

Abstract
We present a novel ensemble-based computational framework, EnsembleExpr, that achieved the best performance in the Fourth Critical Assessment of Genome Interpretation (CAGI4) “eQTL-causal SNPs” challenge for identifying eQTLs and prioritizing their gene expression effects. Expression quantitative trait loci (eQTLs) are genome sequence variants that result in gene expression changes and thus are prime suspects in the search for contributions to the causality of complex traits. When EnsembleExpr is trained on data from massively parallel reporter assays (MPRA) it accurately predicts reporter expression levels from unseen regulatory sequences and identifies sequence variants that exhibit significant changes in reporter expression. Compared with other state-of-the-art methods, EnsembleExpr achieved competitive performance when applied on eQTL datasets determined by other protocols. We envision EnsembleExpr to be a resource to help interpret non-coding regulatory variants and prioritize disease-associated mutations for downstream validation.
1 Introduction
Genome-wide association studies (GWAS) have identified thousands of variants relevant to complex traits or diseases (Hindorff et al., 2009; Manolio, 2010; McCarthy et al., 2008; Stranger et al., 2011). However, as most of these variants reside in non-coding regions of the genome (Frazer et al., 2009; Hindorff et al., 2009), distinguishing the causal variants from variants simply in strong linkage disequilibrium (LD) remains challenging. Expression quantitative trait loci (eQTL) analysis has been widely used to assist in fine-mapping the causal mutations and can provide immediate insight into their biological basis (Cookson et al., 2009). Like GWAS, the statistical power of eQTL analysis is constrained by SNP linkage in the human genome and the statistical burden from the large number of variant-gene pairs to investigate that requires multiple hypothesis correction.
Massively parallel reporter assays (MPRA) are an efficient way to systematically dissect transcriptional regulatory elements (Melnikov et al., 2012) in a manner that approximates their native context behavior. In MPRA, synthesized DNA elements and corresponding sequence tags are cloned into plasmid reporter constructs, transferred into cells, sorted for expression, and assayed by high-throughput sequencing. Tewhey et al. improved the efficiency and reproducibility of MPRA (Tewhey et al., 2016) to interrogate the expression level of reference and alternate alleles of 9,116 variants linked to 3,157 eQTLs. They discovered hundreds of variants with allele-specific expression, which is defined as significantly different expression between two alleles tested in the same genomic context. In the Fourth Critical Assessment of Genome Interpretation (CAGI4), this dataset was used as the training and test sets in the “eQTL-causal SNPs” challenge to identify the best computational approaches to predict reporter expression level from DNA sequence and to classify which sequence variants will lead to allele-specific expression.
We present a computational framework, EnsembleExpr, that prioritizes genetic variants that modulate gene expression. EnsembleExpr outperformed competing methods in both parts of the CAGI4 “eQTL-causal SNPs” challenge in all of the evaluation metrics used. As an ensemble model, EnsembleExpr achieves performance superior to any single component by integrating complementary features with different properties and from different sources. Although trained on MPRA datasets, EnsembleExpr produces competitive performance in prioritizing eQTLs determined by other protocols, demonstrating its capacity to serve as a general eQTL-prioritization framework. We also demonstrate how a sufficient range of sequence-based functional element annotations is crucial to achieving accurate prediction of gene expression levels.
2 Background
2.1 Datasets in CAGI4 eQTL challenge
Tewhey et al. (Tewhey et al., 2016) identified all of the variants (range = 1 to 205, mean = 2.87, median = 1) in perfect LD with 3,157 eQTLs drawn from the Geuvadis RNA-seq dataset of lymphoblastoid cell lines (LCLs) from individuals of European ancestry (Consortium et al., 2012; Lappalainen et al., 2013). For each variant, a 150-bp flanking sequence of each of the two alleles was synthesized with the corresponding allele centered at the middle of the synthesized oligonucleotide. With these sequences as the library, MPRA experiments were carried out in two lymphoblastoid cell lines.
