

Abstract
Genetic data consists of a wide range of marker types, including common, low frequency, and rare variants. Multiple genetic markers and their interactions play central roles in the heritability of complex disease. In this study, we propose an algorithm that uses a stratified variable selection design by genetic architectures and interaction effects, achieved by a data-set adaptive W-test. The polygenic sets in all strata were integrated to form a classification rule. The algorithm was applied to the Critical Assessment of Genome Interpretation 4 bipolar challenge sequencing data. The prediction accuracy was 60% using genetic markers on an independent test set. We found that epistasis among common genetic variants contributed most substantially to prediction precision. However, the sample size was not large enough to draw conclusions for the lack of predictability of low frequency variants and their epistasis.
Keywords: Classification of complex disorder, bipolar, polygenic risk stratification, W-test, epistasis, interaction effect, mutation, disease prediction
Introduction
The risk of psychiatric disorders is largely attributed to genetic factors and their interactions with the environment (Smoller and Finn 2003). The estimated heritability of bipolar disorder (BPD) is as high as 90%; however, few large main effect genes have been identified (Craddock and Sklar 2013). In recent years, exome sequencing studies identified a number of rare or de novo variants associated with bipolar disease risk (Goes, Pirooznia et al. 2016, Kataoka, Matoba et al. 2016). While these finding are very interesting and important, it is unclear how much they contribute to the prediction of complex disorders. The prediction challenge also lies in the diverse genetic marker compositions including common variants with minor allele frequencies (MAFs) greater than 5%, low frequency variants (1% < MAF < 5%), and rare variants (MAF < 1%). Disease classification models typically do not distinguish these different variant types and use machine learning approaches to conduct variable selection and phenotype prediction (Touw, Bayjanov et al. 2013). The distinct nature of these genetic markers requires specialized statistical models to evaluate their risk effect. Therefore, in this study, we developed a stratified polygenic risk model: from simple to complex, the model is gradually built based on the effect of common and low-frequency variants and their respective epistasis. When the sample size is sufficiently large, the model may include rare variants. Variable selection is conducted using the W-test, which estimates null probability distributions of each stratum. The polygenic risk sets from all strata are finally integrated to form a unified classification rule through boosting. The method was applied to the Critical Assessment of Genome Interpretation 4 (CAGI 4) bipolar challenge, which contains exome sequencing data for 500 subjects with the objective of predicting an independent test set. Context is challenging for complex disease predictions, as rare variation association tests require a large sample size to have enough power; furthermore, rare mutations may not reappear in another sampling group of modest size. Therefore, we focused on common to low-frequency variables and their epistasis effect in the challenge. Using the proposed model, the prediction accuracy for the independent test set was 60%, mainly because of common variant polygenic epistasis.
Method
Data set and quality control
The data set included whole exome sequencing data consisting of 500 samples and 501,253 single-nucleotide polymorphisms (SNPs), sequenced using the Illumina HiSeq 2000 platform (San Diego, CA, USA). Variants with more than 5% missing or Hardy-Weinberg-Equilibrium test p-value < 10−6 were filtered first. Principle component analysis based on the remaining SNPs identified a cluster of 3 outlier subjects, which were excluded from further analysis (Price, Patterson et al. 2006). The remaining data consisted of 226 bipolar disorder subjects and 221 healthy controls. From the total of 497 subjects, 50 individuals were randomly drawn as the independent test set, while the remaining 447 subjects were used as the training set. Missing SNP data were imputed using SHAPEIT2 (Delaneau, Zagury et al. 2013). SNPs with MAF above 1% were analyzed. The total number of low to common frequency SNPs evaluated was 75,288, among which 21,339 SNPs had MAFs between 1% and 5% and 53,949 SNPs had MAFs > 5%. Feature selection was performed without using SNP location information. The final selected SNPs were mapped to genes using the UCSC Genome Browser on Human within a 10 kb genome distance (Kent, Sugnet et al. 2002).
