturation induced by guanidine hydrochloride. J Biol Chem. 1988;263:2787–93. [PubMed] [Google Scholar]
- Consler TG, Uberbacher EC, Bunick GJ, Liebman MN, Lee JC. Domain interaction in rabbit muscle pyruvate kinase. II. Small angle neutron scattering and computer simulation. J Biol Chem. 1988;263:2794–801. [PubMed] [Google Scholar]
- Consler TG, Woodard SH, Lee JC. Effects of primary sequence differences on the global structure and function of an enzyme: a study of pyruvate kinase isozymes. Biochemistry. 1989;28:8756–64. doi: 10.1021/bi00448a012. [DOI] [PubMed] [Google Scholar]
- Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44:9417–29. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- Fenton AW. Allostery: an illustrated definition for the ‘second secret of life’. Trends Biochem Sci. 2008;33:420–5. doi: 10.1016/j.tibs.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW, Alontaga AY. The impact of ions on allosteric functions in human liver pyruvate kinase. Methods Enzymol. 2009;466:83–107. doi: 10.1016/S0076-6879(09)66005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW, Hutchinson M. The pH dependence of the allosteric response of human liver pyruvate kinase to fructose-1,6-bisphosphate, ATP, and alanine. Arch Biochem Biophys. 2009;484:16–23. doi: 10.1016/j.abb.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW, Johnson TA, Holyoak T. The pyruvate kinase model system, a cautionary tale for the use of osmolyte perturbations to support conformational equilibria in allostery. Protein Sci. 2010a;19:1796–1800. doi: 10.1002/pro.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW, Paricharttanakul NM, Reinhart GD. Disentangling the web of allosteric communication in a homotetramer: heterotropic activation in phosphofructokinase from Escherichia coli. Biochemistry. 2004;43:14104–10. doi: 10.1021/bi048569q. [DOI] [PubMed] [Google Scholar]
- Fenton AW, Reinhart GD. Isolation of a single activating allosteric interaction in phosphofructokinase from Escherichia coli. Biochemistry. 2002;41:13410–6. doi: 10.1021/bi026450g. [DOI] [PubMed] [Google Scholar]
- Fenton AW, Reinhart GD. Mechanism of substrate inhibition in Escherichia coli phosphofructokinase. Biochemistry. 2003;42:12676–81. doi: 10.1021/bi0349221. [DOI] [PubMed] [Google Scholar]
- Fenton AW, Reinhart GD. Disentangling the Web of Allosteric Communication in a Homotetramer: Heterotropic Inhibition in Phosphofructokinase from Escherichia coli. Biochemistry. 2009 doi: 10.1021/bi901456p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW, Tang Q. An activating interaction between the unphosphorylated n-terminus of human liver pyruvate kinase and the main body of the protein is interrupted by phosphorylation. Biochemistry. 2009;48:3816–8. doi: 10.1021/bi900421f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AW, Williams R, Trewhella J. Changes in small-angle X-ray scattering parameters observed upon binding of ligand to rabbit muscle pyruvate kinase are not correlated with allosteric transitions. Biochemistry. 2010b;49:7202–9. doi: 10.1021/bi100147w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen RH, Castellani RJ, Lee JC, Braun W. Allostery in rabbit pyruvate kinase: development of a strategy to elucidate the mechanism. Biochemistry. 1998a;37:15266–76. doi: 10.1021/bi981273y. [DOI] [PubMed] [Google Scholar]
- Friesen RH, Chin AJ, Ledman DW, Lee JC. Interfacial communications in recombinant rabbit kidney pyruvate kinase. Biochemistry. 1998b;37:2949–60. doi: 10.1021/bi971990c. [DOI] [PubMed] [Google Scholar]
- Friesen RH, Lee JC. The negative dominant effects of T340M mutation on mammalian pyruvate kinase. J Biol Chem. 1998;273:14772–9. doi: 10.1074/jbc.273.24.14772. [DOI] [PubMed] [Google Scholar]
- Holyoak T, Zhang B, Deng J, Tang Q, Prasannan CB, Fenton AW. Energetic coupling between an oxidizable cysteine and the phosphorylatable N-terminus of human liver pyruvate kinase. Biochemistry. 2013;52:466–76. doi: 10.1021/bi301341r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard DR, Cardenas JM. Kinetic properties of pyruvate kinase hybrids formed with native type L and inactivated type M subunits. J Biol Chem. 1975;250:4931–6. [PubMed] [Google Scholar]
- Ishwar A, Tang Q, Fenton AW. Distinguishing the interactions in the fructose 1,6-bisphosphate binding site of human liver pyruvate kinase that contribute to allostery. Biochemistry. 2015;54:1516–24. doi: 10.1021/bi501426w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen TM, Benning MM, Rayment I, Reed GH. Structure of the bis(Mg2+)-ATP-oxalate complex of the rabbit muscle pyruvate kinase at 2.1 A resolution: ATP binding over a barrel. Biochemistry. 1998;37:6247–55. doi: 10.1021/bi980243s. [DOI] [PubMed] [Google Scholar]