The resulting MPRA data were split into three groups of similar sizes: one for training and two for testing. For each variant, only its genomic location and the sequences of its alleles were provided.
The training set consists of 3,044 variants, and each variant is described by its normalized plasmid count, RNA counts, log2 fold expression level (“Log2FC”), expression p-value, multiple-testing corrected p-value, and whether the expression for either of the two alleles is significantly high (Regulatory Hit or “RegHit”). For each variant, the dataset also includes the log2 ratio of the alternative/reference allele of observed RNA expression (“LogSkew”), LogSkew p-value, LogSkew FDR and whether the change in expression is significant enough to be labeled an expression-modulating variant or “emVar”.
The first test set consists of 3,006 variants, and for each variant CAGI4 participants were required to submit a prediction of expression fold change (“Log2FC”) and whether the variant is significant (“RegHit”).
The second test set consists of 3,066 variants, 401 of which have at least one allele with strong expression (“RegHit”). For these 401 variants, the participants were asked to predict allelic change of expression (“LogSkew”) and whether it is significant (“emVar”).
2.2 Tasks in CAGI4 eQTL challenge
Expression Prediction
In this task, the participants needed to submit predictions and confidence estimates for the expression level (“Log2FC”, real value) and whether the expression is significant (“RegHit”, binary label) for the first test set.
Allele-specific Expression Prediction
In this task, the participants needed to submit predictions and confidence estimates for the change of expression between two alleles of a variant (“LogSkew”, real value) and whether the change is significant (“emVar”, binary label) for the second test set.
3 Methods
3.1 Features
Sequence-based features were generated for the regulatory regions in the CAGI4 challenge (Figure 1A). First, 150-bp model input sequences were obtained for both studied alleles as described in the challenge input files. Then we applied several computational approaches to analyze this set of sequences, including Kmer-Set Motif (KSM, in submission), DeepSEA (Zhou and Troyanskaya, 2015), DeepBind (Alipanahi et al., 2015) and ChromHMM (Ernst and Kellis, 2012) to derive sets of functional features that we hoped would help us predict expression levels.
Figure 1.
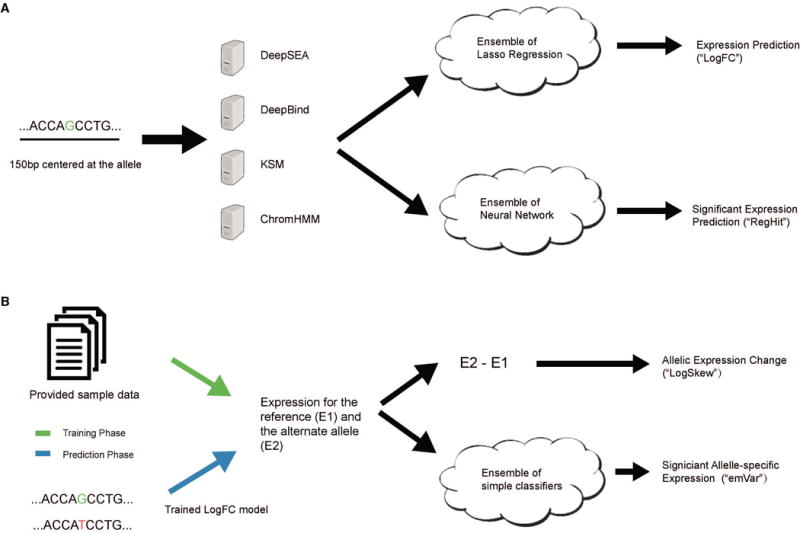
The schematic of EnsembleExpr. (A) The 150-bp sequence centered at the queried allele is taken as input to four computational models to generate functional features that are used by two ensemble models to make expression predictions (“log2FC”) and significance estimates (“RegHit”). (B) During training the provided expression levels of the two alleles of each variant are used to train an ensemble model of significant allele-specific expression (ASE). During testing we first apply the trained expression model in (A) to generate expression predictions, which are then given to the significant ASE model to make predictions (“emVar”). The difference of the predicted expression levels is directly output as the prediction for allelic expression change (“LogSkew”).