Stratified variable selection via W-test
We used the W-test to select important variables and construct epistasis sets (Wang, Sun et al. 2016). The W-test measures the distributional differences between cases and controls in a contingency table and follows a chi-square distribution with data-set adaptive degrees of freedom. This feature makes the test robust to small sample sizes and complex genetic architectures. The test statistic takes the following form:
| Equation 1 |
where k is the number of categories formed by a marker set. For a single SNP, k = 3 and for an SNP-pair, k = 9. p̂1i is the proportion of subjects in cell-i in cases, and p̂0i is the proportion of subjects in cell-i among total controls. SEi is the standard error of the log odds ratio of cell-i, in which n1i and n0i are the number of cases and controls in the ith cell; N1 and N0 are the total number of cases and controls, respectively. The statistic follows a chi-squared distribution of f degrees of freedom. The scalar h and degrees of freedom f were obtained by estimating the covariance matrix from bootstrapped samples under the null hypothesis. The W-test was performed using the wtest package in R. Genetic risk variables were selected in a stratified manner by evaluating the: 1. main effect of common variants; 2. epistasis effect among common variants; 3. main effects of low-frequency variants; and 4. epistasis effect among low-frequency variants. The W-test adaptively estimates the probability distribution according to the genetic architecture of each stratum and provides an accurate evaluation of association effects. The procedure is illustrated in Diagram 1.
Diagram 1.

Stratified Polygenic Risk Prediction
Classification algorithm
The top genetic markers were candidates for the adaptive-boosting (ada-boost) algorithm (Schapire 1999). Each SNP or SNP-pair forms a classifier through logistic regression. The ada-boost recursively selects the next best classifier from the remaining classifiers list, and each time reweights all samples based on the prediction error rate in the training set, with samples that are more difficult to classify given heavier weights. The algorithm is most suitable for aggregating multiple modest effect classifiers to form a stronger rule. Before submitting the classifiers to boosting, a filtering method is applied to remove the dependency among the pairs: First, all pairwise interactions were evaluated among SNPs with main effect p-values < 0.1; second, these pairs were evaluated using the W-test and ranked by p-value in an ascending order; third, an SNP-pair will be removed if it contains an overlapping SNP in a set (Wang, Tsoi et al. 2015). This screening method was used for two reasons: (1) When an SNP has a very strong main effect, it can couple with a large number of SNPs to form significant pairs, most of which are redundant and do not help the prediction. Filtering can remove most of these main effect-driven pairs and allows new epistasis that reveals additional information for classification. (2) Filtering can reduce the correlation among classifiers and improve prediction accuracy. In the adaboost algorithm, heavier weights were assigned to rules that have predictive power for a more difficult training case. Adaboost shows better results if the predictive powers from many rules are complementary and are more effective for different subsets of cases. This logic of boosting favors the classifiers to exhibit low correlation among them that is achieved by removing overlapping pairs. The final vote is the classification rule formed by the boosting algorithm (Wang, Lo et al. 2012). Standard deviation of the vote was calculated by taking the sigmoid function on the boosting rule R: S.D = 1/(1+e−R), −∞ < R < ∞.
Results
Classification results
For common SNPs, 21 variants with main effect p-values < 1 × 10−5 were selected. The classification error rate was 31% for the training set and 48.0% for the independent test set (Supp. Figure S1). For common variants epistasis, variants with main effect p-values smaller than 0.1 were used to calculated pair-wise interactions. The number of SNPs with main effect p-values < 0.1 was 5521. The Bonferroni-corrected significance level at a nominal alpha 0.05 was 3.28 × 10−9. The top 24 pairs with p-values smaller than 1 × 10−5 were used in the classification algorithm. The final error rate for the training set was 24.1% and for the independent test set was 40.0%. No prior biological knowledge or clinical characteristics has been used, and the classification was purely based on SNP data. Figure 1 shows the adaptively improved boosting error; the algorithm updates voting rule in each iteration and decreased the overall error rates as more classifiers are added. The error of the test set decreased with fluctuation and stabilized at 40.0% for the final 6 iterations, indicating that the classifier was not over-trained in the training set (Figure 1).
Figure 1.