- Larsen TM, Benning MM, Wesenberg GE, Rayment I, Reed GH. Ligand-induced domain movement in pyruvate kinase: structure of the enzyme from rabbit muscle with Mg2+, K+, and L-phospholactate at 2.7 A resolution. Arch Biochem Biophys. 1997;345:199–206. doi: 10.1006/abbi.1997.0257. [DOI] [PubMed] [Google Scholar]
- Larsen TM, Laughlin LT, Holden HM, Rayment I, Reed GH. Structure of rabbit muscle pyruvate kinase complexed with Mn2+, K+, and pyruvate. Biochemistry. 1994;33:6301–9. doi: 10.1021/bi00186a033. [DOI] [PubMed] [Google Scholar]
- Lee JC. Modulation of allostery of pyruvate kinase by shifting of an ensemble of microstates. Acta Biochim Biophys Sin (Shanghai) 2008;40:663–9. doi: 10.1111/j.1745-7270.2008.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Herman P. Structural and functional energetic linkages in allosteric regulation of muscle pyruvate kinase. Methods Enzymol. 2011;488:185–217. doi: 10.1016/B978-0-12-381268-1.00008-2. [DOI] [PubMed] [Google Scholar]
- Morgan HP, O'Reilly FJ, Wear MA, O'Neill JR, Fothergill-Gilmore LA, Hupp T, Walkinshaw MD. M2 pyruvate kinase provides a mechanism for nutrient sensing and regulation of cell proliferation. Proc Natl Acad Sci U S A. 2013;110:5881–6. doi: 10.1073/pnas.1217157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberfelder RW, Barisas BG, Lee JC. Thermodynamic linkages in rabbit muscle pyruvate kinase: analysis of experimental data by a two-state model. Biochemistry. 1984a;23:3822–6. doi: 10.1021/bi00312a005. [DOI] [PubMed] [Google Scholar]
- Oberfelder RW, Consler TG, Lee JC. Measurement of changes of hydrodynamic properties by sedimentation. Methods Enzymol. 1985;117:27–40. doi: 10.1016/s0076-6879(85)17004-7. [DOI] [PubMed] [Google Scholar]
- Oberfelder RW, Lee JC. Measurement of ligand-protein interaction by electrophoretic and spectroscopic techniques. Methods Enzymol. 1985;117:381–99. doi: 10.1016/s0076-6879(85)17023-0. [DOI] [PubMed] [Google Scholar]
- Oberfelder RW, Lee LL, Lee JC. Thermodynamic linkages in rabbit muscle pyruvate kinase: kinetic, equilibrium, and structural studies. Biochemistry. 1984b;23:3813–21. doi: 10.1021/bi00312a004. [DOI] [PubMed] [Google Scholar]
- Prasannan CB, Artigues A, Fenton AW. Monitoring allostery in D2O: a necessary control in studies using hydrogen/deuterium exchange to characterize allosteric regulation. Anal Bioanal Chem. 2011;401:1083–6. doi: 10.1007/s00216-011-5133-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasannan CB, Tang Q, Fenton AW. Allosteric Regulation of Human Liver Pyruvate Kinase by Peptides that Mimic the Phosphorylated/Dephosphorylated N-Terminus. Methods Mol Biol. 2012;796:335–49. doi: 10.1007/978-1-61779-334-9_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart GD. The determination of thermodynamic allosteric parameters of an enzyme undergoing steady-state turnover. Arch Biochem Biophys. 1983;224:389–401. doi: 10.1016/0003-9861(83)90225-4. [DOI] [PubMed] [Google Scholar]
- Reinhart GD. Quantitative analysis and interpretation of allosteric behavior. Methods Enzymol. 2004;380:187–203. doi: 10.1016/S0076-6879(04)80009-0. [DOI] [PubMed] [Google Scholar]
- Valentini G, Chiarelli LR, Fortin R, Dolzan M, Galizzi A, Abraham DJ, Wang C, Bianchi P, Zanella A, Mattevi A. Structure and function of human erythrocyte pyruvate kinase. Molecular basis of nonspherocytic hemolytic anemia. J Biol Chem. 2002;277:23807–14. doi: 10.1074/jbc.M202107200. [DOI] [PubMed] [Google Scholar]
- Weber G. Ligand binding and internal equilibria in proteins. Biochemistry. 1972;11:864–78. doi: 10.1021/bi00755a028. [DOI] [PubMed] [Google Scholar]
- Williams R, Holyoak T, McDonald G, Gui C, Fenton AW. Differentiating a Ligand's Chemical Requirements for Allosteric Interactions from Those for Protein Binding. Phenylalanine Inhibition of Pyruvate Kinase. Biochemistry. 2006;45:5421–9. doi: 10.1021/bi0524262. [DOI] [PubMed] [Google Scholar]
- Wooll JO, Friesen RH, White MA, Watowich SJ, Fox RO, Lee JC, Czerwinski EW. Structural and functional linkages between subunit interfaces in mammalian pyruvate kinase. J Mol Biol. 2001;312:525–40. doi: 10.1006/jmbi.2001.4978. [DOI] [PubMed] [Google Scholar]
- Yu S, Lee LL, Lee JC. Effects of metabolites on the structural dynamics of rabbit muscle pyruvate kinase. Biophys Chem. 2003;103:1–11. doi: 10.1016/s0301-4622(02)00146-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.