We used the DeepSEA probabilistic model (v0.94, downloaded from http://deepsea.princeton.edu/help/) to predict 919 different measurements, including DNase-seq based chromatin accessibility, transcription factor ChIP-seq, and histone mark ChIP-seq experiments, for each of the 150-bp input sequences. Each 150bp input sequence was padded with 425 unknown nucleotides (“N”) to match DeepSEA’s input format. Similarly, we applied the DeepBind model to the same sequences and generated allele-specific predictions for the binding affinities of 538 distinct transcription factors. In addition, a KSM model trained on 57 ENCODE ChIP-seq experiments for a lymphoblastoid cell line (GM12878) was used to produce predictions for transcription factor binding affinities. Chromatin state annotations from the NIH Roadmap Epigenomics (Kundaje et al., 2015) project were also compiled for all regions and used as one-hot encoded binary features.
3.2 Computational model
Expression Prediction Task
Armed with this set of potentially predictive features for expression levels, many of which are sequence-specific, we used an ensemble of regularized regression and classification models to predict expression values for both alleles and regulatory hit status based on the provided training data (Figure 1A). We trained multiple LASSO regression models to predict the normalized log expression levels for each allele using the DeepSEA features alone, the DeepBind features alone, DeepSEA and KSM features combined, and DeepSEA along with KSM and chromatin state annotations. All learning algorithms were tuned by cross-validation within the training set, and the various feature sets were chosen using a heuristic manual analysis. We averaged the LASSO model predictions to produce the final predictions and took the standard deviation of their separate predictions as confidence estimates. For the binary prediction task (“RegHit”), we trained a one-layer neural network with 400 neurons on the same four sets of features described previously and aggregated their predictions in the same manner.
Allele-specific Expression Prediction Task
For allelic expression change (“LogSkew”) prediction, given that “LogSkew” is defined and calculated as the expression difference between the two alleles, we decided to directly utilize the “Log2FC” expression model we trained in the previous section instead of training a new model. We applied our trained “Log2FC” model to generate expression predictions for each allele in the held-out test set to submit. Then for each variant, we took the difference in predicted expression levels between the reference and the alternate alleles as our “LogSkew” prediction (Figure 1B).
For predicting “emVar” labels (allele-specific expression status), we trained on the actual expression levels for the reference and alternate alleles provided in the sample data. An ensemble of binary classification models was considered, with all regularization parameters tuned by cross-validation (Figure 1B). Models used in the final ensemble included linear regularized logistic regression, kernel regularized logistic regression, k-nearest neighbors, support vector machine (SVM) with linear kernel and SVM with radial basis function kernel. The predictions of all models were combined to form the final probability estimate, along with a measure of confidence in the prediction. After training, we first ran our prediction module in the previous task (Expression Prediction) to generate expression predictions for each allele of the held-out test set. Then we applied the expression-to-emVar model trained here to make predictions of significant allele-specific expression (“emVar” hits).
4 Results
4.1 EnsembleExpr outperforms competing approaches in CAGI eQTL challenge
We assessed the predictions from EnsembleExpr and other competing methods in the challenge. For predicting log (normalized) expression levels (“Log2FC”) and expression level differences between two alleles (“LogSkew”), both of which are regression tasks, we used Spearman’s rank correlation coefficient which is non-parametric and stable with value scaling. For predicting significant expression (“RegHit”) and significant allele-specific expression (“emVar”), both of which are binary classification tasks, we chose two benchmarks: the receiver operating characteristic (ROC) and the precision recall curve (PRC). ROC evaluates how the true positive rate changes with the false positive rate, where a random prediction would be along the diagonal with an area under curve (AUC) of 0.5 and a better model would have larger AUC. PRC shows how the precision changes with increasing recall (true positives), where the desired model should maintain high precision for large recall.