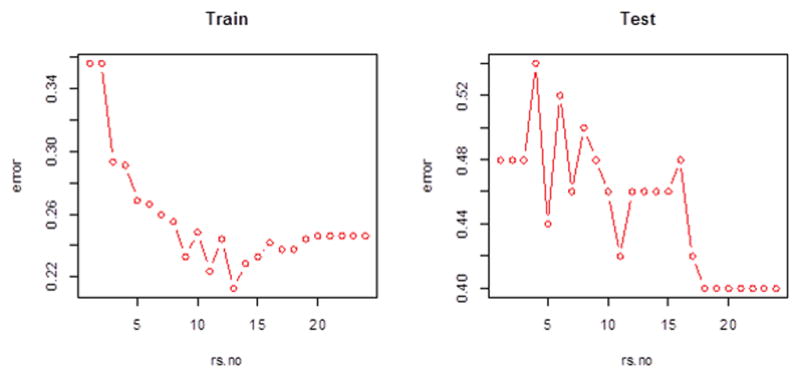
Prediction error of bipolar phenotype using common SNPs in epistasis
For low-frequency variants, the same procedure was used to select main effect SNPs and epistasis pairs. However, when these variables were added to the previous common variants sets, the test set error rates increased to 46%. This demonstrates that the low-frequency variant did not improve prediction of the disease phenotype, although the results may be limited because of the small sample size.
Top SNP-pairs are biologically relevant
The top 24 common pairs consisted of 48 unique SNPs, 45 of which were located in protein coding genes; pairs located in known psychiatric genes are listed in Table 1 (The full table can be found in Supplement Table S1). One of the most frequently appearing genes is GAS7. SNPs in this gene show strong epistasis with SDF4, TPO, PLK2, and others (Figure 2). Because none of the SNPs in GAS7 were marginally significant, the epistasis did not result from a large main effect. GAS7 is also known as MLL and plays a putative role in brain development by regulating neuronal cell morphology (Chao, Chang et al. 2005, Gotoh, Hidaka et al. 2013), and is known to be related to Alzheimer’s disease (Hidaka, Koga et al. 2012). Another significant epistasis pair was found between CPNE5-CRLF1. Both genes play roles in neuronal disorder. CPNE5 is a calcium-dependent membrane-binding protein and was reported to be associated with alcohol dependence and obesity (Wang, Zuo et al. 2015); CRLF1 encodes a protein complex that acts on cells expressing ciliary neurotrophic factor receptors and promotes the survival of neuronal cells (NCBI, Herholz, Meloni et al. 2011). SDF4 also encodes proteins containing calcium-binding motifs. The top genes identified are consistent with the findings that the etiology of bipolar disorder involves calcium-channel.
Table 1.
Top epistasis pairs with known psychiatric genes
| Num | SNP1 | SNP2 | Gene1 | Gene2 | OR1 | P-value2 |
|---|---|---|---|---|---|---|
| 1 | chr17:9816390 | chr17:9816452 | GAS7 | GAS7 | 1.5 | 6.91E-11 |
| 2 | chr19:47278859 | chr1:208197785 | SLC1A5 | PLXNA2 | 1.3 | 5.45E-09 |
| 3 | chr10:43127326 | chr3:184289152 | ZNF33B | EPHB3 | 3.4 | 1.30E-08 |
| 4 | chr17:9816671 | chr1:1163804 | GAS7 | SDF4 | 0.7 | 2.90E-08 |
| 5 | chr17:9817874 | chr5:57754005 | GAS7 | PLK2 | 0.6 | 1.36E-07 |
| 6 | chr19:18707878 | chr6:36733132 | CRLF1 | CPNE5 | 2.1 | 1.47E-07 |
| 7 | chr11:18240645 | chr20:19951534 | -- | RIN2 | 1.0 | 1.85E-07 |
| 8 | chr17:9816226 | chr2:1437163 | GAS7 | TPO | 0.7 | 1.24E-06 |
: OR are calculated assuming dominate genetic model of the two SNPs
: P-value is calculated using the W-test. Bonferroni corrected significance level at 5% alpha = 3.28×10−9
Figure 2.