EnsembleExpr outperformed all the competing methods in both the expression prediction task and the allele-specific expression prediction task. In the first part of the challenge, expression predictions from EnsembleExpr correlate the best with the experimental observations (Table 1, a Spearman correlation of 0.485 for the reference allele and 0.470 for the alternate allele). In predicting significant expression (“RegHit”), EnsembleExpr is the only model with an auROC > 0.8 and an auPRC > 0.5 (Table 1, Figure 2A).
Table 1.
Performance comparison for Expression Prediction task (sorted by RegHit auROC).
| Participant (Lab-Submission) | Ref. Spearman corr. | Alt. Spearman corr. | RegHit auPRC | RegHit auROC |
|---|---|---|---|---|
| 4 (EnsembleExpr) | 0.484936 | 0.470176 | 0.528288 | 0.807690 |
| 6-1 | 0.290971 | 0.399997 | 0.461099 | 0.786722 |
| 2-2 | 0.278613 | 0.262536 | 0.402448 | 0.777035 |
| 2-4 | 0.260072 | 0.245915 | 0.437067 | 0.774688 |
| 2-5 | 0.261064 | 0.245595 | 0.432639 | 0.772747 |
| 6-2 | 0.290971 | 0.399997 | 0.433406 | 0.771472 |
| 6-3 | 0.433043 | 0.399116 | 0.426697 | 0.767268 |
| 2-6 | 0.199247 | 0.171989 | 0.353660 | 0.728643 |
| 2-1 | 0.173587 | 0.169082 | 0.385369 | 0.723336 |
| 1-5 | 0.295519 | 0.272904 | 0.304145 | 0.719242 |
| 1-1 | 0.251873 | 0.248376 | 0.329400 | 0.716045 |
| 1-3 | 0.251027 | 0.248319 | 0.328427 | 0.713983 |
| 1-6 | 0.318642 | 0.300630 | 0.312914 | 0.713570 |
| 1-4 | 0.254598 | 0.236243 | 0.311588 | 0.709843 |
| 5-1 | 0.252023 | 0.223952 | 0.369462 | 0.693357 |
| 1-2 | 0.174655 | 0.168176 | 0.293471 | 0.683512 |
| 7 | 0.208036 | 0.194298 | 0.437487 | 0.670681 |
| 3 | 0.304933 | 0.236940 | 0.242830 | 0.652059 |
| 5-2 | 0.352951 | 0.353493 | 0.189516 | 0.578095 |
| 2-3 | 0.000766 | −0.002387 | 0.126747 | 0.513558 |
Figure 2.
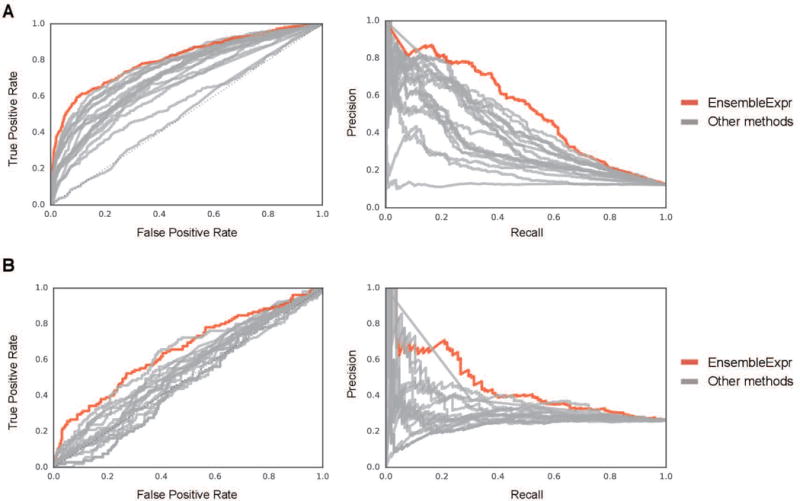
EnsembleExpr outperforms all CAGI4 competing methods. (A) The area under ROC (auROC, left) and area under precision-recall curve (auPRC, right) for EnsembleExpr (red) and other methods (grey) in predicting significant expression. (B) The auROC (left) and auPRC (right) for EnsembleExpr (red) and other methods (grey) in predicting significant allele-specific expression.
In the second part of the challenge, EnsembleExpr accurately predicted the LogSkew change in expression with a Spearman correlation better than other submissions, many of which had correlations close to zero (Table 2). Prioritizing variants that give rise to significant change of expression (“emVar”) was the hardest among all four tasks. For emVar EnsembleExpr also demonstrated better performance than other methods, yielding an auROC of 0.655 and an auPRC of 0.452 (Table 2, Figure 2B).
Table 2.
Performance comparision for Allele-specific Expression Prediction task (sorted by emVar auROC)
| Participant (Lab-Submission) |
LogSkew Spearman corr. | emVar auPRC | emVar auROC |
|---|---|---|---|
| 4 (EnsembleExpr) | 0.449760 | 0.452561 | 0.655261 |
| 5-1(deltaSVM) | 0.333893 | 0.409730 | 0.626850 |
| DeepSEA | Not Applicable | 0.389 | 0.589 |
| 5-2(deltaSVM) | 0.342004 | 0.369083 | 0.577220 |
| 7 | 0.007343 | 0.431639 | 0.562854 |
| 6-1 | 0.217845 | 0.345064 | 0.561953 |
| 6-2 | 0.190123 | 0.354726 | 0.561776 |
| 1-3 | NaN* | 0.311243 | 0.556499 |
| 1-1 | NaN* | 0.305258 | 0.550820 |
| 1-2 | 0.030243 | 0.295886 | 0.550048 |
| 2-3 | −0.015476 | 0.303051 | 0.545206 |
| 1-5 | 0.056143 | 0.284863 | 0.541216 |
| 1-4 | 0.079049 | 0.293321 | 0.530856 |
| 3 | 0.030049 | 0.284356 | 0.511181 |
| 1-6 | 0.105376 | 0.286584 | 0.510103 |
| 2-2 | −0.007377 | 0.249473 | 0.479746 |
| 2-1 | −0.024347 | 0.234723 | 0.477301 |
| 2-5 | −0.023092 | 0.233144 | 0.472651 |
| 2-6 | −0.023092 | 0.233144 | 0.472651 |
| 2-4 | −0.023092 | 0.233144 | 0.472651 |
every variant was assigned the same score, leading to incalculable Spearman correlations
EnsembleExpr also outperforms state-of-the-art methods in the eQTL-prioritization literature. Zhou and Troyanskaya, 2015 reported that an L2-regularized logistic regression model trained on DeepSEA-derived features and evolutionary conservation scores achieved predictive performance that surpasses existing approaches for eQTLs. We trained this DeepSEA plus conservation model on the DeepSEA-derived features and conservation scores for the variants in the CAGI training set, and predicted “emVar” labels of variants in the test set. While ranking third among all the submissions, the DeepSEA plus conservation model achieved a performance inferior to that of EnsembleExpr (auROC=0.589, auPRC=0.389, Table 2). We also note that submissions 5-1 and 5-2 solely use deltaSVM (Lee et al. 2015), the state-of-the-art method in predicting dsQTLs (variants modulating DNase-hypersensitivity). deltaSVM achieved a performance better than DeepSEA, but inferior to EnsembleExpr (Table 2). This comparison shows that EnsembleExpr not only excels among all the submitted methods, but outperforms the state-of-the-art in the literature.
Thus EnsembleExpr modeled the diversity of expression well and demonstrated unmatched capacity as a predictive model for eQTL prioritization. More importantly, the consistently high performance of EnsembleExpr across different tasks and evaluation metrics proves the robustness of the predictions.