Epistasis network in bipolar disorder common variants
Discussion
In this study, an algorithm for stratified variable selection by genetic architecture and classification was developed. We used main and epistasis effects in common variants and low-frequency variants to perform bipolar disorder phenotype prediction. Our results showed that the classification algorithm utilizing common variants interactions reached 60% accuracy in disease classification.
To design a risk prediction model, we first conducted stratification, followed by consolidation. Genetic marker selection was stratified by the genetic architectures of common and low frequencies, and then by main and interaction effects. The advantage of stratification is that it improves the inference of variable selection. Therefore, the statistical method that is most suitable for that stratum’s genetic architecture can be applied. For the W-test, a different set of h and f parameters for the data-adaptive probability distribution was estimated for each stratum. In the prediction step, all risk effects were combined through the boosting algorithm to produce a final vote. The stratification effect is clear: when only main effect common variants were included in the model, the independent test error rate was 48% (Supp. Table S1). By including epistasis within the common variants, the test error improved to 40%. Low-frequency variables in this dataset did not improve the prediction accuracy, likely because of the limited sample size. In a recent review, Chatterjee et al. demonstrated the necessity of understanding the relative contribution of common, low-frequency, and rare variants towards absolute risk estimation for genetic disease diagnosis (Chatterjee, Shi et al. 2016). Our findings are also consistent with the current understanding of bipolar disorder, a highly complex disease triggered by polygenic effects and the interplay of environmental factors.
In the feature selection part, the W-test was used to test the main effects and epistasis effects. The method has three distinct characteristics that differentiate it from other statistical methods: First, the test is data-set adaptive; it contains a degree of freedom parameter and scalar that are adjusted according to the data structure of each dataset, and therefore produces data-set adaptive null distributions that allows for more accurate p-value calculation. Second, the W-test is model-free; it is constructed by directly testing the distributional differences between the cases and control groups. Thus, the method is not restricted by assumptions such as linearity and may capture effects arising from different types of associations, linear or non-linear. Third, the W-test has a closed statistical form and calculates a p-value from a probability distribution. These properties make the W-test a practical and interpretable method applicable for large genetic dataset analysis.
One of the limitations of the study is the sample size, which prevented us from fully determining the effect of low-frequency and rare variants effect in disease classification. Clinical and environmental variables were also unavailable and thus could not be incorporated into the algorithm. However, using the CAGI project bipolar challenge dataset, we demonstrated the important contribution of common variants to complex disorder risk prediction, with the most substantial improvement of accuracy made by including epistasis among the SNPs. This CAGI project data set was generated from a single batch, and thus there was no systematic difference in the training and testing set. In real-life scenarios, when applying the prediction algorithm to a distant data set, the classification model should be calibrated for the test set to ensure unbiased estimation. The identified polygenic markers used in the final prediction model must also be validated in future studies.
Supplementary Material
Acknowledgments
Supporting grants: National Science Foundation of China [81473035, 31401124], Health and Medical Research Fund (Project reference no: CU-16-C11) of Food and Health Bureau of the Hong Kong Special Administrative Region Government. The CAGI experiment coordination is supported by NIH U41 HG007346 and the CAGI conference by NIH R13 HG006650.
This work is supported by the National Science Foundation of China [81473035, 31401124 to MHW].
Footnotes
Conflict of Interests
The authors declare no conflict of interests.