4.2 Ensemble demonstrated competitive performance on public eQTL dataset
We next evaluated the ability of EnsembleExpr to predict eQTLs determined by protocols other than MPRA to test its generality beyond the CAGI4 competition. Recent work (Zhou and Troyanskaya, 2015) collected 78,613 non-coding eQTLs from the GRASP (Genome-Wide Repository of Associations between SNPs and Phenotypes) databases (Leslie et al 2014) and constructed several size-matched negative sets sampled from different subsets of 1000 Genomes Project non-coding variants. We classified these held-out positive and negative eQTL examples using our MPRA trained EnsembleExpr model, DeepSEA, deltaSVM, GWAVA (Ritchie et al 2014), CADD (Kircher et al 2014) and FunSeq2 (Fu et al 2014). For deltaSVM, we used the same model in submission 5-1 (using parameters trained on GM12878 DNase-hypersensitive region), given its good performance in CAGI4. For DeepSEA, we used the same model trained on CAGI4 data as discussed in the previous section. To make the comparison fair, we did not directly use DeepSEA’s performance reported in Zhou et al (Zhou and Troyanskaya, 2015), where the classifier was trained on the same eQTL data and evaluated with a cross-validation scheme. For CADD, we used the provided web server (http://cadd.gs.washington.edu/, v1.0). We downloaded the GWAVA software from http://www.sanger.ac.uk/resources/software/gwava/ and downloaded the FunSeq2 software from http://info.gersteinlab.org/Funseq2. To avoid test set contamination, we excluded any variants that were included in CAGI4.
We found that EnsembleExpr outperformed all the competing methods in classifying eQTLs from the three negative sets (Figure 3), although the AUROC for all methods was low. This demonstrates that although trained on MPRA datasets, EnsembleExpr can serve as a general eQTL-prioritization framework that is applicable on other datasets. We attribute the generalizability to EnsembleExpr’s use of features that were predicted for different cell lines. To examine whether the unsatisfactory performance of detalSVM primarily resulted from cell line mismatch, we further retained only the eQTLs discovered from lymphoblastoid cell lines, and classified this set of eQTLs from the negative sets using the cell-line-matched deltaSVM model. However, we observed no notable improvement in performance (Supplementary Figure 2). On the other hand, we do note that the parameters of deltaSVM model we used, which performed well on CAGI dataset, were originally optimized for predicting dsQTLs instead of eQTLs.
Figure 3.
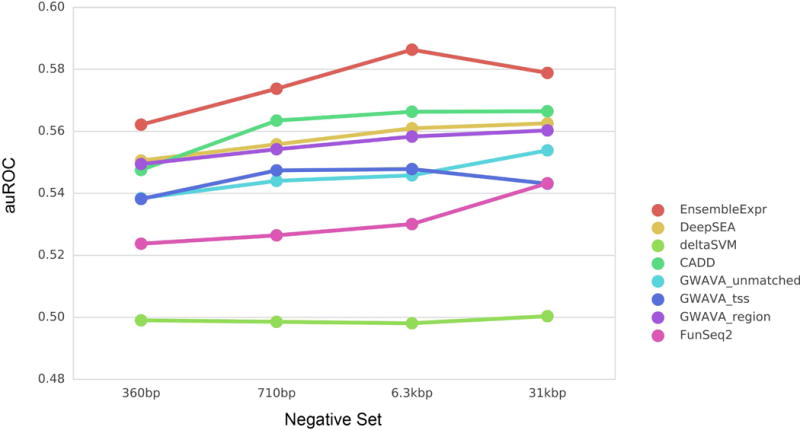
EnsembleExpr achieved competitive performance compared with published methods in predicting GRASP eQTLs from various size-matched negative sets sampled from 1000 Genomes Project non-coding variants. The x-axis denotes the different negative sets by the average distance to the paired eQTL.