References
- Chao CC, Chang PY, Lu HH. Human Gas7 isoforms homologous to mouse transcripts differentially induce neurite outgrowth. Journal of neuroscience research. 2005;81(2):153–162. doi: 10.1002/jnr.20552. [DOI] [PubMed] [Google Scholar]
- Chatterjee N, Shi J, Garcia-Closas M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nature reviews Genetics. 2016;17(7):392–406. doi: 10.1038/nrg.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, Sklar P. Genetics of bipolar disorder. Lancet. 2013;381(9878):1654–1662. doi: 10.1016/S0140-6736(13)60855-7. [DOI] [PubMed] [Google Scholar]
- Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nature methods. 2013;10(1):5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- Goes FS, Pirooznia M, Parla JS, Kramer M, Ghiban E, Mavruk S, Chen YC, Monson ET, Willour VL, Karchin R, Flickinger M, Locke AE, Levy SE, Scott LJ, Boehnke M, Stahl E, Moran JL, Hultman CM, Landen M, Purcell SM, Sklar P, Zandi PP, McCombie WR, Potash JB. Exome Sequencing of Familial Bipolar Disorder. JAMA psychiatry. 2016;73(6):590–597. doi: 10.1001/jamapsychiatry.2016.0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh A, Hidaka M, Hirose K, Uchida T. Gas7b (growth arrest specific protein 7b) regulates neuronal cell morphology by enhancing microtubule and actin filament assembly. The Journal of biological chemistry. 2013;288(48):34699–34706. doi: 10.1074/jbc.M113.513119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herholz J, Meloni A, Marongiu M, Chiappe F, Deiana M, Herrero CR, Zampino G, Hamamy H, Zalloum Y, Waaler PE, Crisponi G, Crisponi L, Rutsch F. Differential secretion of the mutated protein is a major component affecting phenotypic severity in CRLF1-associated disorders. European Journal of Human Genetics. 2011;19(5):525–533. doi: 10.1038/ejhg.2010.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidaka M, Koga T, Gotoh A, Sanada M, Hirose K, Uchida T. Alzheimer’s disease-related protein hGas7b interferes with kinesin motility. Journal of biochemistry. 2012;151(6):593–598. doi: 10.1093/jb/mvs038. [DOI] [PubMed] [Google Scholar]
- Kataoka M, Matoba N, Sawada T, Kazuno AA, Ishiwata M, Fujii K, Matsuo K, Takata A, Kato T. Exome sequencing for bipolar disorder points to roles of de novo loss-of-function and protein-altering mutations. Molecular Psychiatry. 2016;21(7):885–893. doi: 10.1038/mp.2016.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Research. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI. NCBI Gene CRLF1. from http://www.ncbi.nlm.nih.gov/gene/9244.
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Schapire RE. A brief introduction to boosting. Ijcai-99: Proceedings of the Sixteenth International Joint Conference on Artificial Intelligence; 1999. pp. 1401–1406. [Google Scholar]
- Smoller JW, Finn CT. Family, twin, and adoption studies of bipolar disorder. American Journal of Medical Genetics Part C-Seminars in Medical Genetics. 2003;123C(1):48–58. doi: 10.1002/ajmg.c.20013. [DOI] [PubMed] [Google Scholar]
- Billy Sun RC, Zee Benny Chung-Ying, Wang Maggie Haitian. wtest: an R package for testing main and interaction effect in genotype data with binary traits 2016 [Google Scholar]
- Touw WG, Bayjanov JR, Overmars L, Backus L, Boekhorst J, Wels M, van Hijum SA. Data mining in the Life Sciences with Random Forest: a walk in the park or lost in the jungle? Briefings in bioinformatics. 2013;14(3):315–326. doi: 10.1093/bib/bbs034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lo SH, Zheng T, Hu I. Interaction-based feature selection and classification for high-dimensional biological data. Bioinformatics. 2012;28(21):2834–2842. doi: 10.1093/bioinformatics/bts531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KS, Zuo LJ, Pan Y, Xie CC, Luo XG. Genetic variants in the CPNE5 gene are associated with alcohol dependence and obesity in Caucasian populations. Journal of Psychiatric Research. 2015;71:1–7. doi: 10.1016/j.jpsychires.2015.09.008. [DOI] [PubMed] [Google Scholar]
- Wang MH, Sun R, Guo J, Weng H, Lee J, Hu I, Sham PC, Zee BC. A fast and powerful W-test for pairwise epistasis testing. Nucleic acids research. 2016;44(12):e115. doi: 10.1093/nar/gkw347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MH, Tsoi K, Lai X, Chong M, Zee B, Zheng T, S-HLo S-H, Hu I. Two Screening Methods for Genetic Association Study with Application to Psoriasis Microarray Data Sets. IEEE International Congress on Big Data.2015. pp. 324–326. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.