4.3 Components of the ensemble provide complementary functional information
We benchmarked EnsembleExpr and each of the single models included in the ensemble to understand the major sources of improvement. Through ten-fold cross-validation, for each model we evaluated the median R2 when predicting log expression level (“Log2FC”) and the median auROC and auPRC when predicting significant expression (“RegHit”). We observed that with the DeepSEA-predicted functional features, including TF binding, histone marks and DNase hypersensitivity, we could already reach decent accuracy in both tasks (Table 3). However, models with only TF binding-based features from either deep learning (DeepBind) or k-mer based models (KSM) are much less satisfactory. But we did observe that incorporating DeepSEA with features from KSM and ChromHMM led to better performance, suggesting that these two models provide complementary information despite the comprehensiveness of the DeepSEA output.
Table 3.
Performance of each component in the ensemble.
| Features included | Task1-a | Task1-b | |
|---|---|---|---|
| R2 | auROC | auPRC | |
| Ensemble | 0.3976 | 0.8647 | 0.5830 |
| KSM+DeepSEA* | 0.3803 | 0.8515 | 0.5622 |
| KSM+DeepSEA+ChromHMM* | 0.3769 | 0.8462 | 0.5380 |
| DeepSEA* | 0.3728 | 0.8347 | 0.5391 |
| DeepBind* | 0.2508 | 0.8209 | 0.4438 |
| KSM | 0.2393 | 0.7943 | 0.3943 |
models included in the ensemble
4.4 Accurate eQTL prioritization requires a comprehensive panel of functional features
We next sought to understand what sequence-derived functional features, among the hundreds we used, are most predictive of expression and eQTL status. Expression is regulated by sophisticated machinery where numerous regulators and epigenetic marks act in concert. To include a large enough panel of features, we investigated one of the LASSO regression models in the ensemble that was trained to predict expression (“Log2FC”) from sequence-derived prediction of DNase hypersensitivity, histone marks, transcription factor binding and chromatin state (Supplementary Table 1).
We first analyzed the sign of the coefficients in the LASSO model to understand the direction in which each feature affects the expression prediction. As expected, the model assigned large positive weights to DNase hypersensitivity, histone marks known to be associated with promoters (such as H3K4me3) and predicted functional elements (such as H3K27ac) (Creyghton et al, 2010), and transcription initiators (such as IRF1) (Supplementary Table 2). The model also gave large negative weights to chromatin regulators known for repressive effects on transcription (such as EZH2) and histone marks predictive for gene bodies (such as H3K36me3). These observations persisted even when we retrained the model 10 times, and calculated the mean and 95% confidence interval of the coefficients (Supplementary Table 2).
We next analyzed the predictive importance of the features. By design, LASSO models impose sparsity and force the coefficients for non-important features to zero. However, the limitation of such L1-regularization based models is that when faced with a group of highly correlated features, as in our experiments, the model may only pick one feature at random from a correlated set. Thus to fully understand which features are important for expression prediction, instead of directly looking at the coefficients in the LASSO model, we retrained a Randomized Lasso model that performs “stability selection” (Meinshausen and Bühlmann, 2010) by resampling the training data and computing a LASSO model on each resampling. The more often a feature gets selected, the more important it is for the performance of the model. We observed a bi-modal distribution of feature importance (Supplementary Figure 1). Most of the 994 features are considered not very important, while a group of 60 features demonstrate great importance. These top 60 features are highly diverse, including histone marks predictive for enhancer/promoter/repressive regions, important transcription regulators, and chromatin states predictions (Supplementary Table 3). This diversity of useful features suggests that a comprehensive functional annotation of the sequence, rather than one type or two, is essential for accurate expression prediction and eQTL prioritization. We also observed that while many of the important features are predicted for the same type of cell line as the one the MPRA experiment was performed on (lymphoblastoid cell lines), many features predicted for other cell lines, such as K562 and H1-hESC, also proved to be highly informative.
5 Discussion
We have described an ensemble-based framework, EnsembleExpr, that achieved the best performance in both parts of the “eQTL-causal SNPs” challenge in the Fourth Critical Assessment of Genome Interpretation (CAGI4). We also demonstrated that although trained on MPRA datasets, EnsembleExpr can also produce competitive predictions on eQTL datasets determined by other protocols.
We found that each component model of EnsembleExpr provides useful yet complementary information, and when combined they lead to a successful ensemble with performance surpassing any single model that we examined. Through a systematic analysis of feature importance, we demonstrated that features important for accurate prediction by EnsembleExpr are highly diverse, ranging from chromatin state and histone marks to transcription factor binding. Most EnsembleExpr features, except the chromatin state labels from ChromHMM, are obtained from sequence-based computational models that can provide predictions for each allele. This enables precise characterization of how a single base change affects expression levels, which we consider crucial for any model aiming to interpret sequence variants.
With the capacity to accurately predict sequence variants with significant allele-specific expression determined by different protocols, we expect EnsembleExpr will serve as a resource to assist in pinpointing causal mutations for complex traits and diseases, and to help in understand the pathogenic pathways. The power of EnsembleExpr can likely be further improved, as more and larger-scale MPRA data become available. EnsembleExpr is openly available at http://ensembleexpr.csail.mit.edu for researchers to utilize freely for downstream analysis.
Supplementary Material
Acknowledgments
We are grateful to the organizers of the Fourth Critical Assessment of Genome Interpretation for coordinating and hosting the open challenge. We appreciate Ryan Tewhey and Pardis Sabeti from the Broad Institute for providing the MPRA data in the “eQTL-causal SNPs” challenge. We thank Kevin Tian for the help in feature preparation. We thank other members of the Gifford Lab for constructive discussions and feedback. We acknowledge funding from the National Institutes of Health under grants R01 HG008363 and U01 HG007037 to D.K.G. and an equipment grant from NVIDIA. The CAGI experiment coordination is supported by the NIH grant U41 HG007346 and the CAGI conference by the NIH grant R13 HG006650.
Footnotes
Disclosure statement: The authors declare no conflict of interest.
References
- Alipanahi B, Delong A, Weirauch MT, Frey BJ. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nature biotechnology. 2015;33(8):831–838. doi: 10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- Consortium G. P. et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nature Reviews Genetics. 2009;10(3):184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone h3k27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nature Methods. 2012;9(3):215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Murray SS, Schork NJ, Topol EJ. Human genetic variation and its contribution to complex traits. Nature reviews. Genetics. 2009;10(4):241–51. doi: 10.1038/nrg2554. [DOI] [PubMed] [Google Scholar]
- Fu Y, Liu Z, Lou S, Bedford J, Mu XJ, Yip KY, Khurana E, Gerstein M. Funseq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome biology. 2014;15(10):1. doi: 10.1186/s13059-014-0480-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(23):9362–7. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics. 2014;46(3):310. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedländer MR, AC’t Hoen P, Monlong J, Rivas MA, Gonzàlez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Gorkin DU, Baker M, Strober BJ, Asoni AL, McCallion AS, Beer MA. A method to predict the impact of regulatory variants from dna sequence. Nature genetics. 2015;47(8):955–961. doi: 10.1038/ng.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie R, O’Donnell CJ, Johnson AD. Grasp: analysis of genotype–phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics. 2014;30(12):i185–i194. doi: 10.1093/bioinformatics/btu273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA. Genomewide association studies and assessment of the risk of disease. The New England journal of medicine. 2010 doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JPA, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nature reviews. Genetics. 2008;9(5):356–69. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- Meinshausen N, Bühlmann P. Stability selection. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 2010;72(4):417–473. [Google Scholar]
- Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG, Jr, Kinney JB, et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nature biotechnology. 2012;30(3):271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie GR, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nature methods. 2014;11(3):294–296. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Stahl EA, Raj T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011;187(2):367–83. doi: 10.1534/genetics.110.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, et al. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell. 2016;165(6):1519–1529. doi: 10.1016/j.cell.2016.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nature methods. 2015;12(10):931–9. